
Abstract
Background
Both haemophagocytic lymphohistiocytosis and acute necrotizing encephalopathy are life-threatening condition. It presents major diagnostic difficulties, since it may have a diversity in clinical picture and with many conditions leading to the same clinical presentation. So it is key important to understand the disorders.
Case presentation
We report a pediatric case of haemophagocytic lymphohistiocytosis with specific presentation which predominantly featured as acute necrotizing encephalopathy of childhood. We discuss the diagnosis and differential diagnosis, and speculate the etiology of haemophagocytic lymphohistiocytosis is due to hypersensitivity.
Conclusion
Haemophagocytic lymphohistiocytosis and brain damage in this case may be induced by hypersensitivity, which have good clinical outcome if diagnosed and treated early.
Similar content being viewed by others


Background
Hemophagocytic lymphohistiocytosis (HLH) was first reported by Risdall et al. [1]. It is a life-threatening condition that can rapidly deteriorate and lead to multiple organ failure and death [2]. Cardinal symptoms of HLH are prolonged fever, hepatosplenomegaly and cytopenias, and characteristic laboratory findings in HLH include hyperferritinemia, hypertriglyceridemia, cytopenia, hypoalbuminemia, decreased fibrinogen, hypercholesterolemia, and abnormal liver function [3].
Acute necrotizing encephalopathy of childhood (ANEC) represents an entity of acute encephalopathy which manifests with symptoms of coma, convulsions, and hyperpyrexia [4]. This disease is associated with severe neurological sequelae and profoundly high mortality [5]. ANEC is characterized by multiple and symmetrical lesions with edema and necrosis in the thalamus, the cerebral and cerebellar medulla, and the brainstem tegmentum [4].
Haemophagocytic lymphohistiocytosis and acute necrotizing encephalopathy of childhood are life-threatening conditions with severe consequences and high mortality. It is key important to understand the disorders. The present paper describes the case of a 3-month-old boy presenting with characteristic manifestation resembling acute necrotizing encephalopathy of childhood associated with hemophagocytic lymphohistiocytosis, which was thought due to allergic reaction.
Case presentation
The 3-month-old boy, the only child of non-consanguineous parents, was hospitalized in Shenzhen Children's Hospital because of persistent fever and skin rash for more than two weeks, resistant to conventional therapy. He had no cough, vomiting, lethargy or irritability. His past history included allergic rashes, and his mother suffered allergic reaction. There was no HLH patient in his family.
On admission, physical examination found rashes mainly in face, trunk and limbs, liver and spleen just palpable, no edema of palms, no periungual or perianal desquamation, no cervical lymphadenopathy, no conjunctival congestion, no strawberry tongue, erythema of the oropharyngeal mucosa or cracking of the lip. No positive neurological sign was found. Laboratory tests showed an increase in leukocytes (20.1 × 109/L) and neutrophils (12.82 × 109/L), erythrocyte sedimentation rate (33 mm/h), positive antinuclear antibody and antineutrophil cytoplasmic antibody, mild anemia (hemoglobin 89 g/L) and slightly elevation of C-reactive protein (19.5 mg/L). Liver function, lymphocyte subtypes, immunoglobulin isotypes, serum ferritin, coagulation function test and blood fat level were normal; Serology for human immunodeficiency virus; widal test; bacterial culture of peripheral blood, bone marrow and urine; hepatitis A, B, and C; mycoplasma pneumoniae (IgM and DNA); tests for tuberculosis; cytomegalovirus (IgM, IgG, and DNA); Human Herpes Virus; Toxopasma (IgM); Rubella (IgM); influenza virus A and B; parainfluenza virus; respiratory syncytial virus; adenovirus; legionella; rickettsia; and Epstein Barr Virus (IgM, IgG and DNA) were all negative. Echocardiography was normal. A diagnosis of acute inflammatory response syndrome and autoimmune vasculitis was entertained on clinical grounds and laboratory findings. Bacterial infection could not be ruled out, then antibiotics (linezolid and cefuroxime) and methylprednisolone was used for 3 days;the high fever subsided and the skin rash disappeared thereafter. 4 days later, high fever recurred and generalized skin eruption was noted. Intravenous immunoglobulin (IVIG) with a dosage of 400 mg/Kg was administered. 8 hours later, his condition deteriorated rapidly to present with a decrease of oxygen saturation, hypotension, tachycardia, tachypnea, irritability and cyanosis. He was transferred to the pediatric intensive care unit (PICU), and administration of fluids, inotropes (dopamine, norepinephrine) and mechanical ventilation was started. Blood routine test showed leukocyte count 10 x 109/L (neutrophil 3 %; eosinophil 40.1 %), erythrocyte count 3.21 x 1012/L, hemoglobin 83 g/L, platelet count 46 x 109/L. Total IgE (104.4 KIU/L) was elevated significantly (Table 1). Hypersensitive to IVIG was considered and high-dose intravenous methylprednisolone (30 mg/Kg) was given. During his hospitalization in PICU, the patient experienced several shocks each time after transfusion of blood and blood products, including IVIG, albumin, plasma, washed red blood cells. He developed progressive hepatosplenomegaly, severe pancytopenia (leukocyte count 0.2 x 109/L, erythrocyte count 1.11 x 1012/L, hemoglobin 29 g/L, platelet count 9 x 109/L). He presented with neck stiffness, convulsion and light coma, and his eyes couldn't follow light. Serum ferritin (18699 ng/mL), lactate dehydrogenase (1685 IU/L) and triglycerides (4.53 mmol/L) were elevated significantly, but lactic acid was normal and plasma fibrinogen was decreased apparently (0.49 g/L). Both the prothrombin time and the activated partial thromboplastin time were above limit of detection. Lumbar puncture revealed the initial pressure of 125 mmH2O and clear cerebrospinal fluid containing white blood cells 0 cells/L, red blood cells 28 cell/L, protein 513 mg/L. The fifth bone marrow aspiration displayed large amount of hemophagocytic histiocytes (Fig. 1). Brain magnetic resonance imaging (MRI) suggested ANEC (Fig. 2). DNA samples were obtained from the peripheral blood of the patient by standard procedures, and the sequences of HLH-associated genes were analyzed. The result verified 1 variation in the PRF1 gene, 10 variations in the UNC13D gene, 8 variations in the STXBP2 gene, 2 variations in the XIAP gene and no variations in STX11 and SH2D1A (Table 2).

Hemophagocytic histiocytes in bone marrow. Bone marrow aspiration showing a normal histiocyte (a), a hemophagocytic histiocyte containing aphagocytosed neutrophil and platelets (b), a hemophagocytic histiocyte containing aphagocytosed erythroblast and platelets (c). (Wright staining; ×1000)

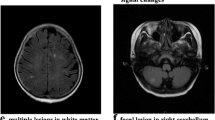
Neurological images. Brain magnetic resonance imaging (MRI) on admission revealed extensive edema (a); after 4 weeks, it showed long T2 signal (b) and hyperintensity on fluid-attenuated inversion recovery (FLAIR, c) mainly in thalamus, basal ganglia, brainstem. 1 year later, the neurological image (MRI) showed slightly abnormal shape of bi-lateral ventricular, not any other obvious abnormal changes noticed (d)
The diagnosis of hemophagocytic lymphohistiocytosis and acute necrotizing encephalopathy was made based on the above findings. The HLH-2004 treatment protocol was used once diagnosis made. The clinical symptoms were rapidly improved. Followed up to 3½ years old, he was well. The neurological image (MRI) showed slightly abnormal shape of bi-lateral ventricular, not any other obvious abnormal noticed.
Discussion
HLH may occur either as a primary condition with a genetic mutation or as a secondary condition associated with an infection, immunologic disorder, malignancy, or metabolic disease [6–8]. This patient presented with persistent fever, skin rash, ensuing hypersensitive to all blood products, hepatosplenomegaly, pancytopenia, and central nervous symptoms. He fulfilled 5 of the 8 criteria for acquired HLH [9]. Hemophagocytosis is one of the criteria and reported to be negative at diagnosis in about 20-25 % of cases as reported before and more recently in a case series [10, 11]. As some researcher reported that the repetitive bone marrow aspirations may reveal hemophagocytosis in the further course [12]. No hemophagocytosis was detected in the first four bone marrows in this case, but the fifth bone marrow biopsy displayed large amount of hemophagocytic histiocytes. It is important to note that the diagnosis of HLH does not depend on this morphological finding. The genes relevant to familial HLH (FHLH) including PRF1, UNC13D, STX11, STXBP2 [3], were analyzed to confirm the mutations, as well the genes related to immune deficiency syndromes associated with HLH: XIAP and SH2D1A [3, 13]. Sequencing results showed that all these variations were synonymous or in intron region and untranslated region. Analysis of the genetic data revealed that no variation was likely to produce modified amino acid sequence, thus to cause a severe impairment of protein function in this patient, with addition of the fact that there was no HLH patient or consanguinity in his family. This information is therefore relevant in that it is not likely to be genetic HLH.
This patient presented with neck stiffness, convulsion and light coma; and the severe lesions of brain MRI showed changes mainly in thalamus, basal ganglia, brainstem and white matter as described in patients with ANEC [14, 15], which is characterized by multiple and symmetrical lesions with edema and necrosis in the thalamus, the cerebral and cerebellar medulla, and the brainstem tegmentum [4, 16], which evoked the diagnosis of acute necrotizing encephalopathy of childhood. The clinical feature of typical ANEC is fulminant, with severe neurologic sequelae and profoundly high mortality [17]. However, the discrepancy between the severe MRI findings, the relatively mild neurological manifestations, and the unexpected clinical outcome was not consistent with ANEC. We speculated that the brain damage was not true ANEC, but it was caused by transient ischemia induced by allergic vasculitis due to hypersensitivity. The extensive allergic vasculitis involves central nerve system, and extravasation of plasma takes place secondary to local vascular lesions. This is followed by destruction and necrosis of glial cells and neurons [18]. The neurological signs were improved promptly with the subsiding of hypersensitivity, which gave a good interpretation to our speculation.
The central nervous system (CNS) involvement was reported in about a third of the children with HLH, which could easily be misdiagnosed in many children because its radiologic manifestations are nonspecific and overlap with various infectious and inflammatory disorders. Therefore, differential diagnosis of resembling diseases is very important for this patient. A wide range of disorders should be considered in the differential diagnosis, including Leigh disease, hypoxic-ischemic encephalopathy, viral encephalitis, hemorrhagic encephalitis, acute disseminated encephalomyelitis, and Reye syndrome. First of all, the brain damages focused on thalami and brainstem was not consistent with CNS involvement in HLH [18]. Absence of inflammatory cells in cerebrospinal fluid and the symmetric brain lesions in this case differentiates from the entities of acute disseminated encephalomyelitis and acute hemorrhagic encephalitis. Reye syndrome can be excluded by hyperammonemia, hypoglycemia, and lactic acidosis absent in this case [19], as well as Leigh disease. The differentiation from hypoxic-ischemic encephalopathy presented much more difficulty in this patient. In spite of shocks, the oxygen saturation, heart rate and blood pressure were corrected quickly, which did not influence cerebral perfusion severely. Moreover, the imaging findings could not explain hypoxic-ischemic brain damage which focuses on obscuration of grey-white matter junctions and widespread laminar necrosis of the cortex [20]. Furthermore, certain viral encephalitis may be difficult to exclude clinically e.g. Japanese encephalitis may also involve the deep gray matter symmetrically. However, the involvement of the brain stem is uncommon in Japanese encephalitis, and thalamic involvement is not necessarily symmetrical [21].
The etiologies of acquired HLH and ANEC were suggested to be associated with infectious, metabolic, immune related causes [6, 7, 15], and hypersensitivity was more recently reported to be complicated by hemophagocytic lymphohistiocytosis in a case series [22–24]. This boy did not meet the diagnostic criteria of systemic lupus erythematosus [25], and there was no arthritis during the disease course so he was not consistent with the diagnosis of juvenile idiopathic arthritis [26]; moreover, there was not any evidence of scleroderma and mixed connective tissue disease, though he was detected anti-nuclear and anti-neutrophil auto-antibodies and severe pancytopenia. He suffered allergy in his past and had positive allergy family history; eosinophils and IgE significantly elevated after IVIG and blood transfusion then gradually declined followed steroids; furthermore, he was hypersensitive to all kinds of blood and blood products, and subsequently, developed cytopenias. All above, it led us to speculate that allergic reaction was involved in the pathogenesis of HLH and brain lesions in this patient. This was never reported previously.
Increase of eosinophils and reaction to blood products suggests type I, II and III hypersensitivity was involved in the pathogenesis in this case. Hypersensitivity and HLH appear somewhat similar, being characterized by activated lymphocytes and hypercytokinemia [27]. Hypersensitivity results in hypercytokinemia, leading to uncontrolled activation of benign scavenger macrophages and development of hemophagocytosis [22]. The pathogenesis of HLH involves the dysfunction of natural killer (NK) cells and cytotoxic T cells leading to prolonged and intense activation of antigen-processing cells (macrophages and histiocytes) [28] and CD8+ T cells, and excessive proliferation and ectopic migration of T cells, which triggers overproduction of proinflammatory cytokines, and unrestrained hemophagocytosis [29]. It is suggested that cell destruction by cytotoxic antibodies and a reversible depression of stem cell activity with myeloid maturation blockade contribute to the pathophysiology of the cytopenias. The deposited immune complexes can trigger neutrophils to release free radicals and enzymes causing tissue destruction, which can lead to endothelial cell necrosis [30]. Hypercytokinemia and the hyperpermeability of both the blood-brain barrier and the capillary walls of the central nervous system might be essential in the pathogenesis of acute necrotizing encephalopathy [31].
Hemophagocytic lymphohistiocytosis has a poor prognosis [32]. Patients with secondary HLH have had only a 55 % survival at 3 years [33]. This boy was followed up regularly after continuation therapy. Followed up to 3½ years old, he is doing well; there is not any symptom that could be due to central nervous system dysfunction; the routine blood count, blood lipid level, liver function and ferritin were normal, and the bone marrow cytology demonstrated sustained remissions. The neurological image (MRI) showed no obvious abnormal.
Conclusions
This is a rare case of hemophagocytic lymphohistiocytosis, with specific presentation resembling ANEC. Hypersensitivity may involve in the etiologies of HLH and brain damage, which have good clinical and neurologic outcome if treated early. From this study, we should have the awareness of such a rare disease as delay of diagnosis and treatment is common with subsequent poor outcome. Early diagnosis is crucial to reduce mortality rates. It is of importance to know that hypersensitivity may result in HLH and brain damage resembling ANEC from the findings in this study.
Abbreviations
ANEC, acute necrotizing encephalopathy of childhood; CNS, central nervous system; DNA, deoxyribose nucleic acid; HLH, hemophagocytic lymphohistiocytosis; Ig, immunoglobulin; IVIG, intravenous immunoglobulin; MRI, magnetic resonance imaging; NK, natural killer; PICU, pediatric intensive care unit
References
Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour Jr HH, Simmons RL, Brunning RD. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 1979;44:993–1002.
Karapinar B, Yilmaz D, Balkan C, Akin M, Ay Y, Kvakli K. An unusual cause of multiple organ dysfunction syndrome in the pediatric intensive care unit: hemophagocytic lymphohistiocytosis. Pediatr Crit Care Med. 2009;10:285–90.
Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233–46.
Mizuguchi M, Abe J, Mikkaichi K, Noma S, Yoshida K, Yamanaka T, Kamoshita S. Acute necrotising encephalopathy of childhood: a new syndrome presenting with multifocal, symmetric brain lesions. J Neurol Neurosurg Psychiatry. 1995;58:555–61.
Khan MR, Maheshwari PK, Ali SA, Anwarul H. Acute necrotizing encephalopathy of childhood: a fatal complication of swine flu. J Coll Physicians Surg Pak. 2011;21:119–20.
Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041–52.
Filipovich AH. Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am. 2008;28:293–313. viii.
Ishii E. Hemophagocytic lymphohistiocytosis in children: pathogenesis and treatment. Front Pediatr. 2016;4:47.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
Arico M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, Martinetti M, Rusca MP. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10:197–203.
Ferreira DG, do Val Rezende P, Murao M, Viana MB, de Oliveira BM. Hemophagocytic lymphohistiocytosis: a case series of a Brazilian institution. Rev Bras Hematol Hemoter. 2014;36:437–41.
Bode SF, Lehmberg K, Maul-Pavicic A, Vraetz T, Janka G, Stadt UZ, Ehl S. Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis. Arthritis Res Ther. 2012;14:213.
Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, Kanegane H, Lopez-Granados E, Mejstrikova E, Pellier I, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. 2011;117:1522–9.
Wang HS, Huang SC. Acute necrotizing encephalopathy of childhood. Chang Gung Med J. 2001;24:1–10.
Salehiomran MR, Nooreddini H, Baghdadi F. Acute necrotizing encephalopathy of childhood; a case report. Iran J Child Neurol. 2013;7:51–4.
Yoganathan S, Sudhakar SV, James EJ, Thomas MM. Acute necrotising encephalopathy in a child with H1N1 influenza infection: a clinicoradiological diagnosis and follow-up. BMJ Case Rep. 2016;2016.
Mizuguchi M. Acute necrotizing encephalopathy. Nihon Rinsho. 2011;69:465–70.
Matsushita E, Takita K, Shimada A. Suspected acute encephalopathy with symmetrical abnormal signal areas in the basal ganglia, thalamus, midbrain and pons diagnosed by magnetic resonance imaging. Acta Paediatr Jpn. 1997;39:454–8.
Mizuguchi M. Acute necrotizing encephalopathy of childhood: a novel form of acute encephalopathy prevalent in Japan and Taiwan. Brain Dev. 1997;19:81–92.
Back T, Hemmen T, Schuler OG. Lesion evolution in cerebral ischemia. J Neurol. 2004;251:388–97.
Verma R. MRI features of Japanese encephalitis. BMJ Case Rep. 2012;2012.
Gauchan D, Shaaban H, Parikh N, Chang NL, Altheeb Z, Maroules M. Severe hemophagocytic lymphohistiocytosis as a complication of drug-induced hypersensitivity syndrome. Int J Crit Illn Inj Sci. 2015;5:60–1.
Picard M, Fernandez MI, Des Roches A, Begin P, Paradis J, Paradis L, Le Deist F. Ceftazidime-induced drug reaction with eosinophilia and systemic symptoms (DRESS) complicated by hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol Pract. 2013;1:409–12.
Koinuma T, Nunomiya S, Wada M, Koyama K. [Case report: Hemophagocytic syndrome developed after drug eruption: report of two cases]. Nihon Naika Gakkai Zasshi. 2014;103:1931–4.
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, He X, Maldonado-Cocco J, Orozco-Alcala J, Prieur AM, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–2.
Filipovich AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr Opin Allergy Clin Immunol. 2011;11:512–6.
Ohga S, Matsuzaki A, Nishizaki M, Nagashima T, Kai T, Suda M, Ueda K. Inflammatory cytokines in virus-associated hemophagocytic syndrome. Interferon-gamma as a sensitive indicator of disease activity. Am J Pediatr Hematol Oncol. 1993;15:291–8.
Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814–22.
Ng WF, Chiu SC, Lam DS, Wong YC, Tam S, Kwong NS, Loo KT, Yuen KY. A 7-year-old boy dying of acute encephalopathy. Brain Pathol. 2010;20:261–4.
Akiyoshi K, Hamada Y, Yamada H, Kojo M, Izumi T. Acute necrotizing encephalopathy associated with hemophagocytic syndrome. Pediatr Neurol. 2006;34:315–8.
Elyamany G, Alzahrani A, Elfaraidi H, Alsuhaibani O, Othman N, Al Mussaed E, Alabbas F. Hemophagocytic Lymphohistiocytosis: Single-Center Series of 12 Cases from Saudi Arabia. Clin Med Insights Pediatr. 2016;10:21–6.
Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D, Ladisch S, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–73.
NCBI Assay, U.S. National Library of Medicine. http://www.ncbi.nlm.nih.gov/projects/SNP/. Accessed 13 Jun 2016.
Acknowledgments
The authors wish to thank the patient and their families for allowing us to use the medical documentation and information leading to the present article, and we wish to thank Dr Philippa Mills (PhD, Lecturer in Metabolic pediatrics, Biochemistry Research Group, Clinical & Molecular Genetics Unit, Institute of Child Health & Great Ormond Street Hospital for Sick Children, University College London) for assistance to gene analysis, Gang Xu (M.D, Clinical Laboratory Department of Shenzhen Children's Hospital) for marrow cytology inspection, technical and material support, and Dr Weiguo Cao (M.D, Radiologist, Radiology Department of Shenzhen Children's Hospital) for imaging technical assistance. There is not any conflict of interest for both Dr Philippa Mills, Gang Xu and Weiguo Cao.
Funding
This study was funded by Shenzhen Innovation and Technology Committee (Grant number CXZZ20130320172336579).
Availability of data and materials
The dataset(s) supporting the conclusions of this article is (are) included within the article.
Authors’ contributions
DD: writing of manuscript, critical revision of the manuscript. DD and SL: caring for the patients, performing the treatment protocol, and data collection. FW: study concept and design. SZ: analysis and interpretation of data. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Signed consent was obtained from the parents for analysis of DNA samples from their child, for publication of the case report and any accompanying images.
Ethics approval and consent to participate
This study was approved by Research Ethics Committee of Shenzhen Children’s Hospital (Reference Number: SCH ethical (article) 2016 (002)).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dai, D., Wen, F., Liu, S. et al. Brain damage resembling acute necrotizing encephalopathy as a specific manifestation of haemophagocytic lymphohistiocytosis - induced by hypersensitivity. Ital J Pediatr 42, 79 (2016). https://doi.org/10.1186/s13052-016-0286-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-016-0286-z