
Abstract
As our understanding of the mechanisms of cancer treatment has increased, a growing number of studies demonstrate pathways through which DNA damage repair (DDR) affects the immune system. At the same time, the varied response of patients to immune checkpoint blockade (ICB) therapy has prompted the discovery of various predictive biomarkers and the study of combination therapy. Here, our investigation explores the interactions involved in combination therapy, accompanied by a review that summarizes currently identified and promising predictors of response to immune checkpoint inhibitors (ICIs) that are useful for classifying oncology patients. In addition, this work, which discusses immunogenicity and several components of the tumor immune microenvironment, serves to illustrate the mechanism by which higher response rates and improved efficacy of DDR inhibitors (DDRi) in combination with ICIs are achieved.
Similar content being viewed by others

Background
Since its development, immunotherapy has played an important role in the treatment of melanoma [1,2,3], breast cancer [4], prostate cancer [5], small cell lung cancer [6], and many other types of malignancies [7, 8]. However, the application of an immune checkpoint blockade (ICB) is only provided to a few patients due to its limited efficacy [9,10,11,12,13]. At present, the effect of ICB for cancer patients is primarily predicted by the measurements of programmed cell death 1 ligand 1 (PD-L1) expression levels [14], microsatellite instability (MSI) or mismatch repair-deficient (dMMR) [15], and the tumor mutational burden (TMB) [16,17,18,19], to name a few. Apart from the disadvantages associated with the heterogeneity of expression and the uniformities of test platforms [20], evidence has shown that patients may present insensitivity to immunotherapy when PD-L1 expression is high [14, 21, 22]. In addition, although microsatellite instability-high (MSI-H)/dMMR is an acceptable prognostic measure [23, 24], the incidence of dMMR is very low in certain cancers [25,26,27]. Moreover, the use of TMB values as prognostic markers requires improvement through the continued refinement of subcategories and standard values. Hence, given the shortcomings of current intervention approaches, it is necessary to identify more markers to accurately diagnose and stratify cancer patients of all types.
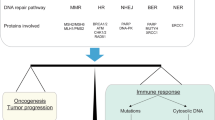
DNA damage repair (DDR) is a response of cells to DNA damage. Based on the different types of DNA damage, DDR initiates the repair process via different pathways, including mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER), homologous recombination repair (HRR), and non-homologous end joining (NHEJ) [28,29,30]. The high frequency of the DDR gene and pathway alterations exhibited in the samples in the Cancer Genome Atlas (TCGA) PanCanAtlas identifies opportunities to improve cancer therapy [31].
Several studies have recognized a correlation between DDR and ICB [10, 32,33,34]. The underlying mechanism pertaining to the DDR pathway that affects immune infiltration has also garnered increasing attention. An increase in neoantigen accumulation and induction of the anti-tumor immune response, initiated through the enlargement of somatic mutations and intracellular DNA fragment accumulation, have been recognized to play an active role in DDR dysfunction [35]. In addition to enhancing immunogenicity, the DDR pathways related to tumor cells affects both immune surveillance and the immune response. These can be assessed through both the DNA damage signaling pathway and the accumulation of cytoplasmic DNA subsequently activating the interferon pathway, which is related to the change of T cell infiltration and programmed cell death protein 1 (PD-1)/PD-L1 expression [9, 36]. The negative regulation of DDR inhibition on Treg cells also promotes T cell infiltration [37, 38]. Meanwhile, the role of the DDR pathway in immunotherapy is being evidenced in increasing tumor types [39]. At present, although several reviews have demonstrated the influence of DDR on ICB and immune infiltration [16, 40,41,42,43,44], we illustrate this relationship using specific tumor microenvironment (TME) components. Hence, this work aims to identify, summarize and explore the existing mechanisms and effects of DDR on ICB, so as to facilitate an improved understanding of these specific interactions and the efficacy of related therapies.
The effect of altered DDR pathway interactions on ICIs
The predictive effect of altered DDR pathways in ICIs
Change in the tumor DDR pathway is significantly correlated with the response to immune checkpoint inhibitors (ICIs) [24, 45,46,47] and serves to directly affect patient survival [35, 39, 47]. Among the patients treated with ICB, those with homologous recombination deficiency (HRD) or DDR mutation exhibited better response [13, 47]. For patients with gastrointestinal cancer, advanced urothelial cancer, and metastatic prostate cancer treated with anti-PD-(L)1, DDR changes can lead to the prolongation of overall survival (OS) and progression-free survival (PFS) [47, 48]. Furthermore, as the number of DDR changes increases, the objective response rate (ORR) and the durable clinical benefit (DCB) increased significantly [47, 49]. Therefore, DDR changes can be used as a reliable biomarker in ICI clinical applications [50].
Specific DDR changes can be used to predict different effects. For one, MSI-H/dMMR, which is listed as a predictive biomarker [51, 52], is directly related to greater benefits observed in ICB patients [24, 27, 38, 53]. A well-established close positive relationship between MMR defects and MSI exists [54, 55]. Interestingly, DDR mutations have also been identified in microsatellite stable (MSS) patients [56, 57]. These findings reflect the potential predictive value of DDR mutation as a tool capable of identifying the responder group [56]. In some cases, patients with the homologous recombination (HR) pathway defect have received increased benefits from ICIs [38]. Patients with BAP1 mutant malignant mesothelioma exhibit a significant increase in tumor-infiltrating lymphocytes (TILs) and high expression of immune checkpoint receptors [58]. However, clinical observation of the BRCA1/2 mutation has revealed contradictory results [59, 60]. In an analysis of whole exome sequences recorded in 38 patients diagnosed with melanoma and previously treated with PD-1 inhibitors, the ICB responders expressed augmented states in BRCA2 mutations [61]. However, a large phase 1b study investigating patients with recurrent or refractory ovarian cancer demonstrated that BRCA status was not associated with response to anti-PD-L1 treatment [62]. Cancer presenting with a defective BER pathway may increase the response to ICB. XRCC1 gene expression can be used to divide PD-L1+ and PD-1+ breast cancer patients into groups with different prognoses [63]. POLD1/POLE mutation is applicable to multi-tumor states and is a promising target for achieving accurate stratification [59, 64, 65]. Further evidence can be obtained from a related Phase II clinical trial (NCT03810339) [66]. Lastly, ataxia-telangiectasia mutated (ATM) protein kinase is known to improve the ORR of patients, although it may be related to a shorter OS [22, 49], and combined mutation of TP53 and ATM also constitutes a potential biomarker [67]. The relevant DDR mutations and DDR deficiencies involved in the predication of ICI efficacy are listed in Tables 1 and 2 below, respectively.
However, the use of individual DDR pathway changes in guiding ICIs treatment remains to be explored [20, 47, 69]. Researchers have established reliable DDR scoring models and effectively analyzed the predictive effect of co-mutation, the DDR gene models of various researchers can effectively predict specific response to ICIs treatment [69,70,71,72]. In one case, Yi-Ru Pan et al. selected 18 DDR genes as the defined gene panel and achieved a predicted response rate of about 60% in patients with DDR mutations [73]. Among 102 melanoma patients, 34 patients (33.3%) were classified as the mutated group, and 22 of 34 (64.7%) patients responded to ICIs [73]. The mutant group had significantly longer PFS and OS than the wild-type group [73].
Patients with a co-mutation in multiple DDR pathways demonstrate a good response to ICIs [59, 74]. HRR-MMR, or co-mutation in the HRR-BER pathway, is regarded as a potential biomarker [74,75,76]. In a study on pembrolizumab, 11 of 34 non-small cell lung cancer (NSCLC) patients were assigned to the co-mutation positive (co-mut+) group [74], in which ORR was 63.6% compared with 21.7% of the co-mutation negative (co-mut-) group. There was also significant PFS improvement in the co-mut + group (median, 4.1 months vs. not reached (NR), P = 0.006) [74]. Accordingly, 429 NSCLC patients were treated with atezolizumab, in whom the proportion of DCB in co-mut + patients was twice as high as that in co-mut- patients (56.7% vs. 28.8%), while the ORR was 11.9% higher (26.7% vs. 14.8%) [20]. In addition, even for patients with low PD-L1 expression, co-mut + status improved the overall results [20]. One hundred seventy-four patients presenting with metastatic melanoma and treated with the CTLA-4 antibody exhibited a significantly longer OS in the co-mut + group (median, 32.4 months vs. 10.8 months, P = 0.04) [74]. However, no significant difference was observed in the tumor response rate (43.8% vs. 29.5%, P = 0.15) [74].
In addition, simultaneous changes in the NOTCH signaling pathway and at least two DDR pathways [77], as well as the combination of TMB and an HR-DDR-positive status [78] may also represent effective biomarkers. Given this, DDR mutations should be considered a useful biomarker for patients receiving ICI treatment.
With regard to the activation level of the DDR pathway, in an analysis of 348 patients with metastatic urothelial carcinoma (UC) who received ICI treatment, the DSSH (DDR ssGSEA enrichment score-high) group was associated with longer OS (hazard ratio = 0.67), whereby a high activation level of the DDR pathway was identified as effective in improving the outcome and prognosis of patients that received ICI treatment [34]. Other studies have also revealed that high expression of the DDR pathway can predict an improved ICI response in patients with gastric cancer (GC) and advanced UC [79, 80]. In addition, the characteristics used to divide patients with hepatocellular carcinoma (HCC) into the DDR activation or inhibition subgroup can effectively help distinguish the clinical and molecular features of HCC, and predict different immunotherapy responses and prognoses [71, 72].
Indeed, even when compared to existing predictive markers such as TMB and PD-L1 expression, DDR mutation demonstrates an excellent prognostic effect [20, 22, 47, 48]. In addition, DDR status predicts different clinical outcomes between immunotherapy and non-immunotherapy [20, 72].
The combination of DDRi with ICIs in clinical trials
Although a limited number of clinical trials implementing combination therapy have been completed, many researchers have recognized the promising potential of combining DDR inhibitors (DDRi) and ICIs. As a result, a number of trials are currently recruiting, with a majority of them using anti-PD-(L)1 antibodies including durvalumab, pembrolizumab, dostarlimab, and nivolumab, with poly (ADP-ribose) polymerase (PARP) inhibitors (Table 3). In addition, the use of combination therapy seems quite promising for many cancer types. Combination therapy trials for solid tumors have most commonly been conducted on gynecologic tumors, breast cancer, and lung cancer (Table 3), while several clinical trials have also been conducted on gastrointestinal cancers, melanoma, urologic tumors, nasopharyngeal cancer, and osteosarcoma (Table 3).
PARP inhibitors (PARPi) are primarily used in the treatment of HRD tumors [38, 115, 116]. The synthetic lethality strategy entails a simultaneous inactivation of two functional genes leading to cell death. It is generally understood that PARPi functions by blocking the BER pathway, or other repair factors, through its inhibition of the core DNA damage sensors and signal transducers, PARP1/2 enzymes [84, 116]. As a result, cells are unable to repair single-stranded DNA damage. After the transformation of single-strand damage into double-strand damage, unlike normal cells, HR-deficient cells cannot be repaired through the HR pathway [117, 118]. Moreover, PARP activity inhibition can cause the NHEJ repair process to be more error prone [119]. Now, PARPi can be used in patients with BRCAness beyond germline BRCA1/2 mutations [116, 120]. In addition, some PARPi have been found to trap PARP1 on DNA-PARP complexes, which stalls the DNA replication fork and interferes with the DNA replication process [84]. Indeed, the trapping potency of many PARPi is roughly comparable to their related cytotoxic effects [118, 120].
PARPi can enhance anti-tumor immunity induced by anti-PD-L1 therapy, and its non-model specificity has been scientifically proven [9, 121,122,123]. The overall tolerance of 35 patients diagnosed with metastatic castration-resistant prostate cancer after combined treatment with olaparib and durvalumab was good, with a reported overall response rate of 14% and a disease control rate (PR + SD) of 71% [124]. A similar type of PARPi named niraparib combined with pembrolizumab, was observed in the treatment of triple-negative breast cancer (TNBC) and ovarian cancer, which also proved efficient with regard to anti-tumor activity in Phase I and Phase II trials [125]. Moreover, the ORR of patients with a BRCA1/2 mutation is significantly higher [125]. Further, the 1a/b Phase trial of oral PARPis, pamiparib and tislelizumab, demonstrated good tolerance [126]. Therefore, when compared individually with PARPi or a PD-(L)1 monoclonal antibody, the combination of the two can enhance tumor control. In another study, Konstantinopoulos et al. showed that, for patients who lack a BRCA mutation and have platinum-resistant ovarian carcinoma, the combination of an anti-PD-1 antibody and niraparib (ORR, 19%) appears to improve efficacy when compared with a single-agent PARP inhibitor (ORR, about 5%) or an anti-PD-1 antibody (ORR, 4–10%) [125].
Also, targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), PARPi combined with anti-CTLA-4 antibodies has demonstrated similar effects [127, 128]. Apart from immune checkpoint blockade therapy, chimeric antigen receptor T cell (CAR-T) immunotherapy has not been applied to solid tumors. However, olaparib has been shown to enhance CAR-T in the treatment of renal cancer and breast cancer models [129, 130].
At present, several other DDR inhibitors are also being actively implemented in clinical trials (Table 3). It has been shown that the combination of checkpoint kinase (CHK) inhibition and ICB is effective in the treatment of various cancers [9, 113, 131]. The WEE1 inhibitor, when combined with both radiation and a PD-1 blockade, can enhance the cytotoxic activity mediated by CD8 + T cells [132]. In addition, to further aggravate DNA damage and other mechanisms affiliated with the killing of tumor cells, some clinical trials have investigated triple therapy of two DDR inhibitors combined with ICIs, which have also yielded positive results [112, 133].
The mechanism of altered DDR pathway affecting the efficacy of ICIs
The success of immunotherapy is based on the characteristics of tumor cells and the ability to initiate an anti-tumor immune response and, likewise, the efficacy of ICIs is directly related to the functions of the immune microenvironment [134, 135]. The altered DDR pathway in tumors, which results from DDR gene defects or DDRi, directly affects the efficacy of ICIs by affecting immunogenicity (Fig. 1), immune cell infiltration, and the related regulating molecules (Fig. 2) [34, 38, 51, 136].

DDR pathway disorders contribute to immune recognition and tumor killing by increasing tumor immunogenicity. DDR mutation/inhibition impedes damaged DNA repair, enlarges chromosomal variation, and thus increases the levels of the tumor mutational burden (TMB) and neoantigen load (NAL). This subsequently activates CTLs and NK cells to exert anti-tumor activity via upregulating MHC-I and the antigen presentation process of APCs. This includes DCs and TAMs. At the same time, more tumor infiltrating lymphocytes (TILs) are recruited. (APC: Antigen-presenting cell; CTL: Cytotoxic T lymphocyte; DC: Dendritic cell; DDR: DNA damage repair; DSB: DNA double strand break; GZMA: Granzyme A; IL-2: Interleukin-2; NK cells: Natural killer cells; PRF1: Perforin 1; SSB: DNA single strand break; TAM: Tumor-associated macrophage)

The mechanism by which DDR affects PD-L1 expression and TME in tumors. In tumors, the defectiveness or inhibition of the DDR can lead to the accumulation of DNA damage, whereby double-stranded DNA and single-stranded DNA both accumulate in cytoplasm. Cytoplasmic DNA activates the cGAS/STING and RIG-I/MAVS pathways and eventually the type I interferon (IFN) pathway, ultimately recruiting both chemokines and immune cells (such as T cells, NK cells, and DCs). Specifically, STING promotes the phosphorylation and nuclear translocation of type I IFN transcriptional regulatory factors TBK1 and IFN regulator 3 (IRF3), while also activating the NF-κB pathway that interacts with IRF3. RIG-I can be considered an important participant in the immune activation of cancers presenting with genomic instability, as it can be activated via DNA, and combined with the adapter molecule MAV, which then activates IKK and/or TBK1 when stimulated, and finally activates the type I IFN pathway through downstream transcription factors. The activated TIL releases IFNγ, which acts on tumor cells and mediates STAT1/3-dependent PD-L1 upregulation. The ATM/ATR/Chk1 pathway can also induce PD-L1 expression. ATM can directly activate and participate in STING-mediated downstream pathways, and PARPi can promote PD-L1 expression by downregulating GSK3β. The release of the HMGB1 protein from dying tumor cells can bind TLR-4 on the surface of both DCs and macrophages in order to induce INF-β (TRIF) signal transduction, which subsequently activates IRF3 and NF-κB pathways. In addition, TLR4 recruits MyD88 and activates the NF-κB pathway to promote the transcription and secretion of various pro-inflammatory factors. Following this sequence of events, these factors serve to promote DC activation and trigger an immune response. HMGB1 can also upregulate PD-L1 expression in adjacent surviving tumor cells via TLR4/MyD88/TRIF signaling. In addition, ATM inhibitors can inhibit the induction of Tregs by tumor cell-derived small extracellular vesicles (sEV). (ATM: Ataxia telangiectasia mutated protein; ATR: Ataxia telangiectasia and Rad3-related protein; CCL5: C-C motif chemokine ligand 5; cGAMP: Cyclic GMP-AMP; cGAS: Cyclic GMP-AMP synthase; CHK1: Checkpoint kinase 1; CTL: Cytotoxic CD8+ T cell; CXCL10: C-X-C motif chemokine ligand 10; DDR: DNA damage response; DNAM-1: DNAX accessory molecule 1; DSB: Double-strand break; GSK-3β: Glycogen synthase kinase-3β; HMGB1: High mobility group box 1; IFNγ: Interferon-γ; IFNGR: Interferon gamma receptor; IKK: IκB kinase; IRF1: Interferon regulatory factor 1; IRF3: Interferon regulatory factor 3; MAVS: Mitochondrial antiviral signaling protein; MyD88: Myeloid differentiation factor 88; NF-κB: Nuclear factor kappa-B; NKG2D: Natural-killer group 2, member D; NKG2DL: NKG2D ligand; PARP: Poly-ADP-ribose polymerase; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand 1; RIG-I: Retinoic acid-inducible gene I; sEV: Small extracellular vesicles; SSB: Single-strand break; STAT1/3: Signal transducer and activator of transcription 1/3; STING: Stimulator of interferon genes; T1IFN: Type I interferon; TBK1: TANK binding kinase-1; TLR4: Toll-like receptor 4; TNFα: Tumor-necrosis factorα; Tregs: Regulatory T cells; TRIF: TIR-domain-containing adaptor inducing interferon-β)
DDR pathway dysfunction in tumors is capable of activating an immune response, reshaping the immune environment, and is ultimately beneficial to the effectiveness of ICIs. The primary mechanisms involved can be described as follows: (1) increasing the level of neoantigen and tumor immunogenicity, which is beneficial to the immune recognition process in antigen presentation; (2) the activated cyclic GMP-AMP synthase (cGAS) stimulator of interferon genes (STING) pathway triggers innate immunity and enhances the recruitment and tumor infiltration of T cells; (3) ATM/ataxia telangiectasia and Rad3-related protein (ATR)/Chk1 signals cascade to regulate the tumor microenvironment (TME), such as up-regulating PD-L1; and (4) other ways to enhance immunogenic cell death that help activate adaptive immunity [51, 76, 136, 137].
Alteration of the DDR pathway affects immunogenicity
The fidelity of DNA repair affects the generation of genome mutation [76]. DDR defects in tumor cells, or DDR inhibitors, can aggravate DNA damage and genomic instability (Fig. 1), leading to an increase in MSI, TMB, and neoantigen load (NAL) [38, 138,139,140]. Previous research has described the ability of DDR alterations in predicting high TMB and high MSI [64, 73, 76, 141,142,143,144]. In a NSCLC and melanoma cohort, a high TMB was the result of a DDR mutation accumulation [74]. Compared to HR wild-type tumors, the average TMB of HR-mutated tumors is higher in subtypes, MSI-H/dMMR is most common, and the result of a higher TMB is independent of microsatellite status [145]. Moreover, tumor samples presenting with different altered DDR pathways are also observed alongside an increase of NAL [47]. Notably, dMMR tumors have displayed the ability to enahnce the expression of mutation-related neoantigens [146]. In the other study, tumor samples with any mutation in NHEJ, HR, or DNA damage signaling genes have shown high NAL expression [76, 147]. Furthermore, combined mutation of multiple pathways indicates a higher level of NAL, which may be related to impaired availability of alternative repair pathways [76]. Tumors with POLE/POLD mutation have shown higher TMB and NAL, alongside improved responses to ICIs [59, 148, 149], while the DDR pathway characteristics established by Lou et al. are positively correlated with both the TMB and NAL [80]. Meanwhile, in addition to dMMR tumors, tumors with low BER/single-strand break repair (SSBR) gene expression have shown high MSI and neoantigen production [76, 150].
A high TMB can predict MSI-H and vice versa [64, 69, 151]. It should be noted that although MSI-H/dMMR is associated with TMB-H, DDR genes are still associated with a high TMB after MSI-H/dMMR is excluded [69]. As for the relationship between NAL and TMB, previous studies have proven that high TMB can cause an increase in neoantigens [137].
The elevation of MSI, TMB, and NAL are all beneficial to therapeutic immune recognition, and thus affect the response of the tumor to ICIs (Fig. 1). DDR inhibitors, such as PARPi, may increase both the mutation load and the number of neoepitopes, thereby increasing the response rate to ICIs [152]. The underlying mechanism of action is that a high TMB can increase the expression of immunogenic peptides [69, 73, 118, 153,154,155,156]. Specifically, an increased mutation load enhances CD8 + T cell infiltration (Fig. 1) [69, 101, 144], and the intracellular accumulation of DNA fragments furthers induces innate and adaptive immune activation [78]. An increase of NAL can also lead to active immune stimulation of the TME. For example, NAL can actively stimulate TILs to release IFN-γ (Fig. 1) [52]. dMMR tumors are highly sensitive to ICIs, and the objective response rate in CRC is recorded to be between 30 and 50% [24, 157, 158]. In examining tumor tissue, the effective rate of ICB was found to be higher in the TMB-H and MSI-H state than in the TMB-low or MSI-low/MSS state [56, 154].
However, certain cancer cells can also escape immune surveillance [84, 159]. This evasive ability may be attributed to the fact that mutation accumulation caused by DDR defects can also increase the level of PD-L 1[160, 161]. A dMMR/MSI-H tumor or HR-mutated tumor exhibits a strong expression of immune checkpoint ligand [15, 145, 146]. Much like the mechanism behind neoantigen production, the PD-L1 upregulation in tumor cells is related to both the TMB and the MSI. In addition, DDR deficiency upregulates the release of interferons (IFN) by neoantigen-activated T cells, which also promotes the expression of PD-L1 in the tumor environment [162].
DDR mutation can also affect the response to ICIs in ways that are independent of the TMB and MSI [47, 56]. For instance, the increase of the TMB is only one of the ways in which DDR defects lead to genomic instability. The genome of small cell lung cancer (SCLC), specifically characterized by the joint deletion of RB1 and TP53, is known to potentially cause inherent genomic instability, which is independent of the mutation burden [121]. In breast cancer, BRCA1 inactivation is accompanied by an increase of T cells [4, 147, 152, 163]. However, defects observed in TILs were detected in BRCA2-mutated breast cancers [163, 164]. Furthermore, in addition to the above regarded antigenicity, the adjuvanticity of immunogenic cell death (ICD) induction remains an important feature [165]. In this regard, before tumor-specific antigens elicit adaptive immunity, specific microorganism-associated molecular patterns (MAMPs) already activate pattern recognition receptors (PRRs) and initiate the cancer killing effect of the innate immune system. Such signals can be delivered to PRRs via the damage-associated molecular patterns (DAMPs) of dead cells [165, 166].
Alteration of the DDR pathway and immune infiltration
The poor response of some patients to ICIs is directly related to insufficient production and activity of anti-tumor CD8 + T cells. The aggravation of tumor hypoxia is recognized as another major obstacle in improving the efficacy of ICIs, which is related to the regulation of the TME via immunosuppressive microenvironment-related macrophages, regulatory T cells (Tregs), and so on [75, 93, 167,168,169]. Therefore, the effect of DDR on immune cell infiltration is likely to facilitate effective ICI treatments.
The effect of the DDR pathway activation level on immune infiltration is complex. The increased immune cell infiltration of gastric cancer is unexpectedly correlated with a low DDR characteristic score. However, insight into the relative abundances of immune cell subpopulations suggests that TILs are positively correlated with DDR pathway signature scores. Therefore, patients with a high score demonstrate a higher proportion of TIL, which corresponds to a better prognosis [80]. In HCC, the proportion of activated immune cells increases significantly in patients with a high expression of DDR-related genes (DDR2) [71, 72]. The subtype of activated DDR has demonstrated significantly increased expression of the immune checkpoint, and its immune score was significantly higher than that of the low expression group [72]. In addition, it is important to note that mutations or defects in the DDR signaling pathway genes are associated with altered immune cell infiltration.
T cell
DDR inhibitors, such as PARPi and CHK1 inhibitor (CHK1i) enhance T-cell infiltration [113]. Olaparib alone has been shown to increase the number of intratumoral effector CD4+ and CD8+ T cells, it can also decrease the expression of PD-1/Tim-3 and PD-1/Lag-3 co-inhibitory receptors on CD8+ T cells [170]. ICIs alone do not significantly alter the abundance and function of effector/memory T cells within BRCA1-deficient models [128]. When PARPi was used in combination with CTLA-4 mabs (but not with PD-1 mabs), CD4+ and CD8+ T cells, in addition to IFN-γ levels, were all significantly elevated [128]. However, another study demonstrated no significant change in CD8+ CTL infiltration with olaparib alone, while the combination of olaparib and PD-L1 blockade increased CTL infiltration [9].
Moreover, mutations in the DDR signaling pathway are associated with an increase of T cell infiltration in the TME [56], although the infiltration components are heterogeneous in different tumors [22, 61, 63, 143, 147, 152, 153, 171,172,173,174]. To name a few, gastrointestinal cancer patients with more than 2 DDR mutations display a greater infiltration of CD4+/CD8+ T cells [47], while DDR gene somatic mutations in ovarian cancer patients are related to higher levels of Th1 cell infiltration [143].
Alterations of the DDR pathway in tumors promote effector T cell and other cell infiltration in the TME through activating the IFN pathway. DNA damage exacerbated by altered DDR pathways can activate cGAS/STING, ATR/SRC/TBK1, ATM/ATR/Chk1, and other pathways, all of which then activate the downstream IFN pathway (Fig. 2) [33, 171]. In tumor cells, STAT1 and/or STAT3 can initiate typical IFN1 signals. This promotes systemic immune responses and regulates various anti-tumor immune components including T cells, NK cells, and DC, as well as effector molecules (Fig. 2) [38, 44, 84, 171]. The downstream target TIL then collects chemokine genes CXCL9, CXCL10, CXCL11, and CD8 + T cell activation markers (Fig. 2) [171], all of which contribute to a successful anti-tumor immune response. Thus, aside from its beneficial effect on adaptive immunity, IFN1 demonstrates an important functional role in driving the adjuvanticity of immunogenic cell death [165]. PARPi treatment has also been shown to increase the level of chemokines through the cGAS/STING pathway, induce the activation and function of both CD4 + T cells and CD8 + T cells, and increase the level of tumor necrosis factor α (TNF-α) in the TME (Fig. 2) [38, 124, 170, 175].
However, IFN-γ produced by activated TIL often acts as a double-edged sword in anti-tumor immune response. On the one hand, IFN-γ displays the positive anti-tumor immune effect of up-regulating the MHC, which in turn contributes to antigen presentation [52]. On the other hand, IFN-γ exposure increases PD-L1 expression through the JAK-STAT pathway, thus inducing negative feedback and adaptive resistance [52, 176]. IRF1 combined with the PD-L1 promoter enhances PD-L1 transcription through IFN-γ induction (Fig. 2) [162].
CD8+ T cell
DDR inhibition enhances CTL function. For example, in one study, PARP/CHK1 inhibition in SCLC patients increased CTL infiltration [131]. While augmented production of IFN-γ and TNF-α by CD8+ T cells was observed in patients after olaparib treatment [170]. Moreover, the active inhibition of ATM prevented Treg-induced CD8+ T cell senescence [177].
A non-neoantigen-dependent increase in CTL infiltration may be associated with the activation of the ATM gene [178]. Phosphorylated ATM was identified as a driving factor of cytokine production that can increase tumor cell-derived levels of CCL5, CXCL9, CXCL10, and IL16, leading to increased CD8+ CTL infiltration [178]. Moreover, in various cancer types, positive CTL score correlation with ATM protein, and neoantigens levels have been identified as being largely mutually exclusive [178]. Ciriello et al. appropriately classified cancers into either class C clusters or class M clusters [179]. Class M tumor profiles exhibit neoantigen-dependent CTL, whereas class C tumor profiles often indicate dependence on ATM [178].
CD4+ T cell
DDR inhibitors promote Th1 infiltration. In tumors, continuous low-level DNA stimulation incites the infiltration of inhibitory immune cells [38]. This transformation promotes chronic inflammation, immunosuppression, and tumor progression [180, 181]. Notably, a PARP inhibitor may turn this chronic, low-level DNA stimulation into a more significant Th1 immune response [38, 182].
DDR inhibition may create a more sensitive TME by inhibiting Tregs. ATM activation is vitally important for tumor cell-derived small extracellular vesicles (sEV) to effectively induce Tregs (Fig. 2) [37]. ATM inhibitor KU-60019 treatment can reverse the phosphorylation of ATM and reduce the Treg ratio during sEVs stimulation [37]. However, olaparib administered in TNBC increases the level of Tregs, thus causing the CD4+/CD8+ ratio to increase [183]. However, cases have been reported where no significant change in Treg expression was observed when administering olaparib and a PD-1 antibody [170].
Innate immune cell
In addition to the aforementioned IFN pathway increasing NK cell and DC infiltration, several other mechanisms can also affect intrinsic cell infiltration. These processes involve changes on the surface of and within the immune cells.
In addition to NK cells that kill cancer cells [184, 185], innate immunity is inextricably involved in the antitumor effects associated with adaptive immunity. The upregulation of innate immunity promotes neoantigen presentation and enhances immune stimulation [185, 186]. From another perspective, intrinsic immune cells contribute to the adjuvanticity (as related to the antigenicity) of ICD and help form an activated immune microenvironment. Therefore, it is essential to consider the role of innate immune cell presentation in ICI responses.
NK cell
DDR inhibition affects NK and CD8 + T cell functions by regulating NKG2D/NKG2DL. The NKG2D receptor, which exists on NK cells and CD8+ CTL, combined with NKG2DL, can help drive innate immunity and support adaptive immunity (Fig. 2) [17]. NKG2D-CAR-T cell therapy targeting NKG2D ligands is at an early clinical stage [187]. The DNA damage observed leads to the up-regulation of the ligands of the NKG2D and DNAM-1 receptors. This damage also promotes the increase of IFN-γ secretion and stimulates both NK cells and CD8 + CTL [17, 44]. ATM inhibition prevents the expression of stress molecules on tumor cells [188]. Moreover, DDRi can delay NK cell failure and benefit NK cell proliferation, survival, and function [188]. The expression of NKG2DL in tumor cells may be the result of cGAS/STING pathway-induced IFN-1 expression (Fig. 2) [115].
Aging-related secretory phenotypes function to recruit innate immune cells, including NK cells and macrophages, as well as CD8 + T cells [127]. Instability of aneuploid accumulation after chromosome separation into the micronucleus can directly up-regulate senescence-associated secretory phenotypes (SASP) [189,190,191]. Since a large subset of SASP cytokines are dependent on DDR signaling for their production, SASP is considered to be the extracellular extension of DDR which affects the microenvironment via the paracrine signal [192,193,194]. Notably, IL-6 plays an important role in the promotion of cancer cell invasion by SASP [193]. Moreover, chronic signaling of cGAS/STING seems to promote the aging phenotype [192, 195,196,197,198].
Dendritic cell
DDR dysfunction-activated STING/IFN regulator 3 (IRF3) can lead to elevated DC levels [17, 44, 84, 199]. Type I interferon promotes the cross-presentation capability of DCs [84, 133]. In addition, compared with patients exhibiting < 2 DDR gene mutations, patients diagnosed with gastrointestinal cancer and ≥ 2 DDR gene mutations have been shown to express increased levels of effective immune cells, including activated dendritic cells, and decreased levels of immunosuppressive cells observed in tumor infiltration [47].
In a study of BRCA1-deficient ovarian cancer, dendritic cells expressed upregulated levels of CD80, CD86, and MHC II expression after olaparib treatment in comparison to the study’s vehicle control and the PD-1 antibody [170]. Moreover, an increase in CD103 + DC in the tumor tissue was observed, which reportedly stimulated effector T cell trafficking and T cell immune initiation [200].
HMGB1 is regarded as a relevant factor in ICD, and its effective role appears to be dependent on Toll-like receptor 4 (TLR-4) involvement (Fig. 2) [165]. The HMGB1 protein secreted by damaged apoptotic tumor cells can promote transcription of type I interferons (e.g., INF-β), as well as instigate DC maturation (Fig. 2) [201, 202]. DCs express IL-12, which allows Th1 cells to differentiate into Th1 effector cells and ultimately facilitates T-cell immunity [203].
Macrophage
In melanoma, ATR mutation is related to inhibitory TME features such as increased inhibitory macrophages. In addition, ATR inhibitors are beneficial to the response and effect of ICIs. Compared to the ATR wild-type subjects, the number of macrophages and B cells increased noticeably, and the number of CD3+ T cells decreased in the immune infiltration of ATR mutant melanomas [204]. Macrophages, which have been previously detected in genetically heterogeneous tumors, can promote melanoma invasion and metastasis [204]. In a Phase I trial of ceralasertib combined with paclitaxel, 11 out of 33 patients with melanoma experienced a reversal of the drug resistance to PD-1 therapy [205].
Myeloid-derived suppressor cell
The DDRi olaparib can successfully inhibit the recruitment of myeloid-derived suppressor cells (MDSCs) through the SDF1α/CXCR4 axis to improve the anti-tumor effect of CAR-T in mouse breast cancer [130]. Olaparib reduces the expression of SDF1α released by cancer-associated fibroblasts, thereby reducing CXCR4-regulated MDSC migration and effectively promoting CAR-T cell infiltration [130]. In addition, olaparib treatment in BRCA1-deficient ovarian cancer results in a decrease in granulocyte MDSCs [170].
Innate immune cell
One TCGA cohort analysis of cervical cancer proved that somatic DDR alterations were positively correlated with the characteristics and scores of hypoxia. And the high hypoxia scores were positively correlated with the abundance of resting mast cells, while the abundance of activated mast cells were low [142]. In HCC, the subtype of inhibitory DDR showed that mast cells and neutrophils related to ICB drug resistance were significantly enriched [71].
Alteration of the DDR pathway affects immune regulatory molecules
The correlation between DDR changes, immune gene expression, and immune cycle steps has been verified in previous studies [56, 143, 171]. The level of DDR mutation and activation is related to enrichment of various immune features [34, 56]. These includes chemokines, lysis markers, MHC class II molecules, immune stimulators, and immune inhibitors [56]. High scores in DDR pathway activation were associated with both higher immune activation cell patterns and immune-related gene expression levels [34]. Interestingly, in muscle-invasive urothelial cancer, Thiago et al. analyzed the immune profiles of tumors harboring either biallelic, monoallelic, or wild-type mutations in the DDR genes. The expression of DDR genes and immunoregulatory genes were negatively correlated [171].
Pathway with cGAS/STING as the core
The cGAS/STING/IFNs pathway regulates innate and adaptive immunity. In tumors, defectiveness or inhibition of the DDR can lead to the accumulation of cytoplasmic DNA (Fig. 2) [44, 206, 207]. The sensing of cGAS to tumor-derived double-stranded DNA activates STING by generating cyclic GMP-AMP (cGAMP) (Fig. 2) [44, 208]. STING then induces a type I interferon reaction and consequently regulates the level of other immune and inflammatory factors (Fig. 2) [38, 44, 75, 208, 209]. PARPi, WEE1 inhibitor, ATM deficiency, and BRCA2 deficiency observed in tumors can promote both the recruitment of T cells, as well as the secretion of IFN-γ and TNFα in the TME through the cGAS/STING pathway (Fig. 2) [44, 114, 115, 124, 210,211,212,213]. This interferon-dependent pathway can activate both innate immunity and adaptive immunity, thus benefiting the remodeling of inhibitory TME [44, 212, 214]. In addition, the expression of PD-L1 is up-regulated at the transcription level [209], which is regarded as beneficial to ICIs.
There are several factors that can eliminate cytoplasmic nucleic acids in cancer cells. DDR factors, like RPA/RAD51, SAMHD1, and TREX1, can prevent excessive accumulation of cytoplasmic DNA [44, 76, 208, 215], while MRE11 can promote or inhibit the production of cytoplasmic DNA in different situations [44].
In this regard, STING is considered to be the focal point of the pathway. For example, PARPi promotes CAR-T infiltration in the TME via the cGAS-STING pathway. Specifically, PARPi promotes the upregulation of both IFN-β expression and chemokines via the STING-TBK1-IRF3 pathway, leading to CD8+ CAR-T cell recruitment and the secretion of large amounts of granzyme B [129]. This process ultimately allows low-dose CAR-T cell treatment to induce effective tumor regression in mice with kidney cancer [129]. DDR proteins ATM (Fig. 2), PARP1, DNA sensor (or DNA binding protein) IFI16, and Tp53 can also activate the STING pathway signal [76, 208, 216]. Moreover, MUS81 may help activate STING [44, 213].
Recently, researchers have found that the retinoic acid-inducible gene I (RIG-I)/mitochondrial antiviral signaling protein (MAVS) pathway has notable similarities to the cGAS/STING pathway (Fig. 2) [217, 218]. The interaction between the two pathways has been described in research [217].
ATM/ATR/Chk1 pathway
The ATM/ATR/Chk1 pathway regulates the cell cycle and can be activated by DNA damage signals [219,220,221,222,223,224,225,226], such as HR and NHEJ pathways, which are involved in DSB activation (Fig. 2) [209, 221].
ATM defective tumors can result in an increase in interferon signal transduction, innate immunity, and PD-L1 expression, all of which promote sensitivity to the PD-(L)1 blockade [227]. Specifically, ATM may inhibit SRC tyrosine kinase activity, while related ATM defects up-regulate SRC-promoted TBK1-PRR complex formation and type I interferon production independent of the cGAS/STING pathway [227, 228]. However, certain studies have shown that DSB activates ATM/ATR/Chk1, which can induce the activation of the classic STAT-IRF1 pathway. The inhibition of ATM or Chk1 can reduce IRF1 expression (Fig. 2) [221]. Furthermore, ATM can promote cancer cell migration by regulating the expression of IL-8 independent of its role in the repair of DNA double-strand breaks [229]. Co-inhibition of WEE1 combined with ATM reduced the expression of MMP-9, IL-8, CXCL1, CCL2, and CCL5, thus achieving an anti-migration effect in pancreatic cancer (PC) [230].
ATR mutation can regulate the tumor immune microenvironment in melanoma models and promote tumor growth [112, 204]. In previous studies, in addition to promoting T-cell homing to the epithelium, ATR mutant pigmented nevi also demonstrated a specific CD4 down-regulation effect [204, 231, 232]. Moreover, it is directly related to the increased expression of PD-L1 and CD206 [204], suggesting a relationship to the immune environment affiliated with T cell inhibition [112].
On the other hand, the effect of CHK1 inhibitors on interferon response is divergent. CHK1i can lead to an increase in cytoplasmic DNA, as well as the subsequent activation of the STING-TBK1-IRF3 pathway [9, 51]. As a result, both PD-L1 expression and type I interferon expression are upregulated. This results in the release of chemokines CXCL10 and CCL5 and facilitates the recruitment of CD8+ T cells [9]. CHK inhibitor prexasertib and anti-PD-L1 antibodies combination treatment can induce significant tumor regression in SCLC models [121]. However, Wayne et al. showed that CHK1i increased the phosphorylation of TBK1, but not the activation of IRF or type I interferon responses observed in solid cancer cell lines [131].
Abnormal DDR pathway and immune checkpoint molecules
The mutation of the DDR gene in cancer cells is usually associated with the increased expression of PD-1/PD-L1 genes within the TME [56, 64, 233, 234], such as the DSB repair gene [221], MMR gene [146, 235, 236] and RB1 [171, 237]. The expression of BER/SSBR has been shown to be negatively correlated with the level of PD-L1 observed in cancer cells [150, 235]. In addition, the clinical significance of PD-L1 in advanced GC depends specifically on the mutation of ARID1A and the level of ATM expression [238].
The following example illustrates the mechanism of the influence of DDR on PD-(L)1: the cGAS/STING pathway, activated by PARPi, leads to compensatory up-regulation of the PD-1/PD-L1 pathway [9, 118, 122]. This mechanism may be related to the secretion of IFN-γ, IFN-α, and IFN-β in the TME [211]. IFN-α and IFN-β can induce PD-L1 expression in endothelial cells, monocytes, and DC [209]. Glycogen synthase kinase 3β (GSK3β) subsequently interacts with PD-L1 and regulates its expression by inducing proteasome degradation of PD-L1 (Fig. 2) [38, 239]. PARPi deactivates GSK3β, thus enhancing the up-regulation of PD-L1 (Fig. 2) [33, 38, 122].
The ATM/ATR/CHK pathway is activated by DSB, such as when the BER/SSBR pathway is defective or during PARPi treatment, which in turn can up-regulate PD-L1 expression by phosphorylating STAT1/3 and therefore effectively inducing IRF1. It is also capable of up-regulating PD-L1 in circumstances that are independent of the IFN pathway (Fig. 2) [122, 136, 141, 150, 209, 221, 240,241,242,243,244]. Some studies have shown that the ATM/ATR/Chk1 pathway can up-regulate PD-L1 by activating the STAT1/3-IRF1 pathway in cancer cells (Fig. 2) [221, 245]. Up-regulation of PD-L1 expression induced by activating IR is carried out through transcription and post-translation mechanisms, while the ATM/BCLAF1/PD-L1 axis regulates the stability of PD-L1 in response to IR [246].
The inhibition of specific components in the ATM/ATR/Chk1 pathway can cause various effects on the stability of PD-L1. For instance, increased expression of PD-L1 can be observed in ATM-defective tumors [227]. However, another study investigating pancreatic cancer cells revealed that the inhibition of WEE1/ATM could effectively down-regulate the expression of PD-L1 by blocking GSK-3β and reducing the expression of CMTM6 [230]. ATM can also up-regulate PD-L1 through the JAK-STAT3 pathway, and inhibition of ATM can reduce PD-L1 expression [245]. Furthermore, while ATR inhibitor (ATRi) up-regulated PD-L1 mRNA in cells, it also led to the activation of the CDK1-SPOP axis and subsequent degradation of PD-L1. ATRi/ATR siRNA treatment led to the downregulation of PD-L1 protein expression, while, conversely, olaparib increased the expression of the PD-L1 protein [10, 122]. According to previous studies, post-translational regulation may in fact serve to play a major role in the regulation of PD-L1 by ATR [219]. Similarly, CHK1i activates the STING/TBK1/IRF3 innate immune pathway and increases the expression of PD-L1, as well as other immune infiltration changes [114, 211].
The alteration of the DDR pathway also affects other immune checkpoints, as has been seen when the expression of CTLA-4 and other immune checkpoints in the dMMR tumor increased as a result [146, 235]. IFN1 signaling can increase the molecular level of checkpoints, such as indoleamine 2,3-dioxygenase 1 (IDO1) and lymphocyte activation gene 3 protein (LAG3), in various immune cells [171]. The high expression of BRCA1 in tumor cells is significantly related to the positive expression of the new target Siglec-15 and a favorable prognosis [247, 248].
Prospects
At present, expanding the use of immune checkpoint inhibitors is a matter of great urgency. DDR displays great potential in this regard [249], as the alteration of the DDR pathway in tumors as a predictive marker of ICB presents many advantages [20, 249, 250]. More importantly, the benefits of DDR inhibitors have been extended to other immunotherapies beyond ICB [130]. Although the role of the DDR pathway dysfunction in tumor progression and immune regulation is somewhat uncertain [58, 112, 251] and not always useful [58, 83, 110, 115, 181].
As the anti-tumor benefits generated by DDR functional deficiency are deserving of greater research attention, we have put forward here several research directions and suggestions regarding the application of the DDR pathway in ICB. (1) DDR pathways require improved detection methods. This may entail devising novel strategies that dynamically detect DNA repair function and essentially avoid the associated errors caused by the time heterogeneity of somatic mutations [58]. (2) It is necessary to determine reliable predictive biomarkers, pay more attention to the prediction results of the DDR pathway co-mutation, and determine the appropriate unified labeling threshold [87, 115]. (3) Emphasis should be placed on investigative efforts aimed at defining the predictive role of the overall activation level of the DDR pathway. The first large GC cohort to investigate DDR pathway activity proved the clinical transformation value of the DDR pathway spectrum for patients. Patients with low DDR pathway signature scores might not benefit from anti-PD1 therapy [80]. (4) Increased research is advised regarding combination therapy, including the combination of two DDR inhibitors and immunotherapy. Moreover, the synergistic association of DDR inhibitors with certain defects may achieve more effective and accurate immunomodulation [213]. For example, ATM inhibitors are used in ATM defective tumors with positive effect [252]. Meanwhile, drug selection, dosage, and combination time are all carefully considered during personalized treatment, in addition to toxicity, immune-related adverse events, and drug resistance [112, 115]. Tumor type should also be considered in combination therapy. For example, the characteristics of the TNBC make it advantageous for combined therapy [93], while the determinants of ICB activity in SCLC remain unclear [121]. Ultimately, more randomized clinical trials are required to determine whether these combined treatments are superior to ICI alone, which must also be verified in larger clinical cohorts [58, 115]. (5) While there are limited reports exploring specific genes, such as the NHEJ and NER pathway, the relationship between other DDR pathways and immunotherapy remains to be investigated [58]. (6) To the best of our knowledge, the mechanism of DDR’s influence on the immune microenvironment is primarily focused on tumor cells rather than immune cells, though this particular aspect requires more in-depth study. (7) Further exploration of the relationship between emerging immune checkpoints and DDR pathway alterations is required. New ICIs are one of the primary hopes of patients to improve their OS when they fail to exhibit any response to anti-PD-L1 treatment. However, this topic has considerable space for development [248]. For example, the positive expression of Siglec-15 is significantly correlated with the high expression of BRCA1, but its mechanism has not yet been clarified.
Conclusions
DDR factors are competitive predictive biomarkers of specific ICI responses that have become increasingly relevant in current research. In addition to the more established dMMR, the primary focus of scholarship on this topic thus far has been investigating the predictive effects of combined DDR mutations. The combination of PARPi and anti-PD-L1 mAbs has yielded positive results, though more DDRi combination treatments for ICIs are required in current preclinical trials. Defection or inhibition of the DDR pathway can increase the efficacy of immunotherapy by increasing immunogenicity due to aggravated DNA damage, thus inhibiting the DNA damage sensing pathway and causing the accumulation of cytoplasmic DNA to activate the IFN pathway. However, the effect of DDRi is not consistent. Therefore, the choice of combination therapy should not necessarily be recommended for all cancer types.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- ARID1A:
-
AT-rich interaction domain 1A
- ATM:
-
Ataxia-telangiectasia mutated
- ATR:
-
Ataxia telangiectasia and Rad3-related protein
- ATRi:
-
ATR inhibitor
- BAP1:
-
BRCA1 associated protein 1
- BER:
-
Base excision repair
- BRCA1/2:
-
Breast cancer susceptibility gene 1/2
- CAR-T:
-
Chimeric antigen receptor T cell
- CCL2:
-
C-C motif chemokine ligand 2
- CCL5:
-
C-C motif chemokine ligand 5
- cGAMP:
-
Cyclic GMP-AMP
- cGAS:
-
Cyclic GMP-AMP synthase
- CHK:
-
Checkpoint kinase
- CHK1i:
-
CHK1 inhibitor
- CMTM6:
-
CKLF-like MARVEL transmembrane domain containing 6
- Co-mut-:
-
Co-mutation negative
- Co-mut+:
-
Co-mutation positive
- CTL:
-
Cytotoxic T lymphocyte
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CXCL1:
-
C-X-C motif chemokine ligand 1
- CXCL10:
-
C-X-C motif chemokine ligand 10
- CXCL11:
-
C-X-C motif chemokine ligand 11
- CXCL9:
-
C-X-C motif chemokine ligand 9
- CXCR4:
-
C-X-C motif chemokine receptor 4
- DAMPs:
-
Damage-associated molecular patterns
- DCB:
-
Durable clinical benefit
- DDR:
-
DNA damage repair
- DDR1:
-
The DDR-related gene low expression group
- DDR2:
-
The high expression of DDR-related genes
- DDRi:
-
DDR inhibitors
- dMMR:
-
Mismatch repair-deficient
- DNAM-1:
-
DNAX accessory molecule-1, CD226
- DSB:
-
DNA double strand break
- DSSH:
-
DDR ssGSEA enrichment score-high
- FOXP3:
-
Forkhead box P3
- GC:
-
Gastric cancer
- GSK3β:
-
Glycogen synthase kinase 3β
- HCC:
-
Hepatocellular carcinoma
- HMGB1:
-
High mobility group box 1
- HR:
-
Homologous recombination
- HRD:
-
Homologous recombination deficiency
- HRR:
-
Homologous recombination repair
- ICB:
-
Immune checkpoint blockade
- ICD:
-
Immunogenic cell death
- ICIs:
-
Immune checkpoint inhibitors
- IDO1:
-
Indoleamine 2,3-dioxygenase 1
- IFI16:
-
Interferon-gamma-inducible protein 16
- IFN:
-
Interferon
- IFN1:
-
Interferon type I
- IL-1:
-
Interleukin 1
- IL-6:
-
Interleukin 6
- IL-8:
-
Interleukin-8,C-X-C motif chemokine ligand 8
- IRF1:
-
Interferon regulatory factor 1
- IRF3:
-
IFN regulator 3
- JAK2:
-
Janus kinase 2
- LAG3:
-
Lymphocyte activation gene 3 protein
- MAMPs:
-
Microorganism-associated molecular patterns
- MAVS:
-
Mitochondrial antiviral signaling protein
- mCRPC:
-
Metastatic castration-resistant prostate cancer
- MDSCs:
-
Myeloid-derived suppressor cells
- MHC:
-
Major histocompatibility complex
- MMP-9:
-
Matrix metalloproteinase-9
- MMR:
-
Mismatch repair
- MRE11:
-
Meiotic recombination 11 homolog, double strand break repair nuclease
- MSI:
-
Microsatellite instability
- MSI-H:
-
Microsatellite instability-high
- MSS:
-
Microsatellite stable
- MUS81:
-
Methyl methanesulphonate and ultraviolet-sensitive 81, structure-specific endonuclease subunit
- MyD88:
-
Myeloid differentiation factor 88
- NAL:
-
Neoantigen load
- NER:
-
Nucleotide excision repair
- NHEJ:
-
Non-homologous end joining
- NK cells:
-
Natural killer cells
- NKG2D:
-
Natural-killer group 2, member D
- NKG2DL:
-
Ligands for the receptors NKG2D
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PARP:
-
Poly (ADP-ribose) polymerase
- PARPi:
-
PARP inhibitors
- PC:
-
Pancreatic cancer
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed cell death 1 ligand 1
- PFS:
-
Progression-free survival
- POLD1:
-
DNA Polymerase Delta 1, catalytic subunit
- POLE:
-
DNA Polymerase Epsilon, catalytic subunit
- PRR:
-
Pattern recognition receptor
- RAD51:
-
RAD51 recombinase
- RB1:
-
RB Transcriptional Corepressor 1
- RIG-I:
-
Retinoic acid-inducible gene I
- RPA:
-
Replication protein A
- SAMHD1:
-
Sterile alpha motif and HD domain containing protein 1
- SASP:
-
Senescence-associated secretory phenotype
- SCLC:
-
Small cell lung cancer
- SDF1α:
-
Stromal cell-derived factor 1 alpha, C-X-C motif chemokine ligand 12 alpha
- sEV:
-
Small extracellular vesicles
- Siglec-15:
-
Sialic acid-binding immunoglobulin-like lectin 15
- SIRT1:
-
Sirtuin 1
- SIRT2:
-
Sirtuin 2
- SIRT6:
-
Sirtuin 6
- SRC:
-
SRC tyrosine kinase
- SSBR:
-
Single strand break repair
- STAT1:
-
Signal transducer and activator of transcription 1
- STAT3:
-
Signal transducer and activator of transcription 3
- STING:
-
Stimulator of interferon genes
- TBK1:
-
TANK binding kinase 1
- TCGA:
-
The Cancer Genome Atlas
- TILs:
-
Tumor infiltrating lymphocytes
- TLR-4:
-
Toll-like receptor 4
- TMB:
-
Tumor mutational burden
- TMB-H:
-
Tumor mutational burden-high
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple negative breast cancer
- TNF-α:
-
Tumor necrosis factor α
- TP53:
-
Tumor protein P53
- Treg:
-
Regulatory T cell
- TREX1:
-
Three-prime repair exonuclease 1
- INF-β/TRIF:
-
Toll/IL-1 receptor (TIR) domain-containing adaptor-inducing interferon-β
- UC:
-
Urothelial carcinoma
- WEE1:
-
WEE1 G2 checkpoint kinase
- XRCC1:
-
X-ray cross complementing group 1
References
Marconcini R, Spagnolo F, Stucci LS, Ribero S, Marra E, de Rosa F, et al. Current status and perspectives in immunotherapy for metastatic melanoma. Oncotarget. 2018;9:12452–70 Impact Journals LLC. Available from: https://pubmed.ncbi.nlm.nih.gov/29552325. Accessed 11 Aug 2021.
Si Y, Lin A, Ding W, Meng H, Luo P, Zhang J. CARD11 alteration as a candidate biomarker of skin cutaneous melanoma treated with immune checkpoint blockade. Am J Transl Res. 2021;13:286–300 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7847528/. Accessed 15 June 2022.
Lin W, Lin A, Li Z, Zhou C, Chen C, Chen B, et al. Potential predictive value of SCN4A mutation status for immune checkpoint inhibitors in melanoma. Biomed Pharmacother. 2020;131:110633 Available from: http://www.ncbi.nlm.nih.gov/pubmed/32892029. Accessed 14 June 2022.
Nolan E, Savas P, Policheni AN, Darcy PK, Vaillant F, Mintoff CP, et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci Transl Med. 2017;9 Available from: http://stm.sciencemag.org/. Accessed 9 Sept 2021.
Parker C. A near miss for prostate cancer immunotherapy. Lancet Oncol. 2014;15:669–71 Available from: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(14)70220-7/fulltext. Accessed 7 Sept 2021.
Goetze TO. Immunotherapy: a new era in small-cell lung cancer. Lancet J. 2019;394:1884–5. Available from: https://doi.org/10.1016/S0140-6736(19)32235-4 Accessed 7 Sept 2021.
Souquet P-J, Couraud S. Immune checkpoint inhibitors: a game changer for metastatic non-small-cell lung cancer. Lancet Oncol. 2019;20:1334–5 Available from: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(19)30508-X/fulltext. Accessed 7 Sept 2021.
Lin A, Zhang H, Hu X, Chen X, Wu G, Luo P, et al. Age, sex, and specific gene mutations affect the effects of immune checkpoint inhibitors in colorectal cancer. Pharmacol Res. 2020;159:105028. https://doi.org/10.1016/j.phrs.2020.105028 Accessed 23 Aug 2021.
Sen T, Rodriguez BL, Chen L, della Corte CM, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes anti-tumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–61. Available from: https://doi.org/10.1158/2159-8290.CD-18-1020 American Association for Cancer Research Inc. Accessed 2 Sept 2021.
Tang Z, Pilié PG, Geng C, Manyam GC, Yang G, Park S, et al. ATR inhibition induces CDK1-SPOP signaling and enhances anti-PD-L1 cytotoxicity in prostate cancer. Clin Cancer Res. 2021;27:4898–909 American Association for Cancer Research Inc. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8456453/. Accessed 11 Jan 2022.
Subudhi SK, Vence L, Zhao H, Blando J, Yadav SS, Xiong Q, et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci Transl Med. 2020;12:3577 Available from: https://pubmed.ncbi.nlm.nih.gov/32238575/. Accessed 7 Sept 2021.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54 New England Journal of Medicine (NEJM/MMS). Available from: https://www.nejm.org/doi/full/10.1056/NEJMoa1200690. Accessed 9 Aug 2021.
Sharma P, Pachynski RK, Narayan V, Fléchon A, Gravis G, Galsky MD, et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the CheckMate 650 trial. Cancer Cell. 2020;38:489–499.e3 Cell Press. Available from: https://www.cell.com/cancer-cell/pdf/S1535-6108(20)30418-9.pdf. Accessed 11 Aug 2021.
Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18:345–62 Nature Research. Available from: https://www.nature.com/articles/s41571-021-00473-5. Accessed 11 Sept 2021.
Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res. 2016;22:813–20 American Association for Cancer Research Inc. Available from: https://aacrjournals.org/clincancerres/article/22/4/813/251220/Microsatellite-Instability-as-a-Biomarker-for-PD-1?searchresult=1. Accessed 19 Sept 2021.
Lamberti G, Andrini E, Sisi M, di Federico A, Ricciuti B. Targeting DNA damage response and repair genes to enhance anticancer immunotherapy: rationale and clinical implication. Future Oncol. 2020;16:1751–66 Future Medicine Ltd. Available from: https://www.futuremedicine.com/doi/full/10.2217/fon-2020-0215. Accessed 12 Sept 2021.
Lhuillier C, Vanpouille-Box C, Galluzzi L, Formenti SC, Demaria S. Emerging biomarkers for the combination of radiotherapy and immune checkpoint blockers. Semin Cancer Biol. 2018;52:125–34 Academic Press. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6004231/. Accessed 24 Aug 2021.
Klein O, Kee D, Markman B, Carlino MS, Underhill C, Palmer J, et al. Evaluation of TMB as a predictive biomarker in patients with solid cancers treated with anti-PD-1/CTLA-4 combination immunotherapy. Cancer Cell. 2021;39:592–3 Cell Press. Available from: https://www.sciencedirect.com/science/article/pii/S1535610821002130. Accessed 11 Sept 2021.
Lin A, Zhang J, Luo P. Crosstalk between the MSI status and tumor microenvironment in colorectal cancer. Front Immunol. 2020;11:2039 Available from: http://www.ncbi.nlm.nih.gov/pubmed/32903444. Accessed 14 June 2022.
Xiong A, Nie W, Zhou Y, Li C, Gu K, Zhang D, et al. Comutations in DDR pathways predict Atezolizumab response in non-small cell lung cancer patients. Front Immunol. 2021;12:708558 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34630387. Accessed 12 Jan 2022.
Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, et al. Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2016;34:2980–7 American Society of Clinical Oncology. Available from: https://ascopubs.org/doi/10.1200/JCO.2016.66.9929. Accessed 9 Feb 2022.
de Bono JS, Goh JCH, Ojamaa K, Piulats Rodriguez JM, Drake CG, Hoimes CJ, et al. KEYNOTE-199: Pembrolizumab (pembro) for docetaxel-refractory metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2018;36(Suppl 15):5007. https://doi.org/10.1200/JCO.2018.36.15_suppl.5007 Wolters Kluwer. Accessed 15 Sept 2021.
Ribas A. Releasing the brakes on cancer immunotherapy. N Engl J Med. 2015;373:1490–2 New England Journal of Medicine (NEJM/MMS). Available from: https://www.nejm.org/doi/full/10.1056/NEJMp1510079. Accessed 10 Feb 2022.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20 New England Journal of Medicine (NEJM/MMS). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4481136/. Accessed 14 Jan 2022.
Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun. 2017;8:1–12 Nature Publishing Group. Available from: https://www.nature.com/articles/ncomms15180. Accessed 13 Jan 2022.
Ettrich TJ, Seufferlein T. Systemic therapy for metastatic pancreatic cancer. Curr Treat Options in Oncol. 2021;22:106 Springer. Available from: https://link.springer.com/article/10.1007/s11864-021-00895-4. Accessed 10 Feb 2022.
Marabelle A, Le DT, Ascierto PA, di Giacomo M, de Jesus-Acosta A, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/ mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020;38:1–10 Available from: https://ascopubs.org/doi/10.1200/JCO.19.02105. Accessed 9 Mar 2022.
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. Available from: https://doi.org/10.1038/nrc2342 Accessed 14 Mar 2022.
Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. Available from: https://doi.org/10.1146/annurev.biochem.73.011303.073723 Accessed 14 Mar 2022.
Luo P, Lin A, Li K, Wei T, Zhang J. DDR pathway alteration, tumor mutation burden, and cisplatin sensitivity in small cell lung cancer: difference detected by whole exome and targeted gene sequencing. J Thorac Oncol. 2019;14:e276–9 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31757380. Accessed 14 June 2022.
Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep. 2018;23:239–254.e6. Available from: https://doi.org/10.1016/j.celrep.2018.03.076 Cell Press. Accessed 30 Apr 2022.
Yu EY, Wu H, Schloss C. KEYNOTE-365: phase 1b/2 trial of pembrolizumab combination therapy for metastatic castration-resistant prostate cancer (mCRPC). Eur Urol Suppl. 2017;16:e360 Available from: https://www.sciencedirect.com/science/article/pii/S1569905617302749. Accessed 9 Jan 2022.
Karzai F, Vanderweele D, Madan RA, Owens H, Cordes LM, Hankin A, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6:141. Available from: https://doi.org/10.1186/s40425-018-0463-2 BioMed Central Ltd. Accessed 23 Aug 2021.
Zhou C, Lin A, Cao M, Ding W, Mou W, Guo N, et al. Activation of the DDR pathway leads to the down-regulation of the TGFβ pathway and a better response to ICIs in patients with metastatic urothelial carcinoma. Front Immunol. 2021;12 Frontiers Media S.A. Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2021.634741/full. Accessed 11 Sept 202.
Qing T, Jun T, Lindblad KE, Lujambio A, Marczyk M, Pusztai L, et al. Diverse immune response of DNA damage repair-deficient tumors. Cell Rep Med. 2021;2:100276. Available from: https://doi.org/10.1016/j.xcrm.2021.100276 Accessed 11 Sept 2021.
Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by s-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. 2017;109 Oxford University Press. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5441301/. Accessed 6 Oct 2021.
Shen T, Jia S, Ding G, Ping D, Zhou L, Zhou S, et al. BxPC-3-derived small extracellular vesicles induce FOXP3+ Treg through ATM-AMPK-Sirtuins-mediated FOXOs nuclear translocations. iScience. 2020;23:101431 Available from: http://www.ncbi.nlm.nih.gov/pubmed/32798974. Accessed 8 Dec 2021.
Peyraud F, Italiano A. Combined PARP inhibition and immune checkpoint therapy in solid tumors. Cancers (Basel). 2020;12 Available from: http://www.ncbi.nlm.nih.gov/pubmed/32526888. Accessed 8 Nov 2021.
Ricciuti B, Recondo G, Spurr LF, Li YY, Lamberti G, Venkatraman D, et al. Impact of DNA damage response and repair (DDR) gene mutations on efficacy of PD-(L)1 immune checkpoint inhibition in non–small cell lung cancer. Clin Cancer Res. 2020;26:4135–42 American Association for Cancer Research Inc. Available from: https://aacrjournals.org/clincancerres/article/26/15/4135/82560/Impact-of-DNA-Damage-Response-and-Repair-DDR-Gene. Accessed 12 Sept 2021.
Chabanon RM, Rouanne M, Lord CJ, Soria JC, Pasero P, Postel-Vinay S. Targeting the DNA damage response in immuno-oncology: developments and opportunities. Nat Rev Cancer. 2021;21:701–17 Nature Research. Available from: https://www.nature.com/articles/s41568-021-00386-6. Accessed 15 Mar 2022.
Huang JL, Chang YT, Hong ZY, Lin CS. Targeting DNA damage response and immune checkpoint for anticancer therapy. Int J Mol Sci. 2022;23:3238 Available from: http://www.ncbi.nlm.nih.gov/pubmed/35328658. Accessed 15 June 2022.
Hopkins JL, Lan L, Zou L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. 2022;36:278–93 Available from: http://www.ncbi.nlm.nih.gov/pubmed/35318271. Accessed 15 June 2022.
Wang Y, Duan M, Peng Z, Fan R, He Y, Zhang H, et al. Advances of DNA damage repair-related drugs and combination with immunotherapy in tumor treatment. Front Immunol. 2022;13. Available from: https://doi.org/10.3389/fimmu.2022.854730 Frontiers Media S.A. Accessed 15 June 2022.
Pilger D, Seymour LW, Jackson SP. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev. 2021;35:602–18 Cold Spring Harbor Laboratory Press. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8091970/. Accessed 1 Aug 2021.
Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36:1685–94. Available from: https://doi.org/10.1200/JCO.2017.75.7740 Accessed 22 Aug 2021.
Banchereau R, Leng N, Zill O, Sokol E, Liu G, Pavlick D, et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat Commun. 2021;12:3969. Available from: https://doi.org/10.1038/s41467-021-24,112-w Accessed 11 Oct 2021.
Wang Y, Jiao X, Li S, Chen H, Wei X, Liu C, et al. Alterations in DNA damage response and repair genes as potential biomarkers for immune checkpoint blockade in gastrointestinal cancer. Cancer Biol Med. 2021. Available from: https://doi.org/10.20892/j.issn.2095-3941.2020.0708 China Anti-cancer Association. Accessed 15 Feb 2022.
Fahmy O, Alhakamy NA, Khairul-Asri MG, Ahmed OAA, Fahmy UA, Fresta CG, et al. Oncological response and predictive biomarkers for the checkpoint inhibitors in castration-resistant metastatic prostate cancer: a systematic review and meta-analysis. J Pers Med. 2022;12:8. Available from: https://doi.org/10.3390/jpm12010008 MDPI. Accessed 13 Feb 2022.
Joshi M, Grivas P, Mortazavi A, Monk P, Clinton SK, Sue-Ann Woo M, et al. Alterations of DNA damage response genes correlate with response and overall survival in anti-PD-1/PD-L1-treated advanced urothelial cancer. Cancer Med. 2020;9:9365–72 Blackwell Publishing Ltd. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7774722/. Accessed 19 Aug 2021.
Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7:675–93 American Association for Cancer Research Inc. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5659200/. Accessed 11 Oct 2021.
Sun W, Zhang Q, Wang R, Li Y, Sun Y, Yang L. Targeting DNA damage repair for immune checkpoint inhibition: mechanisms and potential clinical applications. Front Oncol. 2021;11 Frontiers Media S.A. Available from: https://www.frontiersin.org/articles/10.3389/fonc.2021.648687/full. Accessed 11 Aug 2021.
Conway JR, Kofman E, Mo SS, Elmarakeby H, van Allen E. Genomics of response to immune checkpoint therapies for cancer: implications for precision medicine. Genome Med. 2018;10:93 BioMed Central Ltd. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6264032/. Accessed 19 Aug 2021.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (1979). 2017;357:409–13 American Association for the Advancement of Science. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5576142/. Accessed 13 Sept 2021.
Prolla TA, Baker SM, Harris AC, Tsao J-L, Yao X, Bronner CE, et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DMA mismatch repair. Nat Genet. 1998;18:276–9 Available from: https://www.nature.com/articles/ng0398-276. Accessed 14 Mar 2022.
Svrcek M, El-Bchiri J, Chalastanis A, Capel E, Dumont S, Buhard O, et al. Specific clinical and biological features characterize inflammatory bowel disease-associated colorectal cancers showing microsatellite instability. J Clin Oncol. 2007;25:4231–8. Available from: https://doi.org/10.1200/jco.2007.10.9744 Accessed 14 Mar 2022.
Song Y, Huang J, Liang D, Hu Y, Mao B, Li Q, et al. DNA damage repair gene mutations are indicative of a favorable prognosis in colorectal cancer treated with immune checkpoint inhibitors. Front Oncol. 2021;10:549777 Frontiers Media S.A. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7934780/. Accessed 8 Sept 2021.
Keenan BP, van Loon K, Khilnani AD, Fidelman N, Behr SC, Atreya CE, et al. Molecular and radiological features of microsatellite stable colorectal cancer cases with dramatic responses to immunotherapy. Anticancer Res. 2021;41:2985–92 International Institute of Anticancer Research. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8631311/. Accessed 13 Sept 2021.
Jiang M, Jia K, Wang L, Li W, Chen B, Liu Y, et al. Alterations of DNA damage response pathway: biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm Sin B. 2021;11:2983–94 Chinese Academy of Medical Sciences. Available from: http://www.ncbi.nlm.nih.gov/pubmed/34729299. Accessed 9 Nov 2021.
Zhang J, Shih DJH, Lin SY. Role of DNA repair defects in predicting immunotherapy response. Biomark Res. 2020;8:23 BioMed Central. Available from: https://pubmed.ncbi.nlm.nih.gov/32612833/. Accessed 18 Aug 2021.
Cimadamore A, Mazzucchelli R, Lopez-Beltran A, Massari F, Santoni M, Scarpelli M, et al. Prostate cancer in 2021: novelties in prognostic and therapeutic biomarker evaluation. Cancers (Basel). 2021;13:3471 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34298683. Accessed 10 Feb 2022.
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. Available from: https://doi.org/10.1016/j.cell.2016.02.065 Cell Press. Accessed 9 Nov 2021.
Disis ML, Taylor MH, Kelly K, Beck JT, Gordon M, Moore KM, et al. Efficacy and safety of avelumab for patients with recurrent or refractory ovarian cancer: phase 1b results from the JAVELIN solid tumor trial. JAMA Oncol. 2019;5:393–401. Available from: https://doi.org/10.1001/jamaoncol.2018.6258 American Medical Association. Accessed 11 Jan 2022.
Green AR, Aleskandarany MA, Ali R, Hodgson EG, Atabani S, de Souza K, et al. Clinical impact of tumor DNA repair expression and T-cell infiltration in breast cancers. Cancer Immunol Res. 2017;5:292–9. Available from: https://doi.org/10.1158/2326-6066.CIR-16-0195 American Association for Cancer Research Inc. Accessed 6 Feb 2022.
Xiao Y, Lu D, Lei M, Xie W, Chen Y, Zheng Y, et al. Comprehensive analysis of DNA damage repair deficiency in 10,284 pan-cancer study. Ann Transl Med. 2021;9:1661 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34988170. Accessed 19 Jan 2021.
Ying J, Yang L, Yin JC, Xia G, Xing M, Chen X, et al. Additive effects of variants of unknown significance in replication repair-associated DNA polymerase genes on mutational burden and prognosis across diverse cancers. J Immunother Cancer. 2021;9 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34479923. Accessed 6 Jan 2022.
Wang F, Zhao Q, Wang YN, Jin Y, He MM, Liu ZX, et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019;5:1504–6 American Medical Association. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6696731/. Accessed 6 Jan 2022.
Chen Y, Chen G, Li J, Huang YY, Li Y, Lin J, et al. Association of tumor protein p53 and ataxia-telangiectasia mutated comutation with response to immune checkpoint inhibitors and mortality in patients with non-small cell lung cancer. JAMA Netw Open. 2019;2:e1911895 NLM (Medline). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6755545/. Accessed 8 Jan 2022.
Chen Y, Li Y, Guan Y, Huang Y, Lin J, Chen L, et al. Prevalence of PRKDC mutations and association with response to immune checkpoint inhibitors in solid tumors. Mol Oncol. 2020;14:2096–110. Available from: https://doi.org/10.1002/1878-0261.12739 Accessed 2 Sept 2021.
Gong Z, Yang Y, Zhang J, Guo W. Evaluation of 30 DNA damage response and 6 mismatch repair gene mutations as biomarkers for immunotherapy outcomes across multiple solid tumor types. Cancer Biol Med. 2021;18:1080–91 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8610155/. Accessed 11 Jan 2022.
Leng Y, Dang S, Yin F, Gao T, Xiao X, Zhang Y, et al. GDPLichi: a DNA damage repair-related gene classifier for predicting lung adenocarcinoma immune checkpoint inhibitors response. Front Oncol. 2021;11:733533 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34970479. Accessed 19 Dec 2021.
Lin P, Gao RZ, Wen R, He Y, Yang H. DNA damage repair profiles alteration characterize a hepatocellular carcinoma subtype with unique molecular and clinicopathologic features. Front Immunol. 2021;12:715460 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34456923. Accessed 7 Jan 2022.
Chen Y, Wang X, Deng X, Zhang Y, Liao R, Li Y, et al. DNA damage repair status predicts opposite clinical prognosis immunotherapy and non-immunotherapy in hepatocellular carcinoma. Front Immunol. 2021;12:676922 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34335575. Accessed 7 Feb 2022.
Pan YR, Wu CE, Wang YC, Yeh YC, Lu ML, Hung YP, et al. Establishment of a novel gene panel as a biomarker of immune checkpoint inhibitor response. Clin Transl Immunol. 2020;9 John Wiley and Sons Inc. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7326616/. Accessed 11 Jan 2022.
Wang Z, Zhao J, Wang G, Zhang F, Zhang Z, Zhang F, et al. Comutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Cancer Res. 2018;78:6486–96. Available from: https://doi.org/10.1158/0008-5472.CAN-18-1814 American Association for Cancer Research Inc. Accessed 13 Oct 2021.
Kumari S, Sharma S, Advani D, Khosla A, Kumar P, Ambasta RK. Unboxing the molecular modalities of mutagens in cancer. Environ Sci Pollut Res Int. 2021:1–49 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8492102/. Accessed 23 Feb 2022.
Kakoti S, Sato H, Laskar S, Yasuhara T, Shibata A. DNA repair and signaling in immune-related cancer therapy. Front Mol Biosci. 2020;7:205 Frontiers Media S.A. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7506057/. Accessed 19 Sept 2021.
Zhang Z, Gu Y, Su X, Bai J, Guan W, Ma J, et al. Co-occurring alteration of NOTCH and DDR pathways serves as novel predictor to efficacious immunotherapy in NSCLC. Front Oncol. 2021:11 Frontiers Media S.A. Available from: https://www.frontiersin.org/articles/10.3389/fonc.2021.659321/full. Accessed 11 Aug 2021.
Wang H-Y, Deng L, Li Y-Q, Zhang X, Long Y-K, Zhang X, et al. Pan-cancer analysis of tumor mutational burden and homologous recombination DNA damage repair using targeted next-generation sequencing. Cancer Res Treat. 2021;53:973–82. Available from: https://doi.org/10.4143/crt.2020.798 Accessed 11 Sept 2021.
Baek SW, Jang IH, Kim SK, Nam JK, Leem SH, Chu IS. Transcriptional profiling of advanced urothelial cancer predicts prognosis and response to immunotherapy. Int J Mol Sci. 2020:21 MDPI AG. Available from: https://www.mdpi.com/1422-0067/21/5/1850/htm. Accessed 11 Aug 2021.
Lou S, Wang Y, Zhang J, Yin X, Zhang Y, Wang Y, et al. Patient-level DNA damage repair pathway profiles and anti-tumor immunity for gastric cancer. Front Immunol. 2022:12 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8785952/. Accessed 26 Sept 2021.
Knelson EH, Patel SA, Sands JM. Parp inhibitors in small-cell lung cancer: rational combinations to improve responses. Cancers (Basel). 2021;13:1–16. https://doi.org/10.3390/cancers13040727 MDPI AG. Accessed 19 Apr 2022.
Audisio M, Tucci M, di Stefano RF, Parlagreco E, Ungaro A, Turco F, et al. New emerging targets in advanced urothelial carcinoma: is it the primetime for personalized medicine? Crit Rev Oncol Hematol. 2022;174:103682. https://doi.org/10.1016/j.critrevonc.2022.103682 Accessed 21 Mar 2022.
Stewart RA, Pilie PG, Yap TA. Development of PARP and immune-checkpoint inhibitor combinations. Cancer Res. 2018;78:6717–25. https://doi.org/10.1158/0008-5472.CAN-18-2652 American Association for Cancer Research Inc. Accessed 13 Feb 2022.
Li A, Yi M, Qin S, Chu Q, Luo S, Wu K. Prospects for combining immune checkpoint blockade with PARP inhibition. J Hematol Oncol. 2019;12:98 BioMed Central Ltd. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6744711/. Accessed 24 Aug 2021.
Ozaniak A, Vachtenheim J, Lischke R, Bartunkova J, Strizova Z. Novel insights into the immunotherapy of soft tissue sarcomas: do we need a change of perspective? Biomedicines. 2021;9:935. https://doi.org/10.3390/biomedicines9080935 MDPI AG. Accessed 21 Mar 2022.
Moutafi M, Economopoulou P, Rimm D, Psyrri A. PARP inhibitors in head and neck cancer: molecular mechanisms, preclinical and clinical data. Oral Oncol. 2021;117 Elsevier Ltd. Available from: https://www.sciencedirect.com/science/article/pii/S1368837521001159. Accessed 8 Aug 2021.
Xie H, Wang W, Qi W, Jin W, Xia B. Targeting DNA repair response promotes immunotherapy in ovarian cancer: rationale and clinical application. Front Immunol. 2021;12:661115 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34712221. Accessed 17 Jan 2022.
Huang Q, Liang X, Ren T, Huang Y, Zhang H, Yu Y, et al. The role of tumor-associated macrophages in osteosarcoma progression – therapeutic implications. Cell Oncol. 2021;44:525–39. https://doi.org/10.1007/s13402-021-00598-w Springer Science and Business Media B.V. Accessed 21 Mar 2022.
Macchini M, Centonze F, Peretti U, Orsi G, Militello AM, Valente MM, et al. Treatment opportunities and future perspectives for pancreatic cancer patients with germline BRCA1–2 pathogenic variants. Cancer Treat Rev. 2021;100:102262 W.B. Saunders Ltd. Available from: https://www.sciencedirect.com/science/article/pii/S0305737221001109. Accessed 19 Feb 2022.
Wang M, Chen S, Ao D. Targeting DNA repair pathway in cancer: mechanisms and clinical application. MedComm (Beijing). 2021;2:654–91. https://doi.org/10.1002/mco2.103 Wiley. Accessed 20 Mar 2022.
ClinicalTrials.gov. U.S. National Library of Medicine. https://www.clinicaltrials.gov. Accessed 10 Apr 2022.
Jiang M, Jia K, Wang L, Li W, Chen B, Liu Y, et al. Alterations of DNA damage repair in cancer: from mechanisms to applications. Ann Transl Med. 2020;8:1685 AME Publishing Company. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33490197. Accessed 6 Aug 2021.
Clark CA, Yang ES. Harnessing DNA repair defects to augment immune-based therapies in triple-negative breast cancer. Front Oncol. 2021;11:703802 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34631532. Accessed 20 Jan 2022.
Melhem M, Hanze E, Lu S, Alskär O, Visser S, Gandhi Y. Population pharmacokinetics and exposure–response of anti–programmed cell death protein-1 monoclonal antibody dostarlimab in advanced solid tumours. Br J Clin Pharmacol. 2022. https://doi.org/10.1111/bcp.15339 Accessed 19 Apr 2022.
Kang S, El-Rayes BF, Akce M. Evolving role of immunotherapy in advanced biliary tract cancers. Cancers (Basel). 2022;14:1748. https://doi.org/10.3390/cancers14071748 Accessed 18 Apr 2022.
Ding L, Dong HY, Zhou TR, Wang YH, Yan T, Li JC, et al. PD-1/PD-L1 inhibitors-based treatment for advanced renal cell carcinoma: mechanisms affecting efficacy and combination therapies. Cancer Med. 2021;10:6384–401. https://doi.org/10.1002/cam4.4190 John Wiley and Sons Inc. Accessed 21 Mar 2022.
Wang Y, Zheng K, Huang Y, Xiong H, Su J, Chen R, et al. PARP inhibitors in gastric cancer: beacon of hope. J Exp Clin Cancer Res. 2021;40:211. https://doi.org/10.1186/s13046-021-02005-6 BioMed Central Ltd. Accessed 21 Mar 2022.
Passiglia F, Reale ML, Cetoretta V, Parlagreco E, Jacobs F, Listì A, et al. Repositioning PARP inhibitors in the treatment of thoracic malignancies. Cancer Treat Rev. 2021;99. https://doi.org/10.1016/j.ctrv.2021.102256 W.B. Saunders Ltd. Accessed 21 Mar 2022.
Ray-Coquard IL, Bompas E, Cropet C, Donnat M, Bertucci F, Chaigneau L, et al. 824TiP: an open-label, single arm, phase II trial of niraparib in combination with anti-PD1 antibody in recurrent/advanced stage endometrial cancer patients. Ann Oncol. 2021;32(Suppl 5):S772. https://doi.org/10.1016/j.annonc.2021.08.1266 Elsevier BV. Accessed 20 Mar 2022.
Maiorano BA, Maiorano MFP, Lorusso D, Maiello E. Ovarian cancer in the era of immune checkpoint inhibitors: state of the art and future perspectives. Cancers (Basel). 2021;13:4438. https://doi.org/10.3390/cancers13174438 MDPI. Accessed 21 Mar 2022.
Criscuolo D, Morra F, Giannella R, Visconti R, Cerrato A, Celetti A. New combinatorial strategies to improve the PARP inhibitors efficacy in the urothelial bladder cancer treatment. J Exp Clin Cancer Res. 2019;38:91 BioMed Central Ltd. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6385418/. Accessed 12 Aug 2021.
Ricci AD, Rizzo A, Bonucci C, Tober N, Palloni A, Mollica V, et al. PARP inhibitors in biliary tract cancer: a new kid on the block? Medicines (Basel). 2020;7:54. https://doi.org/10.3390/medicines7090054 Accessed 12 Sept 2021.
Kareff SA, Samtani S, Burotto M, Prasad V, Kim C. Current landscape of immunotherapy trials involving the programmed cell death protein 1/programmed death-ligand 1 axis in intrathoracic tumors. JTO Clin Res Rep. 2021;2:100149. https://doi.org/10.1016/j.jtocrr.2021.100149 Elsevier Inc. Accessed 21 Mar 2022.
Tarrar TA, Anwar MY, Ali MA, Saeed M, Rehman S, Bajwa SF, et al. Current status of monoclonal antibodies-based therapies in castration-resistant prostate cancer: a systematic review and meta-analysis of clinical trials. Cureus. 2022;14:e22942. https://doi.org/10.7759/cureus.22942 Cureus, Inc. Accessed 19 Apr 2022.
Sarantis P, Dikoglou Tzanetatou E, Ioakeimidou E, Vallilas C, Androutsakos T, Damaskos C, et al. Cholangiocarcinoma: the role of genetic and epigenetic factors; current and prospective treatment with checkpoint inhibitors and immunotherapy. Am J Transl Res. 2021;13:13246–60 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/pmc8748131/. Accessed 21 Mar 2022.
Verhoeven Y, Quatannens D, Trinh XB, Wouters A, Smits ELJ, Lardon F, et al. Targeting the PD-1 axis with pembrolizumab for recurrent or metastatic cancer of the uterine cervix: a brief update. Int J Mol Sci. 2021;22:1–14. https://doi.org/10.3390/ijms22041807 MDPI AG. Accessed 20 Mar 2022.
Klinakis A, Karagiannis D, Rampias T. Targeting DNA repair in cancer: current state and novel approaches. Cell Mol Life Sci. 2020;77:677–703 Springer. Available from: https://link.springer.com/article/10.1007/s00018-019-03299-8. Accessed 13 Oct 2021.
Brown TJ, Reiss KA. PARP inhibitors in pancreatic cancer. Cancer J (United States). 2021;27:465–75. https://doi.org/10.1097/ppo.0000000000000554 Lippincott Williams and Wilkins. Accessed 20 Mar 2022.
Principe DR. Precision medicine for BRCA/PALB2-mutated pancreatic cancer and emerging strategies to improve therapeutic responses to PARP inhibition. Cancers (Basel). 2022;14:897. https://doi.org/10.3390/cancers14040897 MDPI. Accessed 20 Apr 2022.
Barnieh FM, Loadman PM, Falconer RA. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr Res Pharmacol Drug Discov. 2021;2:100017 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34909652. Accessed 17 Dec 2021.
Chen T, Tongpeng S, Lu Z, Topatana W, Juengpanich S, Li S, et al. DNA damage response inhibition-based combination therapies in cancer treatment: recent advances and future directions. Aging Cancer. 2022;3:44–67. https://doi.org/10.1002/aac2.12047 Wiley. Accessed 4 Apr 2022.
Ngoi NYL, Peng G, Yap TA. A tale of two checkpoints: ATR inhibition and PD-(L)1 blockade. Annu Rev Med. 2022;73:231–50 Available from: https://www.annualreviews.org/doi/full/10.1146/annurev-med-042320-025136#article-denial. Accessed 15 Feb 2022.
Do KT, Manuszak C, Thrash E, Giobbie-Hurder A, Hu J, Kelland S, et al. Immune modulating activity of the CHK1 inhibitor prexasertib and anti-PD-L1 antibody LY3300054 in patients with high-grade serous ovarian cancer and other solid tumors. Cancer Immunol Immunother. 2021;70:2991–3000 Springer Science and Business Media Deutschland GmbH. Available from: https://link.springer.com/article/10.1007/s00262-021-02910-x. Accessed 11 Dec 2021.
Wu Z, Cui P, Tao H, Zhang S, Ma J, Liu Z, et al. The synergistic effect of PARP inhibitors and immune checkpoint inhibitors. Clin Med Insights Oncol. 2021;15. https://doi.org/10.1177/1179554921996288 Accessed 24 Aug 2021.
Zong C, Zhu T, He J, Huang R, Jia R, Shen J. PARP mediated DNA damage response, genomic stability and immune responses. Int J Cancer. 2022;150:1745–59 John Wiley and Sons Inc. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ijc.33918. Accessed 13 Feb 2022.
Pilié PG, Gay CM, Byers LA, O’Connor MJ, Yap TA. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res. 2019;25:3759–71 American Association for Cancer Research Inc. Available from: https://aacrjournals.org/clincancerres/article/25/13/3759/81696/PARP-Inhibitors-Extending-Benefit-Beyond-BRCA. Accessed 21 Aug 2021.
Reguart N, Marin E, Remon J, Reyes R, Teixido C. In search of the long-desired ‘copernican therapeutic revolution’ in small-cell lung cancer. Drugs. 2020;80:241–62 Adis. Available from: https://link.springer.com/article/10.1007/s40265-019-01240-8. Accessed 22 Sept 2021.
Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16:81–104. https://doi.org/10.1038/s41571-018-0114-z Nature Publishing Group. Accessed 13 Dec 2021.
Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108:3406–11. https://doi.org/10.1073/pnas.1013715108 Accessed 15 June 2022.
Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science (1979). 2017;355. https://doi.org/10.1126/science.aam7344 Accessed 15 June 2022.
Drapkin BJ, Farago AF. Unexpected synergy reveals new therapeutic strategy in SCLC. Trends Pharmacol Sci. 2019;40:295–7. https://doi.org/10.1016/j.tips.2019.03.005 American Association for Cancer Research Inc. Accessed 3 Oct 2021.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–20. https://doi.org/10.1158/1078-0432.CCR-16-3215 American Association for Cancer Research Inc. Accessed 12 Dec 2021.
Lee J-M, Cimino-Mathews A, Peer CJ, Zimmer A, Lipkowitz S, Annunziata CM, et al. Safety and clinical activity of the programmed death-ligand 1 inhibitor durvalumab in combination with poly (ADP-ribose) polymerase inhibitor Olaparib or vascular endothelial growth factor receptor 1-3 inhibitor Cediranib in Women’s cancers: a dose-escalation, phase I study. J Clin Oncol. 2017;35:2193–202 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5493052/. Accessed 10 Aug 2021.
Lampert EJ, Zimmer A, Padget M, Cimino-Mathews A, Nair JR, Liu Y, et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase II study. Clin Cancer Res. 2020;26:4268–79 American Association for Cancer Research Inc. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7442720/. Accessed 12 Aug 2021.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5:1141–9 American Medical Association. Available from: https://jamanetwork.com/journals/jamaoncology/fullarticle/2735889. Accessed 10 Oct 2021.
Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019;20:1306–15 Lancet Publishing Group. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1470204519303961. Accessed 10 Oct 2021.
Bröckelmann PJ, de Jong MRW, Jachimowicz RD. Targeting DNA repair, cell cycle, and tumor microenvironment in B cell lymphoma. Cells. 2020;9:2287. https://doi.org/10.3390/cells9102287 Accessed 17 Feb 2022.
Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, et al. CTLA-4 blockade synergizes therapeutically with PARP inhibition in BRCA1-deficient ovarian cancer. Cancer Immunol Res. 2015;3:1257–68. https://doi.org/10.1158/2326-6066.CIR-15-0044 American Association for Cancer Research Inc. Accessed 10 Oct 2021.
Ji F, Zhang F, Zhang M, Long K, Xia M, Lu F, et al. Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. J Hematol Oncol. 2021;14:152 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34556152. Accessed 10 Jan 2022.
Sun R, Luo H, Su J, Di S, Zhou M, Shi B, et al. Olaparib suppresses MDSC recruitment via SDF1α/CXCR4 Axis to improve the anti-tumor efficacy of CAR-T cells on breast cancer in mice. Mol Ther. 2021;29:60–74. https://doi.org/10.1016/j.ymthe.2020.09.034 Cell Press. Accessed 10 Oct 2021.
Wayne J, Brooks T, Landras A, Massey AJ. Targeting DNA damage response pathways to activate the STING innate immune signaling pathway in human cancer cells. FEBS J. 2021;288:4507–40 John Wiley and Sons Inc. Available from: https://febs.onlinelibrary.wiley.com/doi/10.1111/febs.15747. Accessed 11 Jan 2022.
Patel P, Sun L, Robbins Y, Clavijo PE, Friedman J, Silvin C, et al. Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. OncoImmunology. 2019;8 Taylor and Francis Inc. Available from: https://www.tandfonline.com/doi/full/10.1080/2162402X.2019.1638207. Accessed 12 Nov 2021.
Donlon NE, Power R, Hayes C, Reynolds JV, Lysaght J. Radiotherapy, immunotherapy, and the tumour microenvironment: turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett. 2021;502:84–96 Elsevier Ireland Ltd. Available from: https://www.sciencedirect.com/science/article/pii/S030438352100015X. Accessed 15 Oct 2021.
Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–46. https://doi.org/10.1016/s0140-6736(16)00587-0 Lancet Publishing Group. Accessed 12 Nov 2021.
Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. https://doi.org/10.1158/2159-8290.CD-14-0863 American Association for Cancer Research Inc. Accessed 12 Nov 2021.
Varayathu H, Sarathy V, Thomas BE, Mufti SS, Naik R. Combination strategies to augment immune check point inhibitors efficacy - implications for translational research. Front Oncol. 2021;11. https://doi.org/10.3389/fonc.2021.559161 Frontiers Media S.A. Accessed 11 Sept 2021.
Caracciolo D, Riillo C, Arbitrio M, di Martino MT, Tagliaferri P, Tassone P. Error-prone DNA repair pathways as determinants of immunotherapy activity: an emerging scenario for cancer treatment. Int J Cancer. 2020;147:2658–68 Wiley-Liss Inc. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ijc.33038. Accessed 21 Sept 2021.
Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, et al. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res. 2019;25:3392–403. https://doi.org/10.1158/1078-0432.CCR-18-1821 American Association for Cancer Research Inc. Accessed 30 Oct 2021.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–71. https://doi.org/10.1084/jem.20052494 Accessed 30 Oct 2021.
Garnett CT, Palena C, Chakarborty M, Tsang K-Y, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004;64:7985–94. https://doi.org/10.1158/0008-5472.CAN-04-1525 Accessed 30 Oct 2021.
Parikh AR, He Y, Hong TS, Corcoran RB, Clark JW, Ryan DP, et al. Analysis of DNA damage response gene alterations and tumor mutational burden across 17,486 tubular gastrointestinal carcinomas: implications for therapy. Oncologist. 2019;24:1340–7. https://doi.org/10.1634/theoncologist.2019-0034 Oxford University Press (OUP). Accessed 12 Oct 2021.
Wen H, Guo Q-H, Zhou X-L, Wu X-H, Li J. Genomic profiling of Chinese cervical cancer patients reveals prevalence of DNA damage repair gene alterations and related hypoxia feature. Front Oncol. 2021;11:792003 Available from: http://www.ncbi.nlm.nih.gov/pubmed/35071000. Accessed 19 Jan 2022.
Tian W, Shan B, Zhang Y, Ren Y, Liang S, Zhao J, et al. Association between DNA damage repair gene somatic mutations and immune-related gene expression in ovarian cancer. Cancer Med. 2020;9:2190–200. https://doi.org/10.1002/cam4.2849 Accessed 26 Sept 2021.
Mei P, Freitag CE, Wei L, Zhang Y, Parwani AV, Li Z. High tumor mutation burden is associated with DNA damage repair gene mutation in breast carcinomas. Diagn Pathol. 2020;15:50 Available from: http://www.ncbi.nlm.nih.gov/pubmed/32393302. Accessed 8 Jan 2022.
Heeke AL, Xiu J, Elliott A, Korn WM, Lynce F, Pohlmann PR, et al. Actionable co-alterations in breast tumors with pathogenic mutations in the homologous recombination DNA damage repair pathway. Breast Cancer Res Treat. 2020;184:265–75. https://doi.org/10.1007/s10549-020-05849-2 Springer. Accessed 13 Dec 2021.
Singh RR, Goldberg J, Varghese AM, Yu KH, Park W, O’Reilly EM. Genomic profiling in pancreatic ductal adenocarcinoma and a pathway towards therapy individualization: a scoping review. Cancer Treat Rev. 2019;75:27–38. https://doi.org/10.1016/j.ctrv.2019.03.003 W.B. Saunders Ltd. Accessed 13 Dec 2021.
Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7:13587–98. https://doi.org/10.18632/oncotarget.7277 Impact Journals LLC. Accessed 30 Oct 2021.
Heitzer E, Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. 2014;24:107–13. https://doi.org/10.1016/j.gde.2013.12.005 Accessed 30 Oct 2021.
Fakih M, Gong J, Wang C, Lee PP, Chu P. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Cancer Netw. 2017;15:142–7. https://doi.org/10.6004/jnccn.2017.0016 Accessed 11 Sept 2021.
Permata TBM, Hagiwara Y, Sato H, Yasuhara T, Oike T, Gondhowiardjo S, et al. Base excision repair regulates PD-L1 expression in cancer cells. Oncogene. 2019;38:4452–66. https://doi.org/10.1038/s41388-019-0733-6 Accessed 12 Dec 2021.
Zhang H, Wang Y, Ji Q, Cai H, Liang X, Xie J, et al. Clinicopathological and molecular characteristics of patients with hypermutant lung cancer: a retrospective cohort study. Oncol Lett. 2021;21:329. https://doi.org/10.3892/ol.2021.12590 Accessed 11 Sept 2021.
Jenzer M, Keß P, Nientiedt C, Endris V, Kippenberger M, Leichsenring J, et al. The BRCA2 mutation status shapes the immune phenotype of prostate cancer. Cancer Immunol Immunother. 2019;68:1621–33 Springer Science and Business Media Deutschland GmbH. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6805809/. Accessed 12 Sept 2021.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (1979). 2015;348:124–8 American Association for the Advancement of Science. Available from: https://www.science.org/doi/10.1126/science.aaa1348. Accessed 15 Oct 2021.
Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell. 2018;33:853–861.e4 Cell Press. Available from: https://www.cell.com/cancer-cell/fulltext/S1535-6108(18)30172-7. Accessed 12 Oct 2021.
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–20. https://doi.org/10.1016/s0140-6736(16)00561-4 Accessed 18 Oct 2021.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. https://doi.org/10.1056/nejmoa1406498 Accessed 10 Oct 2021.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91. https://doi.org/10.1016/S1470-2045(17)30422-9 Lancet Publishing Group. Accessed 10 Nov 2021.
Overman MJ, Lonardi S, Yeung K, Wong M, Lenz H-J, Gelsomino F, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9. https://doi.org/10.1200/JCO.2017.76.9901 Accessed 10 Nov 2021.
Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018;11. https://doi.org/10.1186/s13045-017-0552-6 BioMed Central Ltd. Accessed 13 Dec 2021.
MacDonald KM, Benguerfi S, Harding SM. Alerting the immune system to DNA damage: micronuclei as mediators. Essays Biochem. 2020;64:753–64. https://doi.org/10.1042/ebc20200016 Portland Press Ltd. Accessed 13 Dec 2021.
Lee CH, Yelensky R, Jooss K, Chan TA. Update on tumor neoantigens and their utility: why it is good to be different. Trends Immunol. 2018;39:536–48. https://doi.org/10.1016/j.it.2018.04.005 Elsevier Ltd. Accessed 1 Nov 2021.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201. https://doi.org/10.1016/j.celrep.2017.04.031 Elsevier B.V. Accessed 4 Nov 2021.
Wen WX, Leong CO. Association of BRCA1- and BRCA2-deficiency with mutation burden, expression of PD-L1/ PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer. PLoS One. 2019;14. https://doi.org/10.1371/journal.pone.0215381 Public Library of Science. Accessed 13 Dec 2021.
Soslow RA, Han G, Park KJ, Garg K, Olvera N, Spriggs DR, et al. Morphologic patterns associated with BRCA1 and BRCA2 genotype in ovarian carcinoma. Mod Pathol. 2012;25:625–36. https://doi.org/10.1038/modpathol.2011.183 Accessed 11 Jan 2022.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. https://doi.org/10.1038/nri.2016.107 Nature Publishing Group. Accessed 11 Feb 2022.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. https://doi.org/10.1146/annurev-immunol-032712-100008 Accessed 1 Feb 2022.
Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255–69. https://doi.org/10.1016/j.immuni.2016.06.001 Cell Press. Accessed 12 Nov 2021.
O’Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81 Available from: https://www.cancertreatmentreviews.com/article/S0305-7372(16)30132-3/fulltext. Accessed 12 Nov 2021.
Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S, et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019;79:795–806. https://doi.org/10.1158/0008-5472.CAN-18-2545 American Association for Cancer Research Inc. Accessed 12 Dec 2021.
Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 2018;25:2972–2980.e5. https://doi.org/10.1016/j.celrep.2018.11.054 Elsevier B.V. Accessed 10 Oct 2021.
Vidotto T, Nersesian S, Graham C, Siemens DR, Koti M. DNA damage repair gene mutations and their association with tumor immune regulatory gene expression in muscle invasive bladder cancer subtypes. J Immunother Cancer. 2019;7:148 BioMed Central Ltd. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6556053/. Accessed 17 Aug 2021.
Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684–9. https://doi.org/10.1126/science.2270482 Accessed 9 Nov 2021.
Nik-Zainal S, van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. https://doi.org/10.1016/j.cell.2012.04.023 Elsevier B.V. Accessed 10 Nov 2021.
Budczies J, Bockmayr M, Denkert C, Klauschen F, Lennerz JK, Györffy B, et al. Classical pathology and mutational load of breast cancer – integration of two worlds. J Pathol Clin Res. 2015;1:225–38. https://doi.org/10.1002/cjp2.25 Wiley-Blackwell Publishing Ltd. Accessed 15 Nov 2021.
Pantelidou C, Sonzogni O, Taveira MDO, Mehta AK, Kothari A, Wang D, et al. Parp inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral sting pathway activation in brca-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9:722–37. https://doi.org/10.1158/2159-8290.CD-18-1218 American Association for Cancer Research Inc. Accessed 13 Dec 2021.
Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, et al. IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci Rep. 2017;7. https://doi.org/10.1038/s41598-017-18132-0 Nature Publishing Group. Accessed 12 Dec 2021.
Liu X, Mo W, Ye J, Li L, Zhang Y, Hsueh EC, et al. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat Commun. 2018;9:249. https://doi.org/10.1038/s41467-017-02689-5 Accessed 12 Sept 2021.
McGrail DJ, Federico L, Li Y, Dai H, Lu Y, Mills GB, et al. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat Commun. 2018;9:1317. https://doi.org/10.1038/s41467-018-03730-x Accessed 12 Aug 2021.
Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–33.
Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol. 2015;12:584–96. https://doi.org/10.1038/nrclinonc.2015.105 Accessed 12 Nov 2021.
Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–34. https://doi.org/10.1038/nrclinonc.2017.101 Accessed 12 Nov 2021.
Yélamos J, Moreno-Lama L, Jimeno J, Ali SO. Immunomodulatory roles of PARP-1 and PARP-2: impact on PARP-centered cancer therapies. Cancers (Basel). 2020;12. https://doi.org/10.3390/cancers12020392. MDPI AG. Accessed 12 Nov 2021.
Schettini F, Corona SP, Giudici F, Strina C, Sirico M, Bernocchi O, et al. Clinical, radiometabolic and immunologic effects of olaparib in locally advanced triple negative breast cancer: the OLTRE window of opportunity trial. Front Oncol. 2021;11:686776 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34262869. Accessed 11 Feb 2022.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18:85–100. https://doi.org/10.1038/s41571-020-0426-7 Nature Research. Accessed 11 June 2022.
Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19:200–18. https://doi.org/10.1038/s41573-019-0052-1 Nature Research. Accessed 11 June 2022.
Burbage M, Amigorena S. A dendritic cell multitasks to tackle cancer. Nature. 2020;584:533–4. https://doi.org/10.1038/d41586-020-02339-9 Accessed 15 June 2022.
Dees S, Ganesan R, Singh S, Grewal IS. Emerging CAR-T cell therapy for the treatment of triple-negative breast cancer. Mol Cancer Ther. 2020;19:2409–21. https://doi.org/10.1158/1535-7163.MCT-20-0385 American Association for Cancer Research Inc. Accessed 11 Mar 2022.
Alvarez M, Simonetta F, Baker J, Pierini A, Wenokur AS, Morrison AR, et al. Regulation of murine NK cell exhaustion through the activation of the DNA damage repair pathway. JCI Insight. 2019;4:e127729 American Society for Clinical Investigation. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6675585/. Accessed 12 Sept 2021.
Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–84. https://doi.org/10.1038/nature14493 Nature Publishing Group. Accessed 12 Nov 2021.
Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–8. https://doi.org/10.1038/nature10802 Accessed 12 Nov 2021.
Santaguida S, Richardson A, Iyer DR, M’Saad O, Zasadil L, Knouse KA, et al. Chromosome Mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev Cell. 2017;41:638–651.e5. https://doi.org/10.1016/j.devcel.2017.05.022 Cell Press. Accessed 12 Nov 2021.
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. https://doi.org/10.1371/journal.pbio.0060301 Accessed 12 Nov 2021.
Rodier F, Coppé JP, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9. https://doi.org/10.1038/ncb1909 Accessed 12 Nov 2021.
Malaquin N, Carrier-Leclerc A, Dessureault M, Rodier F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front Genet. 2015;5. https://doi.org/10.3389/fgene.2015.00094 Frontiers Research Foundation. Accessed 12 Nov 2021.
Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550. https://doi.org/10.1038/nature24050 Nature Publishing Group. Accessed 12 Nov 2021.
Bakhoum SF, Cantley LC. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell. 2018;174:1347–60. https://doi.org/10.1016/j.cell.2018.08.027 Cell Press. Accessed 12 Nov 2021.
Won JK, Bakhoum SF. The cytosolic DNA-sensing cGAS–sting pathway in cancer. Cancer Discov. 2020;10:26–39. https://doi.org/10.1158/2159-8290.CD-19-0761 American Association for Cancer Research Inc. Accessed 23 Nov 2021.
Özcan S, Alessio N, Acar MB, Mert E, Omerli F, Peluso G, et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging. 2016;8:1316–29. https://doi.org/10.18632/aging.100971 Accessed 12 Nov 2021.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–52. https://doi.org/10.1016/j.immuni.2014.10.019 Cell Press. Accessed 15 June 2022.
Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44:924–38. https://doi.org/10.1016/j.immuni.2016.03.012 Cell Press. Accessed 15 June 2022.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9. https://doi.org/10.1038/nm1622 Accessed 10 Sept 2021.
Kashani B, Zandi Z, Pourbagheri-Sigaroodi A, Bashash D, Ghaffari SH. The role of toll-like receptor 4 (TLR4) in cancer progression: a possible therapeutic target? J Cell Physiol. 2021;236:4121–37 John Wiley and Sons Inc. Available from: https://onlinelibrary.wiley.com/doi/10.1002/jcp.30166. Accessed 20 Nov 2021.
Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–45 Available from: http://www.ncbi.nlm.nih.gov/pubmed/11905821. Accessed 11 Jan 2022.
Chen C-F, Ruiz-Vega R, Vasudeva P, Espitia F, Krasieva TB, de Feraudy S, et al. ATR mutations promote the growth of melanoma tumors by modulating the immune microenvironment. Cell Rep. 2017;18:2331–42. https://doi.org/10.1016/j.celrep.2017.02.040 Accessed 12 Nov 2021.
Kim ST, Smith SA, Mortimer P, Loembé AB, Cho H, HKim KM, et al. Phase I study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin Cancer Res. 2021;27:4700–9. https://doi.org/10.1158/1078-0432.CCR-21-0251 American Association for Cancer Research Inc. Accessed 12 Nov 2021.
Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–70. https://doi.org/10.1038/nature23470 Nature Publishing Group. Accessed 12 Nov 2021.
MacKenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. CGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–5. https://doi.org/10.1038/nature23449 Nature Publishing Group. Accessed 12 Nov 2021.
Meng X, Yang S, Camp VJA. The interplay between the DNA damage response, RNA processing and extracellular vesicles. Front Oncol. 2020;9:1538 Frontiers Media S.A. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6978769/. Accessed 12 Aug 2021.
Zhang T, Zheng S, Liu Y, Li X, Wu J, Sun Y, et al. DNA damage response and PD-1/PD-L1 pathway in ovarian cancer. DNA Repair. 2021;102 Elsevier B.V. Available from: https://www.sciencedirect.com/science/article/pii/S1568786421000689. Accessed 11 Oct 2021.
Reisländer T, Lombardi EP, Groelly FJ, Miar A, Porru M, di Vito S, et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat Commun. 2019;10:3143. https://doi.org/10.1038/s41467-019-11048-5 Nature Publishing Group. Accessed 12 Nov 2021.
An D, Banerjee S, Lee JM. Recent advancements of antiangiogenic combination therapies in ovarian cancer. Cancer Treat Rev. 2021;98 W.B. Saunders Ltd. Available from: https://www.sciencedirect.com/science/article/pii/S0305737221000724. Accessed 11 Feb 2022.
Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun. 2019;10:100. https://doi.org/10.1038/s41467-018-07927-y Nature Publishing Group. Accessed 12 Nov 2021.
Li C, Shen Q, Zhang P, Wang T, Liu W, Li R, et al. Targeting MUS81 promotes the anticancer effect of WEE1 inhibitor and immune checkpoint blocking combination therapy via activating cGAS/STING signaling in gastric cancer cells. J Exp Clin Cancer Res. 2021;40:315 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34625086. Accessed 10 Jan 2022.
Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell. 2018;34:361–78. https://doi.org/10.1016/j.ccell.2018.05.013 Cell Press. Accessed 12 Nov 2021.
Frémond M-L, Crow YJ. Chapter 32 - Mendelian disorders of immunity related to an upregulation of type I interferon. In: Sullivan KE, Stiehm ER, editors. Stiehm’s immune deficiencies. 2nd ed: Academic; 2020. p. 751–72. Available from: https://www.sciencedirect.com/science/article/pii/B9780128167687000326. Accessed 12 Dec 2021.
Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell. 2018;71:745–760.e5. https://doi.org/10.1016/j.molcel.2018.07.034 Cell Press. Accessed 12 Dec 2021.
Chen M, Linstra R, van Vugt MATM. Genomic instability, inflammatory signaling and response to cancer immunotherapy. Biochim Biophys Acta Rev Cancer. 2022;1877(1):188661 Elsevier B.V. Available from: https://www.sciencedirect.com/science/article/pii/S0304419X21001591. Accessed 11 Feb 2022.
Feng X, Tubbs A, Zhang C, Tang M, Sridharan S, Wang C, et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. 2020;39. https://doi.org/10.15252/embj.2019104036 EMBO. Accessed 13 Dec 2021.
Sun L-L, Yang R-Y, Li C-W, Chen M-K, Shao B, Hsu J-M, et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res. 2018;8:1307–16 Available from: http://www.ncbi.nlm.nih.gov/pubmed/30094103. Accessed 10 Sept 2021.
Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–28 Available from: http://www.genesdev.org/cgi/doi/10.1101/gad.1067403. Accessed 12 Dec 2021.
Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017;8:1751 Nature Publishing Group. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5701012/. Accessed 11 Sept 2021.
Sato H, Jeggo PA, Shibata A. Regulation of programmed death-ligand 1 expression in response to DNA damage in cancer cells: implications for precision medicine. Cancer Sci. 2019;110:3415–23. https://doi.org/10.1111/cas.14197 Blackwell Publishing Ltd. Accessed 13 Dec 2021.
Weitering TJ, Takada S, Weemaes CMR, van Schouwenburg PA, van der Burg M. ATM: translating the DNA damage response to adaptive immunity. Trends Immunol. 2021;42:350–65 Elsevier Ltd. Available from: https://www.sciencedirect.com/science/article/pii/S1471490621000259. Accessed 12 Nov 2021.
Yan S, Sorrell M, Berman Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell Mol Life Sci. 2014;71:3951–67. https://doi.org/10.1007/s00018-014-1666-4 Accessed 15 June 2022.
Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010. https://doi.org/10.1155/2010/214074 Accessed 15 June 2022.
Hossain MA, Lin Y, Yan S. Single-strand break end resection in genome integrity: mechanism and regulation by APE2. Int J Mol Sci. 2018;19:2389. https://doi.org/10.3390/ijms19082389 MDPI AG. Accessed 15 June 2022.
Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 2019;79:3940–51 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31101760. Accessed 8 Jan 2022.
Li X, Yang M, Yu Z, Tang S, Wang L, Cao X, et al. The tyrosine kinase Src promotes phosphorylation of the kinase TBK1 to facilitate type I interferon production after viral infection. Sci Signal. 2017;10. https://doi.org/10.1126/scisignal.aae0435 Accessed 13 Dec 2021.
Chen W-T, Ebelt ND, Stracker TH, Xhemalce B, van den Berg CL, Miller KM. ATM regulation of IL-8 links oxidative stress to cancer cell migration and invasion. Elife. 2015;4:e07270. https://doi.org/10.7554/eLife.07270.001 Accessed 10 Sept 2021.
Jin MH, Nam A-R, Park JE, Bang J-H, Bang Y-J, Oh D-Y. Therapeutic co-targeting of WEE1 and ATM downregulates PD-L1 expression in pancreatic cancer. Cancer Res Treat. 2020;52:149–66 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31291716. Accessed 7 Sept 2021.
Abeler-Dörner L, Swamy M, Williams G, Hayday AC, Bas A. Butyrophilins: an emerging family of immune regulators. Trends Immunol. 2012;33:34–41. https://doi.org/10.1016/j.it.2011.09.007 Accessed 12 Dec 2021.
Barbee SD, Woodward MJ, Turchinovich G, Mention J-J, Lewis JM, Boyden LM, et al. Skint-1 is a highly specific, unique selecting component for epidermal T cells. Proc Natl Acad Sci. 2011;108:3330–5. https://doi.org/10.1073/pnas.1010890108 Accessed 12 Nov 2021.
Vidula N, Yau C, Rugo HS. Programmed cell death 1 (PD-1) receptor and programmed death ligand 1 (PD-L1) gene expression in primary breast cancer. Breast Cancer Res Treat. 2021;187:387–95. https://doi.org/10.1007/s10549-021-06234-3 Springer. Accessed 13 Jan 2022.
Daemen A, Wolf DM, Korkola JE, Griffith OL, Frankum JR, Brough R, et al. Cross-platform pathway-based analysis identifies markers of response to the PARP inhibitor olaparib. Breast Cancer Res Treat. 2012;135:505–17. https://doi.org/10.1007/s10549-012-2188-0 Accessed 12 Dec 2021.
Minchom A, Aversa C, Lopez J. Dancing with the DNA damage response: next-generation anti-cancer therapeutic strategies. Ther Adv Med Oncol. 2018;10 SAGE Publications Inc. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6047242/. Accessed 1 Aug 2021.
Zhang L, Wang Y, Li Z, Lin D, Liu Y, Zhou L, et al. Clinicopathological features of tumor mutation burden, Epstein-Barr virus infection, microsatellite instability and PD-L1 status in Chinese patients with gastric cancer. Diagn Pathol. 2021;16:38 Available from: http://www.ncbi.nlm.nih.gov/pubmed/33933102. Accessed 8 Feb 2022.
Osoegawa A, Hiraishi H, Hashimoto T, Takumi Y, Abe M, Takeuchi H, et al. The positive relationship between γH2AX and PD-L1 expression in lung squamous cell carcinoma. In Vivo. 2018;32:171–7 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29275316. Accessed 9 Feb 2021.
Buglioni S, Melucci E, Sperati F, Pallocca M, Terrenato I, de Nicola F, et al. The clinical significance of PD-L1 in advanced gastric cancer is dependent on ARID1A mutations and ATM expression. OncoImmunology. 2018;7. https://doi.org/10.1080/2162402X.2018.1457602 Taylor and Francis Inc. Accessed 13 Dec 2021.
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. https://doi.org/10.1038/ncomms12632 Nature Publishing Group. Accessed 12 Dec 2021.
Shevtsov M, Sato H, Multhoff G, Shibata A. Novel approaches to improve the efficacy of immuno-radiotherapy. Front Oncol. 2019;9:156. https://doi.org/10.3389/fonc.2019.00156 Frontiers Media S.A. Accessed 12 Dec 2021.
Huang J, Wang L, Cong Z, Amoozgar Z, Kiner E, Xing D, et al. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1 −/− murine model of ovarian cancer. Biochem Biophys Res Commun. 2015;463:551–6. https://doi.org/10.1016/j.bbrc.2015.05.083 Accessed 12 Dec 2021.
Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer. 2018;118:312–24. https://doi.org/10.1038/bjc.2017.376 Nature Publishing Group. Accessed 12 Dec 2021.
Mouw KW, Konstantinopoulos PA. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br J Cancer. 2018;118:933–5. https://doi.org/10.1038/s41416-018-0017-x Nature Publishing Group. Accessed 12 Dec 2021.
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–9. https://doi.org/10.1038/ni.3558 Accessed 12 Dec 2021.
Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding Q, et al. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 2019;38. https://doi.org/10.1186/s13046-019-1161-8 BioMed Central Ltd. Accessed 12 Dec 2021.
Ma Z, Wang H, Meng F, Han Y, Chen Y, Xiao M, et al. Role of BCLAF-1 in PD-L1 stabilization in response to ionizing irradiation. Cancer Sci. 2021;112:4064–74 Available from: http://www.ncbi.nlm.nih.gov/pubmed/34251713. Accessed 8 Sept 2021.
Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. 2019;25:656–66. https://doi.org/10.1038/s41591-019-0374-x Accessed 12 Dec 2021.
Chen X, Mo S, Zhang Y, Ma H, Lu Z, Yu S, et al. Analysis of a novel immune checkpoint, Siglec-15, in pancreatic ductal adenocarcinoma. J Pathol Clin Res. 2022;8:268–78 John Wiley and Sons Inc. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8977273/. Accessed 13 Jan 2022.
Zhu L, Liu J, Chen J, Zhou Q. The developing landscape of combinatorial therapies of immune checkpoint blockade with DNA damage repair inhibitors for the treatment of breast and ovarian cancers. J Hematol Oncol. 2021;14:206 BioMed Central Ltd. Available from: https://jhoonline.biomedcentral.com/articles/10.1186/s13045-021-01218-8. Accessed 10 Jan 2022.
Permata TBM, Sato H, Gu W, Kakoti S, Uchihara Y, Yoshimatsu Y, et al. High linear energy transfer carbon-ion irradiation upregulates PD-L1 expression more significantly than X-rays in human osteosarcoma U2OS cells. J Radiat Res. 2021;62:773–81 Oxford University Press. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8438258/. Accessed 11 Nov 2021.
Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. 2020;580:517–23. https://doi.org/10.1038/s41586-020-2209-9 Nature Research. Accessed 13 Feb 2022.
Ahrum M, Kyung-Hun L, Seock-Ah I. DNA damage repair inhibitor for breast cancer treatment. In: Dong-Young N, Wonshik H, Masakazu T, editors. Translational research in breast cancer advances in experimental medicine and biology; 2019. p. 163–76. Available from: http://www.springer.com/series/5584. Accessed 7 Sept 2021.
Acknowledgements
Not applicable.
Funding
This work was supported by the Natural Science Foundation of Guangdong Province (Grant No. 2018A030313846 and 2021A1515012593), the Science and Technology Planning Project of Guangdong Province (Grant No. 2019A030317020) and the National Natural Science Foundation of China (Grant No. 81802257, 81871859, 81772457, 82172750 and 82172811).
Author information
Authors and Affiliations
Contributions
PL and JZ conceived the study content and provided constructive guidance. CS and KQ collected and organized the related references, and visualized the figs. CS wrote the manuscript. KQ corrected the manuscript and made the table. AL, AJ, QC and ZL complemented the manuscript and supervised the study. AL, CS, KQ and AJ did the English editing, revised and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shi, C., Qin, K., Lin, A. et al. The role of DNA damage repair (DDR) system in response to immune checkpoint inhibitor (ICI) therapy. J Exp Clin Cancer Res 41, 268 (2022). https://doi.org/10.1186/s13046-022-02469-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02469-0