
Abstract
Pharmacological inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6) are now an established standard of care for patients with advanced hormone receptor-positive breast cancer. The canonical mechanism underlying CDK4/6 inhibitor activity is the suppression of phosphorylation of the retinoblastoma tumor suppressor protein, which serves to prevent cancer cell proliferation. Recent data suggest that these agents induce other diverse effects within both tumor and stromal compartments, which serve to explain aspects of their clinical activity. Here, we review these phenomena and discuss how they might be leveraged in the development of novel CDK4/6 inhibitor-containing combination treatments. We also briefly review the various known mechanisms of acquired resistance in the clinical setting.
Similar content being viewed by others

Background
Cell cycle dysregulation leading to sustained cellular proliferation is a hallmark of cancer [1]. In cancers arising from the luminal mammary epithelium, certain cell cycle regulators—the D-type cyclins and cyclin-dependent kinases 4 and 6 (CDK4/6)—are of particular importance [2,3,4,5]. Targeted and specific inhibitors of CDK4/6 have been developed, and these agents are most effective against cells from the luminal and HER2-amplified subtypes [5, 6]. In recent years, these inhibitors have revolutionized the treatment landscape for advanced hormone receptor (HR)-positive, HER2-negative breast cancer. While the traditional mainstay of treatment for this disease has been endocrine therapy (ET), acquired resistance to ET is a near inevitability, and the addition of CDK4/6 inhibitors markedly improves patient outcomes. Preclinically, it has been shown that CDK4/6 inhibitors act synergistically with ET and can overcome ET resistance [5]. These findings formed the basis of many preclinical and clinical studies of CDK4/6 inhibitors as treatment for ER-positive breast cancer and have ultimately led to their approval for clinical use [7]. Currently, three CDK4/6 inhibitors are approved and available to treat breast cancer: palbociclib, ribociclib, and abemaciclib. Despite widespread usage of these agents in the clinic, we are only just beginning to understand their complex effects within breast cancers, as preclinical studies show that these agents induce numerous phenotypes beyond cell cycle arrest [7]. Deeper insight into the mechanisms by which CDK4/6 inhibition (CDK4/6i) modifies tumor biology will be crucial if the full clinical potential of CDK4/6 inhibitors is to be realized.
Cyclin D-CDK4/6-retinoblastoma pathway in HR-positive breast cancer
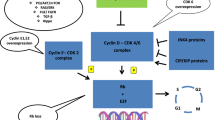
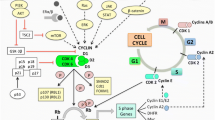
Progression through the four phases of the cell cycle is tightly regulated by a network of cyclin proteins and their partner CDKs. CDK4/6 and their partner D-type cyclins (cyclins D1, D2, and D3) specifically regulate transition from the G1 phase to the S phase. The G1/S transition is driven by E2F transcription factors that promote expression of genes required to support DNA replication in S phase. Importantly, E2F transcriptional activity is repressed by the retinoblastoma (RB) tumor suppressor protein, which (1) directly binds to and blocks the E2F transactivation domain and (2) recruits epigenetic modifiers that install repressive chromatin marks at E2F target gene promoters [8,9,10,11,12].
The RB protein is unphosphorylated in early G1. Exposure to mitogenic growth factors at this point in the cell cycle results in a rapid rise in the level of D-type cyclins, which then bind to CDKs 4 and/or 6 (Fig. 1). The cyclin D-CDK4/6 complex then binds a third protein (either p21 or p27), and the resultant holoenzyme phosphorylates RB [13,14,15]. Under the classical model, CDK4/6 phosphorylates RB, inducing partial de-repression of E2F transcription factors and expression of cyclin E genes [16, 17]. Cyclin E then partners with CDK2 to hyperphosphorylate RB and establish commitment to S phase [8].

The role of cyclin D and CDK4/6 in cell cycle progression in breast cancer
Cyclin D1 and CDK4 play particularly important roles in mammary gland biology and breast cancer. For example, cyclin D1 is required for mammary epithelial proliferation in pregnancy [18, 19], and knockout of either cyclin D1 or CDK4 prevents the development of mammary carcinomas from luminal epithelial cells driven by particular oncogenes, such as Neu or Ras, in mice [2, 3]. Cyclin D1 is also required for the maintained growth of these carcinomas [4]. Furthermore, numerous molecular features suggest that the cyclin D-CDK4/6 pathway can be hyperactivated in human HR-positive breast cancers: (1) At the genomic level, approximately 20 percent of tumors demonstrate CCND1 gene amplification, and a smaller fraction exhibit either CDK4 amplification (2%) or loss of CDKN2A (2%), which encodes for the endogenous inhibitor of CDK4/6 p16INK4A [20,21,22,23]; (2) CCND1 is also a direct transcriptional target gene of the estrogen receptor (ER), a principal driver of proliferation in HR-positive tumors [24]; and (3) activation of certain growth factor signaling pathways (most notably the PI3K-AKT-mTOR pathway) is common—whether by mutation, amplification, or increases in kinase signaling—and can either increase cyclin D levels or enhance its activity through post-translational mechanisms [25,26,27]. Importantly, HR-positive breast cancers also usually retain expression and function of RB, unlike triple-negative breast cancers in which RB is commonly absent or dysfunctional [5, 20, 21, 28]. Collectively, these features render CDKs 4 and 6 as attractive therapeutic targets in HR-positive breast cancer.
It is important to note that although the classical view of G1-to-S phase progression is widely accepted, the precise roles of specific CDKs in this process can be more complex. For example, certain cell types can enter S phase even in the absence of CDKs 4 and 6, including the mammary epithelial cells, and this may be due to the phosphorylation of RB by atypical cyclin D-CDK2 complexes [29, 30]. Indeed, the non-classical model of S phase entry is based upon the idea that the net phosphorylation of RB by CDK4/6 and/or CDK2 ultimately governs the G1/S transition, a concept which supports the hypothesis that CDK2 may facilitate cell cycle progression in the presence of CDK4/6 inhibitors [30, 31].
Mechanisms of action of CDK4/6 inhibition in breast cancer: recent insights
Cytostasis and the senescence-like state
The CDK4/6 inhibitors that are currently approved for treating breast cancer target the ATP-binding domains of CDKs 4 and 6 and are highly selective against these kinases [32, 33]. As one might expect given their mechanism of action, CDK4/6 inhibitors induce cytostasis (G1 cell cycle arrest) in RB-proficient luminal breast cancer cells in vitro (Fig. 2A) [4, 5, 33,34,35,36]. Given that RB is a key mediator of the senescence program, it is also not surprising that pharmacologic CDK4/6 inhibition induces a phenotype resembling senescence in luminal breast cancer cells. Multiple preclinical studies have reported a CDK4/6 inhibitor-induced “senescence-like state,” characterized by cellular enlargement and flattening, and increased β-galactosidase activity [4, 32, 33, 37, 38]. This senescent-like state is largely RB-dependent [39, 40] but might also be linked to reduced phosphorylation of the FOXM1 transcription factor and DNA methyltransferase 1 (DNMT1), both direct CDK4/6 substrates [41, 42]. It is still unclear whether CDK4/6i triggers a senescence-associated secretory phenotype (SASP) in breast cancer, or what the makeup of the SASP might be, and further study is needed to elucidate this.

Inhibition of G1/S cyclin-dependent kinases by CDK4/6 inhibitors. A The traditional model suggests that CDK4/6 inhibitors inhibit active CDK4/6-cyclin D-p21/p27 holoenzymes, preventing RB phosphorylation by CDK4/6. B Two models suggest mechanisms by which CDK4/6 inhibitors indirectly inhibit CDK2 activity. (I) Guiley et al. propose that CDK4/6 inhibitors bind to monomeric CDK4/6, preventing the formation of CDK4/6-cyclin D-p21/p27 trimers. The free p21 then binds to and inhibits cyclin E/CDK2, preventing RB phosphorylation. This model suggests that the CDK4/6i induces cell cycle arrest through an indirect inhibition of CDK2, rather than inhibition of CDK4/6 activity. (II) Pack et al. propose that CDK4/6i inhibitors do inhibit CDK4/6 catalytic activity directly, but also displace p21 from established CDK4-cyclin D-p21 trimers, again leading to indirect inhibition of cyclin E/CDK2
Other phenotypic responses have also been reported in tumor cells, and it has been speculated that the drug effect might differ depending on the inhibitor used. Hafner et al. [43] reported that while palbociclib and ribociclib treatment induced G1 cell cycle arrest, abemaciclib arrested cells in both G1 and G2 phases, particularly at higher in vitro concentrations. They and Torres-Guzman et al. also observed that higher concentrations (greater than 0.3 µM) of abemaciclib induced apoptosis in RB-proficient breast cancer cells [35, 43]. Some of these differences might be explained by differences in the secondary targets of abemaciclib, the best validated of which are PIM kinases [38]. Some reports also describe CDK9, CDK2, and GSK3B as abemaciclib-specific targets, but unique targets of these kinases have not been conclusively shown to be hypo-phosphorylated when treating breast cancer cells at physiologically relevant concentrations [33, 38, 43,44,45,46]. On the other hand, abemaciclib and palbociclib showed similar activity (albeit with different potency) in a large panel of cancer cell lines [6], and all three CDK4/6 inhibitors exhibit greatest effect in the presence of functional RB [5, 6, 47,48,49]. Further study is needed to determine the extent to which the unique kinase inhibitory spectra of these agents underlie any differences in their in vitro or in vivo activity.
Finally, it is important to note that the precise pharmacologic mechanisms by which CDK4/6 inhibitors act have been the subject of recent scrutiny (Fig. 2B). Specifically, a report from Guiley et al. suggested that CDK4/6-induced cell cycle arrest is primarily a result of indirect inhibition of CDK2. Rather than binding to and inhibiting active CDK4/6-cyclin D-p21/27 trimers, CDK4/6 inhibitors were predominantly bound to inactive monomeric CDK4 or 6. As such, the inhibitors did not directly block endogenous CDK4 activity but rather prevented the formation of stable cyclin D-CDK4/6-p21/p27 complexes, leaving p21 to inhibit CDK2 activity and thus induce cell cycle arrest [50]. Pack et al. recently reported that CDK4/6 inhibitor treatment mediates two effects that work together to prevent cell cycle progression: (1) the direct inhibition of CDK4/6-mediated phosphorylation of RB, as the addition of drug leads to a decrease in RB phosphorylation within minutes, and (2) destabilization of CDK4-cyclin D-p21 trimers, allowing non-catalytic inhibition of CDK2 by p21 [51]. The latter effect was reportedly specific to CDK4 and p21 and not CDK6 and p27. Clarification of the exact mechanisms of action of CDK4/6 inhibitors will be critical, and further work is needed to elucidate the precise molecular mechanisms by which these drugs induce cell cycle arrest.
Epigenetic remodeling
In primary fibroblasts, replicative and oncogene-induced senescence is characterized by changes in chromatin organization, including both the formation of senescence-associated heterochromatin foci (SAHF) as well as regions of enhancer activation, leading to alterations in gene transcription [52,53,54,55]. We recently reported that CDK4/6 inhibitors can induce similar changes in luminal breast cancer cells, both in vitro and in vivo (including in clinical specimens). Specifically, CDK4/6i reprograms the active enhancer landscape in an RB-dependent manner [56]. While chromatin at cell cycle gene promoters showed repressive changes upon CDK4/6i, several intergenic and intronic regions showed increased accessibility and gains in H3K27ac. These newly activated enhancers regulate processes including luminal differentiation, resistance to apoptosis, and tumor cell immunogenicity. Mechanistically, these CDK4/6i-activated enhancers are regulated by members of the activator protein-1 (AP-1) transcription factor family, as CDK4/6i increases their expression and phosphorylation [56, 57]. Consistent with this, recent studies have demonstrated that AP-1 drives chromatin accessibility and enhancer activation in benign senescent cells [58, 59]. Importantly, the extent to which ER is involved in the CDK4/6i-induced enhancer activation is yet to be determined. Further investigation is also needed to elucidate how concomitant administration of CDK4/6 inhibitors and ET modifies these epigenetic phenomena.
Apoptotic evasion
While CDK4/6 inhibitors can induce a senescent-like state, it is not clear that these agents can directly kill luminal breast cancer cells. In fact, several preclinical and clinical studies suggest that CDK4/6i suppresses apoptosis, which is consistent with the notion that senescence is an anti-apoptotic state [60, 61]. We have recently demonstrated that in breast cancer, this “apoptosis-resistance” is underpinned in part by activation of a super-enhancer spanning the BCL2L1 locus, which increases intracellular levels of the anti-apoptotic Bcl-2 family protein Bcl-xL. Consistent with this, Bcl-xL inhibitors restore apoptotic sensitivity in CDK4/6i-pretreated cells [56]. Similarly, Bcl-2 inhibitors can induce apoptosis in CDK4/6i-treated cells, and triplet combinations comprising ET, CDK4/6i, and venetoclax (a Bcl-2 inhibitor) are in clinical development (NCT03900884) [62]. Notwithstanding these insights, some studies report that CDK4/6i directly induces apoptosis in ER-positive breast cancer cells [35, 43]. Further work to reconcile these observations is needed to (1) better understand mechanisms of CDK4/6i-induced tumor regression and (2) design rational combinations comprising CDK4/6 inhibitors and BH3 mimetics.
Autophagy
Autophagy and senescence are closely related, often regulated by overlapping signaling pathways. In mammary epithelial cells, kinase-active cyclin D is essential for restraining autophagy [63]. Consistent with this observation, CDK4/6i reportedly elevates various autophagic markers in ER-positive breast cancer cell lines and xenograft models. Interestingly, the addition of various inhibitors of autophagy (e.g., hydroxychloroquine) does not kill CDK4/6 inhibitor-treated breast cancer cells, but rather further enhances the senescent phenotype [64].
Interaction with oncogenic kinase signaling circuits
Most studies searching for effective combination therapies that enhance CDK4/6i efficacy in ER-positive breast cancer have focused on concomitant inhibition of growth factor signaling pathways. Combined inhibition of CDK4/6 and growth factor receptors, such as HER2 and FGFR, or the downstream pathway members, such as PI3K, PDK1, and mTOR, has demonstrated synergy or at least heightened effects [36, 65,66,67,68,69]. In some cases, the effect of combined inhibition of CDK4/6 and a growth factor pathway is enhanced cytostasis or senescence [36, 65, 69], and in others, the effect is apoptosis [30, 66,67,68]. The combination of ET, CDK4/6, and PI3K inhibition results in maximal growth inhibition in ER-positive breast cancer models [30].
Despite the range of effective combination regimens that has been explored preclinically, the molecular mechanisms underlying their additive or synergistic effects have not been clearly delineated. One common theme is a rebound increase in activity of upstream pathways (such as the PI3K pathway) in luminal breast cancer cells treated with CDK4/6i [30, 47, 66,67,68]. We have also reported that CDK4/6i increases the phosphorylation of HER family receptor tyrosine kinases and AKT in luminal HER2-positive cell lines [36]. This might in part be attributed to the fact that cyclin D-CDK4/6 can phosphorylate the canonical mTOR negative regulator TSC2 [36, 70,71,72]. Inhibition of CDK4/6 reduces the phosphorylation of TSC2, leading to a partial reduction in mTOR activity and a rebound in upstream tyrosine kinase receptor activity [73]. While such observations might suggest heightened dependence on upstream growth factor signaling pathways in CDK4/6 inhibitor-treated cells, the downstream effects of this are not yet clear. One consequence of increased growth factor signaling is sustained stimulation of mTORC1 activity, which if uninhibited, could drive S phase progression through numerous mechanisms [74]. Another possibility is an increase in cyclin D protein levels, resulting in the formation of atypical cyclin D/CDK2 complexes that can phosphorylate RB [30].
Given these observations, combination regimens comprising inhibitors of CDK4/6 and certain growth factor pathways has moved into the clinical arena, and for two combinatorial strategies (CDK4/6-PI3K and CDK4/6-HER2), randomized phase 3 trials have already been initiated. Although initial attempts to combine CDK4/6 and PI3K inhibitors led to prohibitive toxicity [75], certain combinations have shown promise in PIK3CA mutant breast cancer [76], ultimately leading to the initiation of an ongoing randomized phase 3 trial exploring the benefit of adding inavolisib to the palbociclib/fulvestrant doublet (NCT04191499). In the case of HER2, numerous randomized trials are exploring the benefits of triple blockade of CDK4/6, HER2, and ER in HR-positive, HER2-positive tumors. The first of these to be reported has shown improved progression-free survival when comparing this approach to a chemotherapy-based regimen in pretreated tumors [36, 77].
Immunogenicity
Numerous preclinical studies have reported that CDK4/6i can boost anti-tumor immune responses in models of breast and other cancers. Several distinct mechanisms underlie this phenomenon, which has been observed with all approved CDK4/6 inhibitors.
In tumor cells, CDK4/6i enhances antigen presentation on major histocompatibility complex (MHC) class I molecules in an RB-dependent manner [37, 78]. Inhibition of CDK4/6 reduces the expression of DNMT1 (encoding DNA methyltransferase 1), an E2F target gene, resulting in hypomethylation and thus transcription of endogenous retroviral (ERV) elements [37, 79, 80]. The resultant intracellular double-stranded RNA triggers a “viral mimicry” response, characterized by interferon production and expression of interferon-stimulated genes (ISG) [37, 78]. Furthermore, we recently proposed that CDK4/6i-induced chromatin remodeling stimulates activity of enhancers overlying ERV sequences that might also drive ISG expression [56]. Recently, it has been reported that CDK4/6i can also induce metabolic stress in tumor cells, leading to expression of chemokines such as CCL5 and CXCL10 that can further enhance anti-tumor immune responses [57].
Treatment with CDK4/6 inhibitors also has a direct effect on T lymphocytes. Numerous CDK4/6 inhibitors potently suppress the proliferation of Foxp3 + regulatory T cells (TReg) in the tumor microenvironment (TME), which is likely an RB-dependent phenomenon [37, 62, 81, 82]. Effector T cell function, on the other hand, can be enhanced by CDK4/6i, evidenced by enhanced effector cytokine production and reduced expression of T cell exhaustion markers [37, 78, 81]. This is attributable, at least in part, to inhibition of CDK6-mediated phosphorylation of nuclear factor of activated T cells (NFAT) transcription factors [78, 81]. Most recently, CDK4/6i has also been demonstrated to promote differentiation of CD8 T cells toward a memory cell fate, which might contribute to enhanced anti-tumor efficacy [83, 84]. Data on whether this memory differentiation effect in CD8 T cells is RB-dependent are mixed.
All told, these phenomena result in an inflamed TME and an increase in effector T cell activity, which independently contributes to the anti-tumor effects of these agents [37, 79, 81]. In an attempt to leverage this, preclinical investigators have combined several CDK4/6 inhibitors with a variety of immunotherapeutics, demonstrating superior control of tumor growth [37, 57, 78, 79, 85, 86], and the generation of T cell memory which engenders resistance to tumor re-challenge [37]. While these preclinical studies were not carried out in ER-positive breast cancer models, their results are relevant and encouraging.
Gene expression analyses of biopsies from the NeoPalAna and NeoMonarch neoadjuvant trials in luminal breast cancer suggest that this immune effect occurs in patients [37, 87], but the extent to which it can be leveraged to improve patient outcomes is still unknown. Reasons for this include that: (1) ER-positive metastatic breast cancer has thus far proven unresponsive to immune-based approaches [88, 89], and (2) early efforts to combine CDK4/6i and immuno-oncology therapy have been complicated by prohibitive toxicity [90, 91].
Acquired resistance: mechanisms and questions
Despite the clinical success of CDK4/6i in treating ER-positive breast cancer, acquired resistance is a major clinical problem. Multiple preclinical studies have described causes for acquired resistance, and these encompass diverse mechanisms, including alterations in components of the cell cycle machinery, increased activity through oncogenic growth factor signaling pathways, metabolic changes within cancer cells, and drug-induced changes in stromal function (recently reviewed in [92]). Many of these mechanisms remain unsupported by clinical evidence, and for the sake of brevity, here we only discuss resistance mechanisms currently supported by both clinical data from breast cancer patients and preclinical evidence.
Loss of RB function
One anticipated mechanism of resistance to CDK4/6i is loss of functional RB. The first examples were reported by Condorelli et al. [93], where acquired RB1 mutations were detected in ER-positive breast cancer patients treated with palbociclib and fulvestrant or ribociclib and letrozole. In the PALOMA-3 study with a larger cohort of patients, whole-exome sequencing of paired circulating tumor (ct)DNA samples definitively confirmed CDK4/6 specificity of acquired RB1 mutations, but the mutations were detected in only 5% of patients who progressed on the palbociclib and fulvestrant combination [94]. Loss of functional RB was subsequently also identified in other studies as both a feature of acquired and de novo resistance to CDK4/6i [95,96,97]. These findings are supported by a wealth of preclinical data showing that many CDK4/6i-mediated effects are RB-dependent.
Other cell cycle machinery proteins
Intriguingly, several preclinical studies suggest that increased levels of CDK6 can drive resistance to CDK4/6i [98, 99]. Whether this relates to incomplete inhibition of CDK6 by the drugs [99, 100] or other kinase-independent effects of CDK6 is unclear. In ER-positive breast cancer patients, FAT1 mutations are associated with CDK4/6i resistance, likely by increasing CDK6 expression [95].
The cyclin E/CDK2 axis has also been implicated in CDK4/6i resistance. CCNE2 amplification has been observed in treatment-resistant tumor specimens, and overexpression of CCNE1 mRNA is associated with poorer response to palbociclib in the metastatic setting [97, 101]. It is possible that elevations in cyclin E result in CDK2-mediated phosphorylation of RB that overcomes CDK4/6i-mediated G1 arrest [30].
Growth factor signaling
The clinical data supporting growth factor signaling as a mechanism of CDK4/6i-resistance remains somewhat limited and is restricted to analysis of genomic alterations within tumors. It has been difficult to interpret the relevance of these data to the CDK4/6 pathway specifically, as they are almost invariably derived from patients treated with combined CDK4/6i and ET, and many of the same alterations have been implicated in ET resistance previously. One rigorous analysis comes from the PALOMA-3 trial, in which patients were randomized to receive fulvestrant with or without palbociclib. Here, ctDNA was assessed (through either whole-exome sequencing or targeted sequencing of hotspot mutation sites) in patients prior to commencing therapy and again at the time of progressive disease. In this analysis, a small number of patients’ tumors acquired mutations in PIK3CA or FGFR2 at the time of progressive disease, but these were seen in both the control and experimental arms of the trial, making it difficult to discern the extent to which they might specifically confer CDK4/6i resistance [94].
Other data sets exploring acquired resistance are derived from cohorts of tumor biopsies studied at the time of progression on CDK4/6i, sometimes with an accompanying analysis of pre-treatment tissue. Collectively, these have shown enrichment of functional hyperactivating alterations in FGFR genes, RAS genes, ERBB2, PTEN, and AKT1 in CDK4/6i resistant tumors [65, 96, 97]. However, these analyses have been limited by (1) small numbers of patients and (2) the lack of comparison to an ET-only treated cohort, and in some cases, (3) confounding by the administration of several other lines of therapy after CDK4/6i prior to acquisition of tumor tissue for analysis. Taken together, these findings suggest that genomic mutations in key growth factor receptors and signal transduction pathway members might mediate resistance to CDK4/6i in the clinic. Exactly how they drive resistance remains an open question and may be related to the ability of these pathways to drive cyclin D, RB phosphorylation, mTOR, or CDK2 [30, 65, 68, 102].
Future directions and unanswered questions
The development of selective, potent CDK4/6 inhibitors has been a major success story for modern breast oncology, and thousands of patients have now benefited from these agents. Moving forward, key unanswered questions must be addressed with the goals of (1) enhancing the efficacy of CDK4/6 inhibitors in HR-positive breast cancer through identification of novel therapeutic combinations; (2) interrogating the plasticity of the cell cycle machinery in breast cancer as a means to understanding acquired resistance; and (3) extending the use of these agents to other breast cancer subtypes. In many cases, this will require the reassessment of assumptions that have formed the basis of much of the research in this field.
First, at a most fundamental level, research is needed to understand the molecular mechanisms of action for CDK4/6 inhibitors. It has been assumed that these agents directly inhibit CDK4/6 enzymatic activity, but this has recently been called into question by work suggesting that by binding to CDK4/6, they in fact operate as indirect CDK2 inhibitors [50]. This claim has, in turn, been refuted by other studies, and it is critical to resolve this issue, the answer to which forms the foundation for all research using these compounds [103]. Indeed, the CDK2 inhibition hypothesis is built upon the notion that CDK4/6 activity requires the formation of trimers which also contain cyclin D and p21/p27, a concept which is also controversial [103,104,105,106].
Second, more studies are needed to better understand the cellular senescence phenotype induced by CDK4/6 inhibitors. To what extent do these agents induce a SASP, and if they do, what are its components and impacts? How does the loss of p53 function, another key senescence mediator, alter the senescence phenotype? Do CDK4/6 inhibitors induce senescence in other proliferative cells within a breast tumor (e.g., fibroblasts, endothelium) and what is the impact of this? Addressing these questions adequately will require multi-omic profiling of CDK4/6 inhibitor-treated cells, sophisticated genetic modeling in vitro and in vivo, and tumor single cell profiling, and the lessons learned will likely inform novel therapeutic combinations and our thinking on drug resistance.
Third, and with specific respect to HR-positive disease, the mechanism(s) underlying synergy between CDK4/6 inhibitors and different endocrine therapies remains a poorly understood topic. In particular, the role of endocrine therapy in either enhancing or modifying the therapy-induced senescence phenotype, versus converting it to apoptosis, requires clarification. Similarly, the molecular determinants of this synergy must be studied in greater depth. Ultimately, this will shape the development of novel endocrine therapy-CDK4/6 inhibitor combinations and inform our understanding of resistance in the clinic which presumably reflects a breakdown of this synergy.
Fourth, the perennial issue of acquired CDK4/6 inhibitor resistance remains a clinical challenge, in large part because the mechanisms underlying it, and their relative frequencies, are not clear. Although preclinical studies have revealed diverse, non-genomic resistance mechanisms including altered kinase signaling, stromal cell senescence, and altered chromatin modifier function, clinical studies have almost exclusively relied on DNA sequencing of resistant cancers [92, 94, 97]. This reflects a major gap between preclinical and clinical research on this subject, which might be addressed through a more comprehensive interrogation of resistant samples including transcriptomic and epigenomic profiling at single cell resolution.
Finally, more work is needed to determine how we might exploit CDK4/6 inhibitor-mediated immunomodulation in tumors. In breast cancer, this phenomenon has gained the most traction in triple-negative disease and the results of ongoing trials in this space are eagerly awaited [107].
Conclusion
Since CDK4/6 inhibitors were approved for use in the clinic in 2015, our understanding of their mechanisms of action has advanced significantly. We now realize that inhibiting CDK4/6 not only restrains cancer cell proliferation, but also elicits numerous diverse biological effects that can be both beneficial or harmful. This understanding should now be leveraged to inform the design of combinatorial strategies that will enhance the efficacy of CDK4/6i and tailor treatments to individual tumors. Acquired resistance remains the most pressing issue: Despite the number of studies published, a complete picture detailing common resistance mechanisms to dual CDK4/6i-ET therapy with clinical validation has yet to be constructed, and prospective, rigorously designed biospecimen collection protocols are needed to acquire pre- and post-treatment tumor samples for further study. Analysis of such samples, together with further trials, will also hopefully address the outstanding question of whether CDK4/6 inhibitors should be used after progression.
Availability of data and materials
Not applicable.
Abbreviations
- CDK:
-
Cyclin-dependent kinase
- RB:
-
Retinoblastoma tumor suppressor protein
- HR:
-
Hormone receptor
- ET:
-
Endocrine therapy
- CDK4/6i:
-
CDK4/6 inhibition
- PI3K:
-
Phosphoinositide-3-kinase
- AKT:
-
Protein kinase B
- mTOR:
-
Mammalian target of rapamycin
- ATP:
-
Adenosine triphosphate
- GSK3B:
-
Glycogen synthase kinase 3 beta
- ER:
-
Estrogen receptor
- FOXM1:
-
Forkhead box protein M1
- DNMT1:
-
DNA methyltransferase 1
- SASP:
-
Senescence-associated secretory phenotype
- SAHF:
-
Senescence-associated heterochromatin foci
- AP-1:
-
Activator protein-1
- FGFR:
-
Fibroblast growth factor receptor
- PDK1:
-
Phosphoinositide-dependent kinase-1
- TSC2:
-
Tuberous sclerosis complex 2
- mTORC1:
-
Mammalian target of rapamycin complex 1
- MHC:
-
Major histocompatibility complex
- ERV:
-
Endogenous retrovirus
- ISG:
-
Interferon stimulated gene
- CCL5:
-
Chemokine ligand 5
- CXCL10:
-
C-X-C motif chemokine ligand 10
- TME:
-
Tumor microenvironment
- Treg :
-
Regulatory T cell
- NFAT:
-
Nuclear factor of activated T cells
- ctDNA:
-
Circulating tumor DNA
- RAS:
-
Rat sarcoma virus
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9(1):23–32.
Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411(6841):1017–21.
Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22(4):438–51.
Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77.
Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, Dowless M, Dempsey J, Zeng Y, Torres R, et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell. 2017;32(6):761-776 e766.
Spring LM, Wander SA, Andre F, Moy B, Turner NC, Bardia A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet. 2020;395(10226):817–27.
Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65(6):1053–61.
Flemington EK, Speck SH, Kaelin WG Jr. E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc Natl Acad Sci U S A. 1993;90(15):6914–8.
Helin K, Harlow E, Fattaey A. Inhibition of E2F–1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13(10):6501–8.
Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, Garcia BA, Bernstein E, Lowe SW. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A. 2012;109(23):8971–6.
Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92(4):463–73.
Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14(8):518–28.
LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11(7):847–62.
Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK “inhibitors” are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18(6):1571–83.
Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98(6):859–69.
Goodrich DW, Wang NP, Qian YW, Lee EY, Lee WH. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell. 1991;67(2):293–302.
Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82(4):621–30.
Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 1995;9(19):2364–72.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163(2):506–19.
Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem. 1997;272(16):10882–94.
Muise-Helmericks RC, Grimes HL, Bellacosa A, Malstrom SE, Tsichlis PN, Rosen N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem. 1998;273(45):29864–72.
Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008;27(8):1106–13.
Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12(22):3499–511.
Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008;10(5):R75.
Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118(4):493–504.
Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76(8):2301–13.
O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417–30.
Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, Keller PR, McNamara DJ, Sherry D, Zhu T, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48(7):2388–406.
Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, Torres R, Ajamie RT, Wishart GN, Flack RS, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014;32(5):825–37.
Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–38.
Torres-Guzman R, Calsina B, Hermoso A, Baquero C, Alvarez B, Amat J, McNulty AM, Gong X, Boehnke K, Du J, et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget. 2017;8(41):69493–507.
Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, Ramm S, Palmer AC, Yuzugullu H, Varadan V, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29(3):255–69.
Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–5.
Litchfield LM, Boehnke K, Brahmachary M, Mur C, Bi C, Stephens JR, Sauder JM, Gutierrez SM, McNulty AM, Ye XS, et al. Combined inhibition of PIM and CDK4/6 suppresses both mTOR signaling and Rb phosphorylation and potentiates PI3K inhibition in cancer cells. Oncotarget. 2020;11(17):1478–92.
Yoshida A, Lee EK, Diehl JA. Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 2016;76(10):2990–3002.
Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, Ozawa T, James CD, Waldman T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70(8):3228–38.
Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20(5):620–34.
Acevedo M, Vernier M, Mignacca L, Lessard F, Huot G, Moiseeva O, Bourdeau V, Ferbeyre G. A CDK4/6-dependent epigenetic mechanism protects cancer cells from PML-induced senescence. Cancer Res. 2016;76(11):3252–64.
Hafner M, Mills CE, Subramanian K, Chen C, Chung M, Boswell SA, Everley RA, Liu C, Walmsley CS, Juric D, et al. Multiomics profiling establishes the polypharmacology of FDA-approved CDK4/6 inhibitors and the potential for differential clinical activity. Cell Chem Biol. 2019;26(8):1067-1080 e1068.
Cousins EM, Goldfarb D, Yan F, Roques J, Darr D, Johnson GL, Major MB. Competitive kinase enrichment proteomics reveals that abemaciclib inhibits GSK3beta and activates WNT signaling. Mol Cancer Res. 2018;16(2):333–44.
Chen P, Lee NV, Hu W, Xu M, Ferre RA, Lam H, Bergqvist S, Solowiej J, Diehl W, He YA, et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol Cancer Ther. 2016;15(10):2273–81.
Knudsen ES, Hutcheson J, Vail P, Witkiewicz AK. Biological specificity of CDK4/6 inhibitors: dose response relationship, in vivo signaling, and composite response signature. Oncotarget. 2017;8(27):43678–91.
O’Brien N, Conklin D, Beckmann R, Luo T, Chau K, Thomas J, Mc Nulty A, Marchal C, Kalous O, von Euw E, et al. Preclinical activity of abemaciclib alone or in combination with antimitotic and targeted therapies in breast cancer. Mol Cancer Ther. 2018;17(5):897–907.
Kim S, Tiedt R, Loo A, Horn T, Delach S, Kovats S, Haas K, Engstler BS, Cao A, Pinzon-Ortiz M, et al. The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget. 2018;9(81):35226–40.
Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, Knudsen ES. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle. 2010;9(20):4153–63.
Guiley KZ, Stevenson JW, Lou K, Barkovich KJ, Kumarasamy V, Wijeratne TU, Bunch KL, Tripathi S, Knudsen ES, Witkiewicz AK, et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science. 2019;366(6471):eaaw2106.
Pack LR, Daigh LH, Chung M, Meyer T. Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin D-CDK4 to inhibit CDK2. Nat Commun. 2021;12(1):3356.
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113(6):703–16.
Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6(6):612–29.
Guan Y, Zhang C, Lyu G, Huang X, Zhang X, Zhuang T, Jia L, Zhang L, Zhang C, Li C, et al. Senescence-activated enhancer landscape orchestrates the senescence-associated secretory phenotype in murine fibroblasts. Nucleic Acids Res. 2020;48(19):10909–23.
Sen P, Lan Y, Li CY, Sidoli S, Donahue G, Dou Z, Frederick B, Chen Q, Luense LJ, Garcia BA, et al. Histone acetyltransferase p300 induces de novo super-enhancers to drive cellular senescence. Mol Cell. 2019;73(4):684-698 e688.
Watt AC, Cejas P, DeCristo MJ, Metzger-Filho O, Lam EYN, Qiu X, BrinJones H, Kesten N, Coulson R, Font-Tello A, et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nat Cancer. 2021;2:34–48.
Uzhachenko RV, Bharti V, Ouyang Z, Blevins A, Mont S, Saleh N, Lawrence HA, Shen C, Chen SC, Ayers GD, et al. Metabolic modulation by CDK4/6 inhibitor promotes chemokine-mediated recruitment of T cells into mammary tumors. Cell Rep. 2021;35(1):108944.
Han R, Li L, Ugalde AP, Tal A, Manber Z, Barbera EP, Chiara VD, Elkon R, Agami R. Functional CRISPR screen identifies AP1-associated enhancer regulating FOXF1 to modulate oncogene-induced senescence. Genome Biol. 2018;19(1):118.
Martinez-Zamudio RI, Roux PF, de Freitas J, Robinson L, Dore G, Sun B, Belenki D, Milanovic M, Herbig U, Schmitt CA, et al. AP-1 imprints a reversible transcriptional programme of senescent cells. Nat Cell Biol. 2020;22(7):842–55.
Rochette PJ, Brash DE. Progressive apoptosis resistance prior to senescence and control by the anti-apoptotic protein BCL-xL. Mech Ageing Dev. 2008;129(4):207–14.
Wang E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995;55(11):2284–92.
Whittle JR, Vaillant F, Surgenor E, Policheni AN, Giner G, Capaldo BD, Chen HR, Liu HK, Dekkers JF, Sachs N, et al. Dual targeting of CDK4/6 and BCL2 pathways augments tumor response in estrogen receptor-positive breast cancer. Clin Cancer Res. 2020;26(15):4120–34.
Brown NE, Jeselsohn R, Bihani T, Hu MG, Foltopoulou P, Kuperwasser C, Hinds PW. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012;72(24):6477–89.
Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, Raghavendra AS, Zhao Y, Bashour SI, Ibrahim NK, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916.
Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, Sudhan DR, Guerrero-Zotano AL, Croessmann S, Guo Y, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10(1):1373.
Zhao M, Scott S, Evans KW, Yuca E, Saridogan T, Zheng X, Wang H, Korkut A, Cruz Pico CX, Demirhan M, et al. Combining neratinib with CDK4/6, mTOR, and MEK inhibitors in models of HER2-positive cancer. Clin Cancer Res. 2021;27(6):1681–94.
Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26(1):136–49.
Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee KM, Hutchinson KE, Witkiewicz AK, Moore PD, Estrada MV, Sanchez V, et al. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 2017;77(9):2488–99.
Michaloglou C, Crafter C, Siersbaek R, Delpuech O, Curwen JO, Carnevalli LS, Staniszewska AD, Polanska UM, Cheraghchi-Bashi A, Lawson M, et al. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long-term growth inhibition in estrogen receptor-positive breast cancer. Mol Cancer Ther. 2018;17(5):908–20.
Romero-Pozuelo J, Figlia G, Kaya O, Martin-Villalba A, Teleman AA. Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep. 2020;31(2):107504.
Romero-Pozuelo J, Demetriades C, Schroeder P, Teleman AA. CycD/Cdk4 and discontinuities in Dpp signaling activate TORC1 in the drosophila wing disc. Dev Cell. 2017;42(4):376-387 e375.
Zacharek SJ, Xiong Y, Shumway SD. Negative regulation of TSC1-TSC2 by mammalian D-type cyclins. Cancer Res. 2005;65(24):11354–60.
Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71.
Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24(1):200–16.
Tolaney SM, Im YH, Calvo E, Lu YS, Hamilton E, Forero-Torres A, Bachelot T, Maur M, Fasolo A, Tiedt R, et al. Phase Ib study of ribociclib plus fulvestrant and ribociclib plus fulvestrant plus PI3K inhibitor (alpelisib or buparlisib) for HR(+) advanced breast cancer. Clin Cancer Res. 2021;27(2):418–28.
Pascual J, Lim JSJ, Macpherson IR, Armstrong AC, Ring A, Okines AFC, Cutts RJ, Herrera-Abreu MT, Garcia-Murillas I, Pearson A, et al. Triplet therapy with palbociclib, taselisib, and fulvestrant in PIK3CA-mutant breast cancer and doublet palbociclib and taselisib in pathway-mutant solid cancers. Cancer Discov. 2020;11:92–107.
Tolaney SM, Wardley AM, Zambelli S, Hilton JF, Troso-Sandoval TA, Ricci F, Im SA, Kim SB, Johnston SR, Chan A, et al. Abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy in women with hormone receptor-positive, HER2-positive advanced breast cancer (monarcHER): a randomised, open-label, phase 2 trial. Lancet Oncol. 2020;21(6):763–75.
Schaer DA, Beckmann RP, Dempsey JA, Huber L, Forest A, Amaladas N, Li Y, Wang YC, Rasmussen ER, Chin D, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 2018;22(11):2978–94.
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984-997 e924.
Haggerty C, Kretzmer H, Riemenschneider C, Kumar AS, Mattei AL, Bailly N, Gottfreund J, Giesselmann P, Weigert R, Brandl B, et al. Dnmt1 has de novo activity targeted to transposable elements. Nat Struct Mol Biol. 2021;28(7):594–603.
Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, Chhabra S, Huang W, Liu H, Aref AR, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018;8(2):216–33.
Lai AY, Sorrentino JA, Dragnev KH, Weiss JM, Owonikoko TK, Rytlewski JA, Hood J, Yang Z, Malik RK, Strum JC, et al. CDK4/6 inhibition enhances antitumor efficacy of chemotherapy and immune checkpoint inhibitor combinations in preclinical models and enhances T-cell activation in patients with SCLC receiving chemotherapy. J Immunother Cancer. 2020;8(2):e000847.
Heckler M, Ali LR, Clancy-Thompson E, Qiang L, Ventre KS, Lenehan P, Roehle K, Luoma A, Boelaars K, Peters V, et al. Inhibition of CDK4/6 promotes CD8 T-cell memory formation. Cancer Discov. 2021;11:2564–81.
Lelliott EJ, Kong IY, Zethoven M, Ramsbottom KM, Martelotto LG, Meyran D, Jiang Zhu J, Costacurta M, Kirby L, Sandow JJ, et al. CDK4/6 inhibition promotes anti-tumor immunity through the induction of T cell memory. Cancer Discov. 2021;11:2582–601.
Teo ZL, Versaci S, Dushyanthen S, Caramia F, Savas P, Mintoff CP, Zethoven M, Virassamy B, Luen SJ, McArthur GA, et al. Combined CDK4/6 and PI3Kalpha inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. 2017;77(22):6340–52.
Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553(7686):91–5.
Hurvitz SA, Martin M, Press MF, Chan D, Fernandez-Abad M, Petru E, Rostorfer R, Guarneri V, Huang CS, Barriga S, et al. Potent cell-cycle inhibition and upregulation of immune response with abemaciclib and anastrozole in neoMONARCH, phase II neoadjuvant study in HR(+)/HER2(−) breast cancer. Clin Cancer Res. 2020;26(3):566–80.
Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, Sachdev J, Le Tourneau C, van Brummelen EMJ, Varga A, et al. Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer. Clin Cancer Res. 2018;24(12):2804–11.
Tolaney SM, Barroso-Sousa R, Keenan T, Li T, Trippa L, Vaz-Luis I, Wulf G, Spring L, Sinclair NF, Andrews C, et al. Effect of eribulin with or without pembrolizumab on progression-free survival for patients with hormone receptor-positive, ERBB2-negative metastatic breast cancer: a randomized clinical trial. JAMA Oncol. 2020;6(10):1598–605.
Rugo HS, Beck JT, Jerusalem G, Wildiers H, Kabos P, Chisamore M, McNaughton R, Chen Y, Hossain A, Tolaney SM. Abstract CT108: A phase 1b study of abemaciclib in combination with pembrolizumab for patients (pts) with hormone receptor positive (HR+), human epidermal growth factor receptor 2 negative (HER2-) metastatic breast cancer (mBC) (NCT02779751): preliminary results. In: AACR annual meeting 2020: 2020; Philadelphia, PA; 2020.
Herold CI, Trippa L, Li T, Do K, Bardia A, Anderson L, Montazeri K, Pittenger J, Andrews C, Mittendorf EA, et al. Abstract P3-14-03: A phase 1b study of the CDK4/6 inhibitor ribociclib in combination with the PD-1 inhibitor spartalizumab in patients with hormone receptor-positive metastatic breast cancer (HR+ MBC) and metastatic ovarian cancer (MOC). In: 2019 San Antonio breast cancer symposium: February 2020 2020; San Antonio, TX; 2020.
Alvarez-Fernandez M, Malumbres M. Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell. 2020;37(4):514–29.
Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, Dubois J, Nagy RJ, Lanman RB, Iafrate AJ, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. 2018;29(3):640–5.
O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, Andre F, Loibl S, Loi S, Garcia-Murillas I, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 2018;8(11):1390–403.
Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, Hsieh W, Sanchez-Vega F, Brown DN, Da Cruz Paula AF, et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the hippo pathway. Cancer Cell. 2018;34(6):893-905 e898.
Costa C, Wang Y, Ly A, Hosono Y, Murchie E, Walmsley CS, Huynh T, Healy C, Peterson R, Yanase S, et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kalpha inhibitors in breast cancer. Cancer Discov. 2020;10(1):72–85.
Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, Kim D, Luo F, Mao P, Helvie K, et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor-positive metastatic breast cancer. Cancer Discov. 2020;10(8):1174–93.
Yang C, Li Z, Bhatt T, Dickler M, Giri D, Scaltriti M, Baselga J, Rosen N, Chandarlapaty S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017;36(16):2255–64.
Cornell L, Wander SA, Visal T, Wagle N, Shapiro GI. MicroRNA-mediated suppression of the TGF-beta pathway confers transmissible and reversible CDK4/6 inhibitor resistance. Cell Rep. 2019;26(10):2667-2680 e2667.
Wu X, Yang X, Xiong Y, Li R, Ito T, Ahmed TA, Karoulia Z, Adamopoulos C, Wang H, Wang L, et al. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat Cancer. 2021;2:429–43.
Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, DeMichele A, Harbeck N, Andre F, Bayar MA, et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2019;37(14):1169–78.
O’Brien NA, McDermott MSJ, Conklin D, Luo T, Ayala R, Salgar S, Chau K, DiTomaso E, Babbar N, Su F, et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Res. 2020;22(1):89.
Pennycook BR, Barr AR. Palbociclib-mediated cell cycle arrest can occur in the absence of the CDK inhibitors p21 and p27. Open Biol. 2021;11(11):210125.
Bagui TK, Mohapatra S, Haura E, Pledger WJ. P27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Mol Cell Biol. 2003;23(20):7285–90.
Sugimoto M, Martin N, Wilks DP, Tamai K, Huot TJ, Pantoja C, Okumura K, Serrano M, Hara E. Activation of cyclin D1-kinase in murine fibroblasts lacking both p21(Cip1) and p27(Kip1). Oncogene. 2002;21(53):8067–74.
Cerqueira A, Martin A, Symonds CE, Odajima J, Dubus P, Barbacid M, Santamaria D. Genetic characterization of the role of the Cip/Kip family of proteins as cyclin-dependent kinase inhibitors and assembly factors. Mol Cell Biol. 2014;34(8):1452–9.
Goel S, Tolaney SM. CDK4/6 inhibitors in breast cancer: a role in triple-negative disease? Lancet Oncol. 2019;20(11):1479–81.
Funding
S.G. is funded by the National Health and Medical Research Council of Australia (GNT1177357) and the US National institutes of Health (P50 CA168504). A.C.W. is funded by the University of Melbourne.
Author information
Authors and Affiliations
Contributions
A.C.W. conducted literature search, wrote the manuscript draft, and generated the figures. S.G. wrote and edited the manuscript and provided overall supervision and coordination of the manuscript preparation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
S.G. reports receipt of laboratory research funding from Eli Lilly and G1 Therapeutics and receipt of honoraria for advisory work from Eli Lilly, G1 Therapeutics, and Pfizer.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Watt, A.C., Goel, S. Cellular mechanisms underlying response and resistance to CDK4/6 inhibitors in the treatment of hormone receptor-positive breast cancer. Breast Cancer Res 24, 17 (2022). https://doi.org/10.1186/s13058-022-01510-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-022-01510-6