
Abstract
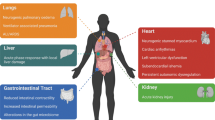
It has been convincingly demonstrated in recent years that isolated acute brain injury (ABI) may cause severe dysfunction of peripheral extracranial organs and systems. Of all potential target organs and systems, the lung appears to be the most vulnerable to damage after ABI. The pathophysiology of the bidirectional brain–lung interactions is multifactorial and involves inflammatory cascades, immune suppression, and dysfunction of the autonomic system. Indeed, the systemic effects of inflammatory mediators in patients with ABI create a systemic inflammatory environment (“first hit”) that makes extracranial organs vulnerable to secondary procedures that enhance inflammation, such as mechanical ventilation (MV), surgery, and infections (“second hit”). Moreover, accumulating evidence supports the knowledge that gut microbiota constitutes a critical superorganism and an organ on its own, potentially modifying various physiological functions of the host. Furthermore, experimental and clinical data suggest the existence of a communication network among the brain, gastrointestinal tract, and its microbiome, which appears to regulate immune responses, gastrointestinal function, brain function, behavior, and stress responses, also named the “gut-microbiome–brain axis.” Additionally, recent research evidence has highlighted a crucial interplay between the intestinal microbiota and the lungs, referred to as the “gut-lung axis,” in which alterations during critical illness could result in bacterial translocation, sustained inflammation, lung injury, and pulmonary fibrosis. In the present work, we aimed to further elucidate the pathophysiology of acute lung injury (ALI) in patients with ABI by attempting to develop the “double-hit” theory, proposing the “triple-hit” hypothesis, focused on the influence of the gut–lung axis on the lung. Particularly, we propose, in addition to sympathetic hyperactivity, blast theory, and double-hit theory, that dysbiosis and intestinal dysfunction in the context of ABI alter the gut–lung axis, resulting in the development or further aggravation of existing ALI, which constitutes the “third hit.”
Similar content being viewed by others

Introduction
ABI is a severe and significant health and socioeconomic problem accompanied by high morbidity and mortality [1]. A plethora of clinical and experimental studies demonstrate that ABI represents a complex biochemical cascade associated with numerous pathophysiological processes, which is not limited to the central nervous system affecting the function of multiple distant organs and systems [1, 2]. Among all potential target organs, the lungs appear to be the most vulnerable, with the development of various clinical syndromes, such as ventilator-associated pneumonia [3], adult respiratory distress syndrome (ARDS), and neurogenic pulmonary edema (NPO) [4]. The pathophysiology of the bidirectional brain–lung interactions is multifactorial and involves inflammatory cascades, immune suppression, and dysfunction of the autonomic system [4]. In addition to the “blast theory” and the “pulmonary venule adrenergic hypersensitivity” theories, which combine hydrostatic and high permeability mechanisms resulting in ALI [4], the “double-hit” theory has also been proposed to explain the pathophysiology of ABI-induced ALI as systemic inflammatory response seems to play an integral role in the pathogenesis of pulmonary injury in patients with ABI [5]. The “double-hit” theory refers to the systemic effects of inflammatory mediators in patients with ABI, which create a systemic inflammatory environment (the “first hit”) that makes extracranial organs susceptible to secondary procedures that amplify inflammation, such as mechanical ventilation (MV), surgery, and infections, that is, the “second hit”[5, 6].
The term “gut microbiota” refers to the highly intricate communities of microorganisms inhabiting the intestinal tract, encompassing more than 1000 types of microorganisms representing at least 4000 distinct species [7,8,9,10]. These microorganisms engage in a hormonal symbiotic relationship with their host [11]. In recent years, there has been a growing interest in biomedical research due to the recognition that gut microbiota constitutes a critical superorganism and an organ on its own that has the potential to influence various physiological functions of the host [12].
Indeed, emerging evidence over the past few decades suggests the existence of an “invisible” bidirectional communication network among the brain, gastrointestinal (GI) tract, and its microbiome [13]. This communication system appears to govern immune responses, GI function, brain function, behavior, and stress responses [14,15,16,17,18]. Furthermore, beyond these processes, interactions within the gut-microbiome–brain (GMB) axis seem to play a pivotal role in the pathophysiology of various medical conditions, including Alzheimer’s disease, depression, anxiety, inflammatory bowel disease, diabetes, and obesity [18]. ABI is also a pathological entity that negatively impacts the gut microbiome and intestinal function [19].
Until recently, the prevailing belief in the sterility of the lungs [20, 21] hindered systematic exploration of the lung microbiome, resulting in delays in research progress [22]. However, multiple studies have now confirmed that microaspiration or gastroesophageal reflux is common even among healthy, asymptomatic individuals, leading to the colonization of alveoli by microbes [23,24,25,26]. The composition of the lung microbiome is closely linked to the host’s immune response and can influence health outcomes, as demonstrated in experimental models and patient cohorts [27]. It is believed that the lung microbiome modulates gene expression in immune cells, leading to the upregulation of various molecules, including interleukin (IL)-5, IL-10, interferon gamma (IFN-γ), C–C motif chemokine ligand 11 (CCL11), and promoting toll-like receptor-4 (TLR4)-dependent responses in lung macrophages [28].
In recent years, emerging experimental and epidemiological evidence has highlighted a crucial interplay between the intestinal microbiota and the lungs, called the “gut–lung axis.” Alterations in the composition of the gut microbiome, brought about by factors such as diet, disease, or medical interventions, are associated with modified immune responses and airway homeostasis. The significance of the gut–lung axis has become more apparent with the identification of several gut microbe-derived components and metabolites, including short-chain fatty acids (SCFAs), which serve as key immune system regulators [29]. Indeed, gut dysbiosis can compromise the integrity of the intestinal barrier, potentially leading to bacterial translocation, sustained inflammation, lung injury and, pulmonary fibrosis [30,31,32,33].
In the present review, we aim to further elucidate the pathophysiology of ALI in patients with ABI by attempting to develop the “double-hit” theory, proposing the “triple-hit” hypothesis based on recent experimental and clinical data supporting the presence of the gut–lung axis. Specifically, we propose, in addition to sympathetic hyperactivity, blast theory, and double-hit theory, that dysbiosis and intestinal dysfunction in the context of ABI activate the gut–lung axis, leading to the development or further exacerbation of existing ALI, which constitutes the “third hit.”
Pathophysiology of ALI in patients with ABI
The blast injury theory
One of the most proposed theories to explain the pathophysiology of ALI in brain-injured patients is the “blast injury” hypothesis, which proposes that the sympathetic surge following a sudden rise in intracranial pressure triggers a temporary increase in intravascular pressure, leading to the disruption of the alveolo-capillary membrane. The development of NPO can be attributed to either hydrostatic forces, as indicated by a low pulmonary/plasma protein ratio, or high permeability mechanisms, supported by an increased accumulation of protein in the extravascular space of the lungs [34]. The connection between the significant sympathetic discharge and NPO is further substantiated by experimental research, which demonstrates that administering alpha-adrenergic antagonists to brain-injured rats before the onset of injury prevents the hypertensive response and mitigates subsequent lung damage (Fig. 1) [4, 34].

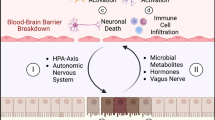
Schematic representation of the “triple-hit” hypothesis. The initial brain injury sets off sympathetic hyperactivity and catecholamine storm, a well-established contributor to ALI. Furthermore, the direct consequences of ABI, characterized by inflammation and oxidative stress (termed the “first hit”), render the lungs susceptible to subsequent interventions like MV, infections, and surgery (referred to as the “second hit”). Notably, the pivotal role of the gut–lung axis in respiratory health reveals that dysbiosis and intestinal dysfunction in ABI patients initiate a sequence of events involving immune dysregulation and microbiome alterations, which subsequently impact the lung tissue. This activation of the gut–lung axis constitutes the “third hit,” culminating in the onset or exacerbation of ALI. ABI: Acute brain injury; ALI: Acute lung injury; ARDS: Acute respiratory distress syndrome; CAP: Cholinergic anti-inflammatory pathway; E/NE: Epinephrine/norepinephrine; HPA: Hypothalamic–pituitary–adrenal; MV: Mechanical ventilation
The double-hit theory
In addition to the “blast theory,” it appears that a systemic inflammatory response plays a crucial role in the development of pulmonary injury in patients with ALI [35]. Indeed, cerebral injury promotes further complex, biochemical, cellular, and metabolic alterations within minutes after the primary event, mainly caused by tissue and cell damage [36], which can persist for even years after the initial injury initiating and maintaining neuroinflammatory and neurodegenerative processes of varying duration leading to a secondary brain injury and damage of distant organs and systems [4, 37]. Both clinical and experimental research in patients and animal models of ABI indicate the existence of an extensive cellular and biochemical cascade initiated within the brain, leading to the production of pro-inflammatory cytokines. Due to the compromised blood–brain barrier (BBB), these cytokines are released into the systemic circulation, triggering inflammatory responses (Fig. 1) [38, 39]. Indeed, similarly to several inflammatory diseases, the dysregulation of BBB has been implicated as central to the pathophysiology of ABI [40, 41], occurs within hours after primary injury, and appears to be biphasic [41,42,43]. BBB dysfunction may be a direct mechanical consequence of the primary cerebral event or the result of sustained maladaptive inflammatory and cellular processes that contribute to secondary injury [44,45,46]. The intracranial production of pro-inflammatory cytokines is likely associated with microglia and astrocyte activation [34]. Microglia, the brain’s resident macrophages, undergo morphological and functional activation shortly after ABI [47, 48]. They produce various pro-inflammatory molecules, including IL-1, IL-6, IL-8, and tumor necrosis factor (TNF)-α [38]. Experimental studies highlight that in models of moderate diffuse traumatic brain injury (TBI), levels of IL-1β, IL-6, and TNF-α peak within the cortex 3–9 h after primary injury [49]. These findings are further supported by clinical studies demonstrating increased levels of IL-6, IL-10, IL-8, TNF-α and c–c motif chemokine ligand 2 within the first two days post-TBI before gradually return to normal over a period of several weeks [50,51,52]. This cytokine cataract has been shown to induce astrogliosis and stimulate further microglial activation and axonal dysfunction, indicating an inseparable association between activated immunity and acute ABI [53]. Furthermore, microglial cell activation significantly contributes to BBB disruption, allowing the release of mediators into the systemic circulation [54, 55]. These processes may explain the extracranial organ dysfunctions observed in patients with isolated ABI [46].
Regarding the consequences of neuroinflammation and systemic inflammation in the lungs, Fisher et al. reported elevated cytokine levels in the bronchoalveolar lavage (BAL) of severe BI patients [56]. In addition, a study of lung transplant patients who had elevated IL-8 levels and who received grafts from brain-dead patients experienced graft dysfunction, early recipient mortality, and poor prognosis [57]. Furthermore, a recent study examining BAL within the first 6–8 h after traumatic brain injury and at days 3 and 7 after admission to the intensive care unit (ICU) demonstrated a significant elevation in the concentration of selected apoptotic factors at admission at day 3 and day 7 after the cerebral event [58]. Several experimental studies further corroborate these findings. In animal models with experimentally induced intracerebral hemorrhage, there was considerable neuroinflammation, characterized by marked expression of intracellular adhesion molecule (ICAM)-1 and tissue factor in both the brain and lungs. The authors noted that pulmonary expression of these mediators was associated with structural changes in the lungs [59]. Kalsotra et al. observed significant migration of inflammatory cells into the airways and alveolar spaces 24 h after initiating BI in animal models, accompanied by a substantial increase in pulmonary leukotriene B4 production [60]. Similarly, in an experimental subarachnoid hemorrhage (SAH) model, the lungs exhibited significant expression of ICAM-1, vascular cell adhesion molecule (VCAM)-1, and E-selectin [61].
In summary, the systemic impact of inflammatory mediators in patients with ABI creates a pro-inflammatory environment, and the “first hit” makes extracranial organs susceptible to secondary factors that amplify inflammation, such as mechanical ventilation, which also induces secondary inflammatory reactions, surgery, and infections, constituting the “second hit” (Fig. 1) [5, 62].
Gut-microbiome–brain interactions
Gut microbiota
The term gut microbiota refers to the highly complex communities of microorganisms inhabiting the intestinal tract and consists of more than 1000 types of microorganisms from at least 4000 distinct species [7,8,9,10] while establishing a hormonal symbiosis with their host [11]. The human GI tract of healthy individuals is inhabited by commensal microbes of all three life domains, that is, bacteria, archaea, viruses, and eukarya, whereas the bacterial representation constitutes the dominant one [63, 64]. The gut microbiota composition appears to show variations depending on the age and genetic factors and parameters, which are modifiable as diet and lifestyle, external stressors, medications including non-steroidal anti-inflammatory drugs, and antibiotics, illness, and sepsis [64,65,66,67,68].
In recent years, there has been an increasing interest in biomedical research, given that gut microbiota represent a crucial superorganism and an organ itself that could potentially modify various physiological functions of the host [12]. Indeed, during the last decades, emerging evidence suggests that there is an “invisible” cross talk between the brain, gastrointestinal tract, and its microbiome [13], which appears to regulate immune responses, GI tract function, brain function, behavior, and stress responses [14,15,16,17,18]. In addition to the processes above, GMB interactions seem to play a pivotal role in the pathophysiology of a variety of pathological conditions, for example, ABI, such as TBI, Alzheimer’s disease, depression, anxiety, inflammatory bowel disease, diabetes, and obesity [18].
Gut microbiome in patients with ABI
The bidirectional communication pathway between the central nervous system and the gut involves the central and enteric nervous systems [7, 69], the hypothalamic–pituitary–adrenal axis (HPA), and immunological pathways (Fig. 1) [70]. The autonomous nervous system is a key communication instrument between the brain and the gut, regulating intestinal homeostasis, gut mobility and permeability, bile secretion, fluid maintenance, mucosal neuroimmune response, and immune cell activation [70, 71].
The pathophysiological consequences of ABI have garnered growing attention in the context of intestinal dysfunction. This communication encompasses various types of signals and pathways, including neuronal, hormonal, and immunological cross talk, and involves both afferent and efferent signals [18]. Indeed, experimental studies in brain and spinal cord injured animal models have convincingly demonstrated that central nervous system injury affects the motility and permeability of the intestinal wall [72, 73] and modifies the composition of the gut microbiome [70, 74], resulting to gut dysbiosis [75]. Numerous investigations have demonstrated that following moderate-to-severe TBI in rodent models, there is an acute surge in mucosal damage and increased permeability within the small intestine, typically occurring within the first 72 h post-injury [72, 76]. Moreover, it has been shown that in the context of TBI, the enterocyte population and its barrier function are severely compromised, resulting in the disruption of tight junctions and an elevation in enterocyte permeability. In the absence of luminal nutrients, gastrointestinal motility is diminished, fostering an environment conducive to bacterial overgrowth. These bacteria are more prone to adhere to enterocytes, provoking apoptosis in these cells and consequently amplifying enterocyte permeability [72]. Recently, Mahajan et al. conducted a similar study, investigating samples for microbial growth from rectal swabs obtained on days 0, 3, and 7 after admission in patients with moderate–severe TBI and found widespread colonization with Proteobacteria phylum with Enterobacteriaceae forming the largest group [77]. Experimental studies further support these findings. Houlden et al. have shown that ABI in the context of experimental stroke influences bacterial communities in the caecum and that brain injury is associated with specific changes in gut microbiota [70]. Moreover, in a mouse model—known as controlled cortical impact (CCI) and designed to simulate TBI—significant alterations in the gut microbiota were observed. Specifically, there was a notable decrease in Lactobacillus gasseri, Ruminococcus flavefaciens, and Eubacterium ventriosum, accompanied by a substantial increase in Eubacterium sulci and Marvinbryantia formatexigens at 24 h post-CCI [78]. In addition, clinical studies in patients with stroke highlight significant gut dysbiosis associated with intestinal dysfunction and intestinal bleeding complications, as well as septicemia of intestinal origin, which has a negative impact on the prognosis [79].
Impact of sympathetic hyperstimulation in GMB axis
Accumulating data highlight that neurotransmitters contribute significantly to GI physiology. In recent years, epinephrine, norepinephrine, dopamine, and serotonin have been the subject of intensive research due to their influence on gut function and their potential participation in gastrointestinal and central nervous system pathophysiology. It has been shown that neurotransmitters ultimately impact gut motility, nutrient absorption, GI innate immune system, and the microbiome (Fig. 1) [80]. Furthermore, catecholamines interface with adrenergic receptors located on the cell membranes of visceral organs and smooth muscles. This interaction triggers the activation of signaling cascades, subsequently inducing modifications in organ function and smooth muscle tone [81]. These observations are further supported by experimental studies demonstrating that autonomic dysregulation and increased sympathetic tone may modify gut permeability in experimental stroke models [82]. Furthermore, it is well documented that the postganglionic sympathetic neurons are of neuralgic importance for the function of the GI system’s lymphatic tissue and immune cells and that adrenergic receptors are expressed by the cells of the innate immune system [83]. Therefore, in case of marked catecholamine secretion, such as in patients with ABI [4], their binding to adrenergic receptors in the gastrointestinal tract could modify inflammatory processes at the local level [84] partly by modifying the migration and function of immune cells, thereby stimulating the production and release of inflammatory mediators [84,85,86]. Furthermore, previous studies have demonstrated the supportive effect of noradrenaline and adrenaline via α and β adrenergic receptors on the growth of gram-negative microbes [87]. In addition, it appears that the presence of catecholamines enhances the growth of Helicobacter pylori and Campylobacter jejuni [88, 89].
Consistent with the sympathetic hyperstimulation observed in patients with ABI [4], an experimental study was able to demonstrate gut dysbiosis and intestinal dysfunction mediated by dysregulated sympathetic nerve signaling within the submucosal plexus of the gut following stroke [82]. These findings are further supported by the observation that pharmacological β-adrenergic receptor blockade with propranolol or metoprolol reestablishes gut permeability in stroke-affected mice to a degree similar to sham-operated mice [82]. In accordance with these findings, a further experimental study in a mouse model of stroke highlighted that noradrenaline excess after stroke results in an imbalance of the host microbiota, mucoprotein, and goblet cell numbers in the cecal [70].
Impact of inflammation on the GMB axis in patients with ABI
As mentioned above, ABI-induced systemic inflammation has been associated with the impairment of peripheral organs and systems, including the gut [56, 90, 91]. Modifications of the physiological interactions between the brain and the enteric and central nervous systems have been implicated in the pathophysiology of peripheral organ damage after TBI [92]. Furthermore, ABI-mediated sympathetic neural stimulation results in the release of inflammatory cytokines in peripheral organs, including the gut [93, 94]. In critically ill trauma patients, the intestinal inflammatory reaction is mediated by the recruitment of neutrophils and monocytes into the intestinal tissue, which in turn results in the release of inflammatory cytokines and superoxide molecules, which have the capability to induce damage to the intestinal mucosa. Studies in patients with TBI have underscored the significance of the localized intestinal inflammatory response, which is mediated by an increase in the concentration of intestinal cytokines, including TNF-α, which potentially can modify the structure and function of tightly bound proteins in the intestinal epithelium [95]. Moreover, experimental studies investigating the effects of the introduction of fecal content from individuals who have experienced a stroke into germ-free mice were associated with meaningful outcomes regarding stroke. This aggravation occurred due to the initiation of a pro-inflammatory immune response mediated by T-helpers (Th) 1 and Th17 cells [96], indicating that stroke-associated dysbiosis mediates an immune response characterized by pro-inflammatory processes, ultimately exacerbating brain damage in the context of stroke. In addition, a recent study investigating the gut microbiota and inflammatory mediators of patients with post-stroke cognitive impairment (PSCI) showed that PSCI patients had significantly higher levels of gut Enterobacteriaceae, lipopolysaccharide (LPS) and peripheral inflammation markers. Moreover, fecal microbiota transplantation from PSCI patients to stroke mice was associated with a higher level of Enterobacteriaceae, intestinal Toll-like receptor-4 (TLR4) expression, circulating LPS, LPS-binding protein and inflammatory cytokines, and a decreased level of fecal butyrate, more intense intestinal damage and cognitive impairment than mice that received microbiota from non-PSCI patients (non-PSCI mice) [97]. Similarly, Roy Sarkar et al. have pointed out that disturbances in the intestinal microbiome can lead to elevated levels of LPS, pro-inflammatory cytokines, Th cells, and monocytes [98]. Furthermore, it is worth noting that ischemic stroke patients who experience post-stroke depression (PSD) exhibit dysregulated intestinal flora and insufficient growth of Bifidobacterium. Conversely, Enterococcus faecalis and Escherichia coli levels are markedly elevated in these patients, and these increases are positively correlated with the levels of interleukin IL-1, IL-2, IL-6, and C-reactive protein in their serum. On the contrary, the content of Bifidobacterium showed a negative association with IL-1 and IL-2 levels in their serum [99].
Gut–lung interactions
Lung microbiome
Until recently, the widely held belief of the sterility of the lungs [20, 21] hindered the systematic exploration of the lung microbiome, resulting in delays in research progress [22]. However, multiple studies have now confirmed that microaspiration or gastroesophageal reflux is common even among healthy, asymptomatic individuals, leading to the colonization of alveoli by microbes [23,24,25,26]. Culture-independent molecular techniques have revealed a diverse bacterial community in the lower airways of healthy individuals, primarily comprising Prevotella, Veillonella, Streptococcus, and Fussobacterium species [100,101,102]. Nevertheless, the microbial biomass in healthy lungs remains low (103 to 105 bacteria per gram of tissue) [102,103,104], primarily due to limited nutrient availability.
The lung microbiome’s composition is closely tied to the host’s immune response and can impact health outcomes, as demonstrated in experimental models and patient cohorts [27]. Evidence from human data and experimental models suggests that the intensity of microbial clearance varies and is related to the specific aspirated microbial species. Even when oral commensals are promptly cleared from the lower airway, such events lead to persistent and dynamic alterations in the lower airway’s immune environment [105]. Experimental models have emphasized the critical role of early-life airway microbiota in shaping a functional immune system, influencing Helios (−) regulatory T cells (Tregs), and reducing susceptibility to allergic respiratory conditions [106]. Furthermore, the early establishment of the microbiome in the airways affects the upper respiratory tract’s microflora, which in turn impacts susceptibility to infectious diseases [107].
Numerous studies consistently show a strong correlation between the upper and lower respiratory tract microbiomes, particularly in cases of acute and chronic inflammation [108,109,110,111]. Microaspiration primarily drives this association, especially in cases involving gastroesophageal reflux disease or impaired airway cleansing mechanisms [108]. The respiratory microflora plays a crucial role in promoting the differentiation of peripheral Tregs, vital for regulating type 2 immune responses. Experimental research also highlights an intricate interplay between the microbiome and natural killer T cells (NKT cells) [112, 113]. Moreover, studies employing models lacking airway microflora have demonstrated increased eosinophils and Th2-lymphocytes in the lungs of animals [112, 113]. The lung microbiome is believed to modulate gene expression in immune cells, influencing the upregulation of various molecules, including IL-5, IL-10, IFN-γ, CCL11, and TLR4-dependent responses in lung macrophages [28].
Gut–lung interactions in critically ill patients with ABI
ABI is a global concern associated with elevated morbidity and mortality rates among adults [4, 114,115,116]. Indeed, nearly 50% of hospitalized TBI patients require intensive care due to secondary brain injury risks [117]. These patients are prone to infections, necessitating mechanical ventilation [118], with approximately 40% receiving prophylactic antibiotics [119]. Moreover, almost 70% of moderate-to-severe TBI patients develop early multi-organ dysfunction [120].
Critical illness and injury diminish microbial diversity, alter bacterial communities, and promote opportunistic microorganisms [121, 122]. The gut microbiome significantly influences distant organs, including the brain, liver, skin, and heart [123], as well as respiratory diseases [124, 125]. The gut–lung axis connects the gut microbiota to lung inflammation, impacting local and systemic immune responses [26, 126, 127] through the release of metabolites and endotoxins [128, 129]. Dysbiosis in the gut microbiota can contribute to respiratory diseases like asthma and COPD by disrupting immune, hormonal, and metabolic homeostasis [26, 130,131,132,133]. The reason why patients with intestinal pathologies are prone to pulmonary inflammation and lung diseases remains uncertain. This could be attributed to the common embryologic origin resulting in inherent similarities between these organs [134, 135].
The gut–lung axis operates through direct and indirect pathways involving substances like peptidoglycan and LPS activating the host’s immune response, SCFA affecting immune cell development, immune cells migrating between the gut and lungs via the bloodstream, and microbial metabolites influencing the host’s type I interferon response [136].
Dysbiosis can compromise the intestinal barrier, potentially leading to bacterial translocation, sustained inflammation, lung injury, and pulmonary fibrosis [31,32,33, 97]. Gut barrier disruption is prevalent in critically ill patients, as the dense mucus layer may be compromised [137, 138]. Lung microbiome disruptions can also result from gut–lung cross talk in critically ill patients [139, 140], with a shift toward gut-associated bacteria in the lungs [140]. Bacterial translocation may occur via gut-draining lymphatics, the portal system, or systemic circulation (Fig. 1) [140,141,142,143].
Increased gut permeability, or “leaky gut,” is common in critically ill patients [144, 145], exacerbated by factors like inflammatory mediators (IFN-γ, IL-6, TNF-α) disrupting tight junctions and intestinal hypoperfusion during stress reactions [145]. Anaerobic bacteria imbalances can further harm the gut immune barrier, promoting pathogenic bacteria growth and the production of inflammatory mediators [97, 146, 147]. In critical illness, luminal components from the small intestine, including bacteria, LPS, and pro-inflammatory molecules, can reach the lungs via the portal circulation or mesenteric lymph vessels, promoting alveolar inflammation and acute lung injury [126, 143, 148].
Moreover, elevated concentrations of both endogenous and exogenous catecholamines have been shown to influence the lung bacterial population [149, 150]. Indeed, previous research has ultimately highlighted an in vitro correlation between catecholamines and the growth of select bacteria, including Pseudomonas aeruginosa (Ps. aeruginosa) and Streptococcus pneumonia, which are classic bacterial representatives of the lung microbiome [87, 151, 152]. Human bronchoalveolar lavage samples have provided evidence that heightened alveolar catecholamine levels are strongly associated with the dominance of a single bacterial species, most frequently Ps. aeruginosa [153]. These findings are consistent with those of an in vitro study by Freestone et al. investigating the association of catecholamines with the growth and virulence of Ps. aeruginosa. The authors reported a strong correlation between catecholamines and Ps. aeruginosa growth, as reflected by 50-fold increases in bacterial numbers. The underlying pathophysiological mechanisms involve inotrope-associated delivery of transferrin-iron, internalization of the inotrope, and upregulation of the key pseudomonal siderophore pyoverdine [151]. It has been suggested that the lung is susceptible to elevated concentrations of neurochemicals associated with stress due to its ample blood supply and dense noradrenergic tissue innervations [87]. Therefore, any source of alveolar injury and inflammation, whether direct—such as aspiration or ventilator-induced lung injury—or indirect—such as sepsis or shock, can initiate a sequence of inflammatory events resulting in elevated intra-alveolar catecholamine concentrations, which, in turn, foster the growth and virulence of specific bacterial community members and contribute to a disrupted bacterial community that sustains alveolar inflammation [154, 155]. Remarkably, Kopin et al. calculated that in non-septic situations, the concentration of norepinephrine at the receptor site is at least three times greater than what can be measured in the bloodstream. Consequently, utilizing plasma measurements to ascertain catecholamine levels during an acute condition reflects a spill-over effect that significantly underestimates the actual catecholamine levels within the organ site [156, 157]. Additionally, bacterial growth promotion is not limited to catecholamines; it is also seen with inflammatory mediators like TNF-α, IL-1, IL-6, and IL-8, as well as glucocorticoids, molecules that are excessively produced in ABI [4, 158,159,160,161].
As previously mentioned, ABI triggers a robust inflammatory response within the brain, which is accompanied by the suppression of the peripheral immune system, a phenomenon referred to as ABI-induced immunosuppression [4]. Some key factors contributing to ABI-induced immunosuppression include the shift of lymphocytes from a Th1 phenotype to a Th2 phenotype, reductions in lymphocytes and NKT cells in both blood and the spleen, as well as impairments in the defensive functions of monocytes and neutrophils [162]. Moreover, one potential factor contributing to ABI-induced immunodeficiency is the activation of the sympathetic nervous system and the HPA axis by the cerebral event [4, 163]. An experimental study in a stroke model further supports this notion, demonstrating that the injection of 200 colony-forming units of Streptococcus pneumoniae into the nasal cavity of stroke-afflicted mice can lead to the development of pneumonia and bacteremia, whereas 200,000 colony-forming units are required in control animals to induce similar conditions. [164]. To complicate things further, experimental research indicates that alterations in gut microbiome have detrimental consequences on pulmonary defense against pathogens. Indeed, an experimental model of antibiotic-treated and germ-free mice showed an enhanced vulnerability to lung infections with Streptococcus pneumoniae and Klebsiella pneumoniae due to low pulmonary levels of IL-17 and granulocyte–macrophage colony-stimulating factor (GM-CSF) in these mice, which are required for an effective lung defense [165].
Lung microbiome in patients with acute lung injury
As mentioned above, critical illness is associated with alterations of the lung microbiome [100, 139, 166, 167], which are significantly related to systemic and local inflammation [139, 166]. Bacterial diversity undergoes a reduction, and commensal microbial populations may be displaced by potential pathogens, often stemming from alternative ecosystems, including the GI tract and the skin [100, 168]. The crucial cross talk between the intestinal microbiota and the lungs receives notable clinical importance as this microbiome shift is associated with remarkable inflammatory processes and lung injury [139, 166, 169]. Specifically, the lung microbiome of patients with ALI is fortified with gut-associated bacteria (e.g., species of the Enterobacteriaceae family [139, 140]), which is associated with the subsequent development of ARDS [139]. Indeed, while the enrichment of lung microbiome with gut-originated bacteria could reflect a generalized state of dysbiosis in these severely injured patients, the study by Panzer et al. indicates a drastic participation of these microbes in the pathophysiology of ARDS [139]. The authors highlight that in critically ill mechanically ventilated patients, lung dysbiosis at the early stages of critical illness is associated with a remarkable increase of inflammatory mediators (IL-6, IL-8), which in turn predisposes these patients to the subsequent development of ARDS [139]. These findings are in accordance with the findings of Dickson et al., who investigated the lung bacterial composition in BAL specimens from ARDS and non-ARDS patients. The authors reported Streptococcaceae, Veillonellaceae, Prevotellaceae, Verrucomicrobiaceae, and Flavobacteriaceae as the dominant species in patients without ARDS, whereas Pasteurellaceae and Enterobacteriaceae are identified in BAL specimens of ARDS patients [170]. Furthermore, an experimental study of lung injury following abdominal sepsis induced by cecal ligation and puncture found that the lung microbiome of rodents was enriched with gut bacteria [171]. These findings are further supported by the study of Schmitt et al., who compared the composition of the lung microbiome in 15 patients with sepsis-induced ARDS undergoing abdominal surgery and 15 patients post-esophagectomy. The authors observed a decreased α-diversity index of the pulmonary microbiome in ARDS patients, which was related to the length of the ICU stay and the need for ventilator support, indicating that an imbalance of the lung microbiome may contribute to the pathogenesis of ALI/ARDS in patients with sepsis [172]. The data, however, in patients with ABI are scarce. A recent study investigating the lung microbiome of patients with TBI receiving specialized (16pts) or standard nutrition reported that the BAL microbiota of patients who develop VAP showed significant differences in beta diversity and high composition of Staphylococcus and Acinetobacter Genera [173].
In contrast to the GI tract, the alveolar space is characterized by an ecologically unfavorable milieu for the majority of bacteria, resulting in minimal bacterial reproduction [166]. A significant factor contributing to this low reproductive capacity is the scarcity of nutrient substrates essential for bacterial metabolism. Unlike the nutrient-rich environment of the gut lumen, the alveolar space is relatively barren, containing only a thin layer of lipid-rich surfactant along the epithelial lining. From the bacterial perspective, healthy alveoli can be considered inhospitable. However, in conditions characterized by alveolar injury, such as ARDS or pneumonia, the environmental conditions undergo a dramatic shift [153, 166, 174, 175]. The pathophysiology of the development of microbial dysbiosis in the lungs of critically ill patients is multifactorial and includes alterations of local physiochemical and metabolic local characteristics such as pH, oxygen concentration, occurrence of free radicals, and nutrients within the alveoli [20, 175, 176]. The emergence of anaerobic zones, attributable to alveolar edema or alveolar collapse and thus atelectasis in injured lungs, creates a more conducive environment for the proliferation of potential pathogens [20]. Furthermore, the presence of an endotracheal tube in mechanically ventilated patients facilitates continuous microaspiration of oropharyngeal flora while impeding the natural clearance mechanisms of the airways [177]. The alveolar airspaces, which were previously devoid of content, become inundated with fluid rich in proteins. This phenomenon is particularly evident in patients with NPO, representing a newfound and ample energy source for proliferating microorganisms [4, 175]. Notably, NPO is defined as the extravasation of protein-rich fluid into the interstitial and alveolar spaces of the lungs following various central nervous system pathologies, including stroke, SAH, subdural hemorrhage, status epilepticus, central nervous system infections, and TBI [178,179,180,181]. Moreover, the bactericidal surfactant layer becomes deactivated, and the process of eliminating microbes is hindered due to impaired mucociliary clearance. From an ecological standpoint, the alveoli, when injured, start to exhibit similarities to the gut environment rather than resembling healthy lung tissue. Consequently, it is not unexpected that a majority of pathogens that emerge during critical illness have their origins in the GI tract. The interplay between the microbiome and alveolar injury can drive a dysregulated feedback loop that extends across the host-microbiome interface [166].
The “triple-hit” hypothesis
The "triple-hit" hypothesis is grounded in the idea that ABI patients experience a threefold impact on their lung pathophysiology. The initial brain injury triggers sympathetic hyperactivity, which has long been recognized as a contributor to ALI. Moreover, the direct effects of ABI, including inflammation and oxidative stress (“first hit”), make the lungs vulnerable to secondary procedures such as mechanical ventilation (“second hit”) [4]. Finally, based on recent research which has uncovered the significance of the gut–lung axis in respiratory health, dysbiosis, and intestinal dysfunction in ABI patients initiates a cascade of events involving immune dysregulation and microbiome alterations, which subsequently affect the lung tissue [21, 166, 170]. This activation of the gut–lung axis forms the “third hit,” leading to the development or worsening of ALI (Fig. 1). Our hypothesis could be used as a theoretical background for research on prophylactic or therapeutic strategies, which will target the GMB axis in order to mitigate the risk for ALI that is exacerbated by systemic inflammation, immunosuppression, and bacterial translocation in this vulnerable patient’s population.
Therapeutic implications
Mechanical ventilation in brain-injured patients with acute lung injury
Managing patients with ALI and severe ABI poses a considerable therapeutic challenge. Traditional approaches to treating ARDS may conflict with the management of elevated intracranial pressure (ICP) and reduced cerebral perfusion pressure (CPP) [182]. The lack of inclusion of brain-injured patients in most clinical trials investigating MV in ARDS has resulted to the lack of substantial and reliable data to instruct clinicians in the management of ARDS in this specific population [4, 183]. Protective MV has demonstrated improved survival in ARDS [183], but it could lead to hypercapnia and hypoxemia, potentially contributing to increased ICP and cerebral hypoxia [184]. For neurocritical care patients, maintaining normal partial pressures of arterial oxygen (PaO2) and preventing hypoxia are of paramount importance. An analysis of the IMPACT study database, encompassing over 9000 TBI patients enrolled in randomized controlled trials and series since the 1980s, has confirmed the impact of hypoxemia on mortality and unfavorable outcomes [185]. A guideline from the European Society of Intensive Care Medicine recommends maintaining PaO2 levels between 80 and 120 mmHg in brain-injured patients [186].
Protective MV has been shown to enhance survival in ARDS [183]. However, it may result in hypercapnia and hypoxemia, potentially contributing to increased ICP and cerebral hypoxia [184]. In ABI patients, the maintenance of sufficient oxygenation and the prevention of hypoxia are of paramount significance. Analyzing the IMPACT study database, which comprises over 9000 patients with TBI enrolled in randomized controlled trials and series dating back to the 1980s, has confirmed the impact of hypoxemia on mortality and unfavorable outcomes [185]. Indeed, recent recommendations regarding the ventilation targets in brain-injured patients with ARDS suggest maintaining PaO2 levels between 80 and 120 mmHg in brain-injured patients [186]. This recommendation contrasts with the ARDSnet practice, which sets oxygenation goals between 55 and 80 mmHg [183], allowing a relatively mild hypoxia. Nevertheless, given that patients with severe ABI face up to a 50% higher likelihood of death when the PaO2 is below 110 mm Hg [187,188,189], it might be justifiable to adopt a strategy targeting a higher PaO2 than the one recommended for ARDS patients without ABI [182, 190]. Indeed, if direct measures of brain physiology, including invasive intracranial pressure and brain tissue oxygen monitoring, are available, an individualized target of PaO2 is strongly recommended. In cases where direct measurement is unavailable, aiming for PaO2 > 110 mm Hg is suggested, considering lung compliance limitations [182]. However, it is crucial to take into account that both hypoxemia and excessive hyperoxemia have been linked to elevated mortality and a reduction in favorable outcomes among patients with ABI [188].
Lung-protective ventilation may induce hypercapnia, which is acceptable as long as oxygenation is secured and the pH level remains above 7.1 [191, 192]. Nevertheless, hypercapnia can negatively impact cerebral hemodynamics and the autoregulation of cerebral blood flow, worsening a preexisting impairment of cerebral autoregulation in neurocritical care patients [193,194,195,196]. Indeed, the cerebral vasculature exhibits high responsiveness to carbon dioxide (CO2) levels, and hypercapnia may lead to vasodilation of cerebral arterioles with subsequent intracranial hypertension and brain tissue hypoxia, which is poorly tolerated after ABI [184]. Nonetheless, a recent meta-analysis indicates that low tidal volumes lead to a modest increase in partial pressure of arterial carbon dioxide (PaCO2), shifting from 38 to 41 mmHg [197]. Based on brain physiology parameters, a tailored strategy targeting normal PaCO2 values (35–45 mm Hg) could be considered [182] by adjusting respiratory rate instead of tidal volume, given that a low tidal volume (7.0 ml/kg of ideal body weight) results in minimal alterations of the PaCO2 level [198].
Positive end-expiratory pressure (PEEP) hinders alveolar collapse, preserving oxygenation, enhancing functional residual capacity, and optimizing ventilation-perfusion matching [199,200,201]. However, the application of PEEP potentially elevates ICP by increasing intrathoracic pressure, thereby compromising venous return from the brain [202]. Moreover, the application of PEEP could induce compensatory vasodilation, potentially leading to intracranial hypertension under conditions of impaired cerebral autoregulation and reduced intracranial compliance. In addition, when cerebral autoregulation is compromised, an elevation in PEEP might decrease CPP, resulting in cerebral ischemia [4]. Concerns regarding elevated ICP lead to the delivery of PEEP at levels not exceeding 5 cm H2O in up to 80% of patients with brain injury. Nevertheless, a retrospective study evaluating the impact of PEEP on intracranial pressure in 20 patients with ABI complicated by ALI highlighted that across the range of 0 to 15 cm H2O, variations in PEEP levels showed no significant influence on ICP or CPP. Moreover, the effect of PEEP on ICP is likely minimal when PaCO2 is adequately controlled [202]. Currently, applying PEEP for ARDS treatment in ABI patients is considered appropriate, with the necessary condition that mean arterial pressure (MAP) is upheld. A consensus panel recommendation suggests using the same PEEP levels for brain-injured patients as those without brain injury unless elevations in ICP are associated with increased PEEP. Monitoring ICP in ABI patients with ALI can aid in the titration of PEEP [186]. However, recruitment maneuvers with high PEEP levels should be avoided as they have the potential to significantly elevate ICP in patients with compromised cerebral autoregulation due to the impairment of jugular blood outflow, elevated intrathoracic pressure, increased central venous pressure (CVP), and impediment of cerebral venous return to the right atrium [203].
Finally, other therapeutic options, including pulmonary vasodilator therapy, steroids, prone positioning, supine chest compression, and extracorporeal membrane oxygenation, might individually be considered [182].
Targeting the brain–gut–lung axis
As mentioned above, extensive research has demonstrated that bacterial metabolites derived from the intestine, for example, SCFAs, exert influence on local and systemic immune responses, regulate the maturation and function of microglia, and modulate the integrity of both the gut and the BBB [204]. Notably, SCFAs have the capacity to traverse the BBB and enhance its integrity, playing a pivotal role in brain homeostasis [215, 216] and might potentially contribute to the limitation of ABI-associated inflammatory injury of peripheral organs, including the gut and the lung. Additionally, SCFAs participate in a crucial way in the immunomodulatory properties of probiotics and prebiotics. Probiotics and prebiotics could modify significantly the pathogenesis of inflammatory processes by modulating the intestinal microbiota. This modulation involves the enhanced proliferation of health-beneficial microorganisms and the decreased presence of pathogenic microorganisms [205]. The impact of probiotics and prebiotics on the development and maturation of innate and adaptive immunity occurs via the secretion of cytokines, including IL-10 and transforming growth factor-β (TGF-β). As stated previously, these cytokines play a pivotal role in controlling the development of Treg cells and influencing the response of Th cells (Th1/Th2). This regulatory process extends to the release of TNF-α, interferons, and chemokines by immune cells [206]. Moreover, clinical studies have shown that the administration of probiotics in patients with ABI results in a decrease in infection rates through the reduction of IL-4 and IL-10 levels and the balance of Th1 and Th2 cytokines. Additionally, the use of probiotics was associated with a decrease in ICU stay and the number of days requiring mechanical ventilation [207, 208].
Fecal microbiota transplantation, characterized by the infusion of feces from a healthy individual into a patient with disrupted gut dysbiosis, has shown therapeutic applications in cases of ABI [209,210,211,212]. This therapeutic approach has been identified as effective in restoring gut microbiota dysbiosis following ABI, demonstrating an additional neuroprotective effect [96, 213]. Ultimately, treatment modalities, including vagus nerve stimulation and administration of gut-derived neuropeptides such as ghrelin, which can potentially modify the inflammatory response seen in patients with ABI, both systemically and locally [74], microbial engineering approaches and bacterial gene therapy significantly improve peripheral organ damage by addressing the dysbiosis of gut microbiota induced by acute CNS injury [214].
Conclusion
The “triple-hit” hypothesis extends our current understanding of the pathogenesis of ALI in ABI patients by incorporating the emerging concept of the gut–lung axis. By considering the interplay of sympathetic hyperactivity, the blast theory, inflammatory cascades, and gut-related pathogenetic mechanisms, this hypothesis provides a more comprehensive framework for investigating and understanding ALI in the context of ABI. Further research in this direction promises to shed light on novel therapeutic strategies that may improve the prognosis of these vulnerable patients.
Availability of data and materials
Not applicable.
References
Robba C, et al. Extracranial complications after traumatic brain injury: targeting the brain and the body. Curr Opin Crit Care. 2020;26(2):137–46.
McDonald SJ, et al. Beyond the brain: peripheral interactions after traumatic brain injury. J Neurotrauma. 2020;37(5):770–81.
Vaporidi K, et al. Clusters of ineffective efforts during mechanical ventilation: impact on outcome. Intensive Care Med. 2017;43(2):184–91.
Ziaka M, Exadaktylos A. Brain–lung interactions and mechanical ventilation in patients with isolated brain injury. Crit Care. 2021;25(1):358.
Mascia L. Acute lung injury in patients with severe brain injury: a double hit model. Neurocrit Care. 2009;11(3):417–26.
Slutsky AS. Lung injury caused by mechanical ventilation. Chest. 1999;116(1 Suppl):9S-15S.
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–48.
Atarashi K, Honda K. Microbiota in autoimmunity and tolerance. Curr Opin Immunol. 2011;23(6):761–8.
Chervonsky AV. Intestinal commensals: influence on immune system and tolerance to pathogens. Curr Opin Immunol. 2012;24(3):255–60.
Paun A, Danska JS. Immuno-ecology: how the microbiome regulates tolerance and autoimmunity. Curr Opin Immunol. 2015;37:34–9.
Ursell LK, et al. Defining the human microbiome. Nutr Rev. 2012;70(Suppl 1):S38-44.
Sommer F, Backhed F. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol. 2013;11(4):227–38.
Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701–12.
Fond G, et al. The “psychomicrobiotic”: targeting microbiota in major psychiatric disorders: a systematic review. Pathol Biol (Paris). 2015;63(1):35–42.
Pirbaglou M, et al. Probiotic supplementation can positively affect anxiety and depressive symptoms: a systematic review of randomized controlled trials. Nutr Res. 2016;36(9):889–98.
Wang Y, Kasper LH. The role of microbiome in central nervous system disorders. Brain Behav Immun. 2014;38:1–12.
Wang H, et al. Effect of probiotics on central nervous system functions in animals and humans: a systematic review. J Neurogastroenterol Motil. 2016;22(4):589–605.
George AK, et al. Rebuilding microbiome for mitigating traumatic brain injury: importance of restructuring the gut-microbiome-brain axis. Mol Neurobiol. 2021;58(8):3614–27.
Baguley IJ, et al. Dysautonomia after severe traumatic brain injury: evidence of persisting overresponsiveness to afferent stimuli. Am J Phys Med Rehabil. 2009;88(8):615–22.
Huffnagle GB, Dickson RP, Lukacs NW. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 2017;10(2):299–306.
Dickson RP, et al. The microbiome and the respiratory tract. Annu Rev Physiol. 2016;78:481–504.
Moffatt MF, Cookson WO. The lung microbiome in health and disease. Clin Med (Lond). 2017;17(6):525–9.
Gleeson K, Eggli DF, Maxwell SL. Quantitative aspiration during sleep in normal subjects. Chest. 1997;111(5):1266–72.
Huxley EJ, et al. Pharyngeal aspiration in normal adults and patients with depressed consciousness. Am J Med. 1978;64(4):564–8.
Hilty M, et al. Disordered microbial communities in asthmatic airways. PLoS ONE. 2010;5(1):e8578.
Budden KF, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63.
Carney SM, et al. Methods in lung microbiome research. Am J Respir Cell Mol Biol. 2020;62(3):283–99.
Segal LN, Rom WN, Weiden MD. Lung microbiome for clinicians. New discoveries about bugs in healthy and diseased lungs. Ann Am Thorac Soc. 2014;11(1):108–16.
Dang AT, Marsland BJ. Microbes, metabolites, and the gut-lung axis. Mucosal Immunol. 2019;12(4):843–50.
Wang H, et al. Gut microbiota is causally associated with poststroke cognitive impairment through lipopolysaccharide and butyrate. J Neuroinflamm. 2022;19(1):76.
Bradley CP, et al. Segmented filamentous bacteria provoke lung autoimmunity by inducing gut-lung axis Th17 cells expressing dual TCRs. Cell Host Microbe. 2017;22(5):697–704.
Wang G, et al. Gut-lung dysbiosis accompanied by diabetes mellitus leads to pulmonary fibrotic change through the NF-kappaB signaling pathway. Am J Pathol. 2021;191(5):838–56.
Boesch M, et al. Local tumor microbial signatures and response to checkpoint blockade in non-small cell lung cancer. Oncoimmunology. 2021;10(1):1988403.
Koutsoukou A, et al. Respiratory mechanics in brain injury: a review. World J Crit Care Med. 2016;5(1):65–73.
Avlonitis VS, et al. Pulmonary transplantation: the role of brain death in donor lung injury. Transplantation. 2003;75(12):1928–33.
Skandsen T, et al. Prevalence and impact of diffuse axonal injury in patients with moderate and severe head injury: a cohort study of early magnetic resonance imaging findings and 1-year outcome. J Neurosurg. 2010;113(3):556–63.
Das M, et al. Lateral fluid percussion injury of the brain induces CCL20 inflammatory chemokine expression in rats. J Neuroinflamm. 2011;8:148.
Ott L, et al. Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma. 1994;11(5):447–72.
McKeating EG, et al. Transcranial cytokine gradients in patients requiring intensive care after acute brain injury. Br J Anaesth. 1997;78(5):520–3.
van Vliet EA, et al. Long-lasting blood–brain barrier dysfunction and neuroinflammation after traumatic brain injury. Neurobiol Dis. 2020;145:105080.
Sweeney MD, et al. Blood–brain barrier: from physiology to disease and back. Physiol Rev. 2019;99(1):21–78.
Shlosberg D, et al. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6(7):393–403.
Baskaya MK, et al. The biphasic opening of the blood–brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett. 1997;226(1):33–6.
McKee CA, Lukens JR. Emerging roles for the immune system in traumatic brain injury. Front Immunol. 2016;7:556.
Jassam YN, et al. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron. 2017;95(6):1246–65.
Habgood MD, et al. Changes in blood–brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur J Neurosci. 2007;25(1):231–8.
Aloisi F. Immune function of microglia. Glia. 2001;36(2):165–79.
Nakajima K, Kohsaka S. Microglia: activation and their significance in the central nervous system. J Biochem. 2001;130(2):169–75.
Bachstetter AD, et al. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci. 2013;33(14):6143–53.
Csuka E, et al. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-alpha, TGF-beta1 and blood–brain barrier function. J Neuroimmunol. 1999;101(2):211–21.
Semple BD, et al. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2-/- mice. J Cereb Blood Flow Metab. 2010;30(4):769–82.
Morganti-Kossman MC, et al. Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol Psychiatry. 1997;2(2):133–6.
Frugier T, et al. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma. 2010;27(3):497–507.
Yenari MA, et al. Microglia potentiate damage to blood–brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37(4):1087–93.
Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43(4):348–64.
Fisher AJ, et al. Enhanced pulmonary inflammation in organ donors following fatal non-traumatic brain injury. Lancet. 1999;353(9162):1412–3.
Fisher AJ, et al. Elevated levels of interleukin-8 in donor lungs is associated with early graft failure after lung transplantation. Am J Respir Crit Care Med. 2001;163(1):259–65.
Siwicka-Gieroba D, et al. Concentration of apoptotic factors in bronchoalveolar lavage fluid, as potential brain-lung oxygen relationship, correspond to the severity of brain injury. J Integr Neurosci. 2023;22(2):49.
Wu S, et al. Enhanced pulmonary inflammation following experimental intracerebral hemorrhage. Exp Neurol. 2006;200(1):245–9.
Kalsotra A, et al. Brain trauma leads to enhanced lung inflammation and injury: evidence for role of P4504Fs in resolution. J Cereb Blood Flow Metab. 2007;27(5):963–74.
Cobelens PM, et al. Interferon-beta attenuates lung inflammation following experimental subarachnoid hemorrhage. Crit Care. 2010;14(4):R157.
Ziaka M, et al. High-tidal-volume mechanical ventilation and lung inflammation in intensive care patients with normal lungs. Am J Crit Care. 2020;29(1):15–21.
Eloe-Fadrosh EA, Rasko DA. The human microbiome: from symbiosis to pathogenesis. Annu Rev Med. 2013;64:145–63.
Wolff NS, Hugenholtz F, Wiersinga WJ. The emerging role of the microbiota in the ICU. Crit Care. 2018;22(1):78.
Turnbaugh PJ, et al. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1(6):6–14.
Claesson MJ, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488(7410):178–84.
Odamaki T, et al. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 2016;16:90.
Makivuokko H, et al. The effect of age and non-steroidal anti-inflammatory drugs on human intestinal microbiota composition. Br J Nutr. 2010;103(2):227–34.
Carabotti M, et al. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28(2):203–9.
Houlden A, et al. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun. 2016;57:10–20.
Wehrwein EA, Orer HS, Barman SM. Overview of the anatomy, physiology, and pharmacology of the autonomic nervous system. Compr Physiol. 2016;6(3):1239–78.
Hang CH, et al. Alterations of intestinal mucosa structure and barrier function following traumatic brain injury in rats. World J Gastroenterol. 2003;9(12):2776–81.
Bansal V, et al. Traumatic brain injury and intestinal dysfunction: uncovering the neuro-enteric axis. J Neurotrauma. 2009;26(8):1353–9.
Sundman MH, et al. The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain Behav Immun. 2017;66:31–44.
Moos WH, et al. Microbiota and neurological disorders: a gut feeling. Biores Open Access. 2016;5(1):137–45.
Jin W, et al. Transcription factor Nrf2 plays a pivotal role in protection against traumatic brain injury-induced acute intestinal mucosal injury in mice. J Surg Res. 2009;157(2):251–60.
Mahajan C, et al. Characteristics of gut microbiome after traumatic brain injury. J Neurosurg Anesthesiol. 2023;35(1):86–90.
Treangen TJ, et al. Traumatic brain injury in mice induces acute bacterial dysbiosis within the fecal microbiome. Front Immunol. 2018;9:2757.
Li N, et al. Change of intestinal microbiota in cerebral ischemic stroke patients. BMC Microbiol. 2019;19(1):191.
Mittal R, et al. Neurotransmitters: the critical modulators regulating gut-brain axis. J Cell Physiol. 2017;232(9):2359–72.
Tank AW, Lee Wong D. Peripheral and central effects of circulating catecholamines. Comp Physiol. 2015;5(1):1–15.
Stanley D, et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat Med. 2016;22(11):1277–84.
Cervi AL, Lukewich MK, Lomax AE. Neural regulation of gastrointestinal inflammation: role of the sympathetic nervous system. Auton Neurosci. 2014;182:83–8.
Lomax AE, Sharkey KA, Furness JB. The participation of the sympathetic innervation of the gastrointestinal tract in disease states. Neurogastroenterol Motil. 2010;22(1):7–18.
Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6(4):318–28.
Yang S, et al. Gut-derived norepinephrine plays a critical role in producing hepatocellular dysfunction during early sepsis. Am J Physiol Gastrointest Liver Physiol. 2000;279(6):G1274–81.
Lyte M, Ernst S. Catecholamine induced growth of gram negative bacteria. Life Sci. 1992;50(3):203–12.
Doherty NC, et al. The effect of the human gut-signalling hormone, norepinephrine, on the growth of the gastric pathogen Helicobacter pylori. Helicobacter. 2009;14(3):223–30.
Cogan TA, et al. Norepinephrine increases the pathogenic potential of campylobacter jejuni. Gut. 2007;56(8):1060–5.
Clifton GL, et al. Cardiovascular response to severe head injury. J Neurosurg. 1983;59(3):447–54.
Kalsotra A, et al. Differential effects of traumatic brain injury on the cytochrome p450 system: a perspective into hepatic and renal drug metabolism. J Neurotrauma. 2003;20(12):1339–50.
Feighery, L., et al., Increased intestinal permeability in rats subjected to traumatic frontal lobe percussion brain injury. J Trauma. 2008;64(1):131–7; discussion 137–8.
Catania A, et al. Detrimental consequences of brain injury on peripheral cells. Brain Behav Immun. 2009;23(7):877–84.
Pavlov VA, Tracey KJ. Neural regulators of innate immune responses and inflammation. Cell Mol Life Sci. 2004;61(18):2322–31.
Khaksari M, et al. The effect of female sexual hormones on the intestinal and serum cytokine response after traumatic brain injury: different roles for estrogen receptor subtypes. Can J Physiol Pharmacol. 2013;91(9):700–7.
Singh V, et al. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J Neurosci. 2016;36(28):7428–40.
Wang, H et al. Gut microbiota is causally associated with poststroke cognitive impairment through lipopolysaccharide and butyrate. J Neuroinflammation. 2022;19:1-15.
Roy Sarkar S, Banerjee S. Gut microbiota in neurodegenerative disorders. J Neuroimmunol. 2019;328:98–104.
Kang Y, et al. Correlation between intestinal flora and serum inflammatory factors in post-stroke depression in ischemic stroke. J Coll Physicians Surg Pak. 2021;31(10):1224–7.
Kelly BJ, et al. Composition and dynamics of the respiratory tract microbiome in intubated patients. Microbiome. 2016;4:7.
Shukla SD, et al. Microbiome effects on immunity, health and disease in the lung. Clin Transl Immunol. 2017;6(3):e133.
Yagi K, et al. The lung microbiome during health and disease. Int J Mol Sci. 2021;22(19):10872.
Kovaleva OV, et al. Human lung microbiome on the way to cancer. J Immunol Res. 2019;2019:1394191.
Weinberg F, et al. The lung microbiome: a central mediator of host inflammation and metabolism in lung cancer patients? Cancers (Basel). 2020;13(1):13.
Natalini JG, Singh S, Segal LN. The dynamic lung microbiome in health and disease. Nat Rev Microbiol. 2023;21(4):222–35.
Gollwitzer ES, et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med. 2014;20(6):642–7.
Biesbroek G, et al. Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children. Am J Respir Crit Care Med. 2014;190(11):1283–92.
Wu BG, Segal LN. The lung microbiome and its role in pneumonia. Clin Chest Med. 2018;39(4):677–89.
Kumpitsch C, et al. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 2019;17(1):87.
Morinaga Y, et al. Exploring the microbiota of upper respiratory tract during the development of pneumonia in a mouse model. PLoS ONE. 2019;14(9):e0222589.
Cuthbertson L, et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome. 2020;8(1):45.
Olszak T, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336(6080):489–93.
Herbst T, et al. Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Respir Crit Care Med. 2011;184(2):198–205.
Gaudêncio TG, de Moura Leão G. Epidemiologia do Traumatismo Crânio- Encefálico. Rev Neurociências. 2013;21(3):427–34.
Oliveira CO, Ikuta N, Regner A. Outcome biomarkers following severe traumatic brain injury. Rev Bras Ter Intensiva. 2008;20(4):411–21.
Bruns J Jr, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia. 2003;44(10):2–10.
Steyerberg EW, et al. Case-mix, care pathways, and outcomes in patients with traumatic brain injury in CENTER-TBI: a European prospective, multicentre, longitudinal, cohort study. Lancet Neurol. 2019;18(10):923–34.
Georgakopoulou VE, et al. Lower respiratory tract infections due to multi-drug resistant pathogens in central nervous system injuries (review). Biomed Rep. 2023;18(4):30.
Dhillon NK, et al. Early antibiotic administration is independently associated with improved survival in traumatic brain injury. J Surg Res. 2022;270:495–502.
Krishnamoorthy V, et al. Association of early multiple organ dysfunction with clinical and functional outcomes over the year following traumatic brain injury: a transforming research and clinical knowledge in traumatic brain injury study. Crit Care Med. 2021;49(10):1769–78.
McDonald D, et al. Extreme dysbiosis of the microbiome in critical illness. mSphere. 2016;1(4):e00199.
Alverdy JC, Laughlin RS, Wu L. Influence of the critically ill state on host-pathogen interactions within the intestine: gut-derived sepsis redefined. Crit Care Med. 2003;31(2):598–607.
Shreiner AB, Kao JY, Young VB. The gut microbiome in health and in disease. Curr Opin Gastroenterol. 2015;31(1):69–75.
Hauptmann M, Schaible UE. Linking microbiota and respiratory disease. FEBS Lett. 2016;590(21):3721–38.
Dumas A, et al. The role of the lung microbiota and the gut-lung axis in respiratory infectious diseases. Cell Microbiol. 2018;20(12):e12966.
Mukherjee S, Hanidziar D. More of the gut in the lung: how two microbiomes meet in ARDS. Yale J Biol Med. 2018;91(2):143–9.
Siwicka-Gieroba D, Czarko-Wicha K. Lung microbiome—a modern knowledge. Cent Eur J Immunol. 2020;45(3):342–5.
Segain JP, et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut. 2000;47(3):397–403.
Lin L, Zhang J. Role of intestinal microbiota and metabolites on gut homeostasis and human diseases. BMC Immunol. 2017;18(1):2.
Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. 2013;14(7):668–75.
Martin FP, et al. Panorganismal gut microbiome-host metabolic crosstalk. J Proteome Res. 2009;8(4):2090–105.
Rapozo DC, Bernardazzi C, de Souza HS. Diet and microbiota in inflammatory bowel disease: the gut in disharmony. World J Gastroenterol. 2017;23(12):2124–40.
Rutten EPA, et al. Disturbed intestinal integrity in patients with COPD: effects of activities of daily living. Chest. 2014;145(2):245–52.
Girosi D, et al. The lung and the gut: common origins, close links. Paediatr Respir Rev. 2006;7(Suppl 1):S235–9.
Tulic MK, Piche T, Verhasselt V. Lung-gut cross-talk: evidence, mechanisms and implications for the mucosal inflammatory diseases. Clin Exp Allergy. 2016;46(4):519–28.
Ma PJ, Wang MM, Wang Y. Gut microbiota: a new insight into lung diseases. Biomed Pharmacother. 2022;155:113810.
Lu Q, et al. The anatomic sites of disruption of the mucus layer directly correlate with areas of trauma/hemorrhagic shock-induced gut injury. J Trauma. 2011;70(3):630–5.
Rupani B, et al. Relationship between disruption of the unstirred mucus layer and intestinal restitution in loss of gut barrier function after trauma hemorrhagic shock. Surgery. 2007;141(4):481–9.
Panzer AR, et al. Lung microbiota is related to smoking status and to development of acute respiratory distress syndrome in critically ill trauma patients. Am J Respir Crit Care Med. 2018;197(5):621–31.
Dickson RP, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. 2016;1(10):16113.
Bingula R, et al. Desired turbulence? Gut-lung axis, immunity, and lung cancer. J Oncol. 2017;2017:5035371.
Stanley D, Moore RJ, Wong CHY. An insight into intestinal mucosal microbiota disruption after stroke. Sci Rep. 2018;8(1):568.
Deitch EA. Gut lymph and lymphatics: a source of factors leading to organ injury and dysfunction. Ann N Y Acad Sci. 2010;1207(Suppl 1):E103–11.
Alsaigh T, et al. In vivo analysis of intestinal permeability following hemorrhagic shock. World J Crit Care Med. 2015;4(4):287–95.
Sertaridou E, et al. Gut failure in critical care: old school versus new school. Ann Gastroenterol. 2015;28(3):309–22.
Hong G, et al. Administration of nicotinamide riboside prevents oxidative stress and organ injury in sepsis. Free Radic Biol Med. 2018;123:125–37.
Zhou X, Liao Y. Gut-lung crosstalk in sepsis-induced acute lung injury. Front Microbiol. 2021;12:779620.
de Jong PR, Gonzalez-Navajas JM, Jansen NJ. The digestive tract as the origin of systemic inflammation. Crit Care. 2016;20(1):279.
Freestone P. Communication between bacteria and their hosts. Scientifica (Cairo). 2013;2013:361073.
Sandrini S, et al. Microbial endocrinology: host-bacteria communication within the gut microbiome. J Endocrinol. 2015;225(2):R21-34.
Freestone PP, et al. Pseudomonas aeruginosa-catecholamine inotrope interactions: a contributory factor in the development of ventilator-associated pneumonia? Chest. 2012;142(5):1200–10.
Marks LR, et al. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. MBio. 2013;4(4):10–1128.
Dickson RP, Erb-Downward JR, Huffnagle GB. Homeostasis and its disruption in the lung microbiome. Am J Physiol Lung Cell Mol Physiol. 2015;309(10):L1047–55.
Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. 2013;369(22):2126–36.
Flierl MA, et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007;449(7163):721–5.
Kopin IJ, et al. Estimation of intrasynaptic norepinephrine concentrations at vascular neuroeffector junctions in vivo. Naunyn Schmiedebergs Arch Pharmacol. 1984;325(4):298–305.
Kovarik MF, Jones SB, Romano FD. Plasma catecholamines following cecal ligation and puncture in the rat. Circ Shock. 1987;22(4):281–90.
Meduri GU, et al. Cytokines IL-1beta, IL-6, and TNF-alpha enhance in vitro growth of bacteria. Am J Respir Crit Care Med. 1999;160(3):961–7.
Kanangat S, et al. Effects of cytokines and endotoxin on the intracellular growth of bacteria. Infect Immun. 1999;67(6):2834–40.
Kaza SK, McClean S, Callaghan M. IL-8 released from human lung epithelial cells induced by cystic fibrosis pathogens Burkholderia cepacia complex affects the growth and intracellular survival of bacteria. Int J Med Microbiol. 2011;301(1):26–33.
Plotkin BJ, et al. Effect of androgens and glucocorticoids on microbial growth and antimicrobial susceptibility. Curr Microbiol. 2003;47(6):514–20.
Faura J, et al. Stroke-induced immunosuppression: implications for the prevention and prediction of post-stroke infections. J Neuroinflammation. 2021;18(1):127.
Dirnagl U, et al. Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke. 2007;38(2 Suppl):770–3.
Prass K, et al. Stroke propagates bacterial aspiration to pneumonia in a model of cerebral ischemia. Stroke. 2006;37(10):2607–12.
Brown RL, Sequeira RP, Clarke TB. The microbiota protects against respiratory infection via GM-CSF signaling. Nat Commun. 2017;8(1):1512.
Dickson RP. The microbiome and critical illness. Lancet Respir Med. 2016;4(1):59–72.
Zakharkina T, et al. The dynamics of the pulmonary microbiome during mechanical ventilation in the intensive care unit and the association with occurrence of pneumonia. Thorax. 2017;72(9):803–10.
Akrami K, Sweeney DA. The microbiome of the critically ill patient. Curr Opin Crit Care. 2018;24(1):49–54.
Dickson RP. The lung microbiome and ARDS. It is time to broaden the model. Am J Respir Crit Care Med. 2018;197(5):549–51.
Dickson RP, et al. Lung microbiota predict clinical outcomes in critically ill patients. Am J Respir Crit Care Med. 2020;201(5):555–63.
Weinstock GM. Genomic approaches to studying the human microbiota. Nature. 2012;489(7415):250–6.
Schmitt FCF, et al. Pulmonary microbiome patterns correlate with the course of the disease in patients with sepsis-induced ARDS following major abdominal surgery. J Hosp Infect. 2020;105:438–46.
Cotoia A, et al. Modifications of lung microbiota structure in traumatic brain injury ventilated patients according to time and enteral feeding formulas: a prospective randomized study. Crit Care. 2023;27(1):244.
Wu H, et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111(10):1589–602.
Gunther A, et al. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med. 1996;153(1):176–84.
Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;7(3):245–57.
Nseir S, et al. Microaspiration in intubated critically ill patients: diagnosis and prevention. Infect Disord Drug Targets. 2011;11(4):413–23.
Mrozek S, Constantin JM, Geeraerts T. Brain-lung crosstalk: implications for neurocritical care patients. World J Crit Care Med. 2015;4(3):163–78.
Rogers, F.B., et al., Neurogenic pulmonary edema in fatal and nonfatal head injuries. J Trauma. 1995;39(5): 860–6; discussion 866–8.
Fontes RB, et al. Acute neurogenic pulmonary edema: case reports and literature review. J Neurosurg Anesthesiol. 2003;15(2):144–50.
Baumann A, et al. Neurogenic pulmonary edema. Acta Anaesthesiol Scand. 2007;51(4):447–55.
Kim JA, et al. Focused management of patients with severe acute brain injury and ARDS. Chest. 2022;161(1):140–51.
Brower RG, et al. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1301–8.
Ropper AH. Hyperosmolar therapy for raised intracranial pressure. N Engl J Med. 2012;367(8):746–52.
McHugh GS, et al. Prognostic value of secondary insults in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24(2):287–93.
Robba C, et al. Mechanical ventilation in patients with acute brain injury: recommendations of the European Society of Intensive Care Medicine consensus. Intensive Care Med. 2020;46(12):2397–410.
Investigators I-R, et al. Conservative oxygen therapy during mechanical ventilation in the ICU. N Engl J Med. 2020;382(11):989–98.
Davis DP, et al. Both hypoxemia and extreme hyperoxemia may be detrimental in patients with severe traumatic brain injury. J Neurotrauma. 2009;26(12):2217–23.
Schjorring OL, et al. Lower or higher oxygenation targets for acute hypoxemic respiratory failure. N Engl J Med. 2021;384(14):1301–11.
Della Torre V, et al. Acute respiratory distress syndrome in traumatic brain injury: how do we manage it? J Thorac Dis. 2017;9(12):5368–81.
Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369(9572):1553–64.
Fanelli V, et al. Acute respiratory distress syndrome: new definition, current and future therapeutic options. J Thorac Dis. 2013;5(3):326–34.
Czosnyka M, et al. Monitoring of cerebral autoregulation in head-injured patients. Stroke. 1996;27(10):1829–34.
Bein T, et al. Lung recruitment maneuver in patients with cerebral injury: effects on intracranial pressure and cerebral metabolism. Intensive Care Med. 2002;28(5):554–8.
Meng L, Gelb AW. Regulation of cerebral autoregulation by carbon dioxide. Anesthesiology. 2015;122(1):196–205.
Severdija EE, et al. Assessment of dynamic cerebral autoregulation and cerebral carbon dioxide reactivity during normothermic cardiopulmonary bypass. Med Biol Eng Comput. 2015;53(3):195–203.
Asehnoune K, et al. Mechanical ventilation in patients with acute brain injury: a systematic review with meta-analysis. Crit Care. 2023;27(1):221.
Asehnoune K, et al. A multi-faceted strategy to reduce ventilation-associated mortality in brain-injured patients. The BI-VILI project: a nationwide quality improvement project. Intensive Care Med. 2017;43(7):957–70.
Schramm P, et al. Influence of PEEP on cerebral blood flow and cerebrovascular autoregulation in patients with acute respiratory distress syndrome. J Neurosurg Anesthesiol. 2013;25(2):162–7.
Modock J. Complex care: ventilation management when brain injury and acute lung injury coexist. J Neurosci Nurs. 2014;46(2):71–8.
Rozet I, Domino KB. Respiratory care. Best Pract Res Clin Anaesthesiol. 2007;21(4):465–82.
Nemer SN, et al. Effects of positive end-expiratory pressure on brain tissue oxygen pressure of severe traumatic brain injury patients with acute respiratory distress syndrome: a pilot study. J Crit Care. 2015;30(6):1263–6.
Nemer SN, et al. Alveolar recruitment maneuver in patients with subarachnoid hemorrhage and acute respiratory distress syndrome: a comparison of 2 approaches. J Crit Care. 2011;26(1):22–7.
Marrocco F, et al. Short-chain fatty acids promote the effect of environmental signals on the gut microbiome and metabolome in mice. Commun Biol. 2022;5(1):517.
Vernocchi P, Del Chierico F, Putignani L. Gut microbiota metabolism and interaction with food components. Int J Mol Sci. 2020;21(10):3688.
Batista KS, et al. Probiotics and prebiotics: potential prevention and therapeutic target for nutritional management of COVID-19? Nutr Res Rev. 2023;36(2):181–98.
Tan M, et al. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care. 2011;15(6):R290.
Falcao de Arruda IS, de Aguilar-Nascimento JE. Benefits of early enteral nutrition with glutamine and probiotics in brain injury patients. Clin Sci (Lond). 2004;106(3):287–92.
Kim KO, Gluck M. Fecal microbiota transplantation: an update on clinical practice. Clin Endosc. 2019;52(2):137–43.
Brandt LJ, Aroniadis OC. An overview of fecal microbiota transplantation: techniques, indications, and outcomes. Gastrointest Endosc. 2013;78(2):240–9.
Gupta S, Allen-Vercoe E, Petrof EO. Fecal microbiota transplantation: in perspective. Therap Adv Gastroenterol. 2016;9(2):229–39.
Du D, et al. Fecal microbiota transplantation is a promising method to restore gut microbiota dysbiosis and relieve neurological deficits after traumatic brain injury. Oxid Med Cell Longev. 2021;2021:5816837.
Spychala MS, et al. Age-related changes in the gut microbiota influence systemic inflammation and stroke outcome. Ann Neurol. 2018;84(1):23–36.
Guha L, et al. Gut microbiota and traumatic central nervous system injuries: insights into pathophysiology and therapeutic approaches. Life Sci. 2023;334:122193.
Oldendorf WH. Carrier-mediated blood-brain barrier transport of short-chain monocarboxylic organic acids. Am J Physiol. 1973;224(6):1450–1453. https://doi.org/10.1152/ajplegacy.1973.224.6.1450
Silva YP, Bernardi A, Frozza RL. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol. 2020;11:25. https://doi.org/10.3389/fendo.2020.00025
Acknowledgements
Figure 1 created with BioRender.com.
Funding
Publication costs for this article were funded by the authors' institutions.
Author information
Authors and Affiliations
Contributions
The study was designed by MZ and AE. MZ searched the articles and drafted the manuscript, to which AE contributed and revised. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ziaka, M., Exadaktylos, A. Pathophysiology of acute lung injury in patients with acute brain injury: the triple-hit hypothesis. Crit Care 28, 71 (2024). https://doi.org/10.1186/s13054-024-04855-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-024-04855-w