
Abstract
Background
Townes-Brocks syndrome (TBS) is a rare genetic disorder characterized by imperforate anus, dysplastic ears, thumb malformations, and other abnormalities. Previous studies have revealed that mutations in the SALL1 gene can disrupt normal development, resulting in the characteristic features of Townes-Brocks syndrome. Spalt-like transcription factors (SALLs) are highly conserved proteins that play important roles in various cellular processes, including embryonic development, cell differentiation, and cell survival. Over 400 different variants or mutations have been reported in the SALL1 gene in individuals with TBS. Most of these variants lead to the formation of premature termination codons (PTCs), also known as nonsense mutations. The majority of these PTCs occur in a specific region of the SALL1 gene called the “hotspot region”, which is particularly susceptible to mutation.
Methods
In this study, we conducted whole-exome sequencing on a three-generation Chinese family with anorectal malformations.
Results
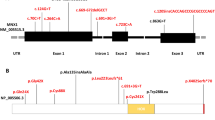
We identified a novel heterozygous mutation (chr16:51175376:c.757 C > T p.Gln253*) in the SALL1 gene. Molecular analysis revealed a heterozygous C to T transition at nucleotide position 757 in exon 2 of the SALL1 (NM_002968) gene. This mutation is predicted to result in the substitution of the Gln253 codon with a premature stop codon (p.Gln253*). The glutamine-rich domain forms a long alpha helix, enabling the mutant protein to interact with the wild-type SALL1 protein. This interaction may result in steric hindrance effects on the wild-type SALL1 protein.
Conclusions
Our findings have expanded the mutation database of the SALL1 gene, which is significant for genetic counseling and clinical surveillance in the affected family. Furthermore, our study enhances the understanding of Townes-Brocks syndrome and has the potential to improve its diagnosis and treatment.
Similar content being viewed by others


Introduction
Townes-Brocks syndrome (TBS, OMIM: #107,480) is a congenital genetic disorder characterized by the triad including malformations of anus, external ears and thumbs. Other features include hearing loss, foot deformity, renal insufficiency with or without associated urogenital abnormalities, and congenital heart disease, including atrial septal defect, ventricular septal defect, tetralogy of Fallot, truncus arteriosus (type A or type B), pulmonary valve atresia, and persistent ductus arteriosus [1,2,3,4]. Dysplastic ears (overfolded superior helices, microtia), thumb abnormalities (preaxial polydactyly, triphalangeal thumbs, and hypoplastic thumbs without hypoplasia of the radius), lower extremities deformities (clubfoot, overlapping toes, syndactyly, and missing toes), vaginal aplasia with bifid uterus, bifid scrotum, cryptorchidism are also observed. Additionally, some individuals with TBS may experience developmental delay or intellectual disability. Currently, the diagnosis of TBS requires the presence of three major or two major and minor features [3]. The major criteria for diagnosing TBS include an imperforate anus (found in 84% of cases), abnormally shaped ears (found in 87% of cases), and thumb malformations (found in 89% of cases). Minor features associated with TBS may include renal abnormalities, hearing loss, heart defects, genitourinary and limb abnormalities, and anal stenosis [5]. Early identification of TBS is fundamental to support child health and development (e.g. hearing aids, early surgical intervention) and to provide genetic counseling [6]. TBS is an autosomal dominant genetic disease due to SALL1 gene mutation [7].
Spalt-like transcription factors, also known as SALLs, belong to a family of zinc finger transcription factors that play important roles in various biological processes, including embryo development, apoptosis, angiogenesis, and metastasis [8]. SALL1 is a zinc finger protein believed to function as a transcription factor. It consists of three exons that encode a zinc finger protein responsible for transcriptional repression. The protein contains four highly conserved double zinc finger domains, with a single C2H2 motif attached to the second domain. SALL1 protein is exclusively found in pericentromeric heterochromatin. Most SALL1 mutations results in alteration in the N-terminal third of the encoded protein, causing truncation mostly at C2H2 and N-terminal transcriptional repressor domains [9]. To date, over 400 different SALL1 variants with overlapping phenotypes have been reported in TBS patients [3]. Indeed, the precise mechanism by which specific variants or mutations in the genes associated with TBS lead to the development of the disorder is not fully understood.
Here, we identified a novel heterozygous mutation (c.757 C > Tp.Gln253*) in SALL1 through whole-exome sequencing of a family presenting a congenital anal agenesis. In addition to this, two family members presented with mild hearing impairment. In silico study was performed to examine the pathogenicity of the variant. The results showed that the glutamine-rich domain forms a long alpha helix, allowing the mutant protein to interact with the wild-type SALL1 protein between the two glutamine-rich domains of both wild-type and mutant SALL1 proteins that might exert steric hindrance effects, altering thus its function. This finding is consistent with previous reports. These results contribute to expanding the spectrum of SALL1 mutations and provide a reference for clinical practice.
Patients and methods
Case presentation
The proband was a 1-day-old female newborn and her two sisters born from twin gestation with known anorectal malformations (ARMs) from a townlet in China. Their mother was 33 years old and previously had a spontaneous birth. During this spontaneous pregnancy, transabdominal ultrasound examination diagnosed triplet gestation. Nuchal Translucency (NT) and amniocentesis were not performed during pregnancy. No abnormalities were found in the Oral Glucose Tolerance Test、quantity of amniotic liquid、fetal heart and blood pressure monitoring.
Due to the mother’s premature rupture of membranes, the triplets were delivered via an emergency cesarean section on the 4th day of the 34th week. During the intervention, it was confirmed that the triplets were dichorionic triamniotic. Following birth, all siblings were diagnosed with anal stenosis (Fig. 1) [10]. Additionally, the third triplet had syndactyly of the second and third left toes. Due to prematurity, they were immediately admitted to the neonatal intensive care unit (NICU). The triplets underwent chest X-ray, as well as abdominal and transfontanellar ultrasound, hearing screening by automated transient otoacoustic emission (aTEOAE), and automatic auditory brainstem response (aABR) which resulted normal. Cardiac ultrasound revealed pulmonary hypertension in the second and the third of the triplet. Abdominal radiography was also performed, revealing the presence of inflated and dilated intestinal loops. Thereafter, we noticed that all the triplets defecate through the vagina so we suspected rectovaginal fistula. Therefore, the posterior rectal advancement anoplasty was not performed at birth. However, limited by the conditions at that time, a barium enema was not performed, which is used to examine the location of the fistula in relation to the normal anal structure. By inquiring about their medical history, we were told that their father was born with imperforate anus, thumb malformations and hearing loss. He underwent anorectoplasty. He had moderate hearing loss and did not need hearing aids. Furthermore, during the family history investigation, another interesting finding emerged: the triplets’s grandmother was diagnosed with deafness and her brother had congenital anal atresia and finger deformities. Unfortunately, contact with him has been lost. Renal and cardiac US were not performed neither in the triplets’s father nor in the grandmother.

The triplets presented clinically as having anal agenesis. Supine photographs showing the clinical presentation of the triplets without anus. The eldest of the triplets (A), the second of the triplets (B), and the third of the triplets (C) in the supine position, red box indicated normal anatomical position without anal appearance
The study describes 3 generations: grandmother affected by moderate hearing loss, father by moderate hearing loss, anal malformation, and thumb deformities, and triplets by anal atresia. The third one (III4) was chosen as the proband (Fig. 2). Peripheral blood samples were collected from all these individuals. Informed consent was obtained from the proband’s parents and all other family members. The study protocol was approved by the Shandong Provincial Hospital Affiliated to Shandong First Medical University Institutional Ethics Committee (SWYX: NO.2023 − 386).

Pedigree of this family. The medical history was inquired about the disease status of family members.Square indicates male, circle indicates female, black indicates TBS patients and arrow indicates the proband (the third of the triplets)
Results
Sequencing data analysis
Although a-CGH is recommended first to detect genomic rearrangements [11,12,13,14], due to various factors we chose the gene sequencing method which is widely used in clinics at present. Genomic DNA was extracted using the QIAamp Blood Midi Kit from QIAGEN, located in Valencia, California. Paired-end reads of 150 bp were generated using an Illumina Novaseq6000 sequencer (Illumina, San Diego, CA, USA) and DNBSEQ (DNBSEQ-T7) (MGI, Shenzhen, China). The raw sequencing data was stored in FASTQ format. Quality control filters were applied to remove low-quality reads. After combining and splicing the clean reads using the second-generation sequencing analysis platform provided by MyGenostics, the coverage and sequencing quality of the target region were assessed. Subsequently, the flash analysis platform was utilized to examine the pathogenicity of the variants, and potential loci for these variants were identified. Furthermore, the pathogenicity of variation loci was evaluated according to the standards and recommendations set by ACMG (American College of Medical Genetics and Genomics). Sanger sequencing was employed to confirm potentially harmful mutations in all family members. An Applied Biosystems ABI 3730 analyzer was used for sequencing the mutations, and the results were compared with those obtained through capture sequencing.
Whole-exome sequencing analysis was performed on all of the family members. Molecular analysis revealed a heterozygous C to T transition at nucleotide position 757 in exon 2 of the SALL1 (NM_002968) gene (Fig. 3). This transition was predicted to result in the substitution of the Gln253 codon with a premature stop codon (p.Gln253*), resulting in truncated SALL1 proteins. The structure of wildtype and mutant SALL1 protein (p.Gln253*) were predicted by AlphaFold (34,265,844). The structures were visualized by PyMol, and shown as orthogonal views (Fig. 4). It shows that the SALL1 (p.Gln253*) is a truncated protein that retains the glutamine-rich domain. Molecular docking, which has proven versatile for identifying protein-ligand interactions (23,508,189), was performed to predict the potential binding sites between wildtype and mutant SALL1 proteins. The results showed that the glutamine-rich domain forms a long alpha helix, through which the mutant protein could interact with wildtype SALL1 protein, with the lowest binding free energy. The binding site (red box in Fig. 4) was between the two glutamine-rich domains of wildtype and mutant SALL1 proteins, which is similar with previous reported [7, 15,16,17,18,19,20]. The interaction might exert steric hindrance effects to the wildtype SALL1 protein. This variant, although not listed in major mutation databases, cosegregated with the disease in six affected family members but was absent in unaffected ones. Following ACMG guidelines, c.757 C > T p.Gln253* is classified as pathogenic (PVS1 + PM2_supporting + PP1).

Sanger sequencing analysis of SALL1 gene in genomic DNA from the family. The arrow indicates the mutation site (chr16:51175376: c.757 C > T p.Gln253* exon2, NM_002968). The proband (III4), her sisters (III2, III3), her father (II1) and her grandmother (I2) carried the mutation. Other family members did not have the mutation

Predicted structures of human wildtype and mutant SALL1 protein (Gln253*). The AlphaFold structure predictions of human wildtype (green, A,D) and mutant (blue, B,E) SALL1 protein (Gln253*). The orthogonal views were visualized by PyMol program. The interaction of the wildtype and mutant SALL1 was highlighted in the red box (C,F)
Clinical prognosis
Follow-up was performed at a pediatric clinic one month after the surgery. The anal appearance of the triplets was satisfactory, and urination and defecation were normal without any signs of constipation [13]. At their corrected age of 1 year, they are thriving, exhibiting good weight gain, and experiencing no feeding or bowel difficulties. There were no reports of urinary tract infections during the outpatient follow-up period. Moreover, no surgical complications, such as anal stenosis, were observed. Clinical evaluation of their growth according to WHO growth charts and their development were followed. The triplets had regular evolution. Clinical laboratory indicators such as hemoglobin, alkaline phosphatase, and 25(OH)D were within normal ranges. Overall, their general growth and living conditions were satisfactory, similar to those of normally developing children [21].
Discussion
As of March 2022, HGMD has documented 116 SALL1 mutations, such as frameshifts, nonsenses, gross deletions, and splice variants [7]. Most of the reported mutations in TBS patients are frameshift or nonsense mutations that reside within a mutational hotspot region spanning from nucleotide 764 to 1565. These particular frameshift or nonsense mutations within the mutation hotspot region usually result in classical or more severe TBS phenotypes. Conversely, clinical manifestations due to haploinsufficiency of SALL1 tend to be relatively mild. (22–23) Furthermore, genetic anticipation has been observed in certain families with Townes-Brocks syndrome, with an increase in clinical severity across generations [24]. The novel mutations (c.757 C > T p.Gln253* exon2) identified in this study were located in the hotspot mutation region of SALL1. The affected individuals in the first generation (the proband’s grandmother) exhibited deafness, while those in the second generation (the proband’s father) manifested polydactyly, deafness, and anal abnormalities. The third generation (comprising the proband and her sisters) displayed syndactyly of the second and third left toes, and anal abnormalities. This clinical presentation is consistent with findings from previous studies [24].
According to earlier studies, SALL1 heterozygous deletions can cause Townes-Brocks syndrome or at least a milder form of the condition [8]. Indeed, the inheritance pattern of TBS follows autosomal dominant inheritance, meaning that a single copy of the mutated gene is sufficient to cause the disorder. In this case, the molecular basis of the disease lies in a specific gene called SALL1, which is located at 16q12.1 and encodes a zinc finger protein involved in transcription regulation [7, 23]. Mutations in the SALL1 gene, typically heterozygous mutations, play a crucial role in the development of TBS. These mutations contribute to the syndrome through a combination of a dominant negative effect, where the mutated protein interferes with normal cellular processes, and haploinsufficiency, where there is an inadequate amount of functional protein produced due to the mutation [25]. Other studies have confirmed that these truncated proteins exert their influence by negatively affecting heart and limb development via a dominant negative effect [26]. In this study, proband’s father and sisters had finger deformity.
Diagnosis of Townes-Brocks syndrome (TBS) involves a combination of clinical evaluation, imaging studies, and genetic testing to identify mutations in the SALL1 gene [7]. During the diagnostic process, it is crucial to exclude other rare syndromes, such as VACTERL/VATER syndrome, cat’s eye syndrome, and Goldenhar syndrome, that have TBS overlapping signs [27,28,29]. Treatment is generally supportive and may involve surgical intervention for certain malformations or the use of hearing aids for hearing loss [6]. In this case, the primary presentation of the triplets was anal agenesis, and timely surgical treatment was performed by experienced medical team. Following surgery for anorectal abnormalities, children may experience constipation and fecal incontinence [30]. Remarkably, the patients described in this study did not experience any negative side effects such as urinary tract infections, fecal incontinence, or constipation after the surgical treatment. Furthermore, our research observed patients with TBS syndrome for more than one year and we intend to continue following up with the patients until they reach maturity. Due to the numerous clinical manifestations of TBS syndrome, triplets need a multidisciplinary follow-up to test their growth, and their neurocognitive and behavioral evolution.
There have been reports of Anorectal malformations (ARMs) in 1 in 2,000 to 5 in 10,000 cases [31]. It is more common among Asians. (32–33) While there appears to be a low rate of inheritance in family members, some individuals exhibited autosomal dominant inheritance patterns [31]. Our subsequent genetic analysis further confirmed that this case represents a typical instance of autosomal dominant inheritance. Reconstructive surgery and ongoing symptom management (e.g., treatment for persistent constipation, incontinence, recurrent infections, and psychosocial support) constitute the major aspects of ARM clinical management [34]. In this case, after a period of conservative treatment, surgery was performed, and the postoperative effect was ideal, allowing the triplets to defecate independently without known sequelae. Multiple pregnancies have become more common in recent years due to the development of reproductive techniques. Spontaneous multiple pregnancies are rarely observed [35]. Their incidence is approximately 1:7000 [36]. It is worth noting that the clinical variability described in the affected subjects may be linked to different types of variants or to potential epigenetic factors (they may play a more relevant role in case of multiple pregnancies). (37–38) Yang et al. [24] reported a case of a 2-month-old child with Townes-Brocks syndrome (TBS). This patient presented clinical symptoms similar to those observed in our reported case. However, unlike us, the child’s parents were consanguineous and had a mutation in the PTPRQ gene, which has been associated with nonsyndromic hearing loss. Innoceta AM, et al. [39] described a case of a 7-year-old girl who not only exhibited symptoms similar to our current case but also presented with a ventrally positioned anus, pantonal sensorineural hearing loss (prevalent in the left ear) of mild-medium severity. Furthermore, she experienced developmental delay and speech delay. It is noteworthy that approximately one third of TBS cases reported in the literature involve some level of postnatal growth retardation [3, 40]. As a result, it is crucial for us to closely monitor the growth and development of these children.
We have successfully identified a novel mutation that has not been previously reported in individuals with TBS, thereby offering new genetic information crucial for genetic counseling and family screening. By reporting this novel variant, we have expanded the existing genetic variation spectrum of TBS and enriched the data available for understanding the molecular mechanisms of the disease. This particular locus has demonstrated an association with the clinical phenotype of TBS, which suggests that our findings could contribute to a better understanding of the disease’s presentation patterns, early diagnosis, and treatment guidance. However, it is important to note that our study focused on a single family, necessitating consideration of factors such as genetic heterogeneity and population differences that may influence the results. Additionally, our study did not take into account the interaction between genetic and environmental factors, including the potential influence of other genetic variants or environmental factors on the development of TBS. Further research is needed to explore these aspects.
Conclusions
In this study a novel heterozygous mutation of SALL1 (chr16:51175376: c.757 C > T p.Gln253* exon2, NM_002968) has been identified in a Chinese family with TBS typical phenotype. These findings enrich knowledge about TBS and contribute to better clinical management and genetic counseling. To gain more specific insights into the outcomes, long-term follow-up is required. Further studies are needed to understand the correlation between genotypes and phenotypes.
Data availability
All data generated or analyzed during this study are included in the article, further inquiries can be directed to the corresponding author.
Abbreviations
- SALLs:
-
Spalt-like transcription factors
- TBS:
-
Townes-Brocks syndrome
- PTCs:
-
Premature termination codons
- ARMs:
-
Anorectal malformations
- NT:
-
Nuchal translucency
- NICU:
-
Neonatal intensive care unit
- ACMG:
-
American college of medical genetics and genomics
- PRAA:
-
Posterior rectal advancement anoplasty
- PH:
-
Pulmonary hypertension
- TRV:
-
Tricuspid regurgitation velocity
- mPAP:
-
Mean pulmonary artery pressure
References
Bozal-Basterra L, Martin-Ruiz I, Pirone L, Liang Y, Sigurethsson JO, Gonzalez-Santamarta M, et al. Truncated SALL1 impedes primary cilia function in Townes-Brocks Syndrome. Am J Hum Genet. 2018;102(2):249–65.
Sudo Y, Numakura C, Abe A, Aiba S, Matsunaga A, Hayasaka K. Phenotypic variability in a family with Townes-Brocks syndrome. J Hum Genet. 2010;55(8):550–1.
Kohlhase J et al. Townes-Brocks Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, editors. GeneReviews((R)). Seattle (WA) 2007 Jan 24 [Updated 2016 Jan 14].
Van Praagh R. What determines whether the great arteries are normally or abnormally related? Am J Cardiol. 2016;118(9):1390–8.
Lugli L, Rossi C, Ceccarelli PL, Calabrese O, Bedetti L, Miselli F, et al. Townes-Brocks syndrome with craniosynostosis in two siblings. Eur J Med Genet. 2022;65(12):104642.
Keegan CE, Mulliken JB, Wu BL, Korf BR. Townes-Brocks syndrome versus expanded spectrum hemifacial microsomia: review of eight patients and further evidence of a hot spot for mutation in the SALL1 gene. Genet Med. 2001;3(4):310–3.
Wang Z, Sun Z, Diao Y, Wang Z, Yang X, Jiang B, et al. Identification of two novel SALL1 mutations in Chinese families with townes-brocks syndrome and literature review. Orphanet J Rare Dis. 2023;18(1):250.
Sills ES, Wood SH. Phenotype from SAMD9 mutation at 7p21.2 appears attenuated by novel compound heterozygous variants at RUNX2 and SALL1. Glob Med Genet. 2022;9(2):124–8.
Botzenhart EM, Bartalini G, Blair E, Brady AF, Elmslie F, Chong KL, et al. Townes-Brocks syndrome: twenty novel SALL1 mutations in sporadic and familial cases and refinement of the SALL1 hot spot region. Hum Mutat. 2007;28(2):204–5.
Amerstorfer EE, Schmiedeke E, Samuk I, Sloots CEJ, van Rooij I, Jenetzky E et al. Clinical differentiation between a normal anus, anterior anus, congenital anal stenosis, and Perineal Fistula: definitions and consequences-the ARM-Net Consortium Consensus. Child (Basel). 2022;9(6).
Serra G, Felice S, Antona V, Di Pace MR, Giuffre M, Piro E, et al. Cardio-facio-cutaneous syndrome and gastrointestinal defects: report on a newborn with 19p13.3 deletion including the MAP 2 K2 gene. Ital J Pediatr. 2022;48(1):65.
Serra G, Memo L, Antona V, Corsello G, Favero V, Lago P, et al. Jacobsen syndrome and neonatal bleeding: report on two unrelated patients. Ital J Pediatr. 2021;47(1):147.
Piccione M, Serra G, Consiglio V, Di Fiore A, Cavani S, Grasso M, et al. 14q13.1-21.1 deletion encompassing the HPE8 locus in an adolescent with intellectual disability and bilateral microphthalmia, but without holoprosencephaly. Am J Med Genet A. 2012;158A(6):1427–33.
Serra G, Antona V, Giuffre M, Piro E, Salerno S, Schierz IAM, et al. Interstitial deletions of chromosome 1p: novel 1p31.3p22.2 microdeletion in a newborn with craniosynostosis, coloboma and cleft palate, and review of the genomic and phenotypic profiles. Ital J Pediatr. 2022;48(1):38.
Halleran DR, Coyle D, Kulaylat AN, Ahmad H, Langer JC, Gasior AC, et al. The cutback revisited - the posterior rectal advancement anoplasty for certain anorectal malformations with rectoperineal fistula. J Pediatr Surg. 2022;57(9):85–8.
Faguer S et al. Nephropathy in Townes-Brocks syndrome (SALL1 mutation): imaging and pathological findings in adulthood. 2009. 24(4): p. 1341–5.
Cassandri M, et al. Zinc-finger Proteins Health Disease. 2017;3:17071.
Basta J et al. Sall1 balances self-renewal and differentiation of renal progenitor cells. 2014. 141(5): p. 1047–58.
Borozdin W et al. Detection of heterozygous SALL1 deletions by quantitative real time PCR proves the contribution of a SALL1 dosage effect in the pathogenesis of Townes-Brocks syndrome. 2006. 27(2): p. 211–2.
Levitt MA, Pena A. Anorectal malformations. Orphanet J Rare Dis. 2007;2:33.
Piro E, Serra G, Schierz IAM, Giuffre M, Corsello G. Neonatal ten-year retrospective study on neural tube defects in a second level University Hospital. Ital J Pediatr. 2020;46(1):72.
Miller EM, Hopkin R, Bao L, Ware SM. Implications for genotype-phenotype predictions in Townes-Brocks syndrome: case report of a novel SALL1 deletion and review of the literature. Am J Med Genet A. 2012;158A(3):533–40.
Botzenhart EM, Green A, Ilyina H, Konig R, Lowry RB, Lo IF, et al. SALL1 mutation analysis in Townes-Brocks syndrome: twelve novel mutations and expansion of the phenotype. Hum Mutat. 2005;26(3):282.
Yang G, Yin Y, Tan Z, Liu J, Deng X, Yang Y. Whole-exome sequencing identified a novel heterozygous mutation of SALL1 and a new homozygous mutation of PTPRQ in a Chinese family with Townes-Brocks syndrome and hearing loss. BMC Med Genomics. 2021;14(1):24.
Liberalesso PBN, Cordeiro ML, Karuta SCV, Koladicz KRJ, Nitsche A, Zeigelboim BS, et al. Phenotypic and genotypic aspects of Townes-Brock syndrome: case report of patient in southern Brazil with a new SALL1 hotspot region nonsense mutation. BMC Med Genet. 2017;18(1):125.
Bozal-Basterra L, Martin-Ruiz I, Pirone L, Liang Y, Sigurethsson JO, Gonzalez-Santamarta M, et al. Truncated SALL1 impedes primary cilia function in Townes-Brocks Syndrome. Am J Hum Genet. 2018;102(2):249–65.
Piro E, Serra G, Giuffrè M, Schierz IAM, Corsello G. 2q13 microdeletion syndrome: report on a newborn with additional features expanding the phenotype. Clin Case Rep. 2021;9:e04289.
Serra G, Giambrone C, Antona V, Cardella F, Carta M, Cimador M, et al. Congenital hypopituitarism and multiple midline defects in a newborn with non-familial Cat Eye syndrome. Ital J Pediatr. 2022;48(1):170.
Infant developmental profile of Crisponi syndrome due to compound heterozygosity for CRLF1 deletion. Schierz Ingrid Anne Mandy, Serra Gregorio, Antona Vincenzo, Persico Ivana, Corsello Giovanni, Piro Ettore. Clinical Dysmorphology. Volume 29, Issue 3, Pages 141–143. 1 July 2020.
Cardamone S, Creighton S. A gynaecologic perspective on cloacal malformations. Curr Opin Obstet Gynecol. 2015;27(5):345–52.
Wood RJ, Levitt MA. Anorectal Malformations. Clin Colon Rectal Surg. 2018;31(2):61–70.
Mahmud AA, Khan N, Islam MS, Islam S, Bari MS, Das SC, et al. Anorectal malformations and Associated anomalies in Children. Mymensingh Med J. 2021;30(1):62–8.
Jun LH, Jacobsen A, Rai R, Case Report. A Case Series of Rare High-Type Anorectal malformations with Perineal Fistula: beware of urethral involvement. Front Surg. 2021;8:693587.
Farquhar C. Avoiding multiple pregnancies in assisted reproductive technologies: transferring one embryo at a time should be the norm. Fertil Steril. 2020;114(4):671–2.
Yildirim E. Spontaneous triplet pregnancy and trap sequence, case report. BMC Pregnancy Childbirth. 2019;19(1):328.
Miller EM, Hopkin R, Bao L, Ware SM. Implications for genotype-phenotype predictions in Townes-Brocks syndrome: case report of a novel SALL1 deletion and review of the literature. Am J Med Genet A. 2012;158A(3):533–40.
Piro E, Serra G, Antona V, Giuffre M, Giorgio E, Sirchia F, et al. Novel LRPPRC compound heterozygous mutation in a child with early-onset Leigh syndrome french-canadian type: case report of an Italian patient. Ital J Pediatr. 2020;46(1):140.
Serra G, Antona V, Schierz M, Vecchio D, Piro E, Corsello G. Esophageal atresia and Beckwith-Wiedemann syndrome in one of the naturally conceived discordant newborn twins: first report. Clin Case Rep. 2018;6(2):399–401.
Innoceta AM, Olivucci G, Parmeggiani G, Scarano E, Pragliola A, Graziano C. Chromosomal microarray analysis identifies a novel SALL1 deletion, Supporting the Association of Haploinsufficiency with a mild phenotype of Townes-Brocks Syndrome. Genes (Basel). 2023;14(2).
Surka WS, Kohlhase J, Neunert CE, Schneider DS, Proud VK. Unique family with Townes-Brocks syndrome, SALL1 mutation, and cardiac defects. Am J Med Genet. 2001;102(3):250–7.
Acknowledgements
Protein structure prediction assistance was provided by Longguang Jiang, PhD (Fuzhou University, China). We appreciate the involvement of all the participants.
Funding
This study was supported by the General Project of Natural Science Foundation of Shandong Province (grant number: ZR2020MH073) and Science and Technology Plan Project of Jinan (grant number: 202328068) .
Author information
Authors and Affiliations
Contributions
YC conducted data management and writing - original draft preparation. YY performed a follow-up telephone call. FS performed the surgical procedure. WZ was responsible for gene sequencing. ZZ completed the protein structure prediction. The completion of data integration by YW facilitated the writing. WH supervised and writing-reviewed and edited. All authors approved the final Manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Shandong Provincial Hospital Affiliated to Shandong First Medical University Institutional Ethics Committee (SWYX: NO.2023 − 386). Informed consent was obtained from all the patients and their families.
Consent for publication
The consent for publication of clinical description and photo was acquired from patients or patients’ parents.
Competing interests
The authors declare no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chi, Y., Yao, Y., Sun, F. et al. A novel SALL1 C757T mutation in a Chinese family causes a rare disease --Townes-Brocks syndrome. Ital J Pediatr 50, 121 (2024). https://doi.org/10.1186/s13052-024-01691-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-024-01691-0