
Abstract
High-throughput sequencing has ushered in a paradigm shift in gastric microbiota, breaking the stereotype that the stomach is hostile to microorganisms beyond H. pylori. Recent attention directed toward the composition and functionality of this 'community' has shed light on its potential relevance in cancer. The microbial composition in the stomach of health displays host specificity which changes throughout a person's lifespan and is subject to both external and internal factors. Distinctive alterations in gastric microbiome signature are discernible at different stages of gastric precancerous lesions and malignancy. The robust microbes that dominate in gastric malignant tissue are intricately implicated in gastric cancer susceptibility, carcinogenesis, and the modulation of immunosurveillance and immune escape. These revelations offer fresh avenues for utilizing gastric microbiota as predictive biomarkers in clinical settings. Furthermore, inter-individual microbiota variations partially account for differential responses to cancer immunotherapy. In this review, we summarize current literature on the influence of the gastric microbiota on gastric carcinogenesis, anti-tumor immunity and immunotherapy, providing insights into potential clinical applications.
Similar content being viewed by others

Introduction
Gastric cancer (GC) is a very aggressive cancer and the third main cause of cancer-related mortality worldwide with more than 1 million diagnosed cases and 770,000 deaths globally in 2020 [1]. Approximately 40% of GC cases are identified at an advanced stage annually, with the 5-year survival rates for patients diagnosed in 2000 and 2014 merely 2.7% and 4.7%, respectively [2]. The infectious agent, Helicobacter pylori (H. pylori), is widely recognized as the most influential contributor to the onset of GC. An estimated 50% of people worldwide are infected with H. pylori [3] and approximately 90% of GC on a global scale result from the inflammation and damage caused by H. pylori infection [4]. Despite the well-established involvement of H. pylori in the process of gastric carcinogenesis, only fewer than 3% of H. pylori-infected patients eventually progress to GC, indicating the initial role of H. pylori rather than its exclusive influence on GC development. The scope of other contributing factors extends to genetic polymorphisms, environmental exposures, age, and gender [1, 5,6,7]. Recent progress has been made in the field, with the emergence of advanced next-generation sequencing technologies that unveiled the variations in diversity and community structure of gastric microbiota between individuals with good health and those afflicted with GC [8, 9]. These distinctions extend beyond the well-known H. pylori and include a broader range of microbial constituents, enlightening the potential role of these non-H. pylori gastric microbiota in the promotion of malignancy.
The microbiota encompasses a diverse array of bacteria, viruses, bacteriophages, fungi, and protozoa collectively shaping a complex ecosystem. These microbial communities, residing in various body habitats, including the oral cavity, gut, lung, skin and genitourinary tract, have been the subject of an extensive investigation regarding their contributions to maintaining homeostasis and their involvement in disease processes [10,11,12,13,14]. In particular, the mounting body of evidence highlights the role of the gut microbiota in driving cancer initiation and progression, orchestrating the intricate landscape of the tumor microenvironment (TME), and providing novel avenues for manipulating the microbiota to modulate anti-tumor immunity [15,16,17,18,19].
Owing to the relatively lower biomass of gastric microbiota, which quantifies merely 101 to 103 bacteria per gram of gastric content—significantly fewer than the 104 to 107 bacteria per gram found in the jejunum and ileum, and the 1011 to 1012 bacteria per gram in the colon, the gastric microbiota has received relatively limited attention in prior research [20]. Until recently, studies have conducted in vivo manipulations of gastric microbiota based on sequencing results, thus providing robust evidence to elucidate the causal relationship between perturbations in the gastric microbial ecosystem (referred to as "gastric dysbiosis") and the onset and progression of GC [21,22,23]. In this comprehensive review, we explore the emerging findings regarding the dynamic influence of gastric microbiota on GC and also address H. pylori as the most thoroughly investigated constituent of the microbiota, but are more inclined to discuss the evidence concerning its influence on the coexistence of other bacterial species in the stomach. We emphasize the pivotal role of gastric microbiota in the processes of gastric carcinogenesis, modulation of anti-tumor immune responses, and its potential implications in clinical application.
Microbial composition in the stomach
Due to low biomass of the microbiota and the unique and challenging growth environment in gastric mucosa, methods for isolation and cultivation of the gastric flora, as well as those for strain identification via morphological, biochemical and serotype characteristics, are greatly constrained. Therefore, in early assessment of gastric microbial composition, cultivation-independent methods such as molecular fingerprinting have been widely used [24]. Among these, temperature gradient gel electrophoresis (TGGE) was the most popular technique, which separate microbial DNA into a series of bands for inter-species comparisons based on differential physical–chemical properties of DNA fragments. Apparently, TGGE's resolution to discriminate between microbial taxa is very limited, leading to marked underestimation of gastric microbiome richness [25]. Additionally, other molecular methods such as fluorescence in situ hybridization (FISH), microarrays, and quantitative PCR exhibit high sensitivity in detecting low-abundance species [26,27,28]. However, the requirement for species-specific oligonucleotide probes or primers prevented large-scale and unbiased analysis of the entire microbial community.
In the past decade, the development of next-generation sequencing (NGS) technologies has dramatically improved the precision and coverage of taxa identification, eventually enabling deep profiling of gastric microbiome, and dissecting their interactions with the host. Among the NGS methods, 16S rRNA sequencing has been widely used, because the 16S gene contains both the conserved regions supporting phylogenetic classification at the phylum level, and the rapidly evolving regions suitable for finer taxonomic resolution. However, 16S rRNA gene sequencing can only identify known species, with a resolution limited to the genus level, and losses all the functional information for each species. By contrast, metagenomics relies on whole genome shotgun sequencing, thus enabling higher resolution for species identification and functional analysis. NGS analysis often synergize with other multi-omics methodologies such as metabolomics and proteomics, providing a comprehensive understanding of the structure and functions of gastric microbiota in health and disease [25, 29].
H. pylori initially identified in the 1980s was regarded as a milestone to investigate gastric microenvironment. Subsequent advancements in detection technologies have enabled the meticulous tracking of the microbial ecosystem and unveiled the microbial profile of the stomach (Table 1). This groundbreaking progress challenges the longstanding stereotype that the gastric mucosa is fundamentally sterile attributed to its inhospitable condition [24, 30].
Initial establishment of gastric microbial community
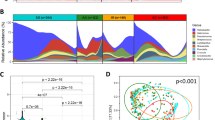
The stomach is not as bacteria-free as expected after birth. Microbes are present in the gastric aspirate from newborn infants, suggesting that the initial establishment of the gastric microbial community may occur at delivery or even earlier. The microbial composition of the neonatal stomach displays variability and potentially bears a connection to the mode of delivery (Fig. 1). Term neonates with vaginal delivery have significantly higher gastric microorganism abundance than those born by cesarean section. Additionally, the identification of the predominant constituent in the neonatal gastric microbial community highlights Lactobacillus Crispatusas the prevailing species within this ecosystem. This bacterium is crucial for maintaining vaginal health and becomes enriched within the vaginal mucosa during pregnancy [31, 39]. Whereas in preterm infants, the initial colonizers in the stomach are instead characterized by a high abundance of Ureaplasma [31, 32]. The detection of Ureaplasm from the maternal placenta and amniotic fluid predicts higher risk of preterm birth and neonatal mortality, and the persistent colonization of Ureaplasm in neonates may be associated with systemic infection and congenital diseases such as bronchopulmonary dysplasia [40]. However, Ureaplasm appeared only in the gastric aspirate of neonates in the first week, but disappeared afterward, probably due to intolerance of the gastric environment and prompt intervention with antibiotics [32]. Subsequent exposure to the environment and breast milk feeding flourish more bacteria species colonization in the stomach. A study involving 13 one-month-old preterm infants collected gastric contents and identified a predominant presence of Staphylococcus, Lactobacillus, and Serratia. These microbial components are recognized as significant constituents of breast milk [33, 41]. In addition, gastric microbial species of these preterm infants with long-term hospitalization were observed to overlap with a large proportion of nosocomial infection-associated bacteria from the hospital environment, such as Staphylococcus aureus, Staphylococcus epidermidis, Enterococcus faecium, Neisseria, Klebsiella pneumoniae, and Escherichia coli [32,33,34]. Environment factors seem to act as an essential selector for inhabitants of the stomach, and even for twins, the initial development of microbial community varies according to different exposure to environmental flora [42].

A multitude of factors influence the colonization of the gastric microbiota from prenatal development through adulthood. In the prenatal stage, the establishment of the initial gastric microbiome is shaped by factors such as the maternal microbiota, duration of gestation, and individual genetic makeup. Subsequently, during the neonatal period, variables including delivery mode, lactation method, and environmental exposures exert additional influence on early-life microbial composition. As individuals’s transition into adulthood, dietary preferences, obesity status, and geographic location persist as pivotal determinants in molding the architecture of the gastric microbiome
The initial gastric colonizers can shape the local and systemic immunity in early life. In animal models, it has been demonstrated that the offspring of mothers who were given antibiotics during pregnancy and breastfeeding had a decrease in microbial diversity in gastric contents accompanied by elevated IgG and IgM [43]. Consistently, Trevisi et al. also indicated that high-quality and complex initial microbial residence in the stomach not only favor oxyntic mucosa maturation and development of acid secretion but also mediate gastric immunity by modulating interferon response, antibodies production and immune cells maturation [44].
Gastric microbial composition in Health
As early as 2000, Monstein et al. initially described gastric “indigenous” microbiota other than H. pylori by temporal temperature gradient gel electrophoresis(TGGE). Enterococcus, Pseudomonas, Streptococcus, Staphylococcus, and Stomatococcuswere detected as major colonized members of genera in the stomach [24]. In 2005, a 16S rDNA sequencing of human stomach bacterial profile in America uncovered 128 phylotypes belonging to five predominant phyla which are Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria and Fusobacteria, accounting for over 90% of the total [30]. Generally, consistent phylum structures were identified in subsequent research in Hong Kong and Japan, whereas the American population showed a higher Firmicutes/Bacteroidetes ratio compared with Asians [35, 36]. High Firmicutes/Bacteroidetes ratio was previously mentioned in both animal study and clinical trial as a biomarker of gut dysbiosis in obesity [45, 46], and mice that were fed high-fiber or high-fat food exhibited overrepresented Bacteroidetes and Firmicutes for each [47, 48]. This inter-ethnic difference in Firmicutes/Bacteroidetes ratios within the gastric microbiota may be influenced by dietary habits. Moreover, a more extensive sequencing was performed in Sweden, an area with low H. pylori prevalence. This study recruited 171 healthy subjects and unveiled the taxonomic composition of their gastric microbial community. Sweden's population showed an unexpectedly low abundance of Proteobacteria [37], which is noteworthy as H. pylori falls within this bacterial clade. Genetic variants and external factors within the Swedish population may contribute to the development of a gastric environment that is less conducive to Proteobacteria colonization, thereby leading to a decreased incidence of H. pylori infections.
The most prevalent gastric mucosa-associated genera frequently described in the scientific literature are Neisseria, Prevotell, Haemophilus, Fusobacterium, Streptococcus, and Veillonella, though the abundance of each varies among subjects [24, 35,36,37]. Neisseria, Haemophilus, Fusobacterium and some species of Streptococcus and Prevotella are typical oral bacteria [8, 36, 49]. The swallowing action allows the oral microbiota to "seed" the lower GI tract. It is noteworthy that these prevailing genera within the stomach are scarcely detected in the lower gastrointestinal (GI) tract, which is primarily characterized by microbial communities dominated by Ruminococcus, Faecalibacterium, and Bacteroides [36, 38]. This large difference highlights the inaccuracy of regarding the lower GI and fecal microbiota as representative of the entire GI microbiota and underscores the importance of studying the GI microbiota by compartmentalization. Nonetheless, the outcomes of sequencing solely capture the configuration of the gastric microbiota at a specific juncture within the digestive process. It remains elusive to ascertain whether these microbial communities represent transient contamination or persistent colonization.
Gastric microbiome, H. pylori and Gastric cancer
GC, a complex and multifactorial disease, is known to develop through a multistep progression involving a series of pathological processes referred to as Correa’s cascade. The sequence of events typically begins with superficial gastritis (SG), followed by chronic atrophic gastritis (CAG), intestinal metaplasia (IM), and dysplasia, ultimately culminating in the occurrence of GC [50]. These sequential pathological changes contribute to the stepwise transformation of normal gastric mucosa into malignant tissue. In addition to H. pylori infection as the main driving factor, characteristic changes in the gastric microbiota concomitant with this process were observed (Table 2).
Gastric microbiome in gastric carcinogenesis
Gastric microbial diversity in gastric carcinogenesis
Recent studies have investigated the variances in gastric microbial diversity across Correa's cascade, ranging from healthy controls (HC) to SG, AG, IM, and ultimately GC [21, 22, 38, 49,50,51,52]. Among these investigations, two studies did not observe significant differences in gastric microbial diversity among these subgroups [22, 51]. Other larger-scale studies reported significantly reduced microbial richness and diversity in GC when compared with SG or HC groups. Whereas the discrimination of gastric flora diversity between GC and precancerous lesions (IM and dysplasia) did not achieve statistical significance in these studies [53, 54]. Microbial diversity may undergo a substantial decrease between the AG and IM stages [67]. As shown in previous studies, H. pylori eradication significantly decreased the risk of GC whereas its impact is constrained in patients with IM and dysplasia [68, 69]. This indicated that the optimal therapeutic window for antibiotic intervention along Correa's cascade lies before the transition from AG to IM and this timepoint may be correlated with the observed changes in the microbial composition and ecosystem dynamics within the gastric environment.
Gastric microbial composition in gastric carcinogenesis
In parallel, the variations in the composition of the microbiota along the spectrum spanning from healthy gastric mucosa to GC were explored in several studies (Table 2). Through inter-comparison between the gastric microbiome of GC, pre-cancerous stages and normal tissue, these investigations unveiled the distinctive dysbiosis that characterizes gastric carcinogenic progress. A noteworthy observation is that gastric microbial composition is significantly influenced by hypochlorhydria, a main pathological manifestation of atrophy. This alteration in gastric acidity may result in increased vulnerability to colonization by microorganisms from other origins, primarily the oral cavity and intestine [33, 43, 48, 50, 53, 54].
For instance, a prospective cohort study undertaken by Sung et al. recruited 587 H. pylori-positive patients. They reported the presence of a specific assembly of oral bacteria, including Prevotella, Rothia, Peptostreptococcus, Parvimonas and Streptococcus, which were implicated in the occurrence and prolonged course of GA and IM [70]. A similar enrichment of oral microbes was found in a case–control study performed on 89 IM patients, where the abundance of Johnsonella ignava, Peptostreptococcus stomatis, Neisseria elongata, and Neisseria flavescens was found to be significantly higher in IM cases [56]. Moreover, an insightful study was conducted on a well-defined cohort, recruiting 34 cases of SC, 20 cases of AG, 5 cases of gastric intraepithelial neoplasia(GIN), and 15 cases of intestinal-type GC. Remarkably, they revealed a noticeable increase in the abundance of specific oral bacteria, including Slackia, Selenomonas, Bergeyella, and Capnocytophaga, which continuously increased from SG to GC. Concurrently, intestinal bacteria including Romboutsia, Fusicatenibacter, Prevotellaceae-Ga6A1-group and Intestinimonas were observed to be overabundant in GIN patients [22].
The predominance of these “non-indigenous colonizers” persists in GC which may significantly characterize malignant transformation. Coker et al. detected distinct Operational Taxonomic Units (OTUs) that were associated with five oral-derived species, including Peptostreptococcus stomatis, Streptococcus anginosus, Parvimonas micra, Lackia exigua, and Dialister pneumosintes, which can significantly distinguish GC from SG [53]. In another investigation where 54 GC patients and 81 chronic gastritis patients were enrolled, GC-representative genera exhibited a remarkable overabundance of intestinal commensals such as Citrobacter, Clostridium, Lactobacillus, Achromobacter and Rhodococcus [57]. Even by autologous comparison, tumor microhabitats were characterized by the discernible increase of oral pathogen, Prevotella melaninogenica, Streptococcus anginosus and skin pathogen Propionibacterium acnes (P.acnes), setting them apart from both paired paracancerous and normal tissues [58].
Gastric microbiota as predictive markers in gastric cancer
The recognition of microbial features during gastric carcinogenesis instigates exploration into the correlation between gastric microbiota and the multifaceted aspects of GC, such as risk, progression and prognosis. A Korean study, comprising a cohort of 268 GC patients and 288 controls, identified H. pylori, P.acnes and Prevotella copri as significant risk factors for GC, as indicated by odds ratios (ORs) of 1.86, 4.7 and 2.54 respectively [9]. Concurrently, another case–control study employed Whole metagenomic shotgun sequencing (WMS) on the gastric biopsy samples, which were obtained from a cohort of Colombian individuals, including both high-risk (n = 10) and low-risk (n = 10) populations for GC. The results revealed that the typical soil bacteria including Bacillus, Actinomyces and Arthrobacter spp., and Keratinibaculum spp. that was over representative in males, as well as skin commensal Staphylococcus and oral microbe Streptococcus, associated with an increased risk of GC [59].
In addition, a prospective longitudinal study was conducted to investigate the predictive value of gastric microbiota in GC progression. This comprehensive investigation involved 43 participants who underwent initial gastroscopy, followed by regular monitoring through 1–2 yearly gastroscopies over a minimum duration of 5 years. The results showed that patients who progressed from IM to early gastric neoplasia (EGN) during the study period had significantly higher relative abundance of Moryella genus and Vibro genus at baseline, compared to those who did not exhibit such progression [51].
Regarding the prognostic assessment of GC based on the gastric microbiome, multiple retrospective studies delved into distinctions in gastric microbial composition between GC patients with favorable and unfavorable prognoses. Several investigations highlighted the association between heightened levels of Fusobacterium nucleatum (F. nucleatum) in tumor samples and unfavorable prognostic outcomes among GC patients [60, 71, 72]. Notably, in Lauren’s diffuse-type GC, F. nucleatum positivity was found to be associated with markedly diminished overall survival rates. The abundance of F. nucleatum demonstrated a positive correlation with patient age, although it exhibited no significant associations with gender, H. pylori infection status, tumor stage, or tumor location [71]. Survival analysis from an additional cohort study has indicated that colonization of F. nucleatum species correlates with poorer prognosis among late-stage GC patients with H. pylori positivity [72]. These findings were further corroborated by an additional study showing that the presence of F. nucleatum and Prevotella species in tumor specimens has most significant impact on prognosis [60]. Furthermore, emerging evidence has also suggested potential correlations between specific bacterial species and favorable prognoses in GC. Using weighted gene co-expression network analysis (WGCNA), a recent study unveiled that GC patients exhibiting higher microbial diversity experienced poorer outcomes compared to those with lower microbial diversity. However, Tissierella enrichment was frequently observed in TP53 wild-type tumors and was associated with a favorable prognosis in various tumor types including GC, breast cancer and lung adenocarcinoma [73]. Another study revealed that GC patients with favorable prognoses displayed elevated H. pylori abundance alongside a decrease in Halomonas and Shewanella abundance [74]. Moreover, the presence of Asinibacterium in non-tumor adjacent tissues correlated with improved overall survival in GC patients, suggesting a potential influence of the gastric microenvironment on the pathophysiology of GC [60].
Role of the gastric microbiota in gastric carcinogenesis
H. pylori in gastric carcinogenesis through the “hit and run” mechanism
H. pylori, a known Class I carcinogen in the development of GC, exerts a significant predisposing effect on carcinogenesis by employing various virulence factors [75]. Its remarkable feature of urease synthesis enables it to thrive within the acidic environment of the stomach. The primary mechanism underlying H. pylori-induced carcinogenesis involves the expression of two oncogenic effector proteins: vacuolating cytotoxin A (VacA) and cytotoxin-associated gene A(CagA) protein [76] (Fig. 2).

The "hit and run" mechanism posits a potential role for H. pylori in gastric carcinogenesis. The virulence factor VacA is internalized upon binding to cell surface receptors on host cells. This interaction initiates cell vacuolation and the subsequent release of cytochrome c from mitochondria within the cells, ultimately leading to cytotoxicity and cell death. The virulence factor CagA is translocated into the host cell through the T4SS. CagA drives cell hyperproliferation through three key mechanisms: It is phosphorylated by Src kinase, leading to the activation of mitogenic signaling pathways. CagA triggers the activation of c-Met and initiates the PI3K/Akt signaling cascade. CagA interacts with E-cadherin, disrupting the E-cadherin/β-catenin complex, thereby activating the Wnt signaling pathway. These pathways converge to induce the accumulation of β-catenin, which in turn may facilitate the overexpression of pro-oncogenic genes. The inhibition of PAR1b by CagA induces BRCAness, leading to double-strand breaks in the host cell. And CagL induces the separation of ADAM17 from α5β1 integrins, which subsequently mediates the inhibition of H, K-ATPase expression through NF-κB inhibitory binding, resulting in hyposecretion of gastric acid. This hypochlorhydria triggers gastric dysbiosis. The genetic instability and dysbiosis may replace H. pylori in maintaining subsequent tumor phenotype and promoting tumor progression
VacA, a potent vacuolating cytotoxin present in all H. pylori strains, is capable of inducing vacuolation in host cells. VacA-forming pores can target mitochondria, causing depolarization of the mitochondrial transmembrane potential and subsequent mitochondrial dysfunction. This event triggers the release of cytochrome c from the mitochondria, which ultimately leads to the initiation of the apoptotic pathway [77]. The cag pathogenicity island (cagPAI) contains genes responsible for the expression of a type IV secretion system (T4SS) and the oncogenic protein CagA [78]. Upon the action of T4SS, H. pylori translocates CagA into the cytosol of host cells, where it serves as a hub protein to disrupt multiple cellular signaling pathways. CagA, when phosphorylated by Src family kinases, can engage with SH2 domain-containing proteins, which are mostly effectors in the mitogenic signaling pathway [79, 80]. Concurrently, unphosphorylated CagA interacts with E-cadherin to disassociate the E-cadherin/β-catenin complex and activate β-catenin-dependent expression of pro-oncogenic genes [81]. In addition to triggering oncogenic signals, CagA also elicits genetic instability, thereby exacerbating the potential for oncogenic transformation. The CagA-mediated sequestration of the polarity-regulating kinase PAR1b, initially known for its disruption of cellular polarity via tight junction perturbation [82], has recently been elucidated to play a role in inhibiting BRCA1 phosphorylation. This inhibition results in the confinement of BRCA1 to the cytoplasm, called BRCAness, where it fails to perform DNA repair in the nucleus, thereby inducing DNA double-strand breaks (DSBs) and genetic instability. The PAR1b kinase inhibited by CagA also activates Hippo signaling, thereby providing DNA-damaged cells with a means to escape apoptosis and facilitating the repair of DSBs via mutagenic repair mechanisms. As the cells with BRCAness expand, the pro-oncogenic role of CagA is eventually replaced by genomic instability which gives rise to cells with cancer predisposition phenotype independent of CagA. Consequently, the neoplastic phenotype no longer relies on the presence of CagA [83].
Moreover, H. pylori seems to assume a pioneering role by establishing a low-acid environment conducive to the colonization of potential pathogenic microorganisms in the stomach. For instance, a longitudinal study showed that patients with successful H. pylori eradication but still with persistent inflammation exhibit an increase in oral bacteria and pathogenic bacterial load [70]. The mechanisms underlying H. pylori-induced hypochlorhydria were partially unveiled. During acute H. pylori infection, T4SS protein CagL plays a dual role: it facilitates the intracellular delivery of CagA by binding to α5β1 integrins and also influences the dissociation of the metalloenzyme ADAM17 from α5β1 integrins. CagL stimulates ADAM17 activation, initiating ADAM17-triggered NF-κB-mediated suppression of H,K-ATPase α subunit (HKα) [84]. Besides, pro-inflammatory cytokines IL-1ß and TNF-α released by immune cells elicited by H. pylori infection are also robust inhibitors on the gastric acid secretion of parietal cells [85].
The “hit and run” theory was initially introduced by Skinner et al. to elucidate how viruses contribute to carcinogenesis by promoting the accumulation of mutations and inducing genomic instability until the virus is no longer necessary for tumor maintenance [86]. Consistently, genetic instability and neutralized PH in the stomach caused by H. pylori infection induces the accumulation of oncogenic insults including driver mutations acquisition in cancer-predisposing cells and pathogenic bacteria colonization in the stomach, which collectively contribute to the establishment and persistence of a neoplastic phenotype. And the actions of H. pylori successively fade in malignancy maintenance [87]. Aforementioned longitudinal study reported that eradicating H. pylori is insufficient to completely reverse the progression of IM [70]. Moreover, a study incorporating 9 gastritis, 7 IM and 11 GC subjects showed a decrease in H. pylori abundance in GC while no significant differences in the abundance of other dominant species between cancer and non-cancer group [55]. Similar conclusions were found in a Portuguese study (54 GC and 81 CG) in which H. pylori load reduced with increased abundance of intestinal commensal in GC versus chronic gastritis [57]. Based on a pathology review conducted by Stewart et al., the prevalence of H. pylori was found to be as high as 90% in cases of active gastritis, but it decreased to a range of 30% to 72% in individuals with atrophic gastritis and to approximately 30% to 35% in those with intestinal metaplasia (IM) and dysplasia. Notably, only 24.6% of GC patients had detectable H. pylori [3]. The “hit and run” model of H. pylori was initially introduced and documented by Hatakeyama. On the one hand, it highlights the action of CagA in cell reprogramming by facilitating genetic and epigenetic alternation, on the other hand, elevated genetic instability evokes host cell protection mechanisms such as cell senescence and apoptosis, impeding the role of CagA in transformed cells, ultimately resulting in the diminishing influence of CagA over time. Likewise, in the context of reduced gastric acid secretion and the absence of H. pylori, invasive microbiota successfully colonizes the stomach, initiating a progressive replacement of H. pylori by other microbial species that eventually come to dominate the gastric environment [87].
The interplay between H. pylori and non-H. pylori microbiota
The presence of H. pylori influences the microecological landscape in the stomach. In turn, gastric microbiota can also modulate the outcome of H. pylori infection. This interplay between H. pylori and the microbiota throughout Correa's cascade is potentially involved in GC development.
H. pylori demonstrates a strong competitive advantage, rendering it the predominant microorganism in the gastric microbiota, constituting 40%–90% of the total gastric microbiota composition [30, 38, 57, 88]. The presence of H. pylori is inversely correlated with the alpha diversity of gastric bacteria [58, 88,89,90]. While the impact of H. pylori on alpha diversity can be reversible after the removal of H. pylori [70, 91]. Evidence from beta diversity analysis suggested that the presence of H. pylori infection leads to significant modifications to microbial composition in the stomach [89, 92]. Individuals infected with H. pylori exhibit a predominant composition of the same phyla as those without H. pylori, but with low levels of Actinobacteria, Bacteroidetes, and the overrepresentation of Proteobacteria, probably attributed to the influence of H. pylori [93]. Co-occurring and co-excluding networks showed the potential interplay between H. pylori and other gastric microbes. In SG, H. pylori manifests a pattern of co-exclusion with Methylobacillus and co-occurrence with Arthrobacter. Within the stomach of subjects with IM, H. pylori demonstrates co-exclusion with various members of the Firmicutes phylum, such as Ruminococcus, Bacillales, SMB53 and Lactobacillus, while concurrently displaying co-occurrence with Prevotella, Moryella and H. ganmani [53]. Among GC patients, a notable negative correlation persists between Lactobacillus and Helicobacter [23].
A study employing germ-free insulin-gastrin (INS-GAS) transgenic mice illuminated the crucial role of gastric microbiota in H. pylori-driven Correa's cascade progression. H. pylori infection in germ-free mice induced milder lesions and delayed progression to gastric intraepithelial neoplasia (GIN), in comparison to H. pylori-infected specific pathogen-free (SPF) INS-GAS mice [94]. Remarkably, it has been documented that gastric colonization involving H. pylori, along with a consortium of commensal microbiota encompassing species such as Clostridium spp., Bacteroides spp., and Lactobacillus murinus, led to the development of GC in INS-GAS mice to a degree on par with that observed in mice harboring a diverse microbiota. Moreover, the colonization of the stomachs of INS-GAS mice with complex microbiota even reduced the level of gastric H. pylori colonization. These findings indicate that the effective colonization with specific commensals seems to exert a greater influence than the overall microbial diversity in the context of H. pylori-associated GC [94, 95].
Several gastric microbiota with potential modulatory effects on the severity and outcomes of H. pylori infection have been identified [96,97,98,99,100,101]. For instance, Staphylococcus epidermidis and Streptococcus salivarius, both urease-positive bacteria, were isolated from individuals in low- and high-cancer-risk regions of Colombia respectively. Co-infection experiments in germ-free mice showed that neither bacterial strain significantly altered H. pylori abundance. However, a more severe pathological lesion was found in H. pylori-S. salivarius dual infection while H. pylori-S. epidermidis co-infection showed alleviation of tissue injury and inhibited gene expression of pro-inflammatory cytokines including IL-22, IL-17A, IL-1β, IFN-γ and TNF-α in contrast to H. pylori mono-infection [96].
Moreover, Weizmannia coagulans (BCF-01), a strain isolated from the healthy gastric mucosa, displayed strong anti-H. pylori activity. This strain could significantly alleviate H. pylori-triggered gastric dysbiosis and reduce inflammation following H. pylori infection. Mechanistically, it enhanced the expression of mucosal barrier proteins and inhibited the TLR4-NFκB-mediated pyroptotic pathway in macrophages [102]. Similarly, Lactobacillus gasseri (Kx110A1), another strain isolated from gastric mucosa, effectively suppressed the synthesis of TNF and IL-6 in macrophages upon H. pylori infection through downregulation of ADAM17 [98]. Also Lactobacillus acidophilus and Lactiplantibacillus plantarum, both indigenous gastric strains, attenuated mucosal inflammation in murine gastritis afflicted with H. pylori [99]. Furthermore, Streptococcus mitis, commonly found in the human stomach, can inhibit the growth of H. pylori and prompt its transformation into a coccoid form when co-cultivated with H. pylori [100, 101]. Variations in the prevalence of these bacteria interacting with H. pylori within the gastric microbial community may underlie the disparate susceptibilities of individuals to H. pylori infection and the development of H. pylori-propelled GC.
The exploration of mechanisms involving non-H. pylori microbiota in gastric carcinogenesis
A recent study by Kwon et al. demonstrated the successful reproduction of premalignant lesions in the stomach by transplanting the gastric microbiota from IM and GC patients to germ-free mice, even in the absence of H. pylori. This discovery underscores the contribution of gastric dysbiosis beyond the involvement of H. pylori to the induction of GC [103]. While a definitive mechanistic elucidation remains insufficient, numerous investigations have indicated the potential implication of specific bacterial species in GC development.
To begin with, Streptococcus anginosus (S. anginosus),a species originating from the oral cavity and known for its marked resilience in acidic conditions (pH 3–5), has shown significant overabundance in the gastric mucosa of GC patients across various human cohorts [53, 58, 104]. Administration of S. anginosus via oral gavage in conventional and germ-free mice induced progressive precancerous lesions, including gastritis, parietal cell atrophy, mucinous metaplasia, and dysplasia. Further, S. anginosus in carcinogen-induced or allograft GC models expedited tumor progression by disrupting gastric barrier function, stimulating cell proliferation and preventing apoptosis. Notably, the influence of S. anginosus on gastric carcinogenesis appeared to be species-autonomous, which relied on the interaction between a bacterial lipoprotein, TMPC, and Annexin A2 (ANXA2) receptor on gastric epithelial cells. This interaction instigated oncogenic MAPK signaling cascades, as evidenced by the phosphorylation of ERK and JNK, as well as activation of downstream oncogenic targets within gastric epithelial cells following S. anginosus infection [105].
Another frequently cited candidate associated with GC, showing significant overabundance in the gastric microbiota of GC patients, is Lactobacillus species [8, 22, 23, 54, 55, 57, 61, 62]. Some hypotheses suggested that Lactobacillus spp. may contribute to GC development by producing lactic acid, which could serve as an energy source for tumor cells, promote reactive oxygen species (ROS) production, and contribute to tumor angiogenesis [67, 106, 107]. Nevertheless, the current body of proofs falls short of providing definitive support for these hypotheses. A converse viewpoint has been proposed by several studies, suggesting that Lactobacillus spp. could play an anti-tumor role in GC. This includes their potential to activate the intrinsic apoptotic pathway in cancer cells [108], prevent H. pylori infection [109, 110], and regulate gastric dysbiosis [111]. Furthermore, recent observations presented an alternative perspective indicating that the overgrowth of Lactobacillus spp. may be a consequence rather than a cause of gastric carcinogenesis [112, 113]. Jin et al. conducted long-term treatment of INS-GAS mice with deoxycholic acid (DCA), a secondary bile acid that increases from chronic gastritis to intestinal metaplasia. DCA treatment accelerated the progression to IM and enriched the population of the Lactobacillus genus [113]. In light of these complexities, further scrutiny is indispensable to elucidate the multifaceted role of Lactobacillus spp. in GC.
Additionally, lipopolysaccharide (LPS)-producing bacteria were found to be present at the high relative abundance in the gastric microenvironment of patients suffering from bile reflux gastritis (BRG) and GC. Among these bacteria, an oral-derived pathogen, Prevotella melaninogenica (P. melaninogenica) was found to be prominently elevated [58, 114, 115]. The abundance of LPS-producing bacteria exhibited a positive correlation with taurodeoxycholic acid (TDCA), a secondary bile acid that can stimulate the growth of gastric epithelial cells by triggering a pro-inflammatory IL-6/JAK1/STAT3 signaling cascade. The gavage instillation of TDCA, LPS, and P. melaninogenica in mice induced gastric inflammation and pre-neoplastic lesions, indicating the potential involvement of LPS-producing microbes in the pathogenesis of GC [114]. Therefore, targeting these microbes through alternative interventions may hold promise as a preventive measure against BRG-associated GC.
Microbial species participating in the metabolism of nitrate and nitrite have been implicated in GC. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) analysis from multiple studies unveiled distinctive microbial functional characteristics in GC that are linked to heightened reduction reactions, converting nitrate to nitrite, a precursor of N-nitroso compounds (NOC) [57, 61, 63]. Indeed, bacteria possessing nitrate reductase enzymes such as Clostridium, Lactobacillus, and Neisseria were found to be significantly overrepresented in GC patients [23, 55, 57, 91]. Conversely, Nitrospira, a bacterium that oxidizes nitrite to nitrate, were found to progressively decrease in individuals with intraepithelial neoplasia (IN), IM and GC. This decrease may indicate the accumulation of nitrite during the progression of neoplastic lesions [52]. The buildup of intragastric nitrite and NOC facilitates malignant transformation by initiating DNA damage through the methylation of purine and guanine with electrophilic species, thereby increasing mutagenicity [116, 117].
A novel investigation unveiled distinctive mucin-microbiome profiles in association with clinical outcomes in GC. The results indicated that tumors displaying an intestinal mucin phenotype with upregulated MUC13 expression are associated with an unfavorable prognosis, whereas tumors characterized by dominant expression of MUC5AC or MUC6 exhibit a more encouraging outcome. The presence of specific oral or intestinal microbes was discovered to be dependent on the mucin phenotype. Notably, oral taxa such as Neisseria, Prevotella, and Veillonella showed a significant association with MUC13 overexpression [118]. In prior studies, MUC13 overexpression was documented in GC [119], and this dysregulated MUC13 signaling was shown to confer protection against apoptosis in colorectal cancer (CRC) cells through the activation of the NF-κB pathway [120], as well as to facilitate the progression of intrahepatic cholangiocarcinoma by activating the EGFR/PI3K/AKT pathways [121]. Nevertheless, its specific involvement in GC has yet to be thoroughly investigated. Whether such interactions between mucin and the microbiome causally contribute to gastric carcinogenesis remains an intriguing area for further exploration.
Microbiota-mediated epigenetic regulation represents a novel mechanism by which gastric bacteria may contribute to carcinogenesis [122]. For instance, tumors harboring F. nucleatum exhibited a trend towards decreased DNA methylation of the long interspersed nuclear elements 1 (LINE1) [71]. Kytococcus sedentarius, Actinomyces oris and Staphylococcus saccharolyticus were implicated in regulating DNA methylation of immune-related genes in gastric adenocarcinoma, which consequently promoted distal metastasis in GC. In an in vitro experiment, the co-culture of GC cells with Staphylococcus saccharolyticus was observed to enhance the proliferation and clonogenicity of GC cells by inducing loss of ZNF215 methylation imprinting [123].
Several microorganisms implicated in other cancers were observed to be also enriched in GC [23, 55, 60, 91]. For instance, F. nucleatum, a periodontal commensal, was reported to be involved in the carcinogenesis of CRC [124]. Two vitro studies provided preliminary insights into the potential role of F. nucleatum in the development of GC. Hsieh et al. revealed that F. nucleatum colonization results in the dysregulation of actin cytoskeletal dynamics, which in turn is likely to alter the motility of GC cells [72]. In another study, it was shown that F. nucleatum is capable of inducing the overexpression of exosomal HOTTIP from GC cells, which subsequently accelerates the progression of GC through the activation of the miR-885-3p/EphB2/PI3K/AKT signaling cascade [125]. However, the impact of F. nucleatum on GC lacks validation from in vivo animal studies, and the molecular mechanisms underlying this effect are not fully understood. In CRC, the carcinogenic effect is primarily mediated by its virulence factors [8, 64, 126, 127]. Fap2 lectin ensures the localization and enrichment of F.nucleatum in cancerous sites [128, 129]. And the interaction of FadA adhesin and E-cadherin of cancer cells over-activate the β-catenin/Wnt signaling pathway [130]. Additionally, LPS of F.nucleatum recognized by the Toll-Like Receptor 4/Myeloid Differentiation Primary Response 88 (TLR4/MYD88) system induces NF-κB signaling pathway activation [131]. Notably, sustained activation and abnormal modulation of the NF-κB signaling pathway play a crucial role in the maintenance of stem-cell-like prosperity of GC cells [131,132,133,134,135,136,137]. Except for F.nucleatum, H. pylori has been demonstrated to stimulate NF-κB signaling pathway activation via its distinctive effector molecule, ADP-glycerol-β-D-mannoheptulose [138, 139], Peptostreptococcus anaerobius and Bacteroides fragilis which were observed to increase in GC, can activate NF-κB signaling pathway via inducing intracellular synthesis of ROS and IL-17 respectively in CRC [140, 141]. However, it remains unclear whether these mechanisms of the bacteria are replicated in the acidic environment of the stomach.
Non-bacterial component in gastric cancer
Although the majority of investigations into the connection between the microbiome and GC have primarily focused on bacterial components, a limited number of studies adopting a pan-biological perspective have yielded insights into the tumorigenic potential of non-bacterial elements, conferring a more comprehensive depiction of the microbiome ecosystem within the context of cancer [142,143,144].
One published by Zhong et al. in 2021 initially characterized fungal features of GC by applying internal transcribed spacer (ITS) rDNA sequencing on 45 gastric cancerous biopsies and peritumor normal biopsies. Candida and Alternaria were detected as representatives of fungi in cancerous tissue whereas Saitozyma and Thermonyces were observed to decrease in GC. Although the causal role of fungus on gastric carcinogenesis has not been proved, they highlighted the role of Candida albicansas an effective biomarker to distinguish GC tissue from control [143]. Candida albicans is a common fungus in human microbiota that normally colonizes but can cause severe issues in immunocompromised patients [145]. Recently bioinformatics analysis of mycobiome performed by Dohlman et al. demonstrated the unique enrichment of Candida in GC tissues. Concerning endosymbiotic crosstalk, authors conducted trans-kingdom network analysis and identified Candida as “Keystone taxa” in cancer which is co-abundant with a cluster of tumor-specific microbiota including Streptococcus, Clostridium, and Lactobacillus in GC. Moreover, they revealed Candida-corresponding gene expression patterns in GC, where pro-inflammatory cytokines such as IL-8, IL-1β, and IL-6 were mainly upregulated, indicating the involvement of Candida in inflammation in GC. However, it is still elusive whether overrepresented Candida is a cause or secondary to the phenotype of GC [144].
The majority of Mycoplasma implicated in GC are Mycoplasma hyorhinis(M. hyorhinis), a recognized pathogen responsible for various swine diseases [127, 146]. Experimental studies involving co-culturing M. hyorhinis with host cells have shed light on its impact on GC cell lines. Lipoprotein P37, expressed on the membrane of M. hyorhinis, serves as the main virulence factor, significantly enhancing the invasiveness of GC cells. This enhancement is strongly correlated with the activation of the EGFR-dependent NF-κB signaling pathway and the activation of matrix metalloproteinase-2(MMP-2) [147, 148]. Besides, Lipid-associated membrane protein(LAMP) of M.hyorhin, recognized by TLR2, induces the monocyte-derived IL-1ß secretion in an NLRP3 inflammasome-mediated manner to enhance cell migration [149]. Furthermore, M.hyorhin-specific methyltransferases selectively act on CG and GATC sites may lead to pro-carcinogenic epigenetic alternation by silencing tumor-suppressing genes [150].
Microbial involvement in immunosurveillance of gastric cancer
In a state of homeostasis, the persistent mutualistic symbiosis between the host and commensal flora not only serves to safeguard the microbial ecosystem but also plays a crucial role in preserving immunological equilibrium. Nonetheless, when the intricate interplay goes awry due to dysbiosis or disruptions in the host system, opportunistic pathogens can seize the opportunity to initiate pathological processes. In this scenario, pathogen-associated molecular patterns (PAMPs), such as flagellin, LPS, peptidoglycan, and formylated peptides recognized by pathogen recognition receptors (PRRs) of host cells may initiate an aberrant immune response. Similarly, within the realm of cancer, the presence of tumor-enriched pathogens harbors the capacity to disrupt the balance between immune defenses and tumor evasion, ultimately facilitating the progression of the disease [75] (Fig. 3).

The impact of gastric microbiota on the modulation of both innate and adaptive immunity within the tumor microenvironment of GC. Innate immunity: Macrophage: Propionibacterium acnes drives the polarization of macrophages into the M2 phenotype by activating TLR4, leading to the secretion of IL-10. pDCs: Stenotrophomonas demonstrates a favorable correlation with the infiltration frequency of pDCs. NK cells: Fusobacterium nucleatum hinders the tumoricidal activity of NK cells by engaging in binding interactions with the inhibitory receptor TIGIT via its Fap2 lectin. ILC2s: H. pylori induces an upregulation in the expression of IL-7R on ILC2s and increases the levels of IL-7 in the gastric environment, thereby promoting the enhanced recruitment of ILC2s. Tumor-activated neutrophils: H. pylori infection induces an elevation in IL-8 secretion, which subsequently attracts the infiltration of neutrophils. F. nucleatum residing in GC cells activates the NF-κB /IL-17 axis, promoting neutrophils recruitment. Adaptive immunity: CD8 + TRM cells: Methobacterium exerts inhibitory effects on TGF-β secretion, leading to a disruption in the residence of CD8 + TRM cells within the gastric epithelium. Effector T cells: H. pylori impedes the recognition of tumor cells by effector T cells by upregulating the expression of PD-L1 within the tumor microenvironment. Treg cells: Selenomonas demonstrates a positive correlation with the frequency of infiltration of Treg cells. H. pylori induces the activation of TLR9 on pDCs, which may contribute to the overexpression of ICOSL and subsequently facilitate the recruitment of Treg cells. Breg cell: H. pylori is associated with the elevation of Breg cells infiltration in the gastric microenvironment
Potential role of gastric microbes on the innate immune system in gastric cancer
Innate immunity encompasses a diverse array of myeloid or lymphoid lineages-derived immune cells, cytokines and complement cascades, all working in concert to coordinate immune balance. The innate immune cells do not possess an intrinsic predisposition to either favor or impede tumors. Instead, their phenotype and function exhibit plasticity, influenced by contextual cues within specialized niches and the signaling molecules they encounter. Among these, the microbial pattern acts as a key component that significantly shapes the plasticity, recruitment, activity, and overall functionality of the innate immune system. In this regard, we present pivotal findings that shed light on the remodeling of innate immune cells by the microbiota within the TME of GC, and how these interactions exert profound effects on the course of cancer.
Macrophages are a significant component of the leukocytic infiltrate and exhibit phenotypic plasticity in GC. The M1 phenotype, triggered by IFN-γ and microbes-derived molecules, has anti-tumor activities. Conversely, the M2 phenotype, induced by IL-4, IL-10, and IL-13 cytokines, exerts an immunosuppressive effect, encouraging the immune escape of cancer cells [151]. A recent study revealed that P. acnes, a skin pathogen prevalent in GC, can drive the polarization of macrophages toward the M2 phenotype, facilitating GC cell migration in vitro. Pre-exposure of macrophages to P. acnes induces the expression of TLR4 on their surface. This, in turn, initiates an interaction between TLR4 and P. acnes, activating the downstream PI3K/AKT signaling pathway, which upregulates IL-10 expression, a canonical stimulus for promoting M2 polarization. Furthermore, co-localization analysis of M2 and TLR4 in GC tissue samples observed sustained expression of TLR4 in the majority of M2 macrophages. This observation implies that the overrepresentation of P. acnes within TME may be involved in a positive-feedback loop for M2 polarization, as self-secreted IL-10 further enhances the M2 phenotype [152].
Group 2 innate lymphoid cells (ILC2s), the principal subset of innate lymphoid cells in the stomach, respond to tissue injury and bacterial exposure to expand with the production of type II cytokines (IL-4, IL-5, IL-9, and IL-13) [153]. The mutualistic relationship between ILC2s cells and the microbiota is implicated in gastric homeostasis maintenance. ILC2s-derived IL-5 efficiently stimulates B cells to secrete IgA, which coats gastric bacteria for elimination. Reciprocally commensal bacteria such as Bacteroidales Family S24-7 actively participate in the preservation of the abundance and functionality of ILC2s within the stomach [154]. However, in TME, the microbiota-ILC2 interaction undergoes dysregulation, leading to a pronounced skew favoring a pro-tumor activity. The ILC2s in the peripheral blood of GC patients were found to exhibit an immunosuppressive phenotype [155]. And Li et al. reported that H. pylori infection induces the overexpression of transcription factor GATA-3 in lymphocytes in GC, contributing to the reprogramming of immune responses towards a dominant type 2 immunity, which is predominantly mediated by immunosuppressive ILC2s [156]. Simultaneously, H. pylori infection elicits an upregulation of the surface expression of interleukin-7 receptor (IL-7R) on ILC2s and induces an elevation in the gastric levels of IL-7, which synergistically enhances the recruitment and propagation of ILC2s in the stomach, potentially exacerbating the immunosuppressive nature of the TME [154, 157].
Tumor-activated neutrophils (TANs), a subset of granulocytes, constitute an immune-inhibitory milieu and are also recruited in response to microbial influences in TME [158]. TANs orchestrate a multifaceted role in promoting tumorigenesis by producing pro-angiogenic factors [159], enhancing the metastatic propensity of cancer cells [160] and suppressing anti-tumor immune responses [161]. Their mobilization in GC can be mediated through chemotaxis along the CXCL6/CXCL8-CXCR1 pathway [162]. Studies highlighted that H. pylori-induced upregulation of hepatocyte growth factor and IL-8 (CXCL8) triggers the infiltration of neutrophils in chronic gastritis and GC [163]. Moreover, IL-17, a cytokine abundant within the mucosa, was also demonstrated to engage in crosstalk with cancer cells, coordinating the recruitment of neutrophils [164, 165]. The intracellular inhabitation of F. nucleatum in GC cells was observed to trigger the NF-κB /IL-17 signaling axis, leading to neutrophil recruitment and facilitating immune evasion by tumor cells [166].
In addition, several bacteria, despite their roles remaining partially understood, were identified in association with specific immune cell populations within TME of GC. For instance, an opportunistic pathogen, Stenotrophomonas was found to exhibit a positive correlation with the infiltration frequency of BDCA2 + plasmacytoid dendritic cells (pDCs) in GC tissues [167]. These pDCs, existing in an immature state, are known to positively regulate the activity of regulatory T cells (Treg), thereby promoting the suppression of anti-tumor immunosurveillance [168, 169]. Likewise, Fusobacterium sp. infection showed a positive association with the lymphocyte influx in GC [126]. The direct immunosuppressive impact of F. nucleatum on human Natural Killer (NK) cells were elucidated in CRC, involving its interaction with the inhibitory receptor TIGIT via the virulence protein Fap2 [170]. However, the applicability of this immunosuppressive mechanism in GC requires further investigations to validate.
Potential role of gastric microbes on the adaptive immune system in gastric cancer
The adaptive immune system, composed of T-cell-mediated and B-cell-mediated immunity, plays an important role in the surveillance and defense against neoplastic growth. The intricate interplay between the gastric microbiota and the adaptive immune system emerges as a significant determinant in shaping the anti-tumor immune response, thus offering the potential to modulate the dynamic process of GC development and progression [22, 65, 126].
The plasticity of T cells is dependent on various stimuli and signals encountered during lineage differentiation and activation [171]. Studies illustrated the impact of gastric microbiota on the balances of effector and Tregs as well as the regulation of co-stimulatory and co-inhibitory signal transmission [75, 172, 173]. For instance, the overabundance of Methylobacterium in GC tissue, highly associated with unfavorable prognostic outcomes in GC patients, was demonstrated to advance GC progression by exhausting CD8 + tissue-resident memory T cells (TRMs) in TME [65, 66]. TRMs, distinguished by their elevated expression of immune checkpoint molecules and effector proteins, serve as a reserve force in anti-tumor immunity and was recognized as a favorable prognostic indicator linked to prolonged patient survival in prior studies [174]. Animal experiments unveiled Methylobacterium's capacity to diminish CD8 + TRMs by downregulating TGFß. This is significant since TGFß plays a pivotal role in the induction of CD103 expression, an essential prerequisite for the TRMs generation and enduring residence within the epithelial tissue [65].
Another bacterium that hampers T cell-mediated anti-tumor immunity is H. pylori. Aydın et al. reported an upregulation of immune-checkpoint inhibitor proteins (PD-1/PD-L1) in H. pylori-positive GC samples [175]. Experimental studies unveiled a putative mechanism underlying H. pylori-mediated PD-L1 expression. Using GC organoids, it was discovered that CagA from H. pylori stimulates increased secretion of the signaling molecule Shh, thereby augmenting the Hedgehog signaling pathway. This intricate cascade of events has the potential to modulate PD-L1 expression by regulating the mTOR pathway [176, 177].
Tregs emerge as pivotal orchestrators of the balance of immune tolerance and immune activation in homeostasis. However, within the complex landscape of cancer, a perturbation unfolds, where with the dominance of Treg cells surpasses that of effector T cells, which skews TME towards a state of immunosuppression. This dysregulation, in turn, allows cancer cells to evade vigilant immune surveillance [178]. Foxp3 + Tregs were reported to be over-represented in gastric tumoral and peritumoral samples compared with normal tissue. Selenomonas, a typical gingival bacterium, enriched in GC, exhibited a positive correlation with the elevated level of the Foxp3 + Tregs as shown in Pearson's correlation analysis. This suggests its potential contribution to Tregs accumulation [22, 167]. Moreover, a strong association was observed between H. pylori infection and the presence of ICOS + Tregs, of which higher level was linked to an unfavorable prognosis in late-stage GC [179]. Within the realm of co-stimulatory receptors belonging to the CD28 family, ICOS stands out as a distinct subtype, characterized by its unique ability to enhance the efficiency of producing immunosuppressive cytokines, including IL-10 and TGF-β. The presence of H. pylori in the gastric environment sets off a chain of events, activating TLR9 and instigating the induction of ICOS ligands on pDCs. This process serves as a compelling signal summoning ICOS + Tregs to infiltrate the TME of GC [180].
It was established that B lymphocytes act as anti-tumor immune agents by producing counterpart antibodies [181]. The immunoglobulin pool and repertoire can be influenced by microbial exposure, which may affect B cell recognition and function of tumor cells [182]. A subtype of B cell identified as B regulatory cells (Breg) exhibit a series of immunosuppressive features including synthesis of IL-10, IL-35 and TGF-β, counteracting the activity of effector T cells and NK cells, which are implicated in the immune evasion mechanisms employed by tumor cells [183, 184]. Evidence showed that the density of IL-10-producing B cells was elevated in the presence of H. pylori [185]. However, how Bregs respond to H. pylori and other microbes is still less well-studied.
The impact of microbiota on cancer immunotherapy
Immunotherapy that aims to boost the body's immune system to target and eliminate cancer cells has been well-established in clinics. However, it only works well for a small subset of cancer patients, underscoring substantial challenges in achieving favorable outcomes, as a significant proportion fails to mount an adequate response or develops treatment resistance [186]. Robust evidence highlighted the regulation of the microbiome on immune surveillance, inspiring the investigation into the impact and application of microbes on immunotherapy [75, 175, 187]. Given considerable inter-individual variability in the microbiome, the patient's distinctive microbial profile may serve as a pivotal factor in predicting and optimizing the therapeutic response to immunotherapy.
Gut microbiota and immunotherapy
The lion's share of experimental investigations exploring microbiota's impact on immunotherapy has revolved around gut microbiota, revealing several microbial taxa associated with immunotherapy efficacy [188,189,190,191,192]. For instance, Bifidobacterium genus, Bacteroides fragilis, Faecalibacterium spp., Akkermansia muciniphila, Ruminococcaceae and Lachnospiraceae were frequently identified as “favorable” bacteria to improve immune checkpoint inhibitors (ICIs) in multiple solid tumors by potentiating anti-tumor immunity [188, 193,194,195,196,197,198], while the high abundance of Bacteroidales was reported to be associated with poor response and weakened tumor-specific immune response [192, 195]. In GC patients, Peng et al. observed an increase in microbial taxa associated with favorable responses to ICIs, including Prevotealla, Bifidobacterium, Bacteroides, Ruminococcaceae, and Lachnospiraceae, whereas non-responders showed a higher abundance of Megamonas, Butyricimonas, Lachnospiraceae_UCG-001, and Agathobacter [199]. This study exclusively concentrated on gut microbial alternation, even though the modulation of the local immune response in the stomach by the gastric microbiota is anticipated to have a potential influence on the efficacy of ICI.
The impact of H. pylori infection and eradication on Immunotherapy
Recently, the impact of H. pylori in gastric microbiota on immunotherapy is increasingly becoming a topic of great interest. Oster et al. found that the responsiveness of ICIs is hampered by H. pylori infection in both MC38 colon adenocarcinoma and B16-OVA melanoma-bearing mice model as well as non-small cell lung cancer (NSCLC) patients. The tumor size in mice with H. pylori infection was much larger than those without infection after anti-CTLA4 or anti-PD-L1 treatment. Likewise, following treatment with anti-PD-1, the median survival time of NSCLC patients with H. pylori seropositivity was found to be significantly shorter in comparison to their seronegative counterparts. The mechanistic investigation in murine models unveiled that the infection causes the hypoactivation of DCs, ultimately leading to compromised activation and proliferation of tumor-specific CD8 + T cells. Concurrently, a decrease in the abundance of monocyte-derived cells and a noteworthy suppression of genes under the regulation of type I IFN, IFNγ, and IL-6 were detected within the TME of NSCLC patients who exhibited resistance to ICIs therapy and had pre-existing H. pylori infection [200]. The other two retrospective cohort studies confirmed the negative impact of H. pylori infection on the response to ICIs. Both ICI-treated melanoma and GC patients with H. pylori seropositivity experienced lower overall survival rates and worse treatment outcomes than H. pylori-seronegative patients [201, 202].
Notably, despite the adverse impact of H. pylori on immunotherapy, Oster et al. were unable to restore the efficacy of immunotherapy even after the successful eradication of H. pylori. They attributed this outcome to the decrease in immune-boosting bacteria resulting from non-specific antibiotics (ATB) treatment [200]. This finding is consistent with many clinical investigations that demonstrated a correlation between the use of antibiotics and decreased clinical efficacy of ICIs [190, 203,204,205]. Regarding the taxonomic composition of the gut microbiome post-eradication of H. pylori, several studies reported a gut dysbiosis characterized by a reduction in alpha diversity [206, 207] and Bifidobacterium abundance [208, 209]. However, longitudinal studies observed that these effects are transient, with a gradual recuperation of alpha diversity over time [210] and even favorable changes in fecal microbiota composition observed in patients, such as a noteworthy enrichment of Bifidobacterium and a decrease in Bacteroidales, after a period of 6 months post-eradication [91]. Moreover, different time windows of exposure to ATB before ICI initiation may exhibit different therapeutic outcomes. A comprehensive meta-analysis conducted by Lurienne et al. found that ATB exposure within 60 days of ICI administration significantly reduced its effectiveness in NSCLC patients, while exposure more than 90 days prior did not result in worse clinical outcomes [211].
Although the impact of antibiotics in H. pylori eradication on immunotherapy needs to be explored in prospective preclinical and clinical studies that include multiple time points, there seems to be agreement that antibiotics lead to transient ICI-unfavorable perturbations of microbial homeostasis, which should be considered in immunotherapy regimens for cancer patients.
The therapeutic potential of H. pylori in immunotherapy
In addition to the immunosuppressive effects of H. pylori infection within the TME, multiple researchers have delved into the strategic harnessing of its immunostimulatory property to augment the therapeutic efficacy of immunotherapy [212,213,214,215].
Helicobacter pylori neutrophil-activating protein (HP-NAP) is a virulence factor in H. pylori, known for its ability to activate neutrophils and induce reactive oxygen species via TLR2 activation [216]. HP-NAP activates neutrophils and monocytes, promoting IL-12 and IL-23 production, which polarizes a CD4 + T cell response into an anti-tumor Th1 response characterized by increased IFN-γ and TNF-α [217]. It also aids DC maturation and shifts macrophages towards an anti-tumoral phenotype [218]. These potent immunogenic properties make HP-NAP a promising candidate for the development of novel cancer immunotherapeutic applications.
Given these, Codolo et al. conducted the first investigation of the therapeutic potential of HP-NAP in bladder cancer-bearing mice. Peritumoral administration of HP-NAP resulted in a reduction of tumorigenesis, promotion of tumor necrosis, and augmentation of CD4 + and CD8 + cell populations secreting IFN-γ within both tumor sites and associated lymph nodes [219]. In subsequent studies, the efficacy of Hp-NAP in various formulations was extensively explored for its anticancer properties. Hp-NAP recombined with the maltose binding protein of Escherichia coli(rMBP-NAP) stimulated TLR2-mediated Th-1-dependent anti-tumor immunity in hepatoma, sarcoma [220], and metastatic lung cancer [221]. Recombinant HP-NAP-embedded Chitosan nanoparticles (Chi-rNap) was reported to reduce breast tumor growth by enhancing cytokine production and shifting immune functionality towards tumor-killing [222]. And the genetic manipulation of oncolytic viruses (OVs) to HP-NAP, harnessing the selective oncolysis of OVs with the immunomodulatory properties of HP-NAP, was demonstrated to amplify anti-tumor immune responses in breast cancer and neuroendocrine tumors [212, 213]. Moreover, a recent study introduced chimeric antigen receptor (CAR) T cell-bearing secretory HP-NAP as a solution for CAR-T's limited effectiveness in solid tumors. CAR(NAP) T cells prompted DC maturation, elicited bystander T-cell responses, and activated cytotoxic CD8 + T cells against multiple tumor antigens, not just the CAR's target. Importantly, CAR(NAP) T cells didn't increase non-specific toxicity or hinder CAR T cell therapy, offering promising clinical potential [214]. These remarkable findings highlight the immense potential of H. pylori in revolutionizing immunotherapy, although practical applications in GC are pending.
Conclusion and prospect
Growing numbers of studies in recent years have uncovered the alternation of gastric microbiota during gastric carcinogenesis, particularly changes in diversity and composition. These changes may serve as promising biomarkers for clinical applications of GC. Additionally, the GC-associated microbial community actively helps tilt TME towards a more immunosuppressive state, contributing to the evasion of cancer cells from immune detection and clearance. Notably, H. pylori possesses an intricate immunomodulatory mechanism. On one hand, its immunosuppressive nature poses a detrimental impact on the effectiveness of immunotherapy. On the other hand, the virulence factors of H. pylori exhibit remarkable immune-stimulating properties, holding promise as adjunctive mediators for immunotherapy.
However, the area is still in its infancy and the tasks trying to elucidate the underlying mechanism through which gastric microbiota influence the carcinogenic process and mediate immunomodulation are challenging. For instance, past retrospective investigations presented intriguing and contentious evidence for linking gastric microbes and cancer. This presumably reflects the co-evolutionary dynamics of the host's immune system, its symbiotic microbiota, and tumorigenic processes, and such dynamic changes are typically unable to be captured by cross-sectional comparison. Concurrently, the variability in sequencing methodologies presents a significant hurdle in attaining a consensus regarding the landscape of the gastric microbiota. Thus, it is necessary to allocate increased research efforts to this area, including the establishment of multi-center longitudinal cohort studies and the application of standardized sequencing tools with the capacity to dissect the entire metagenome at the strain level (e.g., emerging third-generation sequencing technologies [223] and single-cell microbial sequencing [224]).
Furthermore, despite the extensive interest in the involvement of non-H. pylori gastric microbiota in GC, there is limited evidence regarding their causality with GC and their roles in the TME [173]. In light of this, there is a compelling need for in-depth research to monitor the co-evolutionary dynamics within the gastric microbiota. These researches are aimed at achieving a more comprehensive understanding of its multifaceted role in the tumorigenesis process. It is noteworthy to underscore that this exploratory venture should transcend the bacterial taxonomic level and encompass the full spectrum of microorganisms.
Exploring the clinical translation related to microbiota is a pivotal area of future research. Nevertheless, significant challenges persist in the clinical application of gastric microbiota. The invasive nature of endoscopy poses a substantial impediment to early GC screening, potentially limiting the clinical adoption of gastric mucosal microbiota testing as a viable biomarker. Moreover, current studies investigating the influence of gastric bacteria on cancer prevention and therapeutic response are primarily focused on H. pylori, with other strains remaining underexplored. Additionally, the applicability of established microbiota-based therapeutic strategies, including prebiotics, symbiotic microbial consortia, microbiota-derived metabolite therapy, microbiota-targeted interventions, and microbiota engineering, to the gastric microbiota ecosystem remains unclear, particularly given the stomach's unique acidic conditions. Interestingly, despite these challenges, emerging technologies and accumulating evidence indicate some limited progress in this field. For example, the detection of GC-related microbial translocation in the blood [225], oral cavity [56], and feces of GC patients has shown promising predictive significance and offers a non-invasive means for early GC screening [226]. Regarding of microbiota-targed treatment, studies have investigated strategies to target H. pylori, such as developing biomaterials that utilize the gastric acid environment as an activation mechanism to selectively eradicate H. pylori from the stomach [227, 228], and genetically modifying probiotics to release H. pylori-guided antimicrobial peptides [229]. These approaches have the potential to eliminate H. pylori, all while avoiding the induction of gastric dysbiosis. However, extensive clinical trials, underpinned by rigorous preclinical experiments, are essential to assess the safety and efficacy of these strategies and enhance our comprehension of GC prevention and treatment through the modulation of gastric microorganisms, extending beyond H. pylori (Fig. 4).

Advancing the Translation of Gastric Microbial Research: from clinic to clinic. The multicenter longitudinal cohort studies employ standardized strategies crafted to enhance the precision of data collection and analysis related to gastric microbiota. By incorporating a diverse range of genetic and non-genetic methodologies and considering potential influencing factors affecting outcomes, while recognizing inter-individual variability, the initiative aims to advance the comprehension of the intricate interplay between the microbiota and host co-evolution. Through the integration of both in vitro and in vivo studies, the research goes beyond mere identification of associations, correlations, and predictions. Instead, it delves into the exploration of mechanistic underpinnings behind causal relationships, providing profound insights at the molecular level. This concerted effort lays the foundation for personalized diagnostics and microbiota-based synergistic therapies in the intervention of gastric cancer
Going forward, the personalized treatment of cancer will increasingly integrate the microbiome with genomic, transcriptomic, and metabolomic data in the future. This multi-level and multi-dimensional approach will enable a more comprehensive and nuanced understanding of the interplay between the gastric microbiome and GC, facilitating the development of targeted and effective treatment strategies.
Availability of data and materials
Not applicable.
Abbreviations
- GC:
-
Gastric cancer
- TME:
-
Tumor microenvironment
- TGGE:
-
Temperature gradient gel electrophoresis
- FISH:
-
Fluorescence in situ hybridization
- NGS:
-
Next-generation sequencing
- WGCNA:
-
Weighted gene co-expression network analysis
- GI:
-
Gastrointestinal
- HC:
-
Healthy control
- AG:
-
Atrophic gastritis
- SG:
-
Superficial gastritis
- CAG:
-
Chronic atrophic gastritis
- IM:
-
Intestinal metaplasia
- GIN:
-
Gastric intraepithelial neoplasia
- OTUs:
-
Operational Taxonomic Units
- ORs:
-
Odds ratios
- WMS:
-
Whole metagenomic shotgun sequencing
- EGN:
-
Early gastric neoplasia
- NS:
-
Not significant
- VacA:
-
Vacuolating cytotoxin A
- CagA:
-
Cytotoxin-associated gene A
- cagPAI:
-
Cag pathogenicity island
- T4SS:
-
Type IV secretion system
- SH2:
-
SRC homology 2 domain
- PAR1b:
-
Partitioning-defective 1
- BRCA1:
-
Breast cancer susceptibility gene 1
- DSBs:
-
DNA double-strand breaks
- ADAM17:
-
Metalloproteinase domain 17
- NF-κB:
-
Nuclear factor-κB
- HKα:
-
H,K-ATPase α subunit
- IL-1ß:
-
Interleukin-1ß
- TNF-α:
-
Tumor Necrosis Factor-α
- INS-GAS:
-
Insulin-gastrin
- SPF:
-
Specific pathogen-free
- ANXA2:
-
Annexin A2
- IFN-γ:
-
Interferon-γ
- ROS:
-
Reactive oxygen species
- DCA:
-
Deoxycholic acid
- LPS:
-
Lipopolysaccharide
- BRG:
-
Bile reflux gastritis
- TDCA:
-
Taurodeoxycholic acid
- JAK1:
-
Janus kinase 1
- STAT3:
-
Signal transducer and activator of transcription 3
- PICRUSt:
-
Phylogenetic investigation of communities by reconstruction of unobserved States
- NOC:
-
N-nitroso compounds
- MUC:
-
Mucin
- LINE1:
-
Long interspersed nuclear elements 1
- CRC:
-
Colorectal cancer
- HOTTIP:
-
HOXA transcript at the distal tip
- TLR:
-
Toll-Like Receptor
- MYD88:
-
Myeloid Differentiation Primary Response 88
- NOD1:
-
Nucleotide-binding Oligomerization Domain 1
- ITS:
-
Internal transcribed spacer
- MMP-2:
-
Matrix metalloproteinase-2
- LAMP:
-
Lipid-associated membrane protein
- NLRP3:
-
Nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing protein 3
- PAMPs:
-
Pathogen-associated molecular patterns
- PRRs:
-
Pathogen recognition receptors
- ILC2s:
-
Group 2 innate lymphoid cells
- IL-7R:
-
Interleukin-7 receptor
- TANs:
-
Tumor-activated neutrophils
- pDCs:
-
Plasmacytoid dendritic cells
- Treg:
-
Regulatory T cells
- NK:
-
Natural Killer
- TIGIT:
-
T-cell immunoreceptor with Ig and ITIM domains
- TRMs:
-
Tissue-resident memory T cells
- PD-1/PD-L1:
-
Programmed Cell Death Protein 1/Programmed Death-Ligand 1
- shh:
-
Sonic Hedgehog
- ICOS:
-
Inducible T-cell CO-Stimulator
- Foxp3:
-
Forkhead Box P3
- Breg:
-
B regulatory cells
- ICIs:
-
Improve immune checkpoint inhibitors
- NSCLC:
-
Non-small cell lung cancer
- CTLA4:
-
Cytotoxic T-Lymphocyte-Associated Protein 4
- ATB:
-
Antibiotics
- HP-NAP:
-
Helicobacter pylori neutrophil-activating protein
- rMBP-NAP:
-
Hp-NAP recombined with the maltose binding protein
- Chi-rNap:
-
HP-NAP-embedded Chitosan nanoparticles
- OVs:
-
Oncolytic viruses
- CAR:
-
Chimeric antigen receptor
References
Ajani JA, D’Amico TA, Bentrem DJ, Chao J, Cooke D, Corvera C, et al. Gastric cancer, version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2022;20(2):167–92.
Thrift AP, Wenker TN, El-Serag HB. Global burden of gastric cancer: epidemiological trends, risk factors, screening and prevention. Nat Rev Clin Oncol. 2023;20(5):338–49.
Stewart OA, Wu F, Chen Y. The role of gastric microbiota in gastric cancer. Gut Microbes. 2020;11(5):1220–30.
Malfertheiner P, Camargo MC, El-Omar E, Liou JM, Peek R, Schulz C, et al. Helicobacter pylori infection. Nat Rev Dis Primer. 2023;9(1):1–24.
Yeoh KG, Tan P. Mapping the genomic diaspora of gastric cancer. Nat Rev Cancer. 2022;22(2):71–84.
Yin J, Wu X, Li S, Li C, Guo Z. Impact of environmental factors on gastric cancer: a review of the scientific evidence, human prevention and adaptation. J Environ Sci. 2020;89:65–79.
Asaka M, Kobayashi M, Kudo T, Akino K, Asaka Y, Fujimori K, et al. Gastric cancer deaths by age group in Japan: outlook on preventive measures for elderly adults. Cancer Sci. 2020;111(10):3845–53.
Castaño-Rodríguez N, Goh KL, Fock KM, Mitchell HM, Kaakoush NO. Dysbiosis of the microbiome in gastric carcinogenesis. Sci Rep. 2017;7(1):15957.
Gunathilake MN, Lee J, Choi IJ, Kim YI, Ahn Y, Park C, et al. Association between the relative abundance of gastric microbiota and the risk of gastric cancer: a case-control study. Sci Rep. 2019;9(1):13589.
Chen BY, Lin WZ, Li YL, Bi C, Du LJ, Liu Y, et al. Roles of oral microbiota and oral-gut microbial transmission in hypertension. J Adv Res. 2023;43:147–61.
Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol. 2019;16(1):35–56.
O’Dwyer DN, Ashley SL, Gurczynski SJ, Xia M, Wilke C, Falkowski NR, et al. Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199(9):1127–38.
Uberoi A, Bartow-McKenney C, Zheng Q, Flowers L, Campbell A, Knight SAB, et al. Commensal microbiota regulates skin barrier function and repair via signaling through the aryl hydrocarbon receptor. Cell Host Microbe. 2021;29(8):1235–1248.e8.
Adu-Oppong B, Thänert R, Wallace MA, Burnham CAD, Dantas G. Substantial overlap between symptomatic and asymptomatic genitourinary microbiota states. Microbiome. 2022;10(1):6.
Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019;25(6):968–76.
Li R, Zhou R, Wang H, Li W, Pan M, Yao X, et al. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ. 2019;26(11):2447–63.
Overacre-Delgoffe AE, Bumgarner HJ, Cillo AR, Burr AHP, Tometich JT, Bhattacharjee A, et al. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity. 2021;54(12):2812–2824.e4.
Stein-Thoeringer CK, Saini NY, Zamir E, Blumenberg V, Schubert ML, Mor U, et al. A non-antibiotic-disrupted gut microbiome is associated with clinical responses to CD19-CAR-T cell cancer immunotherapy. Nat Med. 2023;29(4):906–16.
Andrews MC, Duong CPM, Gopalakrishnan V, Iebba V, Chen WS, Derosa L, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat Med. 2021;27(8):1432–41.
Sekirov I, Russell SL, Antunes LCM, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90(3):859–904.
Abate M, Vos E, Gonen M, Janjigian YY, Schattner M, Laszkowska M, et al. A novel microbiome signature in gastric cancer: a two independent cohort retrospective analysis. Ann Surg. 2022;276(4):605–15.
Zhang X, Li C, Cao W, Zhang Z. Alterations of gastric microbiota in gastric cancer and precancerous stages. Front Cell Infect Microbiol. 2021;11:559148.
Gantuya B, El Serag HB, Matsumoto T, Ajami NJ, Uchida T, Oyuntsetseg K, et al. Gastric mucosal microbiota in a Mongolian population with gastric cancer and precursor conditions. Aliment Pharmacol Ther. 2020;51(8):770–80.
Monstein HJ, Tiveljung A, Kraft CH, Borch K, Jonasson J. Profiling of bacterial flora in gastric biopsies from patients with Helicobacter pylori-associated gastritis and histologically normal control individuals by temperature gradient gel electrophoresis and 16S rDNA sequence analysis. J Med Microbiol. 2000;49(9):817–22.
Costa M, Weese JS. Methods and basic concepts for microbiota assessment. Vet J. 2019;249:10–5.
Perkins GA, den Bakker HC, Burton AJ, Erb HN, McDonough SP, McDonough PL, et al. Equine stomachs harbor an abundant and diverse mucosal microbiota. Appl Environ Microbiol. 2012;78(8):2522–32.
Narunsky-Haziza L, Sepich-Poore GD, Livyatan I, Asraf O, Martino C, Nejman D, et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell. 2022;185(20):3789–3806.e17.
von Rosenvinge EC, Song Y, White JR, Maddox C, Blanchard T, Fricke WF. Immune status, antibiotic medication and pH are associated with changes in the stomach fluid microbiota. ISME J. 2013;7(7):1354–66.
Wensel CR, Pluznick JL, Salzberg SL, Sears CL. Next-generation sequencing: insights to advance clinical investigations of the microbiome. J Clin Invest. 2022;132(7):e154944.
Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci. 2006;103(3):732–7.
Bajorek S, Parker L, Li N, Winglee K, Weaver M, Johnson J, et al. Initial microbial community of the neonatal stomach immediately after birth. Gut Microbes. 2019;10(3):289–97.
Milisavljevic V, Garg M, Vuletic I, Miller JF, Kim L, Cunningham TD, et al. Prospective assessment of the gastroesophageal microbiome in VLBW neonates. BMC Pediatr. 2013;13(1):49.
Moles L, Gómez M, Jiménez E, Bustos G, de Andrés J, Melgar A, et al. Bacterial diversity of the gastric content of preterm infants during their first month of life at the hospital. Front Nutr. 2017;4:4.
Patel K, Konduru K, Patra AK, Chandel DS, Panigrahi P. Trends and determinants of gastric bacterial colonization of preterm neonates in a NICU setting. Rogers LK, editor. PLoS One. 2015;10(7):e0114664.
Li XX, Wong GLH, To KF, Wong VWS, Lai LH, Chow DKL, et al. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS ONE. 2009;4(11):e7985.
Kashiwagi S, Naito Y, Inoue R, Takagi T, Nakano T, Inada Y, et al. Mucosa-associated microbiota in the gastrointestinal tract of healthy Japanese subjects. Digestion. 2020;101(2):107–20.
Ndegwa N, Ploner A, Andersson AF, Zagai U, Andreasson A, Vieth M, et al. Gastric microbiota in a low–Helicobacter pylori prevalence general population and their associations with gastric lesions. Clin Transl Gastroenterol. 2020;11(7):e00191.
Vasapolli R, Schütte K, Schulz C, Vital M, Schomburg D, Pieper DH, et al. Analysis of transcriptionally active bacteria throughout the gastrointestinal tract of healthy individuals. Gastroenterology. 2019;157(4):1081–1092.e3.
Chee WJY, Chew SY, Than LTL. Vaginal microbiota and the potential of Lactobacillus derivatives in maintaining vaginal health. Microb Cell Factories. 2020;19(1):203.
Sprong KE, Mabenge M, Wright CA, Govender S. Ureaplasma species and preterm birth: current perspectives. Crit Rev Microbiol. 2020;46(2):169–81.
Zimmermann P, Curtis N. Breast milk microbiota: a review of the factors that influence composition. J Infect. 2020;81(1):17–47.
Dong Q, Xin Y, Wang L, Meng X, Yu X, Lu L, et al. Characterization of gastric microbiota in twins. Curr Microbiol. 2017;74(2):224–9.
Nyangahu DD, Lennard KS, Brown BP, Darby MG, Wendoh JM, Havyarimana E, et al. Disruption of maternal gut microbiota during gestation alters offspring microbiota and immunity. Microbiome. 2018;6(1):124.
Trevisi P, Priori D, Motta V, Luise D, Jansman AJM, Koopmans SJ, et al. The effects of starter microbiota and the early life feeding of medium chain triglycerides on the gastric transcriptome profile of 2- or 3-week-old cesarean delivered piglets. J Anim Sci Biotechnol. 2017;8(1):82.
Van Hul M, Cani PD. The gut microbiota in obesity and weight management: microbes as friends or foe? Nat Rev Endocrinol. 2023;19(5):258–71.
Zou Y, Ju X, Chen W, Yuan J, Wang Z, Aluko RE, et al. Rice bran attenuated obesity via alleviating dyslipidemia, browning of white adipocytes and modulating gut microbiota in high-fat diet-induced obese mice. Food Funct. 2020;11(3):2406–17.
Dapa T, Ramiro RS, Pedro MF, Gordo I, Xavier KB. Diet leaves a genetic signature in a keystone member of the gut microbiota. Cell Host Microbe. 2022;30(2):183–199.e10.
Xie Y, Pei F, Liu Y, Liu Z, Chen X, Xue D. Fecal fermentation and high-fat diet-induced obesity mouse model confirmed exopolysaccharide from Weissella cibaria PFY06 can ameliorate obesity by regulating the gut microbiota. Carbohydr Polym. 2023;318:121122.
Sedghi L, DiMassa V, Harrington A, Lynch SV, Kapila YL. The oral microbiome: role of key organisms and complex networks in oral health and disease. Periodontol 2000. 2021;87(1):107–31.
Fann JCY, Chiang TH, Yen AMF, Lee YC, Wu MS, Chen HH. Personalized risk assessment for dynamic transition of gastric neoplasms. J Biomed Sci. 2018;25(1):84.
Png CW, Lee WJJ, Chua SJ, Zhu F, Gastric Consortium5, Yeoh KG, et al. Mucosal microbiome associates with progression to gastric cancer. Theranostics. 2022;12(1):48–58.
Wang Z, Gao X, Zeng R, Wu Q, Sun H, Wu W, et al. Changes of the gastric mucosal microbiome associated with histological stages of gastric carcinogenesis. Front Microbiol. 2020;11:997.
Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. 2018;67(6):1024–32.
He C, Peng C, Shu X, Wang H, Zhu Z, Ouyang Y, et al. Convergent dysbiosis of gastric mucosa and fluid microbiome during stomach carcinogenesis. Gastric Cancer. 2022;25(5):837–49.
Hsieh YY, Tung SY, Pan HY, Yen CW, Xu HW, Lin YJ, et al. Increased abundance of Clostridium and Fusobacterium in gastric microbiota of patients with gastric cancer in Taiwan. Sci Rep. 2018;8(1):158.
Wu F, Yang L, Hao Y, Zhou B, Hu J, Yang Y, et al. Oral and gastric microbiome in relation to gastric intestinal metaplasia. Int J Cancer. 2022;150(6):928–40.
Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, Costa JL, Carneiro F, Machado JC, et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut. 2018;67(2):226–36.
Liu X, Shao L, Liu X, Ji F, Mei Y, Cheng Y, et al. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine. 2019;40:336–48.
Mannion A, Sheh A, Shen Z, Dzink-Fox J, Piazuelo MB, Wilson KT, et al. Shotgun metagenomics of gastric biopsies reveals compositional and functional microbiome shifts in high- and low-gastric-cancer-risk populations from Colombia, South America. Gut Microbes. 2023;15(1):2186677.
Lehr K, Nikitina D, Vilchez-Vargas R, Steponaitiene R, Thon C, Skieceviciene J, et al. Microbial composition of tumorous and adjacent gastric tissue is associated with prognosis of gastric cancer. Sci Rep. 2023;13(1):4640.
Chen XH, Wang A, Chu AN, Gong YH, Yuan Y. Mucosa-associated microbiota in gastric cancer tissues compared with non-cancer tissues. Front Microbiol. 2019;10:1261.
Deng Y, Ding X, Song Q, Zhao G, Han L, Ding B, et al. Alterations in mucosa-associated microbiota in the stomach of patients with gastric cancer. Cell Oncol. 2021;44(3):701–14.
Wu ZF, Zou K, Wu GN, Jin ZJ, Xiang CJ, Xu S, et al. A comparison of tumor-associated and non-tumor-associated gastric microbiota in gastric cancer patients. Dig Dis Sci. 2021;66(5):1673–82.
Dai D, Yang Y, Yu J, Dang T, Qin W, Teng L, et al. Interactions between gastric microbiota and metabolites in gastric cancer. Cell Death Dis. 2021;12(12):1104.
Peng R, Liu S, You W, Huang Y, Hu C, Gao Y, et al. Gastric microbiome alterations are associated with decreased CD8 + tissue-resident memory T cells in the tumor microenvironment of gastric cancer. Cancer Immunol Res. 2022;10(10):1224–40.
Yang Y, Dai D, Jin W, Huang Y, Zhang Y, Chen Y, et al. Microbiota and metabolites alterations in proximal and distal gastric cancer patients. J Transl Med. 2022;20(1):439.
Xi J, Li Y, Zhang H, Bai Z. Dynamic variations of the gastric microbiota: key therapeutic points in the reversal of Correa’s cascade. Int J Cancer. 2023;152(6):1069–84.
Yan L, Chen Y, Chen F, Tao T, Hu Z, Wang J, et al. Effect of Helicobacter pylori eradication on gastric cancer prevention: updated report from a randomized controlled trial with 26.5 years of follow-up. Gastroenterology. 2022;163(1):154–162.e3.
Liou JM, Malfertheiner P, Lee YC, Sheu BS, Sugano K, Cheng HC, et al. Screening and eradication of Helicobacter pylori for gastric cancer prevention: the Taipei global consensus. Gut. 2020;69(12):2093–112.
Sung JJY, Coker OO, Chu E, Szeto CH, Luk STY, Lau HCH, et al. Gastric microbes associated with gastric inflammation, atrophy and intestinal metaplasia 1 year after Helicobacter pylori eradication. Gut. 2020;69(9):1572–81.
Boehm ET, Thon C, Kupcinskas J, Steponaitiene R, Skieceviciene J, Canbay A, et al. Fusobacterium nucleatum is associated with worse prognosis in Lauren’s diffuse type gastric cancer patients. Sci Rep. 2020;10(1):16240.
Hsieh YY, Tung SY, Pan HY, Chang TS, Wei KL, Chen WM, et al. Fusobacterium nucleatum colonization is associated with decreased survival of helicobacter pylori-positive gastric cancer patients. World J Gastroenterol. 2021;27(42):7311–23.
Guan SW, Lin Q, Wu XD, Yu HB. Weighted gene coexpression network analysis and machine learning reveal oncogenome associated microbiome plays an important role in tumor immunity and prognosis in pan-cancer. J Transl Med. 2023;21:537.
Yang J, Xu J, Ling Z, Zhou X, Si Y, Liu X, et al. Prognostic effects of the gastric mucosal microbiota in gastric cancer. Cancer Sci. 2023;114(3):1075–85.
Nasr R, Shamseddine A, Mukherji D, Nassar F, Temraz S. The crosstalk between microbiome and immune response in gastric cancer. Int J Mol Sci. 2020;21(18):6586.
He J, Hu W, Ouyang Q, Zhang S, He L, Chen W, et al. Helicobacter pylori infection induces stem cell-like properties in Correa cascade of gastric cancer. Cancer Lett. 2022;542:215764.
Chauhan N, Tay ACY, Marshall BJ, Jain U. Helicobacter pylori VacA, a distinct toxin exerts diverse functionalities in numerous cells: an overview. Helicobacter. 2019;24(1):e12544.
Toh JWT, Wilson RB. Pathways of gastric carcinogenesis, Helicobacter pylori virulence and interactions with antioxidant systems, vitamin C and phytochemicals. Int J Mol Sci. 2020;21(17):6451.
Zhang X, Soutto M, Chen Z, Bhat N, Zhu S, Eissmann MF, et al. Induction of fibroblast growth factor receptor 4 by Helicobacter pylori via signal transducer and activator of transcription 3 with a feedforward activation loop involving steroid receptor coactivator signaling in gastric cancer. Gastroenterology. 2022;163(3):620–636.e9.
Wang Z, Shan Y, Wang R, Zhou H, Hu R, Li Y, et al. Structural insights into the binding propensity of human SHIP2 SH2 to oncogenic CagA isoforms from Helicobacter pylori. Int J Mol Sci. 2022;23(19):11299.
Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26(32):4617–26.
Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, Saito Y, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447(7142):330–3.
Imai S, Ooki T, Murata-Kamiya N, Komura D, Tahmina K, Wu W, et al. Helicobacter pylori CagA elicits BRCAness to induce genome instability that may underlie bacterial gastric carcinogenesis. Cell Host Microbe. 2021;29(6):941–958.e10.
Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase α subunit. Gastroenterology. 2010;139(1):239–48.
Yao X, Smolka AJ. Gastric parietal cell physiology and Helicobacter pylori–induced disease. Gastroenterology. 2019;156(8):2158–73.
Skinner GR. Transformation of primary hamster embryo fibroblasts by type 2 simplex virus: evidence for a “hit and run” mechanism. Br J Exp Pathol. 1976;57(4):361–76.
Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15(3):306–16.
Gantuya B, El-Serag HB, Matsumoto T, Ajami NJ, Oyuntsetseg K, Azzaya D, et al. Gastric microbiota in Helicobacter pylori-negative and -positive gastritis among high incidence of gastric cancer area. Cancers. 2019;11(4):504.
Miftahussurur M, Waskito LA, El-Serag HB, Ajami NJ, Nusi IA, Syam AF, et al. Gastric microbiota and Helicobacter pylori in Indonesian population. Helicobacter. 2020;25(4):e12695.
Llorca L, Pérez-Pérez G, Urruzuno P, Martinez MJ, Iizumi T, Gao Z, et al. Characterization of the gastric microbiota in a pediatric population according to Helicobacter pylori status. Pediatr Infect Dis J. 2017;36(2):173–8.
Guo Y, Zhang Y, Gerhard M, Gao JJ, Mejias-Luque R, Zhang L, et al. Effect of Helicobacter pylori on gastrointestinal microbiota: a population-based study in Linqu, a high-risk area of gastric cancer. Gut. 2020;69(9):1598–607.
Miao R, Wan C, Wang Z. The relationship of gastric microbiota and Helicobacter pylori infection in pediatrics population. Helicobacter. 2020;25(1):e12676.
Klymiuk I, Bilgilier C, Stadlmann A, Thannesberger J, Kastner MT, Högenauer C, et al. The human gastric microbiome is predicated upon infection with Helicobacter pylori. Front Microbiol. 2017;8:2508.
Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, et al. Lack of commensal flora in Helicobacter pylori–Infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140(1):210–220.e4.
Lertpiriyapong K, Whary MT, Muthupalani S, Lofgren JL, Gamazon ER, Feng Y, et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2014;63(1):54–63.
Shen Z, Dzink-Fox J, Feng Y, Muthupalani S, Mannion AJ, Sheh A, et al. Gastric non-Helicobacter pylori urease-positive Staphylococcus epidermidis and Streptococcus salivarius isolated from humans have contrasting effects on H. pylori-associated gastric pathology and host immune responses in a murine model of gastric cancer. Young VB, editor. mSphere. 2022;7(1):e00772–21.
Chen Z, Tang Z, Li W, Deng X, Yu L, Yang J, et al. Weizmannia coagulans BCF-01: a novel gastrogenic probiotic for Helicobacter pylori infection control. Gut Microbes. 2024;16(1):2313770.
Gebremariam HG, Qazi KR, Somiah T, Pathak SK, Sjölinder H, Sverremark Ekström E, et al. Lactobacillus gasseri suppresses the production of proinflammatory cytokines in Helicobacter pylori-infected macrophages by inhibiting the expression of ADAM17. Front Immunol. 2019;10:2326.
Shen S, Ren F, Qin H, Bukhari I, Yang J, Gao D, et al. Lactobacillus acidophilus NCFM and Lactiplantibacillus plantarum Lp-115 inhibit Helicobacter pylori colonization and gastric inflammation in a murine model. Front Cell Infect Microbiol. 2023;13:1196084.
Khosravi Y, Loke MF, Goh KL, Vadivelu J. Proteomics analysis revealed that crosstalk between Helicobacter pylori and Streptococcus mitis may enhance bacterial survival and reduces carcinogenesis. Front Microbiol. 2016;7:7.
Khosravi Y, Dieye Y, Loke MF, Goh KL, Vadivelu J. Streptococcus mitis induces conversion of Helicobacter pylori to coccoid cells during co-culture in vitro. PLoS ONE. 2014;9(11):e112214.
Chen Z, Tang Z, Li W, Deng X, Yu L, Yang J, et al. Weizmannia coagulans BCF-01: a novel gastrogenic probiotic for Helicobacter pylori infection control. Gut Microbes. 2024;16(1):2313770.
Kwon SK, Park JC, Kim KH, Yoon J, Cho Y, Lee B, et al. Human gastric microbiota transplantation recapitulates premalignant lesions in germ-free mice. Gut. 2022;71(7):1266–76.
Sasaki M, Kodama Y, Shimoyama Y, Ishikawa T, Kimura S. Aciduricity and acid tolerance mechanisms of Streptococcus anginosus. J Gen Appl Microbiol. 2018;64(4):174–9.
Fu K, Cheung AHK, Wong CC, Liu W, Zhou Y, Wang F, et al. Streptococcus anginosus promotes gastric inflammation, atrophy, and tumorigenesis in mice. Cell. 2024;187(4):882–896.e17.
Li Y, Huang X, Tong D, Jiang C, Zhu X, Wei Z, et al. Relationships among microbiota, gastric cancer, and immunotherapy. Front Microbiol. 2022;13:987763.
Vinasco K, Mitchell HM, Kaakoush NO, Castaño-Rodríguez N. Microbial carcinogenesis: lactic acid bacteria in gastric cancer. Biochim Biophys Acta BBA - Rev Cancer. 2019;1872(2):188309.
Hwang CH, Lee NK, Paik HD. The anti-cancer potential of heat-killed Lactobacillus brevis KU15176 upon AGS Cell lines through intrinsic apoptosis pathway. Int J Mol Sci. 2022;23(8):4073.
Chen MJ, Chen CC, Huang YC, Tseng CC, Hsu JT, Lin YF, et al. The efficacy of Lactobacillus acidophilus and rhamnosus in the reduction of bacterial load of Helicobacter pylori and modification of gut microbiota-a double-blind, placebo-controlled, randomized trial. Helicobacter. 2021;26(6):e12857.
Zhao Y, Li Z, Zhao L, Wang J, Wang F, Zhang Q, et al. Two novel lactic acid bacteria, limosilactobacillus fermentum MN-LF23 and Lactobacillus gasseri MN-LG80, inhibited Helicobacter pylori infection in C57BL/6 mice. Food Funct. 2022;13(21):11061–9.
Delgado S, Leite AMO, Ruas-Madiedo P, Mayo B. Probiotic and technological properties of Lactobacillus spp. strains from the human stomach in the search for potential candidates against gastric microbial dysbiosis. Front Microbiol. 2015;5:5.
Xu Z, Xiao L, Wang S, Cheng Y, Wu J, Meng Y, et al. Alteration of gastric microbiota and transcriptome in a rat with gastric intestinal metaplasia induced by deoxycholic acid. Front Microbiol. 2023;14:1160821.
Jin D, Huang K, Xu M, Hua H, Ye F, Yan J, et al. Deoxycholic acid induces gastric intestinal metaplasia by activating STAT3 signaling and disturbing gastric bile acids metabolism and microbiota. Gut Microbes. 2022;14(1):2120744.
Wang S, Kuang J, Zhang H, Chen W, Zheng X, Wang J, et al. Bile acid–microbiome interaction promotes gastric carcinogenesis. Adv Sci. 2022;9(16):2200263.
Gunathilake M, Lee J, Choi IJ, Kim YI, Yoon J, Sul WJ, et al. Alterations in gastric microbial communities are associated with risk of gastric cancer in a Korean population: a case-control study. Cancers. 2020;12(9):2619.
Seyyedsalehi MS, Mohebbi E, Tourang F, Sasanfar B, Boffetta P, Zendehdel K. Association of dietary nitrate, nitrite, and N-Nitroso compounds Intake and gastrointestinal cancers: a systematic review and meta-analysis. Toxics. 2023;11(2):190.
Kraus A, McKeague M, Seiwert N, Nagel G, Geisen SM, Ziegler N, et al. Immunological and mass spectrometry-based approaches to determine thresholds of the mutagenic DNA adduct O6-methylguanine in vivo. Arch Toxicol. 2019;93(2):559–72.
Oosterlinck B, Ceuleers H, Arras W, De Man JG, Geboes K, De Schepper H, et al. Mucin-microbiome signatures shape the tumor microenvironment in gastric cancer. Microbiome. 2023;11(1):86.
Shimamura T, Ito H, Shibahara J, Watanabe A, Hippo Y, Taniguchi H, et al. Overexpression of MUC13 is associated with intestinal-type gastric cancer. Cancer Sci. 2005;96(5):265–73.
Sheng YH, He Y, Hasnain SZ, Wang R, Tong H, Clarke DT, et al. MUC13 protects colorectal cancer cells from death by activating the NF-κB pathway and is a potential therapeutic target. Oncogene. 2017;36(5):700–13.
Tiemin P, Fanzheng M, Peng X, Jihua H, Ruipeng S, Yaliang L, et al. MUC13 promotes intrahepatic cholangiocarcinoma progression via EGFR/PI3K/AKT pathways. J Hepatol. 2020;72(4):761–73.
Cao Y, Xia H, Tan X, Shi C, Ma Y, Meng D, et al. Intratumoural microbiota: a new frontier in cancer development and therapy. Signal Transduct Target Ther. 2024;9:15.
Yue K, Sheng D, Xue X, Zhao L, Zhao G, Jin C, et al. Bidirectional mediation effects between intratumoral microbiome and host DNA methylation changes contribute to stomach adenocarcinoma. Microbiol Spectr. 2023;11(4):e0090423.
Brennan CA, Garrett WS. Fusobacterium nucleatum — symbiont, opportunist and oncobacterium. Nat Rev Microbiol. 2019;17(3):156–66.
Xin Y, Li X, Zhang M, Shang Z, Luo Z, Wang Y, et al. Fusobacterium nucleatum -induced exosomal HOTTIP promotes gastric cancer progression through the microRNA -885-3p/ EphB2 axis. Cancer Sci. 2023;114(6):2360–74.
Nie S, Wang A, Yuan Y. Comparison of clinicopathological parameters, prognosis, micro-ecological environment and metabolic function of gastric cancer with or without Fusobacterium sp. infection. J Cancer. 2021;12(4):1023–32.
do Nascimento Araujo C, Amorim AT, Barbosa MS, Alexandre JCPL, Campos GB, Macedo CL, et al. Evaluating the presence of Mycoplasma hyorhinis, Fusobacterium nucleatum, and Helicobacter pylori in biopsies of patients with gastric cancer. Infect Agent Cancer. 2021;16(1):70.
Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod-Levi T, et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun. 2020;11(1):3259.
Mereiter S, Polom K, Williams C, Polonia A, Guergova-Kuras M, Karlsson NG, et al. The Thomsen-Friedenreich antigen: a highly sensitive and specific predictor of microsatellite instability in gastric cancer. J Clin Med. 2018;7(9):256.
Guo P, Tian Z, Kong X, Yang L, Shan X, Dong B, et al. FadA promotes DNA damage and progression of Fusobacterium nucleatum-induced colorectal cancer through up-regulation of chk2. J Exp Clin Cancer Res. 2020;39(1):202.
Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor − κB, and up-regulating expression of microRNA-21. Gastroenterology. 2017;152(4):851–866.e24.
Kang W, Jia Z, Tang D, Zhang Z, Gao H, He K, et al. Fusobacterium nucleatum facilitates apoptosis, ROS generation, and inflammatory cytokine production by activating AKT/MAPK and NF- κ B signaling pathways in human gingival fibroblasts. Oxid Med Cell Longev. 2019;2019:1–22.
Chen Y, Chen Y, Cao P, Su W, Zhan N, Dong W. Fusobacterium nucleatum facilitates ulcerative colitis through activating IL-17F signaling to NF‐κB via the upregulation of CARD3 expression. J Pathol. 2020;250(2):170–82.
Xu C, Fan L, Lin Y, Shen W, Qi Y, Zhang Y, et al. Fusobacterium nucleatum promotes colorectal cancer metastasis through miR-1322/CCL20 axis and M2 polarization. Gut Microbes. 2021;13(1):1980347.
Kim SY, Park SY, Jang HS, Park YD, Kee SH. Yes-associated protein is required for ZO-1-mediated tight-junction integrity and cell migration in E-cadherin-restored AGS gastric cancer cells. Biomedicines. 2021;9(9):1264.
Peng C, Ouyang Y, Lu N, Li N. The NF-κB signaling pathway, the microbiota, and gastrointestinal tumorigenesis: recent advances. Front Immunol. 2020;11:1387.
Kim JM, Cho SJ, Oh YK, Jung HY, Kim YJ, Kim N. Nuclear factor-kappa B activation pathway in intestinal epithelial cells is a major regulator of chemokine gene expression and neutrophil migration induced by Bacteroides fragilis enterotoxin. Clin Exp Immunol. 2002;130(1):59–66.
Cao L, Zhu S, Lu H, Soutto M, Bhat N, Chen Z, et al. Helicobacter pylori-induced RASAL2 through activation of NF-kB promotes gastric tumorigenesis via β-catenin signaling axis. Gastroenterology. 2022;162(6):1716–1731.e17.
Maubach G, Vieth M, Boccellato F, Naumann M. Helicobacter pylori-induced NF-κB: trailblazer for gastric pathophysiology. Trends Mol Med. 2022;28(3):210–22.
Chung L, Orberg ET, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018;23(2):203–214.e5.
Long X, Wong CC, Tong L, Chu ESH, Ho Szeto C, Go MYY, et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat Microbiol. 2019;4(12):2319–30.
Papon N, Hohl TM, Zhai B. Mycobiota dysbiosis and gastric tumorigenesis. Theranostics. 2021;11(15):7488–90.
Zhong M, Xiong Y, Zhao J, Gao Z, Ma J, Wu Z, et al. Candida albicans disorder is associated with gastric carcinogenesis. Theranostics. 2021;11(10):4945–56.
Dohlman AB, Klug J, Mesko M, Gao IH, Lipkin SM, Shen X, et al. A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell. 2022;185(20):3807–3822.e12.
Lohse MB, Gulati M, Johnson AD, Nobile CJ. Development and regulation of single- and multi-species Candida albicans biofilms. Nat Rev Microbiol. 2018;16(1):19–31.
Zhang Y, Gan Y, Wang J, Feng Z, Zhong Z, Bao H, et al. Dysbiosis of gut microbiota and intestinal barrier dysfunction in pigs with pulmonary inflammation induced by Mycoplasma hyorhinis Infection. mSystems. 2022;7(4):e00282–22.
Duan H, Chen L, Qu L, Yang H, Song SW, Han Y, et al. Mycoplasma Hyorhinis infection promotes NF- κB–dependent migration of gastric cancer cells. Cancer Res. 2014;74(20):5782–94.
Morgan AD, Seely KD, Hagenstein LD, Florey GM, Small JM. Bacterial involvement in progression and metastasis of adenocarcinoma of the stomach. Cancers. 2022;14(19):4886.
Xu Y, Li H, Chen W, Yao X, Xing Y, Wang X, et al. Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PLoS ONE. 2013;8(11):e77955.
Chernov AV, Reyes L, Xu Z, Gonzalez B, Golovko G, Peterson S, et al. Mycoplasma CG- and GATC-specific DNA methyltransferases selectively and efficiently methylate the host genome and alter the epigenetic landscape in human cells. Epigenetics. 2015;10(4):303–18.
Park MD, Silvin A, Ginhoux F, Merad M. Macrophages in health and disease. Cell. 2022;185(23):4259–79.
Li Q, Wu W, Gong D, Shang R, Wang J, Yu H. Propionibacterium acnes overabundance in gastric cancer promote M2 polarization of macrophages via a TLR4/PI3K/Akt signaling. Gastric Cancer. 2021;24(6):1242–53.
Petersen CP, Meyer AR, De Salvo C, Choi E, Schlegel C, Petersen A, et al. A signalling cascade of IL-33 to IL-13 regulates metaplasia in the mouse stomach. Gut. 2018;67(5):805–17.
Satoh-Takayama N, Kato T, Motomura Y, Kageyama T, Taguchi-Atarashi N, Kinoshita-Daitoku R, et al. Bacteria-induced group 2 innate lymphoid cells in the stomach provide immune protection through induction of IgA. Immunity. 2020;52(4):635–649.e4.
Bie Q, Zhang P, Su Z, Zheng D, Ying X, Wu Y, et al. Polarization of ILC2s in peripheral blood might contribute to immunosuppressive microenvironment in patients with gastric cancer. J Immunol Res. 2014;2014:923135.
Li R, Jiang XX, Zhang LF, Liu XM, Hu TZ, Xia XJ, et al. Group 2 innate lymphoid cells are involved in skewed type 2 immunity of gastric diseases induced by Helicobacter pylori infection. Mediators Inflamm. 2017;2017:4927964.
Ohno H, Satoh-Takayama N. Stomach microbiota, Helicobacter pylori, and group 2 innate lymphoid cells. Exp Mol Med. 2020;52(9):1377–82.
Hedrick CC, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. 2022;22(3):173–87.
Li TJ, Jiang YM, Hu YF, Huang L, Yu J, Zhao LY, et al. Interleukin-17–producing neutrophils link inflammatory stimuli to disease progression by promoting angiogenesis in gastric cancer. Clin Cancer Res. 2017;23(6):1575–85.
Li S, Cong X, Gao H, Lan X, Li Z, Wang W, et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J Exp Clin Cancer Res. 2019;38(1):6.
Nie P, Zhang W, Meng Y, Lin M, Guo F, Zhang H, et al. A YAP/TAZ-CD54 axis is required for CXCR2 − CD44 − tumor-specific neutrophils to suppress gastric cancer. Protein Cell. 2022;14(7):515–33.
Shan ZG, Chen J, Liu JS, Zhang JY, Wang TT, Teng YS, et al. Activated neutrophils polarize protumorigenic interleukin-17A-producing T helper subsets through TNF-α-B7-H2-dependent pathway in human gastric cancer. Clin Transl Med. 2021;11(6):e484.
Chu TH, Huang ST, Yang SF, Li CJ, Lin HW, Weng BC, et al. Hepatoma-derived growth factor participates in Helicobacter Pylori-induced neutrophils recruitment, gastritis and gastric carcinogenesis. Oncogene. 2019;38(37):6461–77.
Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, et al. Interleukin-17–induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. 2020;217(12):e20190354.
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8.
Zhang T, Li Y, Chen J, Liu Y, Huang Z, Xu X, et al. IDDF2023-ABS-0058 Fusobacterium nucleatum intracellular parasitism activates the NF-KB/IL-17 signaling pathway induce neutrophil recruitment and promote gastric cancer progression. Gut. 2023;72(Suppl 1):A70.
Ling Z, Shao L, Liu X, Cheng Y, Yan C, Mei Y, et al. Regulatory T cells and plasmacytoid dendritic cells within the tumor microenvironment in gastric cancer are correlated with gastric microbiota dysbiosis: a preliminary study. Front Immunol. 2019;10:533.
Yang J. Roles of plasmacytoid dendritic cells in gastric cancer. Front Oncol. 2022;12:818314.
Liu W, Zhao J, Li Q, Wang Q, Zhou Y, Tong Z. Gastric cancer patients have elevated plasmacytoid and CD1c+ dendritic cells in the peripheral blood. Oncol Lett. 2018;15(4):5087–92.
Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42(2):344–55.
Dutta A, Venkataganesh H, Love PE. New insights into epigenetic regulation of T cell differentiation. Cells. 2021;10(12):533.
Lee K, Hwang H, Nam KT. Immune response and the tumor microenvironment: how they communicate to regulate gastric cancer. Gut Liver. 2014;8(2):131–9.
Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, Knight R. The microbiome and human cancer. Science. 2021;371(6536):eabc4552.
Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, et al. Fatty acid oxidation controls CD8 + tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol Res. 2020;8(4):479–92.
Aydın EM, Demir TD, Seymen N, Said SS, Oktem-Okullu S, Tiftikci A, et al. The crosstalk between H. pylori virulence factors and the PD1:PD-L1 immune checkpoint inhibitors in progression to gastric cancer. Immunol Lett. 2021;239:1–11.
Holokai L, Chakrabarti J, Broda T, Chang J, Hawkins JA, Sundaram N, et al. Increased programmed death-ligand 1 is an early epithelial cell response to Helicobacter pylori infection. Blanke SR, editor. PLOS Pathog. 2019;15(1):e1007468.
Koh V, Chakrabarti J, Torvund M, Steele N, Hawkins JA, Ito Y, et al. Hedgehog transcriptional effector GLI mediates mTOR-Induced PD-L1 expression in gastric cancer organoids. Cancer Lett. 2021;518:59–71.
Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19(1):116.
Urakawa S, Yamasaki M, Makino T, Kurokawa Y, Yamamoto K, Goto K, et al. The impact of ICOS + regulatory T cells and Helicobacter pylori infection on the prognosis of patients with gastric and colorectal cancer: potential prognostic benefit of pre-operative eradication therapy. Cancer Immunol Immunother. 2021;70(2):443–52.
Nagase H, Takeoka T, Urakawa S, Morimoto-Okazawa A, Kawashima A, Iwahori K, et al. ICOS + Foxp3 + TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with H elicobacter pylori: unique expression of ICOS on TREGS in gastric cancer. Int J Cancer. 2017;140(3):686–95.
Engelhard V, Conejo-Garcia JR, Ahmed R, Nelson BH, Willard-Gallo K, Bruno TC, et al. B cells and cancer. Cancer Cell. 2021;39(10):1293–6.
Li H, Limenitakis JP, Greiff V, Yilmaz B, Schären O, Urbaniak C, et al. Mucosal or systemic microbiota exposures shape the B cell repertoire. Nature. 2020;584(7820):274–8.
Catalán D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillón JC, et al. Immunosuppressive mechanisms of regulatory B cells. Front Immunol. 2021;12:611795.
Horii M, Matsushita T. Regulatory B cells and T cell regulation in cancer. J Mol Biol. 2021;433(1):166685.
Nahid-Samiei M, Rahimian G, Shafigh M, Taheri F, Karami-Hurestani M, Sanaei MJ, et al. Enhanced frequency of CD19 + IL-10 + B cells in human gastric mucosa infected by Helicobacter pylori. Am J Med Sci. 2020;359(6):347–53.
Joshi SS, Badgwell BD. Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 2021;71(3):264–79.
Fessler J, Matson V, Gajewski TF. Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer. 2019;7(1):108.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science. 2015;350(6264):1084–9.
Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–84.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7.
Si W, Liang H, Bugno J, Xu Q, Ding X, Yang K, et al. Lactobacillus rhamnosus GG induces cGAS/STING- dependent type I interferon and improves response to immune checkpoint blockade. Gut. 2022;71(3):521–33.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103.
Lee SH, Cho SY, Yoon Y, Park C, Sohn J, Jeong JJ, et al. Bifidobacterium bifidum strains synergize with immune checkpoint inhibitors to reduce tumour burden in mice. Nat Microbiol. 2021;6(3):277–88.
Newsome RC, Gharaibeh RZ, Pierce CM, da Silva WV, Paul S, Hogue SR, et al. Interaction of bacterial genera associated with therapeutic response to immune checkpoint PD-1 blockade in a United States cohort. Genome Med. 2022;14(1):35.
Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol off J Eur Soc Med Oncol. 2019;30(12):2012.
Hakozaki T, Richard C, Elkrief A, Hosomi Y, Benlaïfaoui M, Mimpen I, et al. The gut microbiome associates with immune checkpoint inhibition outcomes in patients with advanced non-small cell lung cancer. Cancer Immunol Res. 2020;8(10):1243–50.
Mao J, Wang D, Long J, Yang X, Lin J, Song Y, et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J Immunother Cancer. 2021;9(12):e003334.
McCulloch JA, Davar D, Rodrigues RR, Badger JH, Fang JR, Cole AM, et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat Med. 2022;28(3):545–56.
Peng Z, Cheng S, Kou Y, Wang Z, Jin R, Hu H, et al. The gut microbiome is associated with clinical response to Anti–PD-1/PD-L1 immunotherapy in gastrointestinal cancer. Cancer Immunol Res. 2020;8(10):1251–61.
Oster P, Vaillant L, Riva E, McMillan B, Begka C, Truntzer C, et al. Helicobacter pylori infection has a detrimental impact on the efficacy of cancer immunotherapies. Gut. 2022;71(3):457–66.
Che H, Xiong Q, Ma J, Chen S, Wu H, Xu H, et al. Association of Helicobacter pylori infection with survival outcomes in advanced gastric cancer patients treated with immune checkpoint inhibitors. BMC Cancer. 2022;22(1):904.
Tonneau M, Nolin-Lapalme A, Kazandjian S, Auclin E, Panasci J, Benlaifaoui M, et al. Helicobacter pylori serology is associated with worse overall survival in patients with melanoma treated with immune checkpoint inhibitors. Oncoimmunology. 2022;11(1):2096535.
Derosa L, Routy B, Fidelle M, Iebba V, Alla L, Pasolli E, et al. Gut bacteria composition drives primary resistance to cancer immunotherapy in renal cell carcinoma patients. Eur Urol. 2020;78(2):195–206.
Pinato DJ, Howlett S, Ottaviani D, Urus H, Patel A, Mineo T, et al. Association of prior antibiotic treatment with survival and response to immune checkpoint inhibitor therapy in patients with cancer. JAMA Oncol. 2019;5(12):1774–8.
Ahmed J, Kumar A, Parikh K, Anwar A, Knoll BM, Puccio C, et al. Use of broad-spectrum antibiotics impacts outcome in patients treated with immune checkpoint inhibitors. Oncoimmunology. 2018;7(11):e1507670.
Hsu PI, Pan CY, Kao JY, Tsay FW, Peng NJ, Kao SS, et al. Helicobacter pylori eradication with bismuth quadruple therapy leads to dysbiosis of gut microbiota with an increased relative abundance of proteobacteria and decreased relative abundances of bacteroidetes and actinobacteria. Helicobacter. 2018;23(4):e12498.
Schulz C, Schütte K, Koch N, Vilchez-Vargas R, Wos-Oxley ML, Oxley APA, et al. The active bacterial assemblages of the upper GI tract in individuals with and without Helicobacter infection. Gut. 2018;67(2):216–25.
Olekhnovich EI, Manolov AI, Samoilov AE, Prianichnikov NA, Malakhova MV, Tyakht AV, et al. Shifts in the human gut microbiota structure caused by quadruple Helicobacter pylori eradication therapy. Front Microbiol. 2019;10:1902.
Kakiuchi T, Mizoe A, Yamamoto K, Imamura I, Hashiguchi K, Kawakubo H, et al. Effect of probiotics during vonoprazan-containing triple therapy on gut microbiota in Helicobacter pylori infection: a randomized controlled trial. Helicobacter. 2020;25(3):e12690.
Liou JM, Chen CC, Chang CM, Fang YJ, Bair MJ, Chen PY, et al. Long-term changes of gut microbiota, antibiotic resistance, and metabolic parameters after Helicobacter pylori eradication: a multicentre, open-label, randomised trial. Lancet Infect Dis. 2019;19(10):1109–20.
Lurienne L, Cervesi J, Duhalde L, de Gunzburg J, Andremont A, Zalcman G, et al. NSCLC immunotherapy efficacy and antibiotic use: a systematic review and meta-analysis. J Thorac Oncol. 2020;15(7):1147–59.
Iankov ID, Allen C, Federspiel MJ, Myers RM, Peng KW, Ingle JN, et al. Expression of immunomodulatory neutrophil-activating protein of Helicobacter pylori enhances the antitumor activity of oncolytic measles virus. Mol Ther. 2012;20(6):1139–47.
Ma J, Jin C, Čančer M, Wang H, Ramachandran M, Yu D. Concurrent expression of HP-NAP enhances antitumor efficacy of oncolytic vaccinia virus but not for Semliki Forest virus. Mol Ther - Oncolytics. 2021;21:356–66.
Jin C, Ma J, Ramachandran M, Yu D, Essand M. CAR T cells expressing a bacterial virulence factor trigger potent bystander antitumour responses in solid cancers. Nat Biomed Eng. 2022;6(7):830–41.
Xue LJ, Mao XB, Liu XB, Gao H, Chen YN, Dai TT, et al. Activation of CD3 + T cells by Helicobacter pylori DNA vaccines in potential immunotherapy of gastric carcinoma. Cancer Biol Ther. 2019;20(6):866–76.
Codolo G, Coletta S, D’Elios MM, de Bernard M. HP-NAP of Helicobacter pylori: the power of the Immunomodulation. Front Immunol. 2022;13:944139.
Ramachandran M, Jin C, Yu D, Eriksson F, Essand M. Correction: Vector-encoded Helicobacter pylori neutrophil-activating protein promotes maturation of dendritic cells with Th1 polarization and improved migration. J Immunol Baltim Md 1950. 2015;194(11):5568.
Codolo G, Facchinello N, Papa N, Bertocco A, Coletta S, Benna C, et al. Macrophage-mediated melanoma reduction after HP-NAP treatment in a zebrafish xenograft model. Int J Mol Sci. 2022;23(3):1644.
Codolo G, Fassan M, Munari F, Volpe A, Bassi P, Rugge M, et al. HP-NAP inhibits the growth of bladder cancer in mice by activating a cytotoxic Th1 response. Cancer Immunol Immunother. 2012;61(1):31–40.
Wang T, Liu X, Ji Z, Men Y, Du M, Ding C, et al. Antitumor and immunomodulatory effects of recombinant fusion protein rMBP-NAP through TLR-2 dependent mechanism in tumor bearing mice. Int Immunopharmacol. 2015;29(2):876–83.
Wang T, Du M, Ji Z, Ding C, Wang C, Men Y, et al. Recombinant protein rMBP-NAP restricts tumor progression by triggering antitumor immunity in mouse metastatic lung cancer. Can J Physiol Pharmacol. 2018;96(2):113–9.
Mohabati Mobarez A, Soleimani N, Esmaeili SA, Farhangi B. Nanoparticle-based immunotherapy of breast cancer using recombinant Helicobacter pylori proteins. Eur J Pharm Biopharm. 2020;155:69–76.
Li Y, Jin Y, Zhang J, Pan H, Wu L, Liu D, et al. Recovery of human gut microbiota genomes with third-generation sequencing. Cell Death Dis. 2021;12(6):569.
Zheng W, Zhao S, Yin Y, Zhang H, Needham DM, Evans ED, et al. High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome. Science. 2022;376(6597):eabm1483.
Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579(7800):567–74.
Zhou CB, Pan SY, Jin P, Deng JW, Xue JH, Ma XY, et al. Fecal signatures of Streptococcus anginosus and Streptococcus constellatus for noninvasive screening and early warning of gastric cancer. Gastroenterology. 2022;162(7):1933–1947.e18.
Zhang W, Zhou Y, Fan Y, Cao R, Xu Y, Weng Z, et al. Metal–organic-framework-based hydrogen-release platform for multieffective Helicobacter pylori targeting therapy and intestinal flora protective capabilities. Adv Mater. 2022;34(2):2105738.
Yu J, Guo Z, Yan J, Bu C, Peng C, Li C, et al. Gastric acid-responsive ROS nanogenerators for effective treatment of Helicobacter pylori infection without disrupting homeostasis of intestinal flora. Adv Sci Weinh Baden-Wurtt Ger. 2023;10(20):e2206957.
Choudhury A, Ortiz PS, Young M, Mahmud MT, Stoffel RT, Greathouse KL, et al. Control of Helicobacter pylori with engineered probiotics secreting selective guided antimicrobial peptides. Microbiol Spectr. 2023;11(5):e02014–23.
Acknowledgements
We thank all our research team members for their invaluable contributions, constructive feedback, and insightful suggestions.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82074074, 82104447 and 81874353).
Author information
Authors and Affiliations
Contributions
Zhiyi Liu and Siyu Chen conceptualized and designed the study; Zhiyi Liu wrote the manuscript, created the figures, and prepared the charts; Dachuan Zhang and Siyu Chen edited and revised the manuscript for critical intellectual content; All authors participated in the final review and approval of the manuscript for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Z., Zhang, D. & Chen, S. Unveiling the gastric microbiota: implications for gastric carcinogenesis, immune responses, and clinical prospects. J Exp Clin Cancer Res 43, 118 (2024). https://doi.org/10.1186/s13046-024-03034-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-024-03034-7