
Abstract
Background
The triad of a presacral mass, sacral agenesis and an anorectal anomaly constitutes the rare Currarino syndrome (CS), which is caused by dorsal–ventral patterning defects during embryonic development. The major causative CS gene is MNX1, encoding a homeobox protein.
Main body
In the majority of patients, CS occurs as an autosomal dominant trait; however, a female predominance observed, implies that CS may underlie an additional mode(s) of inheritance. Often, the diagnosis of CS is established solely by clinical findings, impacting a detailed analysis of the disease. Our combined data, evaluating more than 60 studies reporting patients with CS-associated mutations, revealed a slightly higher incidence rate in females with a female-to-male ratio of 1.39:1. Overall, MNX1 mutation analysis was successful in only 57.4% of all CS patients investigated, with no mutation detected in 7.7% of the familial and 68% of the sporadic patients. Our studies failed to detect the presence of an expressed MNX1 isoform that might explain at least some of these mutation-negative cases.
Conclusion
Aside from MNX1, other genes or regulatory regions may contribute to CS and we discuss several cytogenetic studies and whole-exome sequencing data that have implicated further loci/genes in its etiology.
Similar content being viewed by others


Background
The first report of the syndrome has been published by Roger L. J. Kennedy in 1926 [1]. However, the description of the triad as a unique entity was defined by Guido Currarino and colleagues in 1981 [2] and hence, the term “Currarino syndrome” (CS, OMIM #176450) was coined [3]. Yates and coworkers were the first to describe an autosomal dominant mode of inheritance [4].
Three main features are characteristics of CS: (1) anterior sacral bone defects, known as sacral scimitar or sickle-shaped sacrum, or a complete sacral agenesis below S2; (2) anorectal malformation (often clincically present as chronic constipation); (3) a presacral mass, representing an anterior meningocele, a teratoma or an enteric cyst or any combination of these [2, 5,6,7]. Teratomas in CS are often benign however, a malignant transformation has been observed, even in childhood [6, 8, 9]. There is evidence that the risk of malignant transformation of sacrococcygeal teratoma and of presacral teratoma in CS is different despite similar position and appearance. Dirix et al. have analyzed 205 sacrococcygeal teratoma and 16 CS patients in a retrospective study [10]. Patients with sacrococcygeal teratoma had a lower malignancy-free survival than patients with a presacral teratoma associated with CS (58% versus 100% after two years). However, reliable biomarker that predict the risk of malignancy are lacking. Some authors suggested removal of the tumor, even in asymptomatic patients [11]. In 2008, Crétolle et al. suggested, that neural tube defects should also be considered as a fourth major clinical feature of CS [12]. These defects comprise tethered cord, fatty, thickened filum and/or spinal lipoma and neuroenteric cysts [12, 13].
As a further feature in association with CS, Müllerian duct anomalies (e.g. bicornuate uterus, septate vagina, agenesis or hypoplasia of uterus and vagina) have been found in up to 15% of cases [13, 14]. Additional genitourinary findings in CS comprise horseshoe or duplex kidney, vesicoureteral reflux, neurogenic bladder, recurrent urinary tract infections, urinary incontinence and hydronephrosis [13]. Developmental delay may also be present and is a clue to a cytogenetic anomaly involving 7q36.3 [12]. As CS shows variable expressivity, patients may either be recognized with the complete spectrum or only components of the four major features. Even complete asymptomatic mutation carriers have been reported [13]. Mutations affecting the MNX1 gene on chromosome 7q36.3 [14], encoding the motor neuron and pancreas homeobox protein 1, have been detected in most of the familiar cases and in about one third of sporadic CS patients [13, 16, 17].
In this review, we focus on the current knowledge of CS etiology and the genetic findings in the context of this rare disease.
Main text
Penetrance and prevalence of CS
The clinical phenotype of CS is extremely variable and the classic triad often incompletely present, even within the same family. Even patients with causative heterozygous MNX1 mutations, may present with only one or two classical features and moreover, chronic constipation since early childhood is often noted as the sole clinical symptom [9, 17,18,19,20,21]. Various associated malformations have also been observed, and these were found in such as 84.5% of cases from a series of 45 CS patients, where 23 patients were MNX1 mutation carriers [22].
About 80% of CS cases are at least clinically diagnosed before 16 years of age [7, 23, 24]. However, due to the extensive phenotypic variation, a delayed clinical diagnosis of CS, first revealed in the adult, has been frequently reported [25,26,27,28,29,30]. Even if mutational analysis cannot identify a pathogenic MNX1 variant, the diagnosis of CS can be established clinically. However, the detection of a pathogenic MNX1 mutation is helpful, especially in case of an atypical clinical presentation confirmation of the diagnosis by detection of a pathogenic MNX1 variant would be most helpful.
Many mutation-positive individuals have been regarded as asymptomatic and, as mentioned above, constipation is often noted as the sole finding in relatives of CS patients [31,32,33,34]. Other patients showed only chronic intestinal pseudo-obstruction or müllerian duct anomalies, features that can also be associated with the complete CS spectrum [12, 35]. However, evidence of additional sacral anomalies is often noted on X-ray, CT or MRI in these patients [13, 16, 17]. As an imaging procedure for affected patients and their parents, we recommend first spinal and pelvic ultrasound, then sacral x-ray, and in case of questionable or incomplete findings, MRI. Reviewing the clinical features present in 205 CS patients, Lynch et al. observed only two individuals who were mutation carriers (1%) with no signs of the disease on X-ray [13]. As can be seen from the extensive literature review (Additional file 1: Table S2), a total of 25 cases with a proven MNX1 mutation turned out to be negative for any CS feature following extensive physical examination. However, in two patients, constipation was noted and radiological examinations had not been performed in eight of these individuals. Hence, one must carefully differentiate between asymptomatic and unaffected CS relatives. In any situation, we recommend, that the patient's parents also undergo genetic testing in order to identify mutation carriers among parents. Together with the standardized approach of screening parents for signs of CS, it is possible to obtain true figures of prevalence and penetrance.
The variability of CS has so far prevented an estimation of its true prevalence [15, 31]. However, the OrphaNet entry (ORPHA 1552) for rare diseases lists a prevalence of 1–9/100,000 for CS without referring to the original source.
In several studies, no difference between male and female frequency has been noted [17, 22, 36, 37], whereas other reports listed a higher frequency of CS females [3, 12, 33, 38, 39]. Haga et al. estimated a CS female-to-male ratio in adult cases of 6:1 and of 2:1 in pediatric cases, which may rely, at least in part, on the delayed diagnosis of mildly affected patients [40]. To obtain a more reliable value, we listed the sex of all mutation-positive CS cases and their tested family members from our literature survey (Additional file 1: Table S1). In total, information was given for 308 patients (179 [58.1%] CS females and 129 [41.9%] CS males), resulting in a female-to-male ratio of 1.39:1. This slightly higher incidence rate in females may reflect their gynecological and urological symptoms, possibly representing an ascertainment bias [13]. On the other hand, female predominance would not be expected if CS is exclusively inherited in an autosomal dominant fashion.
Genotype–phenotype correlation
The various different mutations found within the MNX1 gene lead to comparable phenotypes. However, there is not only a wide variability among related patients but also between unrelated patients sharing the same mutation [41]. Consequently, no obvious genotype–phenotype correlation has been noted yet. As suggested by various authors [11, 16, 17, 40], this might be explained by the effects of other genes/proteins modifying MNX1 expression and/or protein function. On the other hand, as among MNX1 mutation carriers, haploinsufficiency is the most likely cause of the disease [16], the degree of transcriptional activity of the non-mutated MNX1 allele may vary between CS patients. Also, sequence variants in the long non-coding MNX1 antisense RNAs, MNX1-AS1 and MNX1-AS2 [42, 43], or differences in their expression, may alter the phenotypic outcome.
Recently, Costanzo et al. analyzed the data of 45 CS patients and their findings indicated a correlation with a more severe phenotype when a MNX1 mutation is present [22]. These authors concluded that MNX1 seems the main gene responsible for the expression and the severity of the CS triad, while the associated anomalies appear to be mainly determined by other genes.
Pathogenic concept
Currarino et al. [2] were the first to speculate, that the features of CS rather represent a syndrome complex and share the same embryonic pathway. In human tailbud, the neural tube, notochord, somites and hindgut arise from pluripotent epiblast cells at around Carnegie stage 12 (CS12) corresponding to an assigned embryonic age of 29–31 days [44]. A primary defect in the caudal notochord may prevent some of these cells to migrate from the primitive node. As proposed by several authors, this will lead to a defect at the caudal most aspect of notochord or splitting of notochord and the formation of a fistula between the gut ventrally and the neural elements dorsally or vice versa [2, 15, 16, 45]. Gupta et al. also suggested that failure of some epiblast cells to migrate from primitive node would lead to remnants at the primitive streak, which may persist in the sacrococcygeal region as a teratoma [45].
Experimental evidence for these theories emerged from the detection of MNX1 in the tailbud of Xenopus laevis embryos at tadpole stages [46, 47]. However, human embryonic expression studies were less convincing. Ross and colleagues detected MNX1 expression in the sacral region at CS19 (embryonic age 45–47 days) but found a pronounced expression in the anterior horn regions along the spinal cord [47]. A more extended analysis was reported by Hagan et al. [16], who investigated embryonic MNX1 expression from CS12 to CS21, corresponding to embryonic age 29–55 days [44]. Here, the authors were consistently unable to detect MNX1 expression in tail bud notochord from CS12 onward or in more anterior regions of the notochord. MNX1 expression was observed in developing human foregut (pharynx, esophagus, stomach), in the basal plate of hindbrain and in developing motor neurons throughout the length of the developing spinal cord. The latter observation supported the findings in Xenopus laevis that MNX1 is a marker for motor neurons [46] and its determined role in cell proliferation and apoptosis in Drosophila neuronal cells [48]. MNX1 has also an essential role in motor neuron differentiation and in the consolidation of motor neuron identity [49], albeit CS patients do not show any motor neuron dysfunction.
Hagan et al. detected MNX1 expression also in early human pancreatic development, at the time before fusion of the dorsal and ventral buds [16]. Contrary to mouse, MNX1 expression was here observed throughout CS12-21, with no obvious differences in intensity between the dorsal and ventral buds after CS15. In mouse, transient Mnx1 expression was seen in dorsal and ventral pancreatic buds from embryonic day (e) 8 to e9-e10 and in mature β-cells of the islets of Langerhans [50, 51]. These observations were verified by Sherwood and colleagues, who revealed, that mouse Mnx1 is expressed throughout the anterior–posterior axis of the endoderm and here, forms a dorsal–ventral gradient at e9.5 [52]. Li and Edlund have also shown that a tight temporal regulation of MNX1 expression is necessary, and that a complete loss of expression resulted in a block of the initiation of the dorsal pancreatic program, while temporally extended MNX1 expression led to a complete impairment of pancreatic development [53]. Mice lacking MNX1 show a selective agenesis of the dorsal pancreas and abnormal islets of Langerhans, whereas a pancreatic agenesis has yet never been observed in CS patients. It has also been speculated that defects in MNX1 may be causally related to the 200–250 times higher prevalence of sacral agenesis in diabetic pregnancies [54, 55] or in cases with failure of pancreatic development [56]. However, in no such case, a mutant MNX1 gene has been identified yet.
The main features in CS patients are frequently associated with urogenital and renal anomalies, e.g. hydronephrosis, horseshoe kidneys, congenital single kidney, vesico-uretral reflux, bicornuate uterus or subseptate vagina [2, 25, 57]. Specific MNX1 caudal mesoderm requirements have so far been detected only in zebrafish. Here, expression of Mnx2b, one of three zebrafish Mnx genes, is restricted to the nephric mesoderm during somite stages and its knockdown led to proximal pronephric tubule dilatation and impaired pronephric excretion [58]. A requirement for Mnx1 in the developing kidney and urogenital system has yet not been observed in mutant mice. However, Mnx1 expression has been found in the mouse genitourinary system at E10.5 [59] and in mouse lower urinary tract [60]. All these Mnx1 gene activities observed in non-human systems should be further investigated and might help to better understand the caudal mesoderm specific agenesis in CS patients.
Further studies also revealed that MNX1 acts in the regulation of progenitor cells into mature hematopoietic cells [61, 62]. The finding of increased MNX1 levels in patients with acute leukemia and colorectal or hepatocellular cancers suggests that MNX1 protein harbors an additional tumor suppressor function [12, 63,64,65].
Although one might suspect genetic factors that define specific subtypes of CS, those factors have not been identified yet. Accordingly, there has no phenotype-genotype correlation been established so far.
Taken together, all these observations demonstrate considerable differences in human and murine development. Factual, none of these models reflects the human situation in terms of timing and intensity of MNX1 expression and the induced mutant phenotype.
Mouse models for CS
As mentioned above, Mnx1 knockout mice described so far showed anomalies in pancreatic and motor-neuron development but did not reflect any feature of CS, in particular disruption of sacral or hindgut development. Moreover, Mnx1−/+ heterozygote mice showed no discernible phenotype [49,50,51, 66]. In view of these interspecies differences Martin Catala proposed, that either mouse Mnx1 protein has different functions compared with its human ortholog or that human MNX1 mutations lead to a gain of function instead of being true null mutations [55].
A sole mouse model, resembling CS, was established by Liu and coworkers in 2003 [67]. Here, administration of the highly lipophilic, long-acting synthetic aromatic retinoid etretinate, a potent teratogen affecting the tail bud, on gestation day (E) 9, proved to be capable to induce a CS phenotype. All treated embryos exhibited a presacral mass (which turned out to be an anterior sacral meningocele) and a sacral defect, with anorectal malformations observed in about ¾ of the fetuses. These authors provided evidence, that anorectal malformations, anterior sacral meningocele and sacral defects share the same embryonic pathway. However, the gene(s) targeted by etretinate remain(s) to be elucidated.
Pcsk5
Szumska and colleagues identified an ethylnitrosourea-induced recessive p.Cys470Arg mutation in the proprotein convertase subtilisin/kexin type 5 (Pcsk5) gene [68]. This mutation induced a pleiotropic effect resulting in cardiac, anorectal, tracheoesophageal and anteroposterior patterning defects, as well as exomphalos, hindlimb hypoplasia, a presacral mass, renal and palatal agenesis, and pulmonary hypoplasia. Since these phenotypic features resembled those seen in VATER/VACTERL association (OMIM% 192350), caudal regression syndrome (OMIM #600145) and CS, the authors coined the term Vcc for this Pcsk5 mutation. It could also been shown, that Vcc caused ectopic Mnx1 expression in the ventral aspect of the murine tail bud and a reduced expression of several Hox genes [68].
Gdf11
As growth/differentiation factor 11 (Gdf11) was shown to be cleaved and activated by Pcsk5, Szumska et al. also investigated Gdf11-deficient mouse embryos [68]. At E15.5 all embryos showed an absent tail, palatal and renal agenesis and abnormal anorectal anatomy. Moreover, these embryos had either an intra-abdominal or an extra-abdominal mass arising from the spinal cord and some showed exomphalos. Hence, Gdf11-deficiency phenocopies the Pcsk5 Vcc variant to a large extent. In this context, Tsuda et al. administered teratogenic doses of all-trans retinoic acid at E9 to pregnant mice [69]. This treatment inhibited Pcsk5 and Gdf11 expression in the hindgut at E12 and E18 and also resulted in a phenotype resembling caudal regression syndrome and CS. Most of these embryos showed anorectal anomalies and a short tail, as well as sacral malformations, tethered spinal cords and presacral masses.
T-box transcription factor brachyury (T)
The idea of the T- (Brachyury) gene as a candidate for sacral agenesis emerged from the observation of tail anomalies in the T-deficient mouse, resembling human spinal defects [70]. As homozygous T−/− mice die between E9.5 and E10.5 with an overall loss of mesoderm, Pennimpede et al. used in vivo knockdown experiments [71]. Here, at E12.5, the embryos displayed axial skeletal defects and urorectal malformations resembling murine uro-rectal-caudal syndrome and human caudal regression syndrome phenotypes.
MNX1, the major human CS disease gene
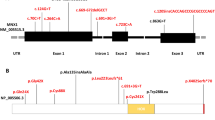
The locus for CS was initially mapped to chromosome 7q36 [15] and here, the MNX1 gene (earlier termed HLXB9), encoding motor neuron and pancreas homeobox protein 1 (MNX1, earlier termed HB9), was first described by Harrison et al. [62]. The gene locates to 7q36.3, consists of three exons and, according to UniProtKB entry P50219, the resultant protein comprises 401 amino acids. The use of an alternate 5′-exon may result in a shorter isoform 2 (189 residues), displaying a distinct aminoterminus (see below). MNX1 functions through sequence-specific DNA binding and transcription factor activity, with its homeodomain encoded by amino acids 241–300 (PROSITE annotation). A further significant feature of MNX1 is a polyalanine stretch of variable length (12–18 residues) starting at amino acid 121 (see below).
Around 120 different heterozygous pathogenic MNX1 single nucleotide variants have yet been described, whereas a large deletion or a complex rearrangement, involving chromosome 7q36, is less commonly involved in the formation of CS. Here, we performed an intensive literature survey for reports describing CS cases and focused on independent patients with a proven genetic anomaly. Also, patients with apparently multiple listing were excluded (Additional file 1: Table S1). This search revealed, that MNX1 gene mutations or complete gene deletions have been observed in a total of 168 independent CS cases yet. Of these, 108 (64.3%) were found in multiple affected families, whereas 55 (32.7%) represent sporadic cases. For five mutation-positive cases (3.0%), the sporadic or familial occurrence was not mentioned. Altogether, these studies investigated 296 unrelated cases of CS, including 117 familial and 172 sporadic cases; for seven patients no status was given. No mutation was found in nine (7.7%) familial and 117 (68%) sporadic cases. Hence, overall mutation analysis was successful in only 57.4% of all CS patients investigated. Consequently, aside from pathogenic MNX1 mutations residing in introns, promoter or other transcriptional relevant elements that were not detectable by the methods applied, one might expect further genes to be involved in the etiology of CS.
So far however, other chromosomal or gene anomalies were identified in only five patients (see below). Here, a CS phenotype was either associated with partial trisomy of chromosomes 13q and 20p [72], a duplication 3q26.32-q27.2 [73], a duplication 3q26.31-q29 with deletion 9p24.3-9p23 [74], a functional disomy Xp with deletion Xq13.2-q28 and duplication 3q25.33-q29 [75], or excessive heterochromatin in chromosome 9 (9qh) [76].
Homozygous MNX1 mutations
Three different MNX1 point mutations, present in the homozygous state, have been reported yet. Here, two homozygous germline MNX1 mutations (p.Phe248Leu and p.Phe272Leu) were identified in two unrelated consanguineous patients with permanent neonatal diabetes mellitus (PNDM; OMIM #606176) [77, 78]. Both patients showed severe intrauterine growth retardation and one female (p.Phe248Leu) died at early age. Whereas p.Phe272Leu was associated with isolated PNDM without any signs of CS, the female harboring the p.Phe248Leu variant also showed sacral agenesis and high imperforate anus, hence cardinal features of CS, as well as severe neurological complications and hypoplastic lungs. The heterozygote parents of the patient with the p.Phe248Leu showed no phenotype. Functional characterization of the p.Phe248Leu variant revealed a normal localization of the mutant protein in the nucleus but a lack of MNX1 phosphorylation [79]. The phenotypic differences caused by these two homozygous mutations might be due to their type and/or location but again reflect the striking variability caused by MNX1 mutations. As PNDM is not a feature of CS and a normal pancreas is present in heterozygous mice [50], one may speculate, that one functional copy of the MNX1 gene is at least sufficient for normal human pancreatic development and glucose homeostasis.
A third homozygous amino acid substitution, affecting the initiator codon (p.Met1Thr), was reported in a fetus, also born to consanguineous parents [80]. Whereas the heterozygote parents showed no phenotype, the fetus died within minutes of birth with intrauterine growth retardation, cleft palate, and the CS features anal imperforation, scrotal agenesis and agenesis of the lumbar and sacral spine and spinal cord. Three further newborns of these parents also died with a CS phenotype. The authors concluded, that p.Met1Thr represents a hypomorphic variant, being tolerated in heterozygous carriers but causing severe defects in the homozygous state [80].
Noteworthy, the probability of being loss-of-function intolerant (pLI) score of MNX1 is 0.79 (https://gnomad.broadinstitute.org/) indicating haploinsufficiency as a potential mechanism. Although the figure does not meet the generally accepted threshold of > 0.9 for intolerant genes, the lower value of MNX1 might be explained by the low numbers on which the pLI score of MNX1 is based (indicated by the wide 90% confidence interval of the observed/expected score of 0.04–0.5). In regard of homozygous variants, in gnomAD no truncating variant is reported homozygously.
MNX1 polyalanine polymorphism
A part of MNX1 exon 1 encodes a polyalanine stretch, first described to comprise 16 consecutive alanine codons [62]. This coding region however, includes a variable CGC (or GCC) repeat, with e.g. (CGC or GCC)8 resulting in a total length of 13 alanine residues, or (CGC or GCC)12 finally leading to 17 alanines. Belloni et al. determined that the (CGC)11 allele was the most common (90.23%) in the general Caucasian population [3]. The frequencies of other alleles observed by this group was: 0.6% (CGC)8, 7.47% (CGC)9 and 1.7% (CGC)12. Holm et al. found comparable values in a Norwegian CS family and unaffected relatives and reported a further rare allele, (CGC)7 [81]. Significant differences in allele distribution were reported in healthy Chinese individuals, where the frequency of (CGC)11 was “only” 50% [21]. Here, other alleles showed frequencies of 0.4% (CGC)8, 33% (CGC)9, and 16.2% (CGC)12, respectively. These authors also found a (CGC)13 allele, being present on 0.4% of the chromosomes, analyzed from Chinese individuals. Similar frequencies were observed in the Japanese and Yoruba population [21]. Hence, the length of the polyalanine stretch can at least vary from 12 to 18 residues and this length variation has yet not been associated with any phenotype [3, 12, 21, 33]. In that context, the finding of a (CGC)12 allele was neither a novel finding nor seems to be responsible for CS in a Taiwanese female reported by Lin et al. [82].
To our knowledge, only one CS patient with a (CGC)n contraction has been reported [12]. In this case, the genotype (CGC)5/(CGC)11 was observed and the heterozygous (CGC)5 allele, never found in controls, was considered responsible for the disease. However, one may also speculate about a critical threshold beyond which a triplet expansion in MNX1 is pathologic. Disease-causing polyalanine expansion mutations have yet been identified in nine genes (HOXA13, HOXD13, SOX3, FOXL2, PHOXB2, RUNX2, ARX, ZIC2, and PABPN1), all except PABPN1 encoding transcription factors [83, 84]. As outlined in these reviews, polyalanine expansions cause a loss-of-function and/or mild gain-of function via promotion of misfolding and formation of aggregates of the resultant protein. As such mutant alleles have not been identified in CS patients yet, one may assume a more severe pathological impact of MNX1 polyalanine expansions on early developmental processes, thereby leading to early embryonic death.
Expression of an alternative MNX1 transcript?
As mutation analysis failed to detect MNX1 mutations in about 43% of all CS patients investigated thus far, the presence of an expressed isoform might explain at least some of these MNX1 mutation-negative cases. Indeed, UniProtKB lists a second predicted shorter MNX1 isoform (P50219-2), that lacks the N-terminus, present in the 401 amino acids long canonical sequence (P50219-1). This transcript should arise through the use of an alternative exon 1, spliced to the common exons 2 and 3. As this exon 1 encodes a different 19 amino acid long N-terminus, not investigated by the common methods applied, mutations in this stretch may have been missed.
To address this question, we have searched for the presence of the shorter isoform in cDNA samples obtained from blood, saliva and hippocampal tissue. Primers were designed to cover part of the novel exon 1 (5′-CGCTGCCCTCCTCTGGAAGG-3′) and to span the canonical exons 2 and 3 (5′-AATCTTCACCTGGGTCTCGGTGAG-3′). However, these analyses failed to detect mRNA isoform P5019-2 in multiple assays (data not shown), indicating that this mRNA is not substantially expressed at least in these tissues. Hence, it seems to be unlikely, that mutations in this predicted shorter MNX1 isoform contribute to CS.
Analysis of further CS candidate genes
In patients with CS, no mutation in the genes encoding PCSK5 or GDF11 has been reported and, to our knowledge, no study analyzing these genes in a CS cohort, negative for MNX1 mutations has yet been performed. However, some other genes, either affected by a deletion of 7q36, or with known phenotypic consequences in mouse models have been investigated.
SHH
Sonic hedgehog (SHH) is an endoderm-derived signaling molecule. Although mutant mice with various defects in SHH signaling only displayed a spectrum of anorectal malformations [85], it might also be involved in caudal regression. As the SHH gene locates to chromosome 7q36.3, the critical region initially linked to CS [15], Seri et al. investigated SHH in a large CS multiplex family with eight affected members, seven further sporadic CS cases and an additional 15 patients with anorectal malformations and sacral hypodevelopment [86]. No mutation was observed, making an involvement of SHH in these diseases rather unlikely. This assumption has been corroborated by Coutton et al. [87]. These authors reported a patient with CS and a microform of holoprosencephaly, who carried a 2.7 Mb deletion in 7q36.3 without affecting the SHH gene.
T (Brachyury)
A study of 28 patients with sacral agenesis with anorectal atresia revealed, that the coding region of the T-gene is highly polymorphic [88]. However, Papapetrou et al. detected one rare heterozygous p.Ala338Val variant in a female patient that was transmitted by an apparently unaffected mother [88]. This variant was also detected in three further heterozygous patients, presenting with either sacral agenesis, Klippel-Feil syndrome or multiple cervical and thoracic vertebral malformations [89]. Again, the variant had been inherited from a clinically unaffected parent in all three cases. The p.Ala338Val substitution has been deposited with no. rs117097130 in dbSNP (Build 146) and the EXAC_0.3 database lists its occurrence with a minor allele frequency of 0.004835 (A) in a sample of 121.412 individuals. Hence, based on current knowledge, its contribution to the associated phenotypes may not be excluded.
This assumption is supported by another study presenting evidence, that homozygous T-mutations cause a syndrome consisting of sacral agenesis, abnormal ossification of the vertebral bodies and a persistent notochordal canal [90]. The authors identified a homozygous p.His171Arg mutation in three consanguineous families of the same ethnic background. The mutant protein showed reduced DNA binding activity and a decrease in activation activity of T binding sites. As outlined by Postma et al., this observation suggests, that screening for the ossification of the vertebrae is warranted in patients with sacral agenesis to evaluate the possible causal involvement of the T gene [90]. Further support arises from the finding of a multiplex consanguineous Saudi family with four patients born with isolated myelomeningocele [91]. The here identified novel p.Gly156Cys T variant was found in the homozygous state in all four patients but also in three apparently non-penetrant siblings, although MRI/CT examination performed in one of them revealed the presence of spina bifida oculta. These observations allow for potentially reduced penetrance and support the assumption of an involvement of the T gene in the etiology of CS.
Cytogenetic findings and copy number variations
Chromosomal anomalies, not involving region 7q36, have rarely been associated with CS or a CS-like phenotype. In one patient with partial sacral agenesis, Anderson-Shotwell and Wilson found a de novo deletion of chromosome 18p [92]. Nagai et al. reported a CS patient with partial trisomy of chromosomes 13q and 20p [72]. Analyzing six patients with pure interstitial or terminal 6q deletions, Titomanlio et al. mapped a locus for sacral/anorectal malformations to a 0.3 Mb critical region at 6q25.3 [93]. With the four genes SYNJ2, SERAC1, GTF2H5 and TULP4, this region harbors no obvious candidate for CS. However, the MNX1 gene had not been analyzed in all these patients. In a MNX1 mutation negative patient, Bevanda et al. detected an excess of heterochromatin in chromosome 9 (9qh) in the patient and his asymptomatic father [76]. Heterochromatin constitutes highly compacted chromatin with regions of silenced DNA and contains hypoacetylated histones and high levels of DNA methylation. Hence, a more severe imbalance in these DNA modifications might have contributed to CS in the patient compared to his father.
In a more recent report, Dworschak et al. detected a pure 7.9 Mb de novo duplication 3q26.32-q27.2 in an MNX1 mutation-negative CS patient [73]. This male patient not only showed the classical CS triad with anal atresia, presacral teratoma and hemisacrum but presented additional features (developmental delay, CNS malformations, facial dysmorphism), frequently detected in duplication 3q syndrome. Aljeaid and coworkers lately described a CS female patient with additional congenital heart defects, generalized hypotonia, global developmental delay, dysmorphic facial features and dysplastic ears [74]. These authors also detected a de novo terminal duplication 3q. Here, the duplicated region turned out to be larger, comprising 24.6 Mb from 3q26.31-q29, and the patient also carried a de novo 12.5 Mb terminal deletion from 9p24.3-9p23. Moreover, Peterson et al. described a female with a combination of functional disomy Xp, deletion Xq13.2-q28 and duplication 3q25.33-q29 [75]. This patient presented with features of CS (sacral teratoma and neurogenic bladder) as well as primary ovarian insufficiency, sensorineural hearing loss and intellectual disability.
All these latter observations suggest, that a dosis effect of (a) gene(s) in dup(3q) may explain the phenotype in some patients negative for MNX1 mutations or 7q36 deletions involving the MNX1 locus.
Whole exome sequencing data
Whole exome sequencing has been performed in three patients with sporadic CS and their parents [94]. Here, Holm et al. also analyzed a custom-made region on chromosome 7 to detect variants in regulatory elements in non-coding regions around the MNX1 gene and additionally investigated biopsy material from the removed presacral mass in one case [94]. No sequence variant was shared between the three patients in non-coding regions surrounding MNX1 or in other transcription factor genes, known to interact with MNX1. However, the authors prioritized three variants that might be of relevance to CS [94]. One patient carried a de novo p.Val126Ile variant in ETV3L, encoding the transcription factor ETS variant 3 like, implicated in tissue development and the equilibrium between proliferation and differentiation [95]. The second patient also carried one de novo variant, affecting the acceptor splice site of exon 11 of the NCAPD3 gene, coding for condensin-2 complex subunit D3. This protein is a regulatory subunit of the condensin-2 complex, which establishes mitotic chromosome architecture [96]. In the third CS patient, a p.Arg55Leu variant was identified in ARID5A. Here, the encoded protein, AT-rich interactive domain-containing protein 5A, is a sequence-specific transcription factor with recognized promoter targeting functions and important roles in development and differentiation [97]. This variant however, was transmitted from the unaffected mother.
Most recently, Han et al. found four likely pathogenic de novo variants in three sporadic CS patients [39]. A CDH2 missense variant (p.Arg151Ser) was detected in one of these patients. This gene encodes the calcium-dependent cell adhesion protein cadherin-2, a cell surface transmembrane protein with a vital structural and functional role in the intercalated disc, involved in cell-to-cell adhesion. So far, mutations in this gene have been associated with arrhythmogenic right ventricular cardiomyopathy [98]. Another patient carried a 4 bp deletion (p.Ile541Ilefs12) leading to a frameshift variant of the ITIH2 gene. ITIH2 codes for the inter-alpha-trypsin inhibitor heavy chain H2, a protein from a family of structurally related plasma serine protease inhibitors playing a key role in extra-cellular matrix biology and tumor progression [99]. In a third patient, two novel HOXB4 variants (p.Lys16Asn) and TLE4 (p.Ser650Leu) gene were found. Homeobox protein Hox-B4 is a sequence-specific transcription factor with diverse roles in embryonic development and the regulation of adult stem cells. As outlined in Morgan et al., it intriguingly can act in opposite ways when expressed by different cells, promoting the proliferation of stem cells whilst activating the apoptotic pathway in some embryonic structures [100]. The transducin-like enhancer protein 4, encoded by TLE4, binds to various transcription factors and acts as a transcriptional corepressor in regulating e.g. Wnt, Notch and transforming growth factor-ß signaling [101].
At least some of these genes represent apparently interesting candidates for an involvement in CS etiology, however, their involvement remains to be elucidated.
Regulatory sequences
Conserved non-coding sequences (CNEs) are known to act as expression regulators and may function either as an enhancer or repressor of transcription, depending on the regulatory protein(s) bound to it [102]. In that context, Woolfe and colleagues have tested various CNEs for enhancer activity in vertebrate development [103]. Three of these CNEs, located distal or proximal to MNX1, indeed directed gene expression to skin/enveloping layer, skeletal muscle or spinal cord in zebrafish embryos. These findings suggest that a sequence variation in one of these CNEs may contribute to CS in a so far unknown way.
Conclusion
The yet suggested testing strategy for CS only includes MNX1 sequence- and MLPA-analysis [104]. In contrast we suggest whole-exome sequencing and molecular karyotyping (CNV analysis) in order to identify possible further candidate genes involved in the formation of CS. As CS exhibits an extreme variable expressivity with many asymptomatic cases, it is often overlooked or misdiagnosed, a fact that yet opposes the chance to more precisely define its true incidence.
Availability of data and materials
Not applicable.
Abbreviations
- CNEs:
-
Conserved non-coding sequences
- CS:
-
Currarino syndrome
References
Kennedy RLJ. An unusual rectal polyp: anterior sacral meningocele. Surg Gynecol Obstet. 1926;43:803–4.
Currarino G, Coln D, Votteler T. Triad of anorectal, sacral, and presacral anomalies. Am J Roentgenol. 1981;137:395–8.
Belloni E, Martucciello G, Verderio D, et al. Involvement of the HLXB9 homeobox gene in Currarino syndrome. Am J Hum Genet. 2000;66:312–9.
Yates VD, Wilroy RS, Whitington I, Simmons JCH. Anterior sacral defects: an autosomal dominantly inherited condition. J Pediatr. 1983;102:239–42.
Kenefick JS. Hereditary sacral agenesis associated with presacral tumours. Br J Surg. 1973;60:271–4.
Ashcraft KW, Holder TM. Hereditary pre-sacral teratoma. J Pediatr Surg. 1974;9:691–7.
Kirks DR, Merten DF, Filston HC, Oakes WJ. The Currarino triad: complex of anorectal malformation, sacral bony defect, and presacral mass. Pediatr Radiol. 1984;14:220–5.
Ashcraft KW, Holder TM. Congenital anal stenosis with presacral teratoma: case reports. Ann Surg. 1965;162:1091–5.
Köchling J, Pistor G, Märzhäuser Brands S, Nasir R, Lanksch WR. The Currarino syndrome—Hereditary transmitted syndrome of anorectal, sacral and presacral anomalies. Case report and review of the literature. Eur J Pediatr Surg. 1996;6:114–9.
Dirix M, van Becelaere T, Berkenbosch L, et al. Malignant transformation in sacrococcygeal teratoma and in presacral teratoma associated with Currarino syndrome: a comparative study. J Pediatr Surg. 2015;50:462–4.
Rod J, Cretolle C, Faivre L, Jacquot C, Yacoub O, Ravasse P, Cheynel N, Sarnacki S. Malignant transformation of presacral mass in Currarino syndrome. Pediatr Blood Cancer. 2019;66:e27659.
Crétolle C, Pelet A, Sanlaville D, et al. Spectrum of HLXB9 gene mutations in Currarino syndrome and genotype-phenotype correlation. Hum Mutat. 2008;29:903–10.
Lynch SA, Wang Y, Strachan T, Burn J, Lindsay S. Autosomal dominant sacral agenesis: Currarino syndrome. J Med Genet. 2000;37:561–6.
Crétolle C, Zérah M, Jaubert F, Sarnacki S, Révillon Y, Lyonnet S, Nihoul-Fékété C. New clinical and therapeutic perspectives in Currarino syndrome (study of 29 cases). J Pediatr Surg. 2006;41:126–31.
Lynch SA, Bond PM, Copp AJ, et al. A gene for autosomal dominant sacral agenesis maps to the holoprosencephaly region at 7q36. Nat Genet. 1995;11:93–5.
Hagan DM, Ross AJ, Strachan T, et al. Mutation analysis and embryonic expression of the HLXB9 Currarino syndrome gene. Am J Hum Genet. 2000;66:1504–15.
Köchling J, Karbasiyan M, Reis A. Spectrum of mutations and genotype-phenotype analysis in Currarino syndrome. Eur J Hum Genet. 2001;9:599–605.
Samuel M, Hosie G, Holmes K. Currarino triad—Diagnostic dilemma and a combined surgical approach. J Pediatr Surg. 2000;35:1790–4.
Gereige RS, Frias JL. Is it more than just constipation? Pediatrics. 2002;109:961–5.
Martucciello G, Torre M, Belloni E, Lerone M, Pini Prato A, Cama A, Jasonni V. Currarino syndrome: proposal of a diagnostic and therapeutic protocol. J Pediatr Surg. 2004;39:1305–11.
Garcia-Barceló MM, So MT, Lau DK, et al. Population differences in the polyalanine domain and 6 new mutations in HLXB9 in patients with Currarino syndrome. Clin Chem. 2006;52:46–52.
Costanzo S, Spaccini L, Pio L, et al. Currarino syndrome: does the presence of a genetic anomaly correlate with a more severe phenotype? A multicentre study J Pediatr Surg. 2017;52:1591–6.
Riebel T, Maurer J, Teichgraber UK, Bassir C. The spectrum of imaging in Currarino triad. Eur Radiol. 1999;9:1348–53.
Elias P, Zizka J, Balicek P. Currarino triad: concurrent US and MRI diagnosis in the fetus and the mother. Prenat Diagn. 2002;22:1005–10.
Colapinto MN, Vowinckel EAM, Colapinto ND. Complete Currarino syndrome in an adult, presenting as a fecalith obstruction: report of a case. Can J Surg. 2003;46:303–6.
Crétolle C, Sarnacki S, Amiel J, et al. Currarino syndrome shown by prenatal onset ventriculomegaly and spinal dysraphism. Am J Med Genet A. 2007;143A:871–4.
Saberi H, Habibi Z, Adhami A. Currarino’s syndrome misinterpreted as Hirschsprung’s disease for 17 years: a case report. Case J. 2009;2:118.
Ciotti P, Mandich P, Bellone E, et al. Currarino syndrome with pelvic neuroendocrine tumor diagnosed by post-mortem genetic analysis of tissue specimens. Am J Med Genet A. 2011;155A:2750–3.
Kassir R, Kaczmarek D. A late-recognized Currarino syndrome in an adult revealed by an anal fistula. Int J Surg Case Rep. 2014;5:240–2.
Akay S, Battal B, Karaman B, Bozkurt Y. Complete Currarino syndrome recognized in adulthood. J Clin Imaging Sci. 2015;5:10.
Emans PJ, Kootstra G, Marcelis CLM, Beuls EAM, van Heurn LWE. The Currarino triad: the variable expression. J Pediatr Surg. 2005;40:1238–42.
Wang RY, Jones JR, Chen S, Rogers RC, Friez MJ, Schwartz CE, Graham JM Jr. A previously unreported mutation in a Currarino syndrome kindred. Am J Med Genet A. 2006;140A:1923–30.
Merello E, De Marco P, Ravegnani M, Riccipettitoni G, Cama A, Capra V. Novel MNX1 mutations and clinical analysis of familial and sporadic Currarino cases. Eur J Med Genet. 2013;56:648–54.
Duru S, Karabagli H, Turkoglu E, Erşahin Y. Currarino syndrome: report of five consecutive patients. Childs Nerv Syst. 2014;30:547–52.
Kim SH, Paek SH, Kim HY, Jung SU, Park KW. Currarino triad with Müllerian duct anomaly in mother and daughter without MNX1 gene mutation. Ann Surg Treat Res. 2016;90:49–52.
Riebel T, Köchling J, Scheer I, Oellinger J, Reis A. Currarino syndrome: Variability of imaging findings in 22 moleculargenetically identified (HLXB9 mutation) patients from five families. Rofo. 2004;176:564–9.
Monclair T, Lundar T, Smevik B, Holm I, Ørstavik KH. Currarino syndrome at Rikshospitalet 1961–2012. Tidsskr Nor Legeforen. 2013;22:2364–8.
Lee S, Kim EJ, Cho SI, et al. Spectrum of MNX1 pathogenic variants and associated clinical features in Korean patients with Currarino syndrome. Ann Lab Med. 2018;38:242–8.
Han L, Zhang Z, Wang H, Song H, Gao Q, Yan Y, Tao R, Xiao P, Li L, Jiang Q, Li Q. Novel MNX1 mutations and genotype-phenotype analysis of patients with Currarino syndrome. Orphanet J Rare Dis. 2020;15:155.
Haga Y, Cho H, Shinoda S, Masuzawa T. Recurrent meningitis associated with complete Currarino triad in an adult. Neurol Med Chir (Tokyo). 2003;43:505–8.
Garcia-Barceló MM, Lui VC, So MT, et al. MNX1 (HLXB9) mutations in Currarino patients. J Pediatr Surg. 2009;44:1892–8.
Kim T, Jeon YJ, Cui R, et al. Role of MYC-regulated long noncoding RNAs in cell cycle regulation and tumorigenesis. J Natl Cancer Inst. 2015;107:pii: dju505.
Long W, Rui C, Song X, et al. Dictinct expression profiles of IncRNAs between early-onset preeclampsia and preterm controls. Clin Chim Acta. 2016;463:193–9.
O’Rahilly R, Müller F. Developmental stages in human embryos: revised and new measurements. Cells Tissues Organs. 2010;192:73–84.
Gupta R, Sharma SB, Mathur P, Goyal RB. Variants of Currarino syndrome: embryological association and review of pertinent literature. Int J Embryol. 2014. https://doi.org/10.1155/2014/636375.
Saha MS, Miles RR, Grainger RM. Dorsal-ventral patterning during neural induction in Xenopus: assessment of spinal cord regionalization with xHB9, a marker for the motor neuron region. Dev Biol. 1997;187:209–23.
Ross AJ, Ruiz-Perez V, Wang Y, et al. A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet. 1998;20:358–61.
Miguel-Aliaga I, Thor S, Gould AP. Postmitotic specification of Drosophila insulinergic neurons from pioneer neurons. PLoS Biol. 2008;6:e58.
Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identitiy. Cell. 1999;23:659–74.
Harrison KA, Thaler J, Pfaff SL, Gu H, Kehrl JH. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat Genet. 1999;23:71–5.
Li H, Arber S, Jessell TM, Edlund H. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet. 1999;23:67–70.
Sherwood RI, Chen TYA, Melton DA. Transcriptional dynamics of endodermal organ formation. Dev Dyn. 2009;238:29–42.
Li H, Edlund H. Persistent expression of Hlxb9 in the pancreatic epithelium impairs pancreatic development. Dev Biol. 2001;240:247–53.
Jaffe R, Zeituni M, Fejgin M. Caudal regression syndrome Fetus Spinal Anom. 1991;7561:1–3.
Catala M. Genetic control of caudal development. Clin Genet. 2002;61:89–96.
Chen R, Hussain K, Al-Ali M, Dattani MT, Hindmarsh P, Jones PM, Marsh P. Neonatal and late-onset diabetes mellitus caused by failure of pancreatic development: report of 4 more cases and a review of the literature. Pediatrics. 2008;121:e1541–7.
Iyer RS, Khanna PC. Currarino syndrome. Pediatr Radiol. 2010;40(Suppl 1):S102.
Ott E, Wendik B, Srivastava M, Pacho F, Töchterle S, Salvenmoser W, Meyer D. Pronephric tubule morphogenesis in zebrafish depends on Mnx mediated repression of irx1b within the intermediate mesoderm. Dev Biol. 2016;411:101–14.
Yokoyama S, Ito Y, Ueno-Kudoh H, et al. A systems approach reveals that the myogenesis genome network is regulated by the transcriptional repressor RP58. Dev Cell. 2009;17:836–48.
Brunskill EW, Aronow BJ, Georgas K, et al. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev Cell. 2008;15:781–91.
Deguchi Y, Kehrl JH. Selective expression of two homeobox genes in CD-34-positive cells from human bone marrow. Blood. 1991;78:323–8.
Harrison KA, Druey KM, Deguchi Y, Tuscano JM, Kehr JH. A novel human homeobox gene distantly related to proboscipedia is expressed in lymphoid and pancreatic tissues. J Biol Chem. 1994;289:19968–76.
Deguchi Y, Yamanaka Y, Theodossiou C, Najfeld V, Kehrl JH. High expression of two diverged homeobox genes, HB24 and HB9, in acute leukemias: molecular markers of hematopoietic cell immaturity. Leukemia. 1993;7:446–51.
Hollington P, Neufing P, Kalionis B, Waring P, Bentel J, Wattchow D, Tilley WD. Expression and localization of homeodomain proteins DLX4, HB9 and HB24 in malignant and benign human colorectal tissues. Anticancer Res. 2004;24:955–62.
Wilkens L, Jaggi R, Hammer C, Inderbitzin D, Giger O, von Neuhoff N. The homeobox gene HLXB9 is upregulated in a morphological subset of poorly differentiated hepatocellular carcinoma. Virchows Arch. 2011;458:697–708.
Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, Pfaff SL. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron. 1999;23:675–87.
Liu Y, Sugiyama F, Yagami K, Ohkawa H. Sharing the same embryogenic pathway in anorectal malformations and anterior sacral myelomeningocele formation. Pediatr Surg Int. 2003;19:152–6.
Szumska D, Pieles G, Essalmani R, et al. VACTERL/caudal regression/ Currarino syndrome-like malformations in mice with mutation in the proprotein convertase Pcsk5. Genes Dev. 2008;22:1465–77.
Tsuda T, Iwai N, Deguchi E, et al. PCSK5 and GDF11 expression in the hindgut region of mouse embryos with anorectal malformations. Eur J Pediatr Surg. 2011;21:238–41.
Herrmann BG, Labeit S, Poustka A, King TR, Lehrach H. Cloning of the T gene required in mesoderm formation in the mouse. Nature. 1990;343:617–22.
Pennimpede T, Proske J, König A, et al. In vivo knockdown of Brachyury results in skeletal defects and urorectal malformations resembling caudal regression syndrome. Dev Biol. 2012;372:55–67.
Nagai T, Katoh R, Hasegawa T, Ohashi H, Fukushima Y. Currarino triad (anorectal malformation, sacral bony abnormality and presacral mass) with partial trisomy of chromosomes 13q and 20p. Clin Genet. 1994;45:272–3.
Dworschak GC, Crétolle C, Hilger A, Engels H, Korsch E, Reutter H, Ludwig M. Comprehensive review of the duplication 3q syndrome and report of a patient with Currarino syndrome and de novo duplication 3q26.32-q27.2. Clin Genet. 2017;91:661–71.
Aljeaid D, Long P, Walters-Sen L, Hallinan B, Saal H. Characterization of a unique de novo 3q duplication and 9p deletion: Support for the association of 3q duplication with Currarino syndrome. ACMG Annual Clinical Genetics Meeting, Charlotte, NC, 2018.7429(abstr.).
Peterson JF, Basel DG, Bick DP, et al. A rare combination of functional disomy Xp, deletion Xq13.2-q28 spanning the XIST gene, and duplication 3q25.33-q29 in a female with der(X)t(X;3)(q13.2;q25.33). J Pediatr Genet. 2018;7:23–8.
Bevanda K, Memidzan I, Boban-Raguz A. Caudal regression syndrome (Currarino syndrome) with chromosome mutation 9. Radiol Case Rep. 2020;15:1184–8.
Bonnefond A, Vaillant E, Philippe J, et al. Transcription factor gene MNX1 is a novel cause of permanent neonatal diabetes in a consanguineous family. Diabetes Metab. 2013;39:276–80.
Flanagan SE, De Franco E, Lango Allen H, et al. Analysis of transcription factors key for mouse pancreatic development establishes NKX2-2 and MNX1 mutations as causes of neonatal diabetes in man. Cell Metab. 2014;19:146–54.
Kharade SS, Parekh VI, Agarwal SK. Functional defects from endocrine disease-associated mutations in HLXB9 and its interacting partner. NONO Endocrinol. 2018;159:1199–212.
Boissel S, Fallet-Bianco C, Chitayat D, et al. Genomic study of severe fetal anomalies and discovery of GREB1L mutations in renal agenesis. Genet Med. 2018;20:745–53.
Holm I, Monclair T, Lundar T, Stadheim B, Prescott TE, Eiklid KL. A 5.8 kb deletion removing the entire MNX1 gene in a Norwegian family with Currarino syndrome. Gene. 2013;518:457–60.
Lin YH, Huang RL, Lai HC. Presacral teratoma in a Currarino syndrome woman with an unreported insertion in MNX1 gene. Taiwan J Obstet Gynecol. 2011;50:512–4.
Messaed C, Rouleau GA. Molecular mechanisms underlying polyalanine disorders. Neurobiol Dis. 2009;34:397–405.
Hughes JN, Thomas PQ. Molecular pathology of polyalanine expansion disorders: new perspectives from mouse models. Methods Mol Biol. 2013;1017:135–51.
Mo R, Kim JH, Zhang J, Chiang C, Hui CC, Kim PC. Anorectal malformations caused by defects in sonic hedgehog signaling. Am J Pathol. 2001;159:765–74.
Seri M, Martucciello G, Paleari L, et al. Exclusion of the Sonic Hedgehog gene as responsible for Currarino syndrome and anorectal malformations with sacral hypodevelopment. Hum Genet. 1999;104:108–10.
Coutton C, Poreau B, Devillard F, et al. Currarino syndrome and HPE microform associated with a 2.7-Mb deletion in 7q36.3 excluding SHH gene. Mol Syndromol. 2014;5:25–31.
Papapetrou C, Drummond F, Reardon W, Winter R, Spitz L, Edwards YH. A genetic study of the human T gene and its exclusion as a major candidate gene for sacral agenesis with anorectal atresia. J Med Genet. 1999;36:208–13.
Ghebranious N, Blank RD, Raggio CL, et al. A missense T (Brachyury) mutation contributes to vertebral malformations. J Bone Miner Res. 2008;10:1576–83.
Postma AV, Alders M, Sylva M, et al. Mutations in the T (brachyury) gene cause a novel syndrome consisting of sacral agenesis, abnormal ossification of the vertebral bodies and a persistent notochordal canal. J Med Genet. 2014;51:90–7.
Shaheen R, Alshail E, Alaqeel A, Ansari S, Hindieh F, Alkuraya FS. T (brachyury) is linked to a Mendelian form of neural tube defects in human. Hum Genet. 2015;134:1139–41.
Anderson-Shotwell S, Wilson WG. 18p- syndrome with partial sacral agenesis. J Med Genet. 1989;26:70–1.
Titomanlio L, Giurgea I, Baumann C, Elmaleh M, Sachs P, Chalard F, Aboura A, Verloes A. A locus for sacral/anorectal malformations maps to 6q25.3 in a 0.3 Mb interval region. Eur J Hum Genet. 2006;14:971–4.
Holm I, Spildrejorde M, Stadheim B, Eiklid KL, Samarakoon PS. Whole exome sequencing of sporadic patients with Currarino syndrome: a report of three trios. Gene. 2017;624:50–5.
Janesick A, Abbey R, Chung C, Liu S, Taketani M, Blumberg B. ERF and ETV3L are retinoic acid-inducible repressors required for primary neurogenesis. Development. 2013;140:3095–106.
Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell. 2003;115:109–21.
Patsialou A, Wilsker D, Moran E. DNA-binding properties of ARID family proteins. Nucleic Acids Res. 2005;33:66–80.
Mayosi BM, Fish M, Shaboodien G, et al. Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017;10:e001605.
Cuvelier A, Muir JF, Martin JP. Sesboüé [Proteins of the inter-alpha trypsin inhibitor (ITI) family. A major role in the biology of the extracellular matrix]. Rev Mal Respir. 2000;17:437–46.
Morgan R, Pettengell R, Sohal J. The double life of HOXB4. FEBS Lett. 2004;578:1–4.
Kamata T, Bong YS, Mood K, Park MJ, Nishanian TG, Lee HS. EphrinB1 interacts with the transcriptional co-repressor Groucho/xTLE4. BMB Rep. 2011;44:199–204.
McDonald JA, Fujioka M, Odden JP, Jaynes JB, Doe CQ. Specification of motoneuron fate in Drosophila: Integration of positive and negative transcription factor inputs by a minimal eve enhancer. J Neurobiol. 2003;57:193–203.
Woolfe A, Goodson M, Goode DK, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7.
Cuturilo G, Hodge JC, Runke CK, Thorland EC, Al-Owain MA, Ellison JW, Babovic-Vuksanovic D. Phenotype analysis impacts testing strategy in patients with Currarino syndrome. Clin Genet. 2016;89:109–14.
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL. G.C.D. is supported by BONFOR Grant O-120.0001 and by the Herbert-Reeck foundation (2019).
Author information
Authors and Affiliations
Contributions
GD, HR and ML participated in the process of the literature review and in the drafting the final manuscript. In addition, ML supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplemental Table 1. CS patients with reported heterozygous MNX1 mutations or associated gene/chromosomal anomalies. Supplemental Table 2. Cases with heterozygous MNX1 or other mutations negative for CS features.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dworschak, G.C., Reutter, H.M. & Ludwig, M. Currarino syndrome: a comprehensive genetic review of a rare congenital disorder. Orphanet J Rare Dis 16, 167 (2021). https://doi.org/10.1186/s13023-021-01799-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-021-01799-0