
Abstract
Background
Ultrasound-guided intertruncal approach (IA) to the supraclavicular block (SB) is recently proposed as a new approach for local anesthetic (LA) injection in terms of the classical approach (CA) at the level of the first rib. The CA-SB has been proven to result in satisfying sensorimotor block, but associate with a high risk of intraneural injection. The aim of this randomized non-inferiority study is to explore whether IA-SB can obtain similar block dynamics, as the CA-SB, but avoiding an intraneural injection during the whole nerve block procedure.
Methods
The total 122 patients undergoing elective upper extremity surgery will be randomly allocated to receive either an IA-SB or a CA-SB using a double-injection (DI) technique. In the IA-SB group, a portion of LA (15 mL) is injected accurately to the intertruncal plane between the middle and lower trunks under real-time ultrasound guidance; then, the remaining volume (10 mL) is carefully distributed to the other intertruncal plane between the upper and middle trunks. In the CA-SB group, the DI technique will be carried out as described in Tran’s study. The primary outcome is the percentage of patients with a complete sensory blockade at 20 min with a predefined non-inferiority margin of − 5%. The secondary outcomes include the sensory-motor blockade of all 4 terminal nerves, onset times of the individual nerves within 30 min, block-related variables, and adverse events.
Discussion
The results will provide sensory-motor blockade-related parameters and safety of the ultrasound-guided intertruncal approach to the supraclavicular block, thereby promoting clinical practice.
Trial registration
Chinese Clinical Trial Registry ChiCTR2000040199. Registered on 25 November 2020
Similar content being viewed by others


Background
The supraclavicular block (SB) is a popular and approved approach to the brachial plexus because of the greater safety due to real-time ultrasound guidance and better block dynamics known as the “spinal anesthesia of the arm” [1, 2]. Compared with its single- or multiple-injection counterpart, a double-injection (DI) technique has been widely used in the classical approach (CA), which is at the level of the trunks and divisions [3,4,5,6,7,8,9,10,11]. However, all these techniques are associated with a high risk of intraneural injection, which is known as the sub-epineurium or intracluster injection. Recently, Siddiqui et al. [12] have proposed an alternative approach using the DI technique based on the clear identification of the outer boundaries (epineurium) of each trunk (upper, middle, and lower), named the intertruncal approach (IA). The double injection in IA aims to provide satisfying block dynamics of the entire brachial plexus while avoiding intraneural injection, by which local anesthetic (LA) is accurately deposited to adipose tissue planes between the upper and middle, and the middle and lower trunks. Although the DI techniques (CA vs IA) require injection of LA very close to contiguous compartments of the brachial plexus, there are essential differences between the two approaches with respect to the injected location of LA [2, 13]. In addition, different approaches direct toward the same goal for better block dynamics, that is, LA spreads evenly within the entire brachial plexus sheath without any nerve sparing [11].
Given that, the DI technique has been associated with less needle passes and results in satisfying block dynamics for ultrasound-guided CA-SB [7]. However, no clinical study data about block dynamics, including the proportion of patients with complete sensory-motor blockade and onset times, are available for IA-SB until now. Therefore, our hypothesis in this study is that the novel IA-SB will obtain comparable block dynamics when compared with CA-SB; meanwhile, it is a promising method of placing the LA immediately adjacent to the trunks without the risk of intraneural injection.
Methods/design
Trial design and setting
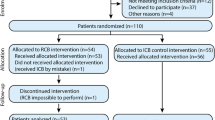
This prospective, non-inferiority, parallel randomized controlled trial will be carried out at the Third Affiliated Hospital of Sun Yat-sen University, China. It is developed according to the Recommendations for Interventional Trials (SPIRIT) 2013 Statements that came from the Standard Protocol Items (Fig. 1, the SPIRIT Checklist is available as Additional file 1) [14]. Besides, the Consolidated Standards of Reporting Trials (CONSORT) flow diagram will be performed in the design period and completing the study to the end. A detailed flowchart of the trial design is shown in Fig. 2.

The standard protocol items of this study

Flow diagram of this study
Informed consent
Written informed consent will be obtained from each patient before enrollment. The ultrasound-guided IA- or CA-SB-related operation sequence, benefits, risks, and data privacy of this study will be explained in detail for the participants during the preoperative visit. We will emphasize that this study is voluntary, and the participants are free to withdraw from the study at any time. If the patient is not willing to participate in the study, he or she will receive other means of brachial plexus block or general anesthesia depending on the patient’s willingness and discretion of the treating anesthesiologist.
Participants and recruitment
The patients scheduled to undergo surgery of the elbow, forearm, wrist, or hand with ultrasound-guided brachial plexus block will be recruited and screened for eligibility in our medical center. An independent researcher will conduct the recruitment when performing the preoperative visit 1 day before surgery.
Inclusion criteria are as follows:
-
Signed written informed consent
-
American Society of Anesthesiologists (ASA) physical status I to III
-
Age 18 to 75 years old
-
Operative site at the elbow, forearm, wrist, or hand
Exclusion criteria are as follows:
-
Patient’s refusal of brachial plexus block
-
Nerve block cannot be performed due to coagulopathy (defined as any coagulation disorders contraindicated to perform peripheral nerve block), pre-existing neuropathy, infection at the supraclavicular fossa, hypersensitivity, or allergy to LA
-
Body mass index (BMI) > 35 kg/m2
-
Pregnancy, severe mental illness, or cognitive dysfunction (unable to communicate or cooperate)
Randomization and blinding
After the enrollment, the participants were randomly allocated to one of the two groups (CA-SB or IA-SB group) in a 1:1 ratio by computer-generated simple randomization. A sealed opaque envelope that contained a card (recorded with a random number and subject number) will be opened by a research assistant who will not be involved in other stages of this study. Ultrasound-guided CA-SB or IA-SB will be carried out by one out of our nerve block team (WF. Y, JQ. G, and HB. X) and supervised by the coauthor (QH. L), in which all have extensive experience with both techniques (over 60 attempts/per technique) before this study. Another anesthesiologist who is blinded to the randomized allocation and intervention will be responsible for recording the research-related variables and anesthesia management based on the conventional scheme. During the study period, research assistants who will be kept blind to the group allocation oversaw postoperative follow-up by face-to-face assessment or telephone. If a serious adverse event (pneumothorax or LA systemic toxicity, etc.) occurs during the nerve block, un-blinding will be permissible, and then emergency measure will be initiated under the supervision of the outcome assessor.
Ultrasound-guided techniques
All participants will be seen on 1 day before surgery and demonstrated on the use of a 3-point scale for evaluating sensory-motor blockade. On arrival to the operating room, standard ASA monitors (non-invasive cuff blood pressure, pulse oxygen saturation, and electrocardiogram) and supplemental oxygen (nasal cannula at 4 L/min) will be applied. An intravenous access (20-gauge) for fluid infusion will be established in the contralateral forearm, and the premedication (midazolam 0.05 mg/kg or combined with fentanyl 0.5 μg/kg) will be given prior to nerve block. Drugs that improve the block effect or duration of the sensory-motor blockade will be not allowed to use in the perioperative period, including dexmedetomidine or dexamethasone or magnesium sulfate. All patients will be received EtCO2 in monitoring during procedure and surgery. For the two approaches, the nerve block will be performed following standard skin disinfection with a portable ultrasound machine (Sonosite M-turbo, SonoSite, Inc., Bothell, WA) and 80-mm short-beveled stimulating needle (B. Braun Melsungen AG, Melsungen, Germany).
The ultrasound-guided CA-SB with the DI technique will be performed in accordance with the method described in Tran’s study [3]. After obtaining a satisfactory image of elliptical hypoechoic trunks and divisions at the supraclavicular fossa, the operators initially orientate the needle tip to the “corner pocket” between the subclavian artery and the lower trunk with the in-plane technique. A part of the LA (15 mL) of 1:1 mixture of 2% lidocaine (Shandong Hualu Pharmaceutical Co., Ltd.) and 1% ropivacaine (Astrazeneca Pharmaceutical Co., Ltd.) will be injected after the accurate position is confirmed by the “water separation” technique under ultrasound guidance. Subsequently, the needle will withdraw and targets the center of the main neural cluster floated upward by the former LA. The remaining volume (10 mL) will be carefully administered into that central position.
For the ultrasound-guided IA-SB, the procedures with the DI technique are replicated from Siddiqui’s study [12] and the optimal order of injections will be followed according to the suggestion in Endersby’s letter [15]. Using a high-frequency pattern, a consecutive scan will be performed initially at the supraclavicular fossa toward the base of the neck in a coronal oblique plane. Once the three trunks of the plexus (upper, middle, and lower) and its epineurium are well defined, the needle is advanced from the lateral end of the probe, and the first part of LA (15 mL) will be accurately injected into the intertruncal plane between the middle and lower trunks. It is worth noting that each trunk is in a different stage of its trajectory and those divisions have not been fused with each other yet in this area. Then, the second placement of LA (10 mL) is carefully distributed to the other intertruncal plane between the upper and middle trunks.
Outcome definitions and evaluations
The definitions and evaluations of the primary and secondary outcomes of this study are summarized in Table 1.
Sample size calculation and statistical analysis
Our working hypothesis is that ultrasound-guided CA- versus IA-SB yield similar block dynamics. Thus, this study will be designed as a non-inferiority trial. In the previous studies using single or multiple injections in SB, we have observed that the proportion of patients with complete sensory blockade reached a plateau starting at 20 min after injection. It fluctuates between 70% and close to 100% within 30 min [3,4,5,6, 16]. In other words, relative to itself, the variation is very subtle from 20 to 30 min. The primary outcome is considered as the proportion of patients with complete sensory blockade of all 4 terminal nerves at 20 min after injection in this study. Based on a pilot study with 15 patients in each group, the proportion of patients with complete sensory blockade achieved was 73% in the CA-SB group and 87% in the IA-SB group (unpublished data). Therefore, we assume that a difference in proportion between the two groups less than − 5%, measured at 20 min after injection, will be considered non-inferiority. The required sample size per group is calculated to be 55 with a statistical power of 80% and a one-sided 95% confidence interval. To account for a possible 10% dropout rate, the total sample size is inflated to 122 participants (n = 61, per group).
Statistical analysis will be performed using SPSS for Windows 18.0 (SPSS Inc., Chicago, IL). For continuous data, normality will be first assessed with the Kolmogorov-Smirnov test and then analyzed using an independent-samples t test. Categorical variables will be summarized as a frequency, n (%), such as the proportion of complete sensory or motor blockade, success rates, and adverse events, etc. The Pearson χ2 test, Fisher’s exact test, or a Mann-Whitney U test will be used for categorical variables as appropriate. A p < 0.05 will be considered statistically significant for all results.
Data collection and retention
The nerve block-related parameters and postoperative follow-up data will be recorded by a research assistant, and a statistics analysis will be carried out by an independent statistician. To enable examination and re-analysis from regulatory authorities, all electronic data will be desensitized and stored securely at the Department of Anesthesiology of the Third Affiliated Hospital of Sun Yat-sen University for 5 years. Preserved paper materials of this study include the original signed informed consents, study protocol and interventions, and case report forms. The project will be monitored by a data monitoring committee composed of specialists in ethics, anesthesiology, and statistics. These data will be kept in our research database and not revealed to other people without appropriate permission.
Adverse events
All adverse events will be monitored and recorded. Once any serious adverse event occurs, it will be immediately reported to the research group, which will determine the causality and therapeutic measures of the adverse events. The chief investigator will be responsible for reporting all adverse events to the Ethics Committee.
Auditing
No formal auditing process is proposed for this trial.
Protocol amendments
In principle, the established study protocol is not to be modified. Any amendments to the study will be first initiated by the principal investigators and then agreed and confirmed by all study participants. Finally, the modified version of the protocol will be submitted to the Ethics Committee for approval.
Trial dissemination
The research results and findings will be disseminated in a peer-reviewed journal or at scientific conferences.
Patient and public involvement
No patients were involved in the design, recruitment, and conduct of the study and were also directly consulted in the development of the research question or outcome measures. An original article will be prepared to present the trial results at the proper time after the end of the study. Results of the final study will be disseminated to all study participants through E-mail recorded at the time of enrolment. The burden of intervention will not be taken by the participants themselves.
Discussion
The supraclavicular brachial plexus block does gain popularity benefited from the application of ultrasonic visualization technology [17, 18]. However, the optimal needling technique for the SB is remaining controversial. Various studies have proposed a series of techniques of where to first inject the LA and how many injections to perform around the brachial plexus, which include single-, double-, triple-, targeted intracluster-, or modified-injection techniques for the last nearly two decades [3,4,5,6, 9, 11, 16]. Up to now, the most common techniques for LA injection described at the first rib level are a single or double injection. The needle intentionally breaches the layer located at the outer border of the entire plexus, and the needle tip is observed as it advances to the “corner pocket” (at the intersection of the first rib and subclavian artery) in real time for a single injection [19]. If a double injection is used, then the needle will be redirected and placed in the middle of hypoechoic structures representing neural tissue [3]. However, these techniques have been proved to be associated with a high risk of intraneural injection. A recent cadaver study has reported that sub-perineural injections can be as high as 24% for a single intracluster injection [20]. As a result, it should be remembered that it cannot exclude direct or indirect nerve injuries even under real-time imaging of the needle-nerve distance [21, 22]. Therefore, in clinical practice, it is eager to carry out a scheme to prevent nerve injuries and finding a more appropriate injection site will be better and safer according to the needs of inexperienced operators.
The development of a new-style approach or needling technique is coming into clinical practice because the advancement in ultrasound guidance allowed for a further understanding of the underlying anatomy of the brachial plexus (trajectory and surrounding fascial sheath) [23]. For instance, the selective trunk block which is a novel brachial plexus block technique is just inspired by accurately identifying the main components of the plexus above the clavicle under real-time visualization and is expected to result in producing surgical anesthesia of the whole upper extremity [24, 25]. Recently, Siddiqui and colleagues [12] have proposed an alternative approach to the classical approach to a SB, in which LA is carefully injected into the investing adipose layers between the trunks. This is a very promising needling technique without intraneural injection for ultrasound-guided SB. However, the specific data of block dynamics is not yet clear compared with the classical approach. The objective of this study was to assess the block dynamics and clinical feasibility with DI in the supraclavicular fossa via IA.
In the previous technique-related trials, the more needle passes seem the shorter onset time and better sensory-motor blockade [7]. However, the shorter onset time associated with the incremental needle passes is counterbalanced by its longer performance time and is accompanied with a high risk of intraneural injection [3,4,5,6, 16]. Nonetheless, the most used and teaching needling technique is the DI technique because, with an acquired satisfactory learning curve, the trainees can implement relatively safe skills while retaining the benefit of quick onset [26, 27]. In addition, we also observed that, in many patients, rather than being a singular entity, the neural cluster is composed of more than one main satellite clusters after the first injection [6, 8]. Finding a better approach for better LA diffusion based on the advancement in ultrasound guidance might be a meaningful technological innovation. Excitingly, the novel IA is just performed between the epineurium of the 3 trunks, and it aims to anesthetize the brachial plexus where the 3 trunks can be visualized as independent of hypoechoic nodules [12]. The authors have proposed that this approach can provide complete blockade of all 4 terminal nerves and satisfying onset times while avoiding intraneural injection and pleural puncture. In our study, the sensory-motor blockade-related clinical parameters including the percentage of patients with complete blockade at all the predetermined intervals, onset times, and adverse events will be recorded. As the IA is closer to the interscalene, an important issue concerning the potential risks of diaphragmatic paralysis is another important dimension of safety [28, 29]. The excursion of the ipsilateral hemidiaphragm will be measured by M-mode ultrasonography in supine position via the anterior subcostal route in centimeters at 35 min after injection [30]. We will also observe the duration of sensorimotor block and potential nerve injury after surgery. The results can help us to determine the efficacy and safety of an IA with the DI technique.
In conclusion, this trial should enable us to better assess the effectiveness of an IA to ultrasound-guided SB, with the potential possibility of avoiding intraneural injection. It may provide us with an ideal inserting approach for optimized risk-benefit at the supraclavicular fossa. It should also advance the understanding of the optimal site of injection and optimization of the diffusion of LA in this inserting approach with the DI technique.
Trial status
The study will be conducted over a period of more than 9 months (from 26 November 2020 to 31 August 2021) with the latest version of the protocol. At the time of manuscript submission, candidates had been included and some patients had participated in the study.
Availability of data and materials
All investigators will have access to the final de-identified study dataset from the corresponding author for the purpose of scientific publications.
Abbreviations
- IA:
-
Intertruncal approach
- CA:
-
Classical approach
- SB:
-
Supraclavicular block
- LA:
-
Local anesthetic
- DI:
-
Double injection
- ASA:
-
American Society of Anesthesiologists
- CONSORT:
-
Consolidated Standards of Reporting Trials
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Trials
References
D'Souza RS, Johnson RL. Supraclavicular block. Treasure Island (FL): StatPearls; 2020.
Pester JM, Varacallo M. Brachial plexus block techniques. Treasure Island (FL): StatPearls; 2020.
Tran DQ, Munoz L, Zaouter C, et al. A prospective, randomized comparison between single- and double-injection, ultrasound-guided supraclavicular brachial plexus block. Reg Anesth Pain Med. 2009;34(5):420–4. https://doi.org/10.1097/AAP.0b013e3181ae733a [published Online First: 2009/11/19].
Roy M, Nadeau MJ, Cote D, et al. Comparison of a single- or double-injection technique for ultrasound-guided supraclavicular block: a prospective, randomized, blinded controlled study. Reg Anesth Pain Med. 2012;37(1):55–9. https://doi.org/10.1097/AAP.0b013e3182367b97 [published Online First: 2011/10/28].
Techasuk W, Gonzalez AP, Bernucci F, et al. A randomized comparison between double-injection and targeted intracluster-injection ultrasound-guided supraclavicular brachial plexus block. Anesth Analg. 2014;118(6):1363–9. https://doi.org/10.1213/ANE.0000000000000224 [published Online First: 2014/05/21].
Luo Q, Yao W, Shu H, et al. Double-injection technique assisted by a nerve stimulator for ultrasound-guided supraclavicular brachial plexus block results in better distal sensory-motor block: a randomised controlled trial. Eur J Anaesthesiol. 2017;34(3):127–34. https://doi.org/10.1097/EJA.0000000000000542 [published Online First: 2016/11/01].
Park SK, Lee SY, Kim WH, et al. Comparison of supraclavicular and infraclavicular brachial plexus block: a systemic review of randomized controlled trials. Anesth Analg. 2017;124(2):636–44. https://doi.org/10.1213/ANE.0000000000001713 [published Online First: 2016/11/10].
Choi JJ, Kwak HJ, Jung WS, et al. Sonographic guidance for supraclavicular brachial plexus blocks: single vs. double injection cluster approach. Pain Physician. 2017;20(6):529–35 [published Online First: 2017/09/22].
Choudhary N, Kumar A, Kohli A, et al. Single-point versus double-point injection technique of ultrasound-guided supraclavicular block: a randomized controlled study. J Anaesthesiol Clin Pharmacol. 2019;35(3):373–8. https://doi.org/10.4103/joacp.JOACP_144_18 [published Online First: 2019/09/24].
Luo Q, Yao W, Chai Y, et al. Comparison of ultrasound-guided supraclavicular and costoclavicular brachial plexus block using a modified double-injection technique: a randomized non-inferiority trial. Biosci Rep. 2020;40(6):BSR20200084. https://doi.org/10.1042/BSR20200084 [published Online First: 2020/05/23].
Lee JY, Park HY, Choi YS, et al. A randomized comparison between two injections from two planes versus two injections with a uniplanar approach for ultrasound-guided supraclavicular block. Pain Physician. 2021;24(1):E15–21 [published Online First: 2021/01/06].
Siddiqui U, Perlas A, Chin K, et al. Intertruncal approach to the supraclavicular brachial plexus, current controversies and technical update: a daring discourse. Reg Anesth Pain Med. 2020;45(5):377–80. https://doi.org/10.1136/rapm-2019-101260 [published Online First: 2020/03/14].
Sivashanmugam T, Ray S, Ravishankar M, et al. Randomized comparison of extrafascial versus subfascial injection of local anesthetic during ultrasound-guided supraclavicular brachial plexus block. Reg Anesth Pain Med. 2015;40(4):337–43. https://doi.org/10.1097/AAP.0000000000000264 [published Online First: 2015/06/13].
Chan AW, Tetzlaff JM, Gotzsche PC, et al. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ. 2013;346:e7586. https://doi.org/10.1136/bmj.e7586 [published Online First: 2013/01/11].
Endersby RVW, Spencer AO, Champagne P, et al. Eight ball, deep side pocket: optimal order of injections for the intertruncal approach to the supraclavicular brachial plexus. Reg Anesth Pain Med. 2021;46(10):930. https://doi.org/10.1136/rapm-2020-102266 [published Online First: 2020/12/05].
Arab SA, Alharbi MK, Nada EM, et al. Ultrasound-guided supraclavicular brachial plexus block: single versus triple injection technique for upper limb arteriovenous access surgery. Anesth Analg. 2014;118(5):1120–5. https://doi.org/10.1213/ANE.0000000000000155 [published Online First: 2014/04/02].
Sadowski M, Tulaza B, Lysenko L. Renaissance of supraclavicular brachial plexus block. Anaesthesiol Intensive Ther. 2014;46(1):37–41. https://doi.org/10.5603/AIT.2014.0008 [published Online First: 2014/03/20].
Schubert AK, Dinges HC, Wulf H, et al. Interscalene versus supraclavicular plexus block for the prevention of postoperative pain after shoulder surgery: a systematic review and meta-analysis. Eur J Anaesthesiol. 2019;36(6):427–35. https://doi.org/10.1097/EJA.0000000000000988 [published Online First: 2019/05/03].
Soares LG, Brull R, Lai J, et al. Eight ball, corner pocket: the optimal needle position for ultrasound-guided supraclavicular block. Reg Anesth Pain Med. 2007;32(1):94–5. https://doi.org/10.1016/j.rapm.2006.10.007 [published Online First: 2007/01/02].
Retter S, Szerb J, Kwofie K, et al. Incidence of sub-perineural injection using a targeted intracluster supraclavicular ultrasound-guided approach in cadavers. Br J Anaesth. 2019;122(6):776–81. https://doi.org/10.1016/j.bja.2019.01.006 [published Online First: 2019/03/28].
Gadsden JC. The role of peripheral nerve stimulation in the era of ultrasound-guided regional anaesthesia. Anaesthesia. 2021;76(Suppl 1):65–73. https://doi.org/10.1111/anae.15257 [published Online First: 2021/01/12].
Reiss W, Kurapati S, Shariat A, et al. Nerve injury complicating ultrasound/electrostimulation-guided supraclavicular brachial plexus block. Reg Anesth Pain Med. 2010;35(4):400–1. https://doi.org/10.1097/aap.0b013e3181e90ec9 [published Online First: 2010/07/09].
Feigl GC, Litz RJ, Marhofer P. Anatomy of the brachial plexus and its implications for daily clinical practice: regional anesthesia is applied anatomy. Reg Anesth Pain Med. 2020;45(8):620–7. https://doi.org/10.1136/rapm-2020-101435 [published Online First: 2020/05/31].
Karmakar MK, Areeruk P, Mok LYH, et al. Ultrasound-guided selective trunk block to produce surgical anesthesia of the whole upper extremity: a case report. A&A Pract. 2020;14(9):e01274. https://doi.org/10.1213/XAA.0000000000001274 [published Online First: 2020/09/11].
Sivakumar RK, Areeruk P, Karmakar MK. Selective trunk block (SeTB): a simple alternative to hybrid brachial plexus block techniques for proximal humeral fracture surgery during the COVID-19 pandemic. Reg Anesth Pain Med. 2021;46(4):376–8. https://doi.org/10.1136/rapm-2020-101733 [published Online First: 2020/06/12].
Kim TE, Tsui BCH. Simulation-based ultrasound-guided regional anesthesia curriculum for anesthesiology residents. Korean J Anesthesiol. 2019;72(1):13–23. https://doi.org/10.4097/kja.d.18.00317 [published Online First: 2018/11/28].
Rueda Rojas VP, Melendez Florez HJ, Orozco GE. Analysis of previous training with simulated models on the success rate of ultrasound-guided supraclavicular block. Prospective cohort study. Rev Esp Anestesiol Reanim. 2019;66(5):241–9. https://doi.org/10.1016/j.redar.2019.01.003 [published Online First: 2019/03/17].
Sivashanmugam T, Maurya I, Kumar N, et al. Ipsilateral hemidiaphragmatic paresis after a supraclavicular and costoclavicular brachial plexus block: a randomised observer blinded study. Eur J Anaesthesiol. 2019;36(10):787–95. https://doi.org/10.1097/EJA.0000000000001069 [published Online First: 2019/08/10].
Kang RA, Chung YH, Ko JS, et al. Reduced hemidiaphragmatic paresis with a “corner pocket” technique for supraclavicular brachial plexus block: single-center, observer-blinded, randomized controlled trial. Reg Anesth Pain Med. 2018;43(7):720–4. https://doi.org/10.1097/AAP.0000000000000795 [published Online First: 2018/05/11].
Tedore TR, Lin HX, Pryor KO, et al. Dose-response relationship between local anesthetic volume and hemidiaphragmatic paresis following ultrasound-guided supraclavicular brachial plexus blockade. Reg Anesth Pain Med. 2020;45(12):979–84. https://doi.org/10.1136/rapm-2020-101728 [published Online First: 2020/10/03].
Acknowledgements
Not applicable.
Funding
This work was supported by the Medical Scientific Research Foundation of Guangdong Province, China (Grant number: A2021047), and a project from the Institute of Science and Technology, National Health Commission of China (Grant number: 2021KYSHX01504).
Author information
Authors and Affiliations
Contributions
Study design: HHS, WY, and QL .Study registration: QL and YC. Patients’ recruitment: WY and QL. Clinical operating: QL, WY, JG, and HX. Manuscript writing and approval: all authors. Data collection and interpretation: GS and YZ. Statistical analysis: YC. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This manuscript reports a study protocol involving human participants and perioperative data. This study has been approved by the Ethics Committee of the Third Affiliated Hospital of Sun Yat-Sen University (No. 2020-02-118-01, protocol version 1.1). The trial has also been registered at http://www.chictr.org.cn/ (ChiCTR2000040199, 25 November 2020). Written informed consent will be obtained from all the participants. Results will be disseminated via an international peer-reviewed publication.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
SPIRIT 2013 Checklist.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Luo, Q., Cai, Y., Xie, H. et al. Intertruncal versus classical approach to the ultrasound-guided supraclavicular brachial plexus block for upper extremity surgery: study protocol for a randomized non-inferiority trial. Trials 23, 91 (2022). https://doi.org/10.1186/s13063-022-06029-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-022-06029-x