
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare life-threatening disease of severe hyperinflammation caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting high amounts of inflammatory cytokines. It is a frequent manifestation in patients with predisposing genetic defects, but can occur secondary to various infectious, malignant, and autoimmune triggers in patients without a known genetic predisposition. Clinical hallmarks are prolonged fever, cytopenias, hepatosplenomegaly, and neurological symptoms, but atypical variants presenting with signs of chronic immunodeficiency are increasingly recognized. Impaired secretion of perforin is a key feature in several genetic forms of the disease, but not required for disease pathogenesis. Despite progress in diagnostics and therapy, mortality of patients with severe HLH is still above 40%. Reference treatment is an etoposide-based protocol, but new approaches are currently explored. Key for a favorable prognosis is the rapid identification of an underlying genetic cause, which has been facilitated by recent immunological and genetic advances. In patients with predisposing genetic disease, hematopoietic stem cell transplantation is performed increasingly with reduced intensity conditioning regimes. Current research aims at a better understanding of disease pathogenesis and evaluation of more targeted approaches to therapy, including anti-cytokine antibodies and gene therapy.
Similar content being viewed by others


Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory disease caused by an uncontrolled and dysfunctional immune response [1]. It is characterized by activation and massive proliferation of T cells and macrophages, leading to marked hypercytokinemia [2, 3]. Low or absent natural killer (NK) cell and CD8+ cytotoxic T lymphocyte (CTL) cytotoxicity is one of the hallmarks of HLH and leads to impaired regulation of the immune response [4]. HLH is fatal when untreated but even with aggressive treatment still has a high mortality rate [1]. When fully developed, HLH has a characteristic phenotype, but different pathways of pathogenesis can lead to this clinical picture. HLH frequently develops in patients with underlying genetic disease (primary or familial HLH), but can also occur secondary to infection, malignancy, metabolic or autoimmune diseases in patients with no known genetic predisposition ('secondary' or acquired HLH).
Primary HLH
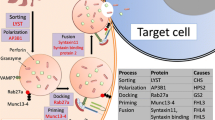
Familial HLH (FHL) has an incidence of approximately 0.12 to 1 cases per 100,000 children per year, even though it may be more common in areas with high consanguinity due to the autosomal-recessive inheritance [5, 6]. Five different forms of FHL have so far been described and four genes, accounting for over 90% of familial cases, have been identified (Table 1) [7–13]. They encode the proteins perforin, MUNC13-4, syntaxin-11 and MUNC18-2, all of which play a key role in lymphocyte cytotoxicity (Figure 1) [14]. Most patients with FHL develop HLH within the first months of life. However, up to 20% of patients present at more than 2 years of age, and in rare cases, patients with FHL remain asymptomatic until adulthood [1, 15–17].

Pathogenesis of hemophagocytic lymphohistiocytosis. The function of LYST, probably important for correct size and function of lytic granules, is not entirely understood. Note the empty granula in perforin deficiency. Adapted from [8, 47, 88]. CHS, Chédiak Higashi syndrome; CTL, CD8+ cytotoxic T lymphocyte; FHL, familial hemophagocytic lymphohistiocytosis; GSII, Griscelli syndrome type II; HPSII, Hermansky-Pudlak syndrome type II; NK, natural killer.
Syndromal immunodeficiencies associated with albinism, including Chédiak Higashi syndrome (CHS), Griscelli syndrome type II (GSII), and Hermansky-Pudlak syndrome type II (HPSII) also predispose to HLH. These patients show variable degrees of (partial) albinism, platelet dysfunction, and immunodeficiency in addition to their risk of developing HLH [18, 19]. The genes affected in CHS, GSII, and HPSII are also involved in granule-dependent lymphocyte cytotoxicity (Figure 1) [20, 21]. Onset of HLH in patients with these diseases tends to be later than in patients with FHL.
Other primary immunodeficiencies predisposing to HLH include XLP1 (SAP (signaling lymphocytic activation molecule associated protein)), XLP2 (XIAP (X-linked inactivator of apoptosis deficiency), ITK (IL-2 inducible T-cell kinase) and CD27 deficiency [22–26]. These defects can present with various signs and symptoms of immunodeficiency and immune dysregulation, but can also manifest primarily with HLH, almost exclusively in association with Epstein-Barr virus (EBV) infection [22–26].
All patients with primary HLH have a high risk of recurrence. Therefore, hematopoietic stem cell transplantation (HSCT) is the only curative option [27].
'Secondary' HLH
Patients manifesting with HLH in the absence of a disease-causing mutation in the known genes and without strong indications for a genetic predisposition, such as familial disease or recurrent episodes of HLH, are currently classified as suffering from 'secondary' HLH. There are sparse data on the incidence, but it is probably more frequent than primary HLH [28]. One source reports an incidence of 0.36 cases of malignancy-associated HLH per 100,000 adults per year [29]. It is highly likely that, due to some overlap with sepsis syndromes, HLH is under-diagnosed, in particular in the adult population. Many patients with 'secondary' HLH manifest beyond infancy, but increasing recognition of late-onset FHL renders the age at onset a poor indicator of disease etiology. Moreover, the genetic basis of HLH remains to be fully defined. Thus, final classification of a patient as suffering from 'secondary' HLH must remain preliminary.
The most common form of 'secondary' HLH is infection-associated HLH. Infectious triggers include viruses (for example, EBV, cytomegalovirus, HHV8, HIV), bacteria (for example, mycobacteria, mycoplasma), parasites (leishmania, plasmodium), and fungi (for example, candida, cryptococcus) [30, 31]. EBV and leishmania infection are the most frequent triggers. Notably, detecting an infectious agent does not help to distinguish between 'secondary' and primary forms of HLH, since also in the latter cases acute episodes are often triggered by infections [1].
Malignancies such as leukemias or lymphomas, particularly T-cell lymphomas and rarely solid tumors, are known to be potential triggers for HLH [1]. Moreover, a number of metabolic disorders, including multiple sulfatase deficiency, lysinuric protein intolerance, Wolman's disease, and disorders of proprionate metabolism, have been associated with HLH [28, 32–34]. Finally, immunosuppressive therapy for malignancies, after organ trans-plantation, or for autoimmune disorders may predispose to 'secondary' HLH [28, 35–37].
Macrophage activation syndrome
Macrophage activation syndrome (MAS) is a potentially life-threatening complication of autoinflammatory and autoimmune diseases and may be classified as a variant of 'secondary' HLH. When fully developed, the clinical features of MAS are indistinguishable from HLH. How-ever, patients with MAS may show distinct findings like neutrophilia or thrombocytosis in early disease stages, which are unusual in patients with primary HLH [38].
MAS is rare, but it is estimated that up to 7 to 30% of patients with active systemic juvenile idiopathic arthritis (Still's disease) experience some form of MAS, ranging from subclinical or mild to full-blown disease, with a mortality of up to 22% [38]. MAS is most frequent in systemic juvenile idiopathic arthritis, but has been observed in Kawasaki's disease, systemic lupus erythematodes, and other rheumatic diseases [38]. There are some reports of MAS in patients after treatment with anti-TNF-α antibodies for rheumatological disease [39].
Specific diagnostic criteria for MAS have been suggested. They include falling leukocyte and platelet counts, hyperferritinemia, hypofibrinogenemia, hemophagocytosis in bone marrow, elevated liver enzymes, elevated erythrocyte sedimentation rate, and hypertriglyceridemia [40, 41]. MAS can be the presenting feature of patients with autoinflammatory and autoimmune diseases and features of such diseases (such as arthritis or rash) should there-fore be carefully looked for in HLH patients in the course of their disease.
Pathophysiology
FHL and immunodeficiencies associated with albinism
One of the main immune defense mechanisms against infections with intracellular pathogens is contact-dependent cytotoxicity mediated by CTLs and NK cells [42]. After recognition of infected cells and formation of an immunological synapse, cytotoxic granules, containing perforin and granzymes, are polarized to the contact site between effector and target cell and are released into the intercellular space, where they can mediate their cytolytic effector function [43]. Importantly, lymphocyte cytotoxicity is not only directed at infected cells, but also against antigen-presenting cells (APCs). Elimination of APCs provides an important negative feedback to limit T-cell-mediated immune responses. There is evidence that CTLs are more important in the pathogenesis of HLH than NK cells, but immune regulation through killing of APCs has also been described for NK cells [44–46]. In the absence of effective cytotoxicity, APCs continue to stimulate CTL [47], leading to ongoing production of cytokines, in particular IFN-γ, which has a key role in macrophage activation. Activated T cells and macrophages infiltrate tissues such as the liver, bone marrow and central nervous system, secrete cytokines and show excessive phagocytic activity. IFN-γ and TNF-α have toxic effects on hematopoietic cells, contributing to cytopenias. TNF-α also inhibits lipoprotein lipase, causing hypertriglyceridemia [1], and IL-1, IL-6, and TNF-α induce fever [1]. Activated macro-phages secrete ferritin as well as plasminogen activator, while activated T and NK cells shed their IL-2 receptor, further contributing to the characteristic laboratory abnormalities of HLH. Data from animal models suggest that IFN-γ is the key cytokine involved in this inflammatory cascade and IFN-γ blockade is an effective treatment for HLH in mice [45].
FHL and the immunodeficiencies associated with albinism are associated with defective lymphocyte cytotoxicity. In FHL2, perforin itself is defective, while the other diseases affect proteins involved in the biogenesis, intracellular transport and exocytosis of perforin-containing lytic granules (Figure 1 and Table 1) [14]. The complex syndromic nature of the albinism disorders can be explained by the similarities in the molecular machineries of vesicle trafficking, including pigment transport in skin and hair or degranulation of platelets and mast cells.
It remains to be determined whether the above stated concept of pathogenesis is sufficient to explain the immune dysregulation of HLH or whether an additional immune regulatory role of perforin - directed at immune cells other than APCs - has to be postulated. Another open question is whether an external triggering factor, such as an infection, is always required (but not always detected) or whether presentation of auto-antigens and APC activation by endogenous inflammatory triggers may be sufficient to trigger disease. In any case, the fulminant nature of this highly inflammatory disease indicates a key role for lymphocyte cytotoxicity in limiting physiological immune reactions.
Other primary immunodeficiencies predisposing to HLH
At least four other primary immunodeficiencies predispose to HLH, the two X-linked disorders SAP and XIAP deficiency and the autosomal-recessive ITK deficiency and CD27 deficiency [22, 23, 25, 26]. In all four diseases, development of HLH is almost exclusively triggered by EBV infection [22, 23, 25, 26]. SAP deficiency is characterized by an impaired T cell-B cell interaction, which also involves an inability of cytotoxic T cells to lyse B cells, which are the main target cells of EBV [48]. ITK and CD27 deficiency are characterized by a poor control of EBV infection, but as in XIAP deficiency, the molecular mechanisms predisposing to HLH so far remain elusive. Of note, NK T-cell development, which may also be relevant for control of EBV infection, is impaired in all four diseases [24–26, 49, 50].
'Secondary' HLH
The pathogenesis of 'secondary' HLH is less well understood. Cytotoxic lymphocyte degranulation and cytotoxicity are not impaired in most cases [51]. Nevertheless, the balance between APC activation and CTL-mediated control may be disrupted through increased APC activation. Intracellular pathogens can activate APC directly - for example, via toll-like receptor (TLR) activation. TLRs could also be stimulated by anti-DNA antibodies in systemic lupus erythematosus [44, 52]. Data from a mouse model indicate an important protective role for IL-10 in 'secondary' HLH induced by TLR9 stimulation [44].
Clinical picture
The full clinical picture of HLH is quite characteristic, but the initial presentation is non-specific. First manifestations of HLH can resemble common infections, malignancy, fever of unknown origin, or autoimmune and autoinflammatory disorders [1]. Typical clinical findings in the further course are prolonged fever, mostly unresponsive to antibiotic therapy, and hepatosplenomegaly. In over 30% of cases, neurologic symptoms, including seizures, meningism, a decreased level of consciousness, irritability, hyper-/hypotonia or ataxia, are present [53, 54]. Lymphadenopathy, different kinds of rash, edema, and jaundice are less frequent.
Laboratory findings include cytopenias, usually beginning with thrombocytopenia evolving into severe pancytopenia, hyperferritinemia, elevated transaminases, hypofibrinogenemia, hypertriglyceridemia, hypoalbuminemia, and hyponatremia [1]. Additional immunological findings include elevated sCD25 and reduced NK cell cytotoxicity. Many patients with HLH show signs of disseminated intravascular coagulation. Hemophagocytosis can be absent in the early stages of the disease [55], but repetitive bone marrow aspirations may reveal hemophagocytosis in the further course. It is important to note that the diagnosis of HLH does not depend on this morphological finding.
There are an increasing number of descriptions of atypical and late-onset cases of primary HLH. Patients may present with different clinical phenotypes, including aplastic anemia, autoimmune lymphoproliferative disease, recurrent fever, hypogammaglobulinemia and infection susceptibility (resembling common variable immunodeficiency), granulomatous lung or liver disease, or sterile encephalitis [16, 17, 56, 57]. In one series up to 14% of adult patients presenting with HLH showed hypomorphic mutations in PRF1, MUNC13-4, and STXBP2, leading to less severe cytotoxicity defects that cause milder and atypical forms of HLH presenting beyond infancy [15].
Prognostic factors for poor outcome
The extent of hyperbilirubinemia, thrombocytopenia, hyperferritinemia and cerebrospinal fluid pleocytosis seem to be key risk factors for early death in HLH, as are lack of improvement in hemoglobin or fibrinogen levels, persisting thrombocytopenia and persistent fever after the start of therapy [58]. In EBV-induced HLH, a high viral load is associated with a poor outcome [31].
Diagnosis
A timely diagnosis is essential in HLH to initiate life-saving treatment. There is no single clinical or laboratory parameter that allows establishing a definite diagnosis. A panel of eight diagnostic criteria proposed by the Histiocyte Society is the current standard for establishing the diagnosis (Table 2) [59].
Five of these eight criteria have to be fulfilled in order to establish the clinical diagnosis. The criteria do not discriminate between primary and 'secondary' forms of the disease. There is some evidence that simplified criteria, including fever, splenomegaly, thrombocytopenia, and elevated ferritin, warrant more detailed immunological diagnostic evaluation. Ferritin >10,000 μg/L and high sCD25 seem to be more specific immunological markers [27, 60]. Nonetheless, their specificity and sensitivity is limited as well and neither normal ferritin nor normal sCD25 values exclude the diagnosis of HLH.
It is relevant to note that currently available additional immunological or genetic findings are not helpful in answering the question of whether or not a patient suffers from the acute clinical syndrome of HLH. They are of key importance, however, to discriminate between the primary and 'secondary' forms of the disease. Moreover, they are relevant for deciding whether symptoms and findings that do not strictly fulfill the criteria for HLH are related to an underlying genetic disease predisposing to HLH. Exclusion of a genetic disease predisposing to HLH is equally important for childhood and adult-onset forms of the disease.
Impaired lymphocyte cytotoxicity is the hallmark of primary HLH and reduced NK cell cytotoxicity has been well established as a diagnostic criterion. However, the test has several practical limitations, in particular in patients with reduced NK cell numbers. Degranulation assays of NK cells or CTLs use the membrane expression of the lysosomal marker protein CD107a upon activation as a marker of exocytosis of lytic granules [61]. These assays can rapidly diagnose patients with a degranulation defect [18, 62, 63] (Figure 2). In a recent large collaborative study involving 494 patients referred for evaluation of HLH, a resting NK degranulation assay below 5% could detect genetic degranulation defects (FHL3-5, GSII, CHS) with a sensitivity of 96% and a specificity of 88% [51]. Blood film and hair shaft microscopy (to identify CHS or GSII) and intracellular stainings for perforin, SAP, and XIAP provide additional simple tests allowing rapid identification of a potential predisposing genetic disease.

Natural killer (NK) cell degranulation assay. X-axis, CD107a; y-axis, CD56. Note that the patient only shows 0.17% degranulation, the control 31.17%. Degranulation >15% is considered normal, 5 to 15% abnormal and possibly pathological, requiring additional testing, and <5% deficient.
Figure 3 gives an overview of a proposed diagnostic approach in patients with suspected primary HLH, and Figure 4 shows additional microscopic findings in HLH. Using these tests, the indication for hematopoietic stem cell transplantation can now be established within 48 hours and before results of genetic tests are known, which will improve the management and prognosis for patients with HLH. On the basis of these immunological tests, a priority list for targeted sequencing of the relevant genes can be established [51, 64, 65]. Of note, deep intronic mutations, genomic inversions or unusual splice site mutations may also predispose to HLH such that conventional exon sequencing is not sufficient to exclude a genetic cause [66, 67].

Diagnostic work-up based on degranulation assays in suspected hemophagocytic lymphohistiocytosis. 2B4, surface molecule on NK cells; AICD, activation-induced cell death; CHS, Chédiak Higashi syndrome; CTL, CD8+ cytotoxic T lymphocyte; FHL, familial hemophagocytic lymphohistiocytosis; GS2, Griscelli syndrome type II; HLH, hemophagocytic lymphohistiocytosis; NK, natural killer; SAP, signaling lymphocytic activation molecule-associated protein; XIAP, X-linked inhibitor of apoptosis. This research was originally published in Blood. Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, Chiang SC, Marcenaro S, Meazza R, Bondzio I, Walshe D, Janka G, Lehmberg K, Beutel K, zur Stadt U, Binder N, Arico M, Moretta L, Henter JI, Ehl S: A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood 2012, 119:2754-63. © The American Society of Hematology [51].

Microscopic findings in hemophagocytic lymphohistiocytosis and conditions predisposing to it. (a) Macrophage phagocytosing erythrocytes and erythroblast (bone marrow). (b,c) Giant granule in monocyte (bone marrow) (b) and small granules in neutrophil (peripheral blood) (c) in Chédiak Higashi syndrome (CHS). (d) Extracellular amastigotes in visceral leishmaniasis (bone marrow). (e) Pigment clumps in a hair shaft in a patient with Griscelli syndrome type II. (f) Evenly distributed melanin granules, larger as in normal hairs, in a patient with CHS. Smears are shown at 600× magnification, hair at 100×.
In parallel to the immunological investigations, a careful microbiological workup is required to identify infectious triggers of HLH, in particular EBV, cytomegalovirus or leishmania infection. Controlling infectious diseases is an important element of HLH therapy [31].
Therapy and outcome
General principles of therapy
Untreated primary HLH is rapidly fatal within a few weeks [5]. Prompt and adequate treatment is of crucial importance for a positive outcome. Therapy should be started in all cases with high suspicion after diagnostic tests have been initiated but regardless of whether the results of all examinations have been obtained. The initial therapy consists of immunosuppressive and/or chemotherapeutic agents and aims at suppressing the hyper-inflammatory component of the disease as well as eliminating activated cytotoxic lymphocytes and macro-phages. Steroids inhibit inflammation by attenuating cytokine responses and inhibiting differentiation of dendritic cells and also have cytotoxic effects on lymphocytes. Cyclosporin A directly affects CTL activation as well as macrophage function. Etoposide induces apoptosis in lymphocytes as well as in APCs. Anti-thymocyte globulin (ATG) directly targets T cells, whereas alemtuzumab, an anti-CD52 antibody, targets lymphocytes in general and APCs. Intrathecal therapy with methotrexate and steroids is targeted at central nervous system disease.
Immunochemotherapy
Immunochemotherapy is widely used to induce and maintain remission until HSCT in primary HLH. The HLH-2004 protocol [59] consists of a two-week induction phase including etoposide, cyclosporin A, dexamethasone, intrathecal methotrexate, and intrathecal prednisone followed by a tapering phase of 6 weeks. If HSCT is planned, patients are placed on continuation therapy consisting of cyclosporine A and biweekly pulses of etoposide and dexamethasone [59]. Remission could be achieved in 78% of all patients treated with the HLH-94 protocol (which included cyclosporin A treatment only in the continuation phase) [68].
Immunotherapy
A more targeted immunotherapeutic protocol has mainly been used by a single center with similar survival rates compared to the HLH-94 protocol [69]. This regimen consists of ATG (rabbit) and methylprednisolone. There-after, cyclosporin A is given until HSCT, generally allowing tapering of methylprednisolone. Patients with central nervous system disease also receive intrathecal methotrexate and corticosteroids. First experiences with alemtuzumab have also yielded promising results [70, 71].
Therapy for 'secondary' HLH
There are not sufficient data to indicate if and which patients with 'secondary' HLH need the full treatment protocol. Although initial treatment with steroids alone or in combination with cyclosporin A may be justified in some patients, the timely use of more aggressive therapy is mandatory for a good outcome. In patients with MAS, immunosuppression with corticosteroids with or without cyclosporin A in most cases leads to a dramatic improvement of disease within days [72, 73]. Etoposide may be added if there is no response or highly active disease [38].
Control of the underlying disease is of key importance in the overall treatment concept. For rheumatological disease this in particular involves the use of IL-1 receptor antagonists and IL-6 antibodies in Still's disease and anti-TNF in some other rheumatological diseases [74, 75]. Whether these agents may also trigger MAS is a matter of debate. Of note, they have been shown to be of limited use in the treatment of primary HLH. Control of infectious disease is of similar importance both in primary as well as 'secondary' HLH. EBV-triggered HLH has been treated successfully with rituximab (anti-CD20 antibody) in addition to conventional therapy [27, 76–78]. Immunoglobulins, which may act against pathological antigens or cytokines, have been used as an adjunct in infection-triggered HLH [1, 27].
Hematopoietic stem cell transplantation
HSCT is recommended in genetic cases, in patients with recurrent HLH and disease progression despite adequate therapy [27]. For genetic cases it is the only curative option. To avoid delays in starting HSCT, HLA typing and donor search should be initiated as soon as the diagnosis of primary HLH is established [27]. Due to the highly inflammatory nature of HLH, myeloablative conditioning has been the standard regimen for many years. However, venoocclusive disease is a well-known complication when using busulfan-based protocols, occurring in up to 25% of HLH patients [79, 80]. Recently, reduced-intensity conditioning regimes have been used successfully and with a much lower incidence of veno-occlusive disease [81, 82]. Mixed chimerism can be a relevant limitation, but murine studies and some observations in patients indicate that a stable long-term level of about 10 to 15% donor chimerism may be sufficient to maintain remission [80, 81].
Outcome
Despite advances in therapy, up to 40 to 60% of children initially do not respond to treatment and die of HLH or die of infections or complications during therapy [27, 83]. Active HLH at the time of HSCT and central nervous system involvement are associated with a worse outcome [84, 85]. The HLH-94 protocol induced remission or allowed the patient to undergo HSCT in 71% of cases [85]. Patients with a positive family history and who received HSCT after induction therapy according to the HLH-94 protocol had a 5-year survival of 50% [85]. Patients with all forms of HLH who were treated according to the HLH-94 protocol had a 5-year survival of 54%. The patient group that underwent HSCT showed a 5-year survival of 66% [85]. The ATG-based therapeutic regimes induced remission of HLH in 73% of patients with FHL in one center, and patients had an overall survival after HSCT of 55% [69]. After induction therapy with varying therapeutic regimes, a survival rate of 58.5% 6 years after HSCT has been reported in a single center study [80]. Survival rates of 86% have been reported in EBV-associated 'secondary' HLH after HSCT in Japan [86]. Malignancy-related HLH had the worst prognosis of all forms of HLH. A Japanese survey on HLH in all age groups showed a 5-year survival of <15% in malignancy-associated HLH [6].
Long-term complications of HLH encompass therapy-related morbidity, especially after HSCT, and neurocognitive deficits. The latter can manifest months to years after HLH, but fortunately most patients can return to normal life [87].
Conclusion
HLH is a clinical syndrome of hyperinflammation that becomes frequently manifest in patients with certain underlying genetic diseases, but may also occur following particular triggers in patients with no known inherited disorder. Initial symptoms may be non-specific and misleading. Therefore, a high index of suspicion and a thorough clinical, immunological, and genetic work-up is required. Prompt initiation of adequate treatment is the key for survival. However, despite important advances in therapy, the overall survival rates remain unsatisfactory.
Outlook
With increased use of whole genome sequencing approaches, it is likely that the genetic bases of the remaining 10% of familial cases of hereditary HLH will soon be elucidated. Moreover, a potential genetic contribution to 'secondary' HLH is being investigated. Several animal models have been established to further elucidate the pathogenetic basis of the different conditions leading to the clinical picture of HLH. New observational clinical trials have been initiated or are being planned in North America and Europe. More targeted treatment approaches, including anti-cytokine therapies and gene therapy, are currently evaluated in mouse models and may be available for patient management in the future.
Note
This article is part of the series on Adult immunodeficiency, edited by Hans-Hartmut Peter. Other articles in this series can be found at http://arthritis-research.com/series/immunodeficiency
Abbreviations
- APC:
-
antigen-presenting cell
- ATG:
-
anti-thymocyte globulin
- CHS:
-
Chédiak Higashi syndrome
- CTL:
-
CD8+ cytotoxic T lymphocyte
- EBV:
-
Epstein-Barr virus
- FHL:
-
familial hemophagocytic lymphohistiocytosis
- GSII:
-
Griscelli syndrome type II
- HLH:
-
hemophagocytic lymphohistiocytosis
- HPSII:
-
Hermansky-Pudlak syndrome type II
- HSCT:
-
hematopoetic stem cell transplantation
- IFN:
-
interferon
- IL:
-
interleukin
- ITK:
-
IL-2 inducible T-cell kinase
- MAS:
-
macrophage activation syndrome
- NK:
-
natural killer
- SAP:
-
signaling lymphocytic activation molecule-associated protein
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor
- XIAP:
-
X-linked inhibitor of apoptosis.
References
Janka GE: Hemophagocytic syndromes. Blood Rev. 2007, 21: 245-253. 10.1016/j.blre.2007.05.001.
Filipovich AH: Hemophagocytic lymphohistiocytosis and related disorders. Curr Opin Allergy Clin Immunol. 2006, 6: 410-415. 10.1097/01.all.0000246626.57118.d9.
Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, Ohta H, Fujisaki H, Kobayashi M, Sakata N, Kawa-Ha K, Okada S, Tawa A: Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997, 89: 4100-4103.
Pachlopnik Schmid J, Cote M, Menager MM, Burgess A, Nehme N, Menasche G, Fischer A, de Saint Basile G: Inherited defects in lymphocyte cytotoxic activity. Immunol Rev. 235: 10-23.
Henter JI, Elinder G, Soder O, Ost A: Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991, 80: 428-435. 10.1111/j.1651-2227.1991.tb11878.x.
Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, Yamamoto K, Horiuchi H, Takada K, Ohshima K, Nakamura S, Kinukawa N, Oshimi K, Kawa K: Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007, 86: 58-65. 10.1532/IJH97.07012.
zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, Kabisch H, Schneppenheim R, Nürnberg P, Janka G, Hennies HC: Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005, 14: 827-834. 10.1093/hmg/ddi076.
zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, Strauss J, Kasper B, Nürnberg G, Becker C, Maul-Pavicic A, Beutel K, Janka G, Griffiths G, Ehl S, Hennies HC: Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009, 85: 482-492. 10.1016/j.ajhg.2009.09.005.
Ohadi M, Lalloz MR, Sham P, Zhao J, Dearlove AM, Shiach C, Kinsey S, Rhodes M, Layton DM: Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. 1999, 64: 165-171. 10.1086/302187.
Cetica V, Pende D, Griffiths GM, Arico M: Molecular basis of familial hemophagocytic lymphohistiocytosis. Haematologica. 2010, 95: 538-541. 10.3324/haematol.2009.019562.
Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A, de Saint Basile G, Kumar V: Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999, 286: 1957-1959. 10.1126/science.286.5446.1957.
Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, Al-Manjomi F, Al-Harbi M, Alangari A, Le Deist F, Gennery AR, Prince N, Cariou A, Nitschke P, Blank U, El-Ghazali G, Ménasché G, Latour S, Fischer A, de Saint Basile G: Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009, 119: 3765-3773. 10.1172/JCI40732.
Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachée-Chardin M, Chedeville G, Tamary H, Minard-Colin V, Vilmer E, Blanche S, Le Deist F, Fischer A, de Saint Basile G: Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003, 115: 461-473. 10.1016/S0092-8674(03)00855-9.
de Saint Basile G, Menasche G, Fischer A: Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010, 10: 568-579. 10.1038/nri2803.
Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, Villanueva J, Risma KA, Wei Q, Klein PS, Filipovich AH: Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial hemophagocytic lymphohistiocytosis. Blood. 2011, 118: 5794-5798. 10.1182/blood-2011-07-370148.
Clementi R, Emmi L, Maccario R, Liotta F, Moretta L, Danesino C, Arico M: Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood. 2002, 100: 2266-2267. 10.1182/blood-2002-04-1030.
Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, Bondzio I, Gross-Wieltsch U, Schündeln M, Schütz B, Woessmann W, Groll AH, Strahm B, Pagel J, Speckmann C, Janka G, Griffiths G, Schwarz K, zur Stadt U, Ehl S: Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. 2010, 95: 2080-2087. 10.3324/haematol.2010.029389.
Enders A, Zieger B, Schwarz K, Yoshimi A, Speckmann C, Knoepfle EM, Kontny U, Müller C, Nurden A, Rohr J, Henschen M, Pannicke U, Niemeyer C, Nurden P, Ehl S: Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood. 2006, 108: 81-87. 10.1182/blood-2005-11-4413.
Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, Wulffraat N, Bianchi D, Fischer A, Le Deist F, de Saint Basile G: Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000, 25: 173-176. 10.1038/76024.
Fischer A, Latour S, de Saint Basile G: Genetic defects affecting lymphocyte cytotoxicity. Curr Opin Immunol. 2007, 19: 348-353. 10.1016/j.coi.2007.04.006.
Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, Brandt SJ, Chotai D, Hodgman C, Solari RC, Lovett M, Kingsmore SF: Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996, 382: 262-265. 10.1038/382262a0.
Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, Kanegane H, Lopez-Granados E, Mejstrikova E, Pellier I, Galicier L, Galambrun C, Barlogis V, Bordigoni P, Fourmaintraux A, Hamidou M, Dabadie A, Le Deist F, Haerynck F, Ouachée-Chardin M, Rohrlich P, Stephan JL, Lenoir C, Rigaud S, Lambert N, Milili M, Schiff C, Chapel H, Picard C, de Saint Basile G, et al: Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. 2011, 117: 1522-1529. 10.1182/blood-2010-07-298372.
Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K, Linka RM, Shaag A, Elpeleg O, Borkhardt A, Resnick IB: IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica. 2011, 96: 472-476. 10.3324/haematol.2010.033910.
Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, Galicier L, Le Deist F, Rieux-Laucat F, Revy P, Fischer A, de Saint Basile G, Latour S: XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006, 444: 110-114. 10.1038/nature05257.
Seidel MG, CD27: A new player in the field of common variable immunodeficiency and EBV-associated lymphoproliferative disorder?. J Allergy Clin Immunol. 2012, 129: 1175-
van Montfrans JM, Hoepelman AI, Otto S, van Gijn M, van de Corput L, de Weger RA, Monaco-Shawver L, Banerjee PP, Sanders EA, Jol-van der Zijde CM, Betts MR, Orange JS, Bloem AC, Tesselaar K: CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. 2012, 129: 787-793. 10.1016/j.jaci.2011.11.013. e786
Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL: How I treat hemophagocytic lymphohistiocytosis. Blood. 2011, 118: 4041-4052. 10.1182/blood-2011-03-278127.
Janka G: Hemophagocytic lymphohistiocytosis: when the immune system runs amok. Klin Padiatr. 2009, 221: 278-285. 10.1055/s-0029-1237386.
Machaczka M, Vaktnas J, Klimkowska M, Hagglund H: Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011, 52: 613-619. 10.3109/10428194.2010.551153.
Maakaroun NR, Moanna A, Jacob JT, Albrecht H: Viral infections associated with haemophagocytic syndrome. Rev Med Virol. 2010, 20: 93-105. 10.1002/rmv.638.
Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C: Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007, 7: 814-822. 10.1016/S1473-3099(07)70290-6.
Duval M, Fenneteau O, Doireau V, Faye A, Emilie D, Yotnda P, Drapier JC, Schlegel N, Sterkers G, de Baulny HO, Vilmer E: Intermittent hemophagocytic lymphohistiocytosis is a regular feature of lysinuric protein intolerance. J Pediatr. 1999, 134: 236-239. 10.1016/S0022-3476(99)70423-3.
Ikeda H, Kato M, Matsunaga A, Shimizu Y, Katsuura M, Hayasaka K: Multiple sulphatase deficiency and haemophagocytic syndrome. Eur J Pediatr. 1998, 157: 553-554. 10.1007/s004310050876.
Gokce M, Unal O, Hismi B, Gumruk F, Coskun T, Balta G, Unal S, Cetin M, Kalkanoglu-Sivri HS, Dursun A, Tokatlı A: Secondary hemophagocytosis in 3 patients with organic acidemia involving propionate metabolism. Pediatr Hematol Oncol. 2011, 29: 92-98.
Lackner H, Urban C, Sovinz P, Benesch M, Moser A, Schwinger W: Hemophagocytic lymphohistiocytosis as severe adverse event of antineoplastic treatment in children. Haematologica. 2008, 93: 291-294. 10.3324/haematol.11704.
Abdelkefi A, Ben Jamil W, Torjman L, Ladeb S, Ksouri H, Lakhal A, Ben Hassen A, Ben Abdeladhim A, Ben Othman T: Hemophagocytic syndrome after hematopoietic stem cell transplantation: a prospective observational study. Int J Hematol. 2009, 89: 368-373. 10.1007/s12185-009-0261-1.
Gurkan A, Yakupoglu U, Yavuz A, Dikici H, Yakupoglu YK, Tuncer M, Demirbas A, Ersoy F: Hemophagocytic syndrome in kidney transplant recipients: report of four cases from a single center. Acta Haematol. 2006, 116: 108-113. 10.1159/000093640.
Ravelli A: Macrophage activation syndrome. Curr Opin Rheumatol. 2002, 14: 548-552. 10.1097/00002281-200209000-00012.
Ramanan AV, Schneider R: Macrophage activation syndrome following initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2003, 30: 401-403.
Davi S, Consolaro A, Guseinova D, Pistorio A, Ruperto N, Martini A, Cron RQ, Ravelli A: An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011, 38: 764-768. 10.3899/jrheum.100996.
Ravelli A, Magni-Manzoni S, Pistorio A, Besana C, Foti T, Ruperto N, Viola S, Martini A: Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005, 146: 598-604. 10.1016/j.jpeds.2004.12.016.
Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H: Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. 1996, 14: 207-232. 10.1146/annurev.immunol.14.1.207.
Stinchcombe JC, Griffiths GM: Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol. 2007, 23: 495-517. 10.1146/annurev.cellbio.23.090506.123521.
Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, Kambayashi T, Koretzky GA: Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest. 121: 2264-2277.
Jordan MB, Hildeman D, Kappler J, Marrack P: An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004, 104: 735-743. 10.1182/blood-2003-10-3413.
van Dommelen SL, Sumaria N, Schreiber RD, Scalzo AA, Smyth MJ, Degli-Esposti MA: Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity. 2006, 25: 835-848. 10.1016/j.immuni.2006.09.010.
de Saint Basile G, Fischer A: Defective cytotoxic granule-mediated cell death pathway impairs T lymphocyte homeostasis. Curr Opin Rheumatol. 2003, 15: 436-445. 10.1097/00002281-200307000-00011.
Palendira U, Low C, Chan A, Hislop AD, Ho E, Phan TG, Deenick E, Cook MC, Riminton DS, Choo S, Loh R, Alvaro F, Booth C, Gaspar HB, Moretta A, Khanna R, Rickinson AB, Tangye SG: Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol. 2011, 9 (11): e1001187-
Huck K, Feyen O, Niehues T, Rüschendorf F, Hübner N, Laws HJ, Telieps T, Knapp S, Wacker HH, Meindl A, Jumaa H, Borkhardt A: Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest. 2009, 119: 1350-1358. 10.1172/JCI37901.
Pasquier B, Yin L, Fondaneche MC, Relouzat F, Bloch-Queyrat C, Lambert N, Fischer A, de Saint-Basile G, Latour S: Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med. 2005, 201: 695-701. 10.1084/jem.20042432.
Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, Chiang SC, Marcenaro S, Meazza R, Bondzio I, Walshe D, Janka G, Lehmberg K, Beutel K, zur Stadt U, Binder N, Arico M, Moretta L, Henter JI, Ehl S: A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012, 119: 2754-2763. 10.1182/blood-2011-08-374199.
Lartigue A, Courville P, Auquit I, Francois A, Arnoult C, Tron F, Gilbert D, Musette P: Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J Immunol. 2006, 177: 1349-1354.
Haddad E, Sulis ML, Jabado N, Blanche S, Fischer A, Tardieu M: Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood. 1997, 89: 794-800.
Horne A, Trottestam H, Aricò M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, Henter JI, Histiocyte Society: Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2008, 140: 327-335. 10.1111/j.1365-2141.2007.06922.x.
Arico M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, Martinetti M, Rusca MP: Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996, 10: 197-203.
Busiello R, Adriani M, Locatelli F, Galgani M, Fimiani G, Clementi R, Ursini MV, Racioppi L, Pignata C: Atypical features of familial hemophagocytic lymphohistiocytosis. Blood. 2004, 103: 4610-4612. 10.1182/blood-2003-10-3551.
Solomou EE, Gibellini F, Stewart B, Malide D, Berg M, Visconte V, Green S, Childs R, Chanock SJ, Young NS: Perforin gene mutations in patients with acquired aplastic anemia. Blood. 2007, 109: 5234-5237. 10.1182/blood-2006-12-063495.
Trottestam H, Berglöf E, Horne A, Onelöv E, Beutel K, Lehmberg K, Sieni E, Silfverberg T, Aricò M, Janka G, Henter JI: Risk factors for early death in children with haemophagocytic lymphohistiocytosis. Acta Paediatr. 2011, 101: 313-318.
Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G: HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007, 48: 124-131. 10.1002/pbc.21039.
Allen CE, Yu X, Kozinetz CA, McClain KL: Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008, 50: 1227-1235. 10.1002/pbc.21423.
Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA: Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003, 281: 65-78. 10.1016/S0022-1759(03)00265-5.
Marcenaro S, Gallo F, Martini S, Santoro A, Griffiths GM, Arico M, Moretta L, Pende D: Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13-4 defect and discriminates between genetic subtypes of the disease. Blood. 2006, 108: 2316-2323. 10.1182/blood-2006-04-015693.
Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, Bechensteen AG, Boelens JJ, Celkan T, Farah RA, Hultenby K, Winiarski J, Roche PA, Nordenskjöld M, Henter JI, Long EO, Ljunggren HG: Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007, 110: 1906-1915. 10.1182/blood-2007-02-074468.
Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH: Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. 2002, 99: 61-66. 10.1182/blood.V99.1.61.
Marsh RA, Bleesing JJ, Filipovich AH: Using flow cytometry to screen patients for X-linked lymphoproliferative disease due to SAP deficiency and XIAP deficiency. J Immunol Methods. 2010, 362: 1-9. 10.1016/j.jim.2010.08.010.
Meeths M, Chiang SC, Wood SM, Entesarian M, Schlums H, Bang B, Nordenskjöld E, Björklund C, Jakovljevic G, Jazbec J, Hasle H, Holmqvist BM, Rajic L, Pfeifer S, Rosthøj S, Sabel M, Salmi TT, Stokland T, Winiarski J, Ljunggren HG, Fadeel B, Nordenskjöld M, Henter JI, Bryceson YT: Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood. 2011, 118: 5783-5793. 10.1182/blood-2011-07-369090.
Santoro A, Cannella S, Trizzino A, Bruno G, De Fusco C, Notarangelo LD, Pende D, Griffiths GM, Arico M: Mutations affecting mRNA splicing are the most common molecular defect in patients with familial hemophagocytic lymphohistiocytosis type 3. Haematologica. 2008, 93: 1086-1090. 10.3324/haematol.12622.
Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D, Ladisch S, Webb D, Janka G, Histocyte Society: Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002, 100: 2367-2373. 10.1182/blood-2002-01-0172.
Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, Neven B, Picard C, Blanche S, Fischer A: Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007, 120: e622-628. 10.1542/peds.2006-3164.
Machaczka M, Vaktnas J, Chiang SC, Bryceson YT: Alemtuzumab treatment for hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol. 2010, 7: doi:10.1038/nrclinonc.2010.40-c1
Strout MP, Seropian S, Berliner N: Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol. 2010, 7: 415-20. 10.1038/nrclinonc.2010.40.
Ravelli A, Viola S, De Benedetti F, Magni-Manzoni S, Tzialla C, Martini A: Dramatic efficacy of cyclosporine A in macrophage activation syndrome. Clin Exp Rheumatol. 2001, 19: 108-
Mouy R, Stephan JL, Pillet P, Haddad E, Hubert P, Prieur AM: Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr. 1996, 129: 750-754. 10.1016/S0022-3476(96)70160-9.
Vastert SJ, Kuis W, Grom AA: Systemic JIA: new developments in the understanding of the pathophysiology and therapy. Best Pract Res Clin Rheumatol. 2009, 23: 655-664. 10.1016/j.berh.2009.08.003.
Nigrovic PA, Mannion M, Prince FH, Zeft A, Rabinovich CE, van Rossum MA, Cortis E, Pardeo M, Miettunen PM, Janow G, Birmingham J, Eggebeen A, Janssen E, Shulman AI, Son MB, Hong S, Jones K, Ilowite NT, Cron RQ, Higgins GC: Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011, 63: 545-555. 10.1002/art.30128.
Milone MC, Tsai DE, Hodinka RL, Silverman LB, Malbran A, Wasik MA, Nichols KE: Treatment of primary Epstein-Barr virus infection in patients with X-linked lymphoproliferative disease using B-cell-directed therapy. Blood. 2005, 105: 994-996.
Imashuku S: Treatment of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis (EBV-HLH); update 2010. J Pediatr Hematol Oncol. 2011, 33: 35-39. 10.1097/MPH.0b013e3181f84a52.
Balamuth NJ, Nichols KE, Paessler M, Teachey DT: Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007, 29: 569-573. 10.1097/MPH.0b013e3180f61be3.
Meresse V, Hartmann O, Vassal G, Benhamou E, Valteau-Couanet D, Brugieres L, Lemerle J: Risk factors for hepatic veno-occlusive disease after high-dose busulfan-containing regimens followed by autologous bone marrow transplantation: a study in 136 children. Bone Marrow Transplant. 1992, 10: 135-141.
Ouachée-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, Neven B, Casanova JL, Tardieu M, Cavazzana-Calvo M, Blanche S, Fischer A: Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006, 117: e743-750. 10.1542/peds.2005-1789.
Marsh RA, Vaughn G, Kim MO, Li D, Jodele S, Joshi S, Mehta PA, Davies SM, Jordan MB, Bleesing JJ, Filipovich AH: Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010, 116: 5824-5831. 10.1182/blood-2010-04-282392.
Cooper N, Rao K, Gilmour K, Hadad L, Adams S, Cale C, Davies G, Webb D, Veys P, Amrolia P: Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. 2006, 107: 1233-1236.
Sung L, King SM, Carcao M, Trebo M, Weitzman SS: Adverse outcomes in primary hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2002, 24: 550-554. 10.1097/00043426-200210000-00011.
Horne A, Janka G, Maarten Egeler R, Gadner H, Imashuku S, Ladisch S, Locatelli F, Montgomery SM, Webb D, Winiarski J, Filipovich AH, Henter JI, Histiocyte Society: Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005, 129: 622-630. 10.1111/j.1365-2141.2005.05501.x.
Trottestam H, Horne A, Aricò M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, Henter JI, Histiocyte Society: Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 118: 4577-4584.
Ohga S, Kudo K, Ishii E, Honjo S, Morimoto A, Osugi Y, Sawada A, Inoue M, Tabuchi K, Suzuki N, Ishida Y, Imashuku S, Kato S, Hara T: Hematopoietic stem cell transplantation for familial hemophagocytic lymphohistiocytosis and Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in Japan. Pediatr Blood Cancer. 2010, 54: 299-306.
Shuper A, Attias D, Kornreich L, Zaizov R, Yaniv I: Familial hemophagocytic lymphohistiocytosis: improved neurodevelopmental outcome after bone marrow transplantation. J Pediatr. 1998, 133: 126-128. 10.1016/S0022-3476(98)70190-8.
Offenhauser C, Lei N, Roy S, Collins BM, Stow JL, Murray RZ: Syntaxin 11 binds Vti1b and regulates late endosome to lysosome fusion in macrophages. Traffic. 2011, 12: 762-773. 10.1111/j.1600-0854.2011.01189.x.
Freeman HR, Ramanan AV: Review of haemophagocytic lymphohistiocytosis. Arch Dis Child. 96: 688-693.
Histiocytosis Association. [https://www.histio.org/]
Acknowledgements
This work was supported by the Bundesministerium für Bildung und Forschung (BMBF 01 EO 0803 to SE) and the EU FP7 project CURE HLH (grant agreement no. 201461).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Bode, S.F., Lehmberg, K., Maul-Pavicic, A. et al. Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis . Arthritis Res Ther 14, 213 (2012). https://doi.org/10.1186/ar3843
Published:
DOI: https://doi.org/10.1186/ar3843