
Abstract
Emerging evidence has demonstrated that radiotherapy (RT) can not only cause direct damage to cancer cells but also lead to immunogenic cell death (ICD), which involves the activation of host antitumor immune response in tumor immune microenvironment (TIME). RT-induced ICD comprises the release of damage-associated molecular patterns (DAMPs) from dying cancer cells that result in the activation of tumor-specific immunity to elicit long-term antitumor efficacy in both original and abscopal tumor sites. Adenosine triphosphate (ATP), as an important DAMP released by irradiated cancer cells and an essential factor within purinergic pathway, can be further hydrolyzed to adenosine (ADO) by two key ectonucleotidases, CD39 and CD73, to further modulate the antitumor immunity in TIME through purinergic signaling via the interaction to its specific receptors such as adenosine 2A receptor (A2AR) and A2BR widely expressed on the surface of the components in TIME, including cancer cells and many immune effector cells. In this review, we first introduced key components in purinergic pathway including ATP, ADO, their receptors, and essential ectonucleotidases. Then we reviewed the regulation of ATP and ADO levels and their main mechanisms by which they promote tumor growth and broadly suppress antitumor immunity through inhibiting the pro-inflammatory response of dendritic cells, cytotoxic T lymphocytes, and natural killer cells, while improving the anti-inflammatory response of regulatory T cells, macrophages, and myeloid-derived suppressor cells in TIME, especially after irradiation. Finally, we presented an overview of dozens of promising therapeutics including pharmacological antagonists and specific antibodies targeting ADO receptors and ectonucleotidases CD39 or CD73 investigated in the clinic for cancer treatment, especially focusing on the preclinical studies and clinical trials being explored for blocking the purinergic signaling to enhance RT as a combination antitumor therapeutic strategy, which has a robust potential to be translated to the clinic in the future.
Similar content being viewed by others

Background
Brief introduction of purinergic signaling pathway
In 1970s, Burnstock, G. and colleagues introduced their finding that adenosine triphosphate (ATP) might be the neurotransmitter involved in both gut and bladder, which initiated the concept and further exploration of purinergic signaling pathway, i.e., extracellular nucleotides signaling [1]. Early studies largely on the biochemistry, physiology, and pharmacology of purinergic signaling pathway revealed its important functions to transmit signals in nervous system and modulate non-neuronal tissues including endothelial, immune, and inflammatory cells [2]. For example, ATP can serve a dual role in the neuro-immune interaction as both neurotransmitter and immunomodulator. ATP can not only relay the signals invoked by resident immune cells including macrophages and T cells (through the release of cytokines, chemokines, or growth factors) that sensed by peripheral nerves and spinal cord to the brain, but also be released by peripheral sensory afferent neurons to modulate local immune cells including macrophages and T cells to maintain their invoked signals to stimulate peripheral nerves [3,4,5]. Nowadays, it is well known that ATP, adenosine diphosphate (ADP), uridine triphosphate (UTP), uridine diphosphate (UDP), and adenosine (ADO) are important cellular messengers from purinergic pathway, which modulate various other signaling pathways and participate in numerous physiological and pathological processes, mainly through specific purinergic receptors [6].
Classification of purinergic receptors
Purinergic receptors have been classified into two families: P1 (sensitive to ADO) and P2 (sensitive to adenine and uridine nucleotides) receptors [6, 7]. P1 receptors (ADO receptors) belong to the G-protein coupled receptor (GPCR) superfamily, which are designated as adenosine 1 receptor (A1R), A2AR, A2BR and A3R. A1R and A3R are mainly coupled to the Gi/o subunit to inhibit adenylate cyclase (AC) and cyclic adenosine monophosphate (cAMP) production, while A2AR and A2BR are mainly coupled to the Gs subunit to increase cAMP synthesis by AC activation [6]. P2 receptors can be further divided into two groups. The first group is P2X receptors (P2XR), which are ligand-gated cation channels receptors (P2X1-P2X7) with ATP as the natural ligand. When activated, P2XRs promote rapid depolarization associated with influx of Ca2+ and Na+ while efflux of K+ [8]. The second group is P2Y receptors (P2YR), which are GPCRs including eight subtypes recognized in mammalian cells: P2Y1, P2Y2, P2Y4, P2Y6 and P2Y11-14. P2YRs can be activated by ATP (P2Y2 and P2Y11), ADP (P2Y1, P2Y12 and P2Y13), UTP (P2Y2 and P2Y4), UDP (P2Y6) and UDP-glucose (P2Y14). P2YRs are coupled via G-proteins (Gq/11, Gs, or Gi/o) to mobilize Ca2+, generate/inhibit cAMP, and stimulate the extracellular signal-regulated kinase 1/2 (ERK) / mitogen-activated protein kinase (MAPK) pathway [9]. For instance, recent studies have revealed that P2X7, P2Y1, and P2Y2 play an important role in immunomodulation and inflammation that contributes to neuroinflammatory disorders including Alzheimer’s Disease, Parkinson’s Disease, and multiple sclerosis [2].
Hydrolysis of nucleotides by ectonucleotidases
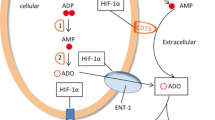
Once in the extracellular space, ATP can either activate P2R or be further dephosphorylated/hydrolyzed by ectonucleotidases, which have four families: ectonucleoside triphosphate diphosphohydrolases (NTPDases, e.g. CD39/NTPDase 1), ecto-5’-nucleotidase (CD73/NT5E), ectonucleotide pyrophosphatase/phosphodiesterase (ENPP), and alkaline phosphatase (ALP) [10]. Besides limiting ATP signaling, these enzymes can also lead to the generation of additional ligands for P2YRs, for example, ADP to P2Y12, and ADO to A2AR [11]. At the end, ADO can either be hydrolyzed by adenosine deaminase (ADA) to inosine or transported intracellularly by nucleoside transporters (NT) [11] (Fig. 1). Purinergic pathway initiates its function by releasing ATP through paracrine and/or autocrine. Its hydrolysis can subsequently generate a cascade of additional signaling molecules including ADP and ADO. Almost every cell type expresses a different combination of purinergic receptors and ectonucleotidases to regulate this pathway. Hence, the comprehensive effect of purinergic pathway on cellular function depends on not only those specific receptors and ectonucleotidases expressed by the cell, but also the dynamic change of extracellular and intracellular concentrations of ATP and ADO [6, 7].

ATP release, receptors, and ectonucleotidases involved in purinergic signaling pathway. ATP is released into the extracellular space via exocytosis, transporters (for example, ABC), channels (for example, PANX-1), P2X7R, or cell lysis (for example, caused by irradiation). Once in the extracellular space, ATP acts at P2XR and P2YR, and is also hydrolyzed to ADP and AMP by ENTPDases such as CD39. ADP can activate P2Y12R and is hydrolyzed to AMP, which can be further hydrolyzed to ADO by CD73. CD73-generated ADO can bind to its P1 receptors (A1R, A2AR, A2BR, and A3R). ADO can be subsequently degraded to inosine by ADA, or transported into the intracellular space via NT. ABC, ATP-binding cassette; ADA, adenosine deaminase; ADO, adenosine; ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; NT, nucleoside transporter; PANX-1, pannexin 1
Purinergic signaling in tumor immune microenvironment (TIME)
ATP in TIME
It is well known in previous studies that both ATP and ADO are strong modulators of tumorigenesis and antitumor immune response in TIME, where ectonucleotidases CD39 and CD73 play a fundamental role in modulating the level of ATP and ADO [12,13,14]. The activation of purinergic receptors is accordingly observed in the setting of inflammation and hypoxia in TIME [13, 15]. First of all, since basic supply of ATP is essential for cancer cell proliferation and tumor growth, TIME contains an elevated level of extracellular ATP comparing to the normal tissue. For example, ATP was within a low nM range in normal tissue, but increased to high µM or mM level in TIME [16]. It has been observed in various lung cancer cell lines that stimulation with high concentrations of ATP (e.g. 0.5–1 mM) favored cancer cell migration and invasion, while ATP in prostate cancer cells can promote the activation of Cdc42 and Rac1 and the expression of matrix metalloproteinases (MMPs) through P2YR activation [17,18,19]. ATP can also favor tumorigenicity by helping tumors escape from host immunosurveillance. Previous studies revealed that regulatory T cells (Tregs) can be activated at higher concentration of extracellular ATP around 1 mM with tighter cell contact, impeding the activation, expansion, and homing of cytotoxic T lymphocytes (CTL) and T helper cells (TH cells) [20, 21]. In addition, AMP can impede the maturation of dendritic cells (DCs) and hence preclude the priming of CD8+ T cells [22]. However, during the process of extracellular release and hydrolysis, ATP can also exert its antitumor function through activating the antitumor immune response, modulating the transition and maturation of DCs to present antigens and migration into TIME via P2X7R and P2Y2R on DCs and other immune cells, stimulating chemotaxis, inducing activation of NLR family pyrin domain-containing protein 3 (NLRP3) inflammasome, triggering intracellular proteolytic transition of pro-interleukin 1β (pro-IL-1β) into IL-1β, and upregulating pro-inflammatory cytokines such as IL-2, IL-12, and interferon γ (IFN-γ), and thus resulting in immunogenic cell death (ICD) with the augment of antitumor immunity [23,24,25].
ADO in TIME
Besides ATP, ADO can also involve in tumorigenesis and immunosuppression in TIME. A2AR-deficient mice were observed to have less tendency of tumorigenesis comparing to the wild type controls, given A2AR’s role of protecting tumors from the attack of CTLs, especially in ADO-rich TIME [26]. The similar effect of ADO has also been observed in prostate cancers, in which prostatic acid phosphatase can generate large amounts of ADO to antagonize the impact of tumor-infiltrating lymphocytes and form an immunosuppressive TIME [27]. A2AR agonists can not only control cytokine secretion related to the function of T cells but also endow long-lasting regulation of effector T cells (Teff), while A2AR antagonists can synergize with immune checkpoint inhibitors (ICI) to revert tumor immunosuppression [28, 29]. Through A2AR, high amounts of ADO can inhibit proliferation, adhesion, migration, cytotoxicity, and differentiation of Teff, for example, suppressing pro-inflammatory cytokine production of IL-2, IFN-γ, and tumor necrosis factor α (TNF-α), downregulating costimulatory receptors such as T-cell receptor (TCR) and CD28, and upregulating immune checkpoints including programmed cell death protein 1 (PD1) and cytotoxic T-lymphocyte associated protein 4 (CTLA4) [30, 31]. Increased ADO reduces the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in CD4+ T cells to modulate the release of a variety of pro-inflammatory cytokines and chemokines [32]. ADO-A2AR signal on the surface of CD4+ T cells can upregulate the expression of forkhead box P3 (FOXP3) to support Tregs [33], which can further induce suppression of immune response via pericellular secretion of ADO to favor the M2 polarization of macrophages [34]. Further, ADO-A2AR/A2BR can hinder the differentiation of monocytes into DCs and the DC-mediated function of Th1 and Th17, while impeding their regulation of the differentiation of Teff and upregulating pro-tumorigenic cytokines including IL-6, IL-8, IL-10, transforming growth factor β (TGF-β), and vascular endothelial growth factor (VEGF) [14, 35]. ADO also impedes macrophages-induced phagocytosis, downregulates the release of pro-inflammatory components including IL-12, TNF-α, nitric oxide, superoxide, stimulates pro-tumorigenic factors including IL-10, arginase-1 and VEGF expressed by tumor-associated macrophages (TAM), and increases the expression of VEGF via A2BR and IL-10 via A2AR on myeloid-derived suppressor cells (MDSC) [36,37,38,39,40,41]. In addition, ADO-A2AR is able to preclude the maturation, activation, proliferation, and cytotoxicity of natural killer (NK) cells to suppress their secretion of pro-inflammatory cytokines including IFN-γ and TNF-α, while the ablation of ADO signaling promotes NK maturation and reduces tumor growth [42]. In neutrophils, ATP and ADO can distinctly function in an autocrine mode where ATP stimulates the oxidative burst and vascular endothelium adhesion for neutrophil activation, whereas ADO reduces tissue injury based on a feedback loop against maintaining consistent inflammatory reaction [43, 44].
CD39 and CD73 in TIME
Moreover, as the key enzymes to regulate the hydrolysis of ATP, CD39 and CD73 are also essential factors from purinergic pathway to modulate TIME. The expression of CD39 is observed on less than 5% of CD8+ T cells and 20–30% of CD4+ T cells in human peripheral blood, and is modulated by pro-inflammatory cytokines in oxidative stress and hypoxia [45]. CD39 and ADO can be activated by various stimuli and provoke oxidative stress and the upregulation of cytokine cascades including TGF-β, hypoxia-induced factor 1 (HIF1), IL-6, IL-18, and TNF-α in chronic inflammatory response [44, 46, 47], whereas CD73 is differentially regulated by cytokines including TGF-β, TNF-α, IL-1β, and prostaglandin E2 (PGE2) [48]. CD73 can help tumors evade immunosurveillance via hindering the expansion and function of CTLs and TH cells, which depends on the generated ADO that affects Treg and Th17 cells [44, 48]. In addition, CD73 can regulate T cell homeostasis and memory T cell survival, differentiation, and function in hypoxia milieu. The active enzymatic format of CD39 and CD73 secreted by Tregs can hydrolyze extracellular ATP to ADO within seconds to increase the pericellular ADO level and mediate Treg-induced immunosuppression and anti-inflammatory reaction [12, 49]. In a pancreatic neuroendocrine tumor model, inhibition of purinergic receptors and CD73 harnessed the proliferation of cancer cell, tumor growth, and metastases of cancer stem cells [50]. In DU145 prostate cancer model, exosomes expressing CD39 and CD73 secreted from prostate cancer cells inhibited DC activities, resulting in an immunosuppressive environment to impede the priming and activation of CD8+ T cells [51]. Further, CD39/CD73 can delicately regulate the differentiation and function of macrophages and their M2 polarization [44]. Among pro-inflammatory M1-polorized macrophages, downregulation of CD39 and CD73 leads to decrease of ATP degradation and increase of its aggregation, while among M2-polarized macrophages, upregulation of CD39 and CD73 can increase ATP hydrolysis into ADO to promote the production of anti-inflammatory cytokines including IL-10 and IL-1 receptor antagonist (IL-1RA) [44]. Furthermore, MDSCs are also ADO-sensitive for physiological function and express CD39 and CD73 to facilitate tumor growth by inhibiting CTLs in colorectal cancers [15, 52]. Although neutrophils widely express CD39 and CD73, exacerbated activation of neutrophils may be attributed to compromised CD39/CD73 function with upregulated chemotaxis and enhanced vascular endothelium adhesion [44].
The role of purinergic pathway in radiotherapy (RT)-induced ICD in cancer
Purinergic pathway and RT-induced ICD
Purinergic pathway is involved in the ICD induced by radiation from multiple layers. RT can trigger cancer cell death followed by elevated extracellular ATP secreted by dying cells, which leads to a variety of complicated downstream cell death processes [53, 54]. Cascades of signaling activate the channels and macropores for ATP penetration and further provoke different manners of cancer cell death including apoptosis mediated by pannexin-1 (PANX-1) channels, necroptosis induced by mixed-lineage kinase domain-like pseudokinase (MLKL) pores, and pyroptosis activated by Gasdermin D (GSDMD) and/or Gasdermin E (GSDME) pores [55]. As a form of mixed cell death, ICD is usually induced by two different types of stimuli. Type I ICD inducers included γ-irradiation, ultraviolet C, and chemotherapy (CT) (e.g. methotrexate, doxorubicin, oxaliplatin), while type II ICD inducers usually refer to hypericin-based photodynamic therapy and oncolytic viruses. Both types of inducers can result in the primary and/or secondary stress of endoplasmic reticulum (ER) that results in apoptosis with caspase-activation and lysosomal exocytosis to finally release damage associated molecular patterns (DAMPs) into TIME [53, 56]. There appears to be an enormously complicated and diverse danger signaling pathways regulating the production of DAMPs in cancer cells. Extracellular DAMPs can then recruit effector cells and initiate tissue repairment and regeneration at the same time [49]. As an essential DAMP, ATP can significantly enhance ICD, which can promote cancer immunogenicity that will effectively stimulate innate and adaptive immune responses. The process involves the recruitment and migration of various immune effector cells, autocrine and/or paracrine of essential cytokines, along with the hydrolysis balance between ATP and ADO modulated by enzymes CD39 and CD73 [53].
Purinergic signaling in post-RT TIME
RT-induced spatiotemporal pattern of ATP distribution is considered pivotal to form a gradient of extracellular ATP for engendering the chemotactic or DAMP function. For example, ATP was found significantly elevated in conditioned culture media of urothelial cancer cells one hour after irradiation [57]. Irradiation can also cause long-lasting stimulation of extracellular ATP release by glioblastoma cells beyond the acute phase of irradiation-induced cell death [58]. On one hand, ATP released by irradiated dying cells acts like a discoverable “find me” signal for chemoattractant and functions as pro-inflammatory signals for further immune stimulation [54, 59]. After irradiation, an in situ vaccine-like effect was induced in TIME that is enriched with components perturbing the initiation of anti-tumor immune response, while the effect can also be reversed by immunosuppressors in the post-RT TIME. RT efficiency relies on citric acid cycle to increase fatty acid and amino acid oxidation, while radiation-related metabolic change of mitochondrial energy involves the participation of adenosine monophosphate-activated kinase (AMPK) signaling, downstream of which is phosphorylated histone H2A family member X (γH2AX) as a marker of DNA damage [60, 61]. Radiation-induced ATP release can result in the activation of purinergic receptors including P2X7, P2Y2, P2Y6, and P2Y12 [61]. P2X7 is widely expressed on almost all immune cells and promotes IL-1β and IL-18 secretion when activated by ATP, while activation of P2Y2 can promote the recruitment of immature DCs, monocytes, macrophages, and neutrophils [62]. P2Y6 and P2Y12 activation is observed following γH2AX formation by γ-irradiation [63]. In irradiated mice, deficiency of P2YRs in hematopoietic stem/progenitors cells are compromised to maintain hematopoiesis [64]. When RT-induced ICD occurs in TIME, tumor antigens from dying tumor cells can be taken up by mature DCs activated by the interaction of ATP and the above receptors expressed on DC surface, which migrate to tumor-draining lymph nodes (TDLNs) to present/cross-present antigen peptides to T cells that will infiltrate the tumor to recognize and eliminate residue tumor cells [15, 62]. Mature DCs can first secret IL-1β, IL-2, IL-6, TNF-α, and IFN-γ to promote the differentiation of T cells into CD8+ subset and then activate those CD8+ T cells to CTLs through the cross-presentation of tumor antigen peptides with major histocompatibility complex class I (MHC I). After that, CTLs can proliferate and expand to enhance their antitumor cytotoxicity by increase the secretion of IFN-γ, perforin-1, and granzyme B and/or with a combination of Fas ligand (FasL) with Fas interaction when infiltrate the irradiated tumor site as well as the abscopal sites to induce ICD [62, 65] (Fig. 2A). Mature DCs can also recruit NK cells with IFN-α, IL-2, and IL-12 and enhance their cytotoxicity to secret IFN-γ through the signaling of the C-X3-C motif chemokine ligand 1 (CX3CL1) from DCs and its receptor CX3C chemokine receptor 1 (CX3CR1) on NK cells [66, 67]. On the other hand, however, induced by repetitive stimuli of radiation, ATP may increase the release of caspase-1 and IL-1β via P2Y2 receptor in RT-resistant breast cancer cells and improved the colony-forming and invasion abilities of these cells during inflammatory process, and further promoted tumor growth and invasion while negatively regulating inflammasome activation [68]. Therefore, inhibition of P2X7R radio-sensitized melanoma, while suppression of ATP storage by targeting CD105 / sirtuin 1 (SIRT1) pathway increased radiosensitivity of prostate cancer cells via G2 cell cycle arrest [69, 70].

The role of purinergic pathway in RT-induced ICD. A ATP in RT-induced T-cell-mediated ICD. Tumor cells release ATP (via cell lysis, channels such as PANX-1, and P2X7R), chemokines, and tumor antigens following RT to recruit immature DCs to TIME. ATP then binds to its receptors (P2XR/P2YR) on immature DCs leading to their activation. These DCs can take up and process tumor antigens released from dying tumor cells to transform into mature DCs. Mature DCs can then migrate to tumor-draining lymph nodes (TDLN), where DCs secret IL-1β, IL-6, TNF-α, and IFN-γ and cross-present tumor antigen peptides by MHC I to stimulate T-cell differentiation to CD8+ CTLs. CTLs can then migrate to the tumor site and maybe remote sites to recognize and kill residue tumor cells by releasing perforin, granzyme B, and/or through the interaction of the Fas/FasL signaling. B Production of ADO leads to a post-RT immunosuppressive TIME. Hypoxia induced by RT upregulates the expression of HIF1-α, leading to the overexpression of CD39 and CD73 of cancer cells to hydrolyze large amounts of ATP to ADO in TIME. ADO is contributed to an immunosuppressive TIME to enable tumor cells to escape immune-surveillance by suppressing the effector immune components including Teff, DCs, NK cells, and neutrophils, while enhancing the activity of immunosuppressive cell subsets including Treg, M2-macrophage, and MDSC, in which pharmacological antagonists against A2AR and/or A2BR as well as blockades inhibiting CD39 and CD73 can reverse ADO-induced immunosuppressive TIME to favor antitumor immunity. A2AR, A2A adenosine receptor; A2BR, A2B adenosine receptor; ADO, adenosine; AMP, adenosine monophosphate; ATP, adenosine triphosphate; CTL, cytotoxic T lymphocyte; CTLA4, cytotoxic T-lymphocyte associated protein 4; DC, dendritic cell; Fas, factor-related apoptosis; FasL, factor-related apoptosis ligand; ICD, immunogenic cell death; IFN-γ, interferon gamma; IL-1β, interleukin-1 beta; IL-2, interleukin-2; IL-6, interleukin-6; IL-10, interleukin-10; IL-12, interleukin-12; MHC I, major histocompatibility complex class I; P2X7R, P2X7 purinergic receptor; PANX-1, pannexin 1; PD1, programmed cell death protein 1; PGE2, prostaglandin E2; RT, radiotherapy; TCR, T-cell receptor; TDLN, tumor-draining lymph node; TGF-β, transforming growth factor beta; TNF-a, tumor necrosis factor alpha; VEGF, vascular endothelial growth factor. Partly created by Figdraw (www.figdraw.com)
After RT, intratumoral hypoxia promotes radio-resistance where an immunosuppressive TIME can be further intensified. After ATP is released by dying tumor cells during RT-induced ICD, it can be hydrolyzed to ADO by CD39 and CD73 upregulated by hypoxia-induced HIF1-α in TIME. HIF1-α, the key factor functioning in hypoxia and excessive inflammation in post-RT TIME, can lead to a cascade of immunosuppressive signaling through the proliferation of Treg, the M2 polarization of TAMs, and the enrichment of MDSC [13, 54, 62]. High level of ADO, both expressed in intracellular reservoir and hydrolyzed from extracellular ATP following RT-induced ICD, has been observed in hypoxic TIME, which intervenes with other key signaling pathways to regulate TIME [33, 49]. The aggregated extracellular ADO can inhibit DC and Teff, favor the proliferation of Tregs, and polarize suppressive M2 TAMs, which can produce substantial immunosuppression in post-RT high oxidative stress to increase reactive oxygen species (ROS)-induced resistance to ICD [15, 53, 71]. TGF-β expressed by tumor cells stimulates the mammalian target of rapamycin (mTOR) pathway and induces the CD73 production while maintaining the stability of HIF1-α. Wnt pathway upregulates the promoter of CD73 via β-catenin and pro-inflammatory cytokines including TNF-α and hepatocyte growth factor (HGF), with the transcription factor c-Jun / activator protein 1 (AP1) for CD73 transcription accompanied by MAPK signaling pathway, whereas CD73 promoter CpG islands can also be methylated under certain circumstances [72, 73]. Exosomes from cancer may stimulate DCs to express CD73 induced by PGE2 [74]. Regulated by mTOR/HIF1-α pathway, CD39 and CD73 are heterogeneously expressed on MDSCs when exposed to TGF-β, which cooperates with IL-6 to increase the expression of CD73 on Th17 cells during its differentiation [75, 76]. Further, A2AR stimulation by ADO can not only restrict the NK maturation and proliferation, but also their function to produce IFN-γ and TNF-α, which impairs their capacity to eliminate cancer. ADO can also exert various inhibitory effects on neutrophils via A2AR signaling to deteriorate their ability of cytokine secretion (e.g. TNF-α), adhesion, degranulation, and phagocytosis [30, 41] (Fig. 2B).
Targeting purinergic pathway to enhance RT for cancer treatment
The summary of novel antitumor therapeutics targeting purinergic signaling pathway
Targeting individual component within purinergic signaling pathway such as ADO, P1 receptors, CD39, or CD73 may be promising with efficacy and safety for cancer immunotherapy. There have been two popular methods to inhibit ADO-induced signaling: (1) direct blockade of the binding of ADO to P1 receptors including A2AR and A2BR to suppress its function, mainly through pharmacological inhibitors/antagonists that are usually orally delivered; and (2) specific blockades of those enzymes including CD39 and CD73 to inhibit ADO production, mainly by specific antibodies that are usually intravenously delivered [41, 77] (Fig. 2B). Nowadays, more than thirty such pharmacological therapeutics including antagonists and antibodies have been or are currently investigated in the clinic for cancer treatment, alone or combined with other therapeutics including CT, RT, and/or ICI, whose benefits have already been reported at the preclinical level in various tumor models [13, 30, 33, 41, 74, 77, 78] (Table 1).
The rationale for combining RT with inhibition of purinergic pathway to improve cancer therapy
Especially for the combination therapeutic strategy with RT, inhibition of purinergic pathway has its unique essence to enhance the efficacy of RT to treat malignancies. For instance, A2BR antagonist PSB603 or A2BR siRNA increased the efficacy of RT in human lung cancer cells by blocking epidermal growth factor receptor (EGFR) translocation and DNA repair response, and reducing radio-resistance [79]. Pretreatment of PSB603 combined with irradiation also significantly suppressed tumor growth both in vitro and in vivo compared to either single-arm treated group in mouse B16 melanoma model [80]. In addition, only the combination of anti-CD73 antibody and RT could significantly delay subcutaneous tumor growth and suppress the lung metasteses through abscopal effect compared to either single treatment option in murine LuM-1 rectal cancer model. This combination also revealed to enhance the cytotoxicity and IFN-γ production of splenocytes in those treated mice [81]. Similar efficacy was also observed in a mouse breast cancer model, in which CD73 blockade with RT restored cDC1 infiltration of TIME under the condition of suboptimal type I IFN induced by RT. Even without RT-induced type I IFN, CD73 blockade was essential for the rejection of the irradiated tumor and remote tumor control as abscopal effect when combined with a CTLA4 blockade [82]. Further, in the human glioblastoma cell line A172, antagonists or siRNA of A2BR and CD73 promoted γ-irradiation-induced cell death while suppressed γ-irradiation-induced cell migration and actin remodeling [83]. In human pancreatic cancer cell line MIA PaCa-2, knockdown of CD73 using shRNA also re-sensitized the radioresistant cells to irradiation and restored irradiation-induced apoptosis [84]. Currently, there are several clinical trials registered to investigate a combination of inhibition of purinergic pathway, RT, and other therapies to treat cancer: PANTHEoN [A Study of Concurrent Chemoradiation in Combination With or Without PD1 Inhibitor (AB122) A2AR/A2BR Inhibitor AB928 Therapies in Locally Advanced Head and Neck Cancers. Phase I, NCT04892875] and PANTHER [A Phase II Study to Test the Efficacy of AB928 (a Dual Adenosine Receptor Antagonist) and AB122 (a PD1 Checkpoint Inhibitor) in Combination With Short Course Radiotherapy and Consolidation Chemotherapy for Rectal Cancer. Phase II, NCT05024097], in which a dual-specific A2AR/A2BR antagonist, AB928, will be combined with RT, CT, and zimberelimab, an anti-PD1 antibody, to treat head and neck cancer and rectal cancer; as well as Neo-CheckRay (Neo-adjuvant Chemotherapy Combined With Stereotactic Body Radiotherapy to the Primary Tumour ± Durvalumab, ± Oleclumab in Luminal B Breast Cancer. Phase II, NCT03875573), in which an anti-CD73 antibody, oleclumab, will be combined with stereotactic body radiotherapy (SBRT) and an anti- programmed cell death protein ligand 1 (PD-L1) antibody, durvalumab, for the treatment of luminal B breast cancer.
Etrumadenant (AB928) is the first dual A2AR/A2BR antagonist investigated in the clinic. AB928 is developed to inhibit the ADO-induced impairment of lymphocytes (CTLs and NK cells) and myeloid cells (DCs and macrophages) in TIME, mediated by A2AR and A2BR, respectively. AB928 inhibits A2AR and A2BR with similar high potencies [equilibrium binding constant (Kb) of 1.4 and 2 nM, respectively]. AB928 has already demonstrated a favorable and well-tolerable safety profile with other reagents and exhibits consistent pharmacokinetics (PK) / pharmacodynamics (PD) by oral dosing [85, 86]. AB928 is currently being evaluated in combination with CT, RT, ICI, and targeted therapeutics in several Phase I, I/II, and II clinical trials among multiple malignancies including advanced or metastatic head and neck cancer, lung cancer, colorectal cancer, pancreatic cancer, and prostate cancer (Table 1).
Oleclumab (MEDI9447) is a human IgG1λ monoclonal antibody (mAb) that inhibits the function of CD73, upregulation of which has been shown to increase extracellular ADO level and lead to subsequent immunosuppressive TIME in multiple cancers [87, 88]. Additive antitumor immunity has been reported when oleclumab is combined with other immunotherapeutics such as ICI in preclinical cancer models [89]. Although the phase I clinical trial of oleclumab in combination with durvalumab only provided marginally improved objective response rate (ORR) with a tolerable safety profile in patients with advanced EGFR–mutated non-small cell lung cancer (NSCLC) [90], recent results from the randomized phase II COAST trial in stage III NSCLC revealed a promising ORR of 30.0% in oleclumab + durvalumab arm (vs. 17.9% in durvalumab alone arm) with a statistically improved 12-month progression-free survival (PFS) rate (62.6% in oleclumab + durvalumab arm vs. 33.9% in durvalumab alone arm), whereas all-cause grade ≥ 3 treatment-emergent adverse events occurred in 40.7% and 39.4% with durvalumab + oleclumab and durvalumab, respectively [91]. Oleclumab has already been investigated or is going to be examined in various studies combined with CT, SBRT, ICI, and targeted therapeutics in several Phase I, I/II, and II clinical trials among multiple malignancies including breast cancer, lung cancer, colorectal cancer, pancreatic cancer, ovarian cancer, and bladder cancer, as well as a Phase III clinical trial in stage III unresectable NSCLC following the aforementioned successful Phase II COAST trial.(Table 1).
Future directions
Purinergic pathway is critically involved in a range of pathological processes including cancer. ADO concentration can be significantly elevated in a variety of malignancies, predominantly due to stress-induced ATP release, for example, irradiation, along with the overexpression of ectonucleotidases including CD39 and CD73 that contribute to its hydrolysis. Primarily by binding to A2AR and A2BR, also often overexpressed in the TIME due to hypoxia and inflammation, ADO impedes the activity of protective immune infiltrated effectors including DCs, Teff cells, and NK cells, whereas promotes immunosuppressive components including Tregs, M2-TAMs, and MDSCs. In addition, ADO also stimulates neo vessel formation to further support tumor growth and metastasis. Administration of pharmacological antagonists or antibodies to block purinergic signaling, either by its binding to their receptors or limiting its production, has achieved significant antitumor efficacy in various pre-clinical cancer models, leading to dozens of trials to investigate them in the clinic.
Furthermore, since the synergy of such modulators for purinergic signaling has already been shown with CT or RT that is known to promote ICD, as well as with other immunotherapies such as ICI, increasing numbers of clinical trials combining ADO blockades with ICIs and/or conventional treatment approaches such as RT and CT have been registered for investigation, although blockades of CD39 and CD73 as well as A2AR/A2BR antagonists are also being tested in the clinic as monotherapies. In addition, simultaneous inhibition of adenosine production (e.g. by an anti-CD73 antibody, mupadolimab) and receptor binding (e.g. by an A2AR antagonist, ciforadenant) has also demonstrated its potential synergy, and is under investigation among multiple advanced cancers in the clinic (NCT03454451) (Fig. 3). Despite the novelty of targeting this pathway with aforementioned strategies, an overall understanding of purinergic molecules and their detailed rationales in terms of modulation and effect, autocrine and/or paracrine, positive and negative feedbacks, in RT-induced ICD in TIME are crucial aspects to be further investigated in the future, which also endow pillar effects in downstream reaction and are reflected eventually in the development of innovative efficient combination therapeutic strategies based on RT for cancer treatment.

The strategy to combine inhibition of purinergic pathway and other antitumor therapies. Several components within purinergic signaling pathway including A2AR, A2BR, CD39 and CD73 can be targeted together to achieve synergy in antitumor efficacy by modulating both tumor cells and immune cells, for example, DC and Teff, by pharmacological antagonist and/or antibodies. In addition, targeting purinergic pathway in combination with other therapies such as ICIs, CT, TT, and RT can also develop potential robust strategies to enhance therapeutic benefit in various cancers, which is currently examined and will be further demonstrated in many important clinical trials. A2AR, A2A adenosine receptor; A2BR, A2B adenosine receptor; ADO, adenosine; ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; CT, chemotherapy; CTLA4, cytotoxic T-lymphocyte associated protein 4; DC, dendritic cell; ICI, immune checkpoint inhibitor; MHC I, major histocompatibility complex class I; PD1, programmed cell death protein 1; PD-L1, programmed cell death protein ligand 1; RT, radiotherapy; TCR, T-cell receptor; Teff, effector T cell; TT, targeted therapy. Partly created by Figdraw (www.figdraw.com)
Conclusions
In conclusion, purinergic pathway functions as a key factor in RT-induced ICD along with downstream immune responses in TIME. Research data in the combination therapeutic strategies targeting purinergic pathway with RT and/or other emerging immunotherapeutic modalities such as ICIs are accumulating with promising results, which have already led to dozens of clinical trials in advanced and metastatic cancers. Further, extensive and thorough investigation for comprehensive understanding of the rationale, regulation, and modulation of purinergic pathway in TIME and its combination with RT and other novel antitumor therapeutics in larger population and various malignancies is warranted.
Availability of data and material
Not applicable.
Abbreviations
- A1R:
-
Adenosine 1 receptor
- A2AR:
-
Adenosine 2A receptor
- A2BR:
-
Adenosine 2B receptor
- A3R:
-
Adenosine 3 receptor
- AC:
-
Adenylate cyclase
- ADA:
-
Adenosine deaminase
- ADO:
-
Adenosine
- ADP:
-
Adenosine diphosphate
- ALP:
-
Alkaline phosphatase
- AMPK:
-
Adenosine monophosphate-activated kinase
- ATP:
-
Adenosine triphosphate
- cAMP:
-
Cyclic adenosine monophosphate
- CT:
-
Chemotherapy
- CTL:
-
Cytotoxic T lymphocyte
- CTLA4:
-
Cytotoxic T-lymphocyte associated protein 4
- CX3CL1:
-
C-X3-C motif chemokine ligand 1
- CX3CR1:
-
CX3C chemokine receptor 1
- DAMP:
-
Damage-associated molecular pattern
- DC:
-
Dendritic cell
- EGFR:
-
Epidermal growth factor receptor
- ENPP:
-
Ectonucleotide pyrophosphatase / phosphodiesterase
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- FasL:
-
Fas ligand
- FOXP3:
-
Forkhead box P3
- GPCR:
-
G-protein coupled receptor
- GSDMD:
-
Gasdermin D
- GSDME:
-
Gasdermin E
- HGF:
-
Hepatocyte growth factor
- HIF1:
-
Hypoxia-induced factor 1
- ICD:
-
Immunogenic cell death
- ICI:
-
Immune checkpoint inhibitor
- IFN-α:
-
Interferon α
- IFN-γ:
-
Interferon γ
- IL-10:
-
Interleukin 10
- IL-12:
-
Interleukin 12
- IL-18:
-
Interleukin 18
- IL-1β:
-
Interleukin 1β
- IL-1RA:
-
IL-1 receptor antagonist
- IL-2:
-
Interleukin 2
- IL-6:
-
Interleukin 6
- IL-8:
-
Interleukin 8
- Kb :
-
Equilibrium binding constant
- mAb:
-
Monoclonal antibody
- MAPK:
-
Mitogen-activated protein kinase
- MDSC:
-
Myeloid-derived suppressor cell
- MHC I:
-
Major histocompatibility complex class I
- MLKL:
-
Mixed-lineage kinase domain-like pseudokinase
- MMP:
-
Matrix metalloproteinase
- mTOR:
-
Mammalian target of rapamycin
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NK:
-
Natural killer cell
- NLRP3:
-
NLR family pyrin domain-containing protein 3
- NSCLC:
-
Non-small cell lung cancer
- NT:
-
Nucleoside transporter
- ORR:
-
Objective response rate
- P2XR:
-
P2X receptor
- P2YR:
-
P2Y receptor
- PANX-1:
-
Pannexin-1
- PD:
-
Pharmacodynamics
- PD1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed cell death protein ligand 1
- PFS:
-
Progression-free survival
- PGE2:
-
Prostaglandin E2
- PK:
-
Pharmacokinetics
- ROS:
-
Reactive oxygen species
- RT:
-
Radiotherapy
- SBRT:
-
Stereotactic body radiotherapy
- shRNA:
-
Short-hairpin RNA
- siRNA:
-
Small interfering RNA
- SIRT1:
-
Sirtuin 1
- TAM:
-
Tumor-associated macrophage
- TCR:
-
T-cell receptor
- TDLN:
-
Tumor-draining lymph node
- Teff :
-
Effector T cell
- TGF-β :
-
Transforming growth factor β
- TH :
-
T helper cell
- TIME:
-
Tumor immune microenvironment
- TNF-α:
-
Tumor necrosis factor α
- Treg:
-
Regulatory T cell
- UDP:
-
Uridine diphosphate
- UTP:
-
Uridine triphosphate
- VEGF:
-
Vascular endothelial growth factor
- γH2AX:
-
Phosphorylated histone H2A family member X
References
Burnstock G. Purinergic signalling. Br J Pharmacol. 2006;147(Suppl 1):S172–81.
Burnstock G. Purinergic Signalling: Therapeutic Developments. Front Pharmacol. 2017;8:661. https://doi.org/10.3389/fphar.2017.00661.
Shouman K, Benarroch EE. Peripheral neuroimmune interactions: selected review and some clinical implications. Clin Auton Res. 2021;31(4):477–89. https://doi.org/10.1007/s10286-021-00787-5.
Kabata H, Artis D. Neuro-immune crosstalk and allergic inflammation. J Clin Invest. 2019;129(4):1475–82. https://doi.org/10.1172/JCI124609.
Baral P, Udit S, Chiu IM. Pain and immunity: implications for host defence. Nat Rev Immunol. 2019;19(7):433–47. https://doi.org/10.1038/s41577-019-0147-2.
Campos-Contreras ADR, Diaz-Munoz, M, Vazquez-Cuevas, FG. Purinergic Signaling in the Hallmarks of Cancer. Cells. 2020;9(7). https://doi.org/10.3390/cells9071612
Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36(3):293–303. https://doi.org/10.1038/onc.2016.206.
Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev. 2011;63(3):641–83. https://doi.org/10.1124/pr.110.003129.
Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58(3):281–341. https://doi.org/10.1124/pr.58.3.3.
Yegutkin GG. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit Rev Biochem Mol Biol. 2014;49(6):473–97. https://doi.org/10.3109/10409238.2014.953627.
Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29(39):5346–58. https://doi.org/10.1038/onc.2010.292.
Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. 2017;276(1):121–44. https://doi.org/10.1111/imr.12528.
Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17(12):709–24. https://doi.org/10.1038/nrc.2017.86.
Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. 2016;29:7–16. https://doi.org/10.1016/j.coph.2016.04.001.
Wennerberg E, Lhuillier C, Vanpouille-Box C, Pilones KA, Garcia-Martinez E, Rudqvist NP, et al. Barriers to Radiation-Induced In Situ Tumor Vaccination. Front Immunol. 2017;8:229. https://doi.org/10.3389/fimmu.2017.00229.
Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased Level of Extracellular ATP at Tumor Sites: In Vivo Imaging with Plasma Membrane Luciferase. PLOS ONE. 2008;3(7):e2599. https://doi.org/10.1371/journal.pone.0002599.
Cao Y, Wang X, Li Y, Evers M, Zhang H, Chen X. Extracellular and macropinocytosis internalized ATP work together to induce epithelial-mesenchymal transition and other early metastatic activities in lung cancer. Cancer Cell Int. 2019;19:254. https://doi.org/10.1186/s12935-019-0973-0.
Zhang Y, Gong LH, Zhang HQ, Du Q, You JF, Tian XX, et al. Extracellular ATP enhances in vitro invasion of prostate cancer cells by activating Rho GTPase and upregulating MMPs expression. Cancer Lett. 2010;293(2):189–97. https://doi.org/10.1016/j.canlet.2010.01.010.
Chen L, He HY, Li HM, Zheng J, Heng WJ, You JF, et al. ERK1/2 and p38 pathways are required for P2Y receptor-mediated prostate cancer invasion. Cancer Lett. 2004;215(2):239–47. https://doi.org/10.1016/j.canlet.2004.05.023.
Krysko O, Love Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013;4:e631. https://doi.org/10.1038/cddis.2013.156.
Trabanelli S, Ocadlikova D, Gulinelli S, Curti A, Salvestrini V, Vieira RP, et al. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J Immunol. 2012;189(3):1303–10. https://doi.org/10.4049/jimmunol.1103800.
Lecciso M, Ocadlikova D, Sangaletti S, Trabanelli S, De Marchi E, Orioli E, et al. ATP Release from Chemotherapy-Treated Dying Leukemia Cells Elicits an Immune Suppressive Effect by Increasing Regulatory T Cells and Tolerogenic Dendritic Cells. Front Immunol. 2017;8:1918. https://doi.org/10.3389/fimmu.2017.01918.
Rapoport BL, Anderson R. Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. Int J Mol Sci. 2019;20(4):959. https://doi.org/10.3390/ijms20040959.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–8. https://doi.org/10.1038/nm.2028.
Adinolfi E, De Marchi E, Orioli E, Pegoraro A, Di Virgilio F. Role of the P2X7 receptor in tumor-associated inflammation. Curr Opin Pharmacol. 2019;47:59–64. https://doi.org/10.1016/j.coph.2019.02.012.
Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103(35):13132–7. https://doi.org/10.1073/pnas.0605251103.
Stultz J, Fong L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021;24(3):697–717. https://doi.org/10.1038/s41391-021-00340-5.
Bastid J, Cottalorda-Regairaz A, Alberici G, Bonnefoy N, Eliaou JF, Bensussan A. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene. 2013;32(14):1743–51. https://doi.org/10.1038/onc.2012.269.
Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol Res. 2015;3(5):506–17. https://doi.org/10.1158/2326-6066.CIR-14-0211.
Sek K, Molck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting Adenosine Receptor Signaling in Cancer Immunotherapy. Int J Mol Sci. 2018;19(12):3837. https://doi.org/10.3390/ijms19123837.
Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. 2020;17(10):611–29. https://doi.org/10.1038/s41571-020-0382-2.
Romio M, Reinbeck B, Bongardt S, Huls S, Burghoff S, Schrader J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am J Physiol Cell Physiol. 2011;301(2):C530-539. https://doi.org/10.1152/ajpcell.00385.2010.
Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. 2018;6(1):57. https://doi.org/10.1186/s40425-018-0360-8.
Zanin RF, Braganhol E, Bergamin LS, Campesato LF, Filho AZ, Moreira JC, et al. Differential macrophage activation alters the expression profile of NTPDase and ecto-5’-nucleotidase. PLoS ONE. 2012;7(2):e31205. https://doi.org/10.1371/journal.pone.0031205.
Wilson JM, Kurtz CC, Black SG, Ross WG, Alam MS, Linden J, et al. The A2B adenosine receptor promotes Th17 differentiation via stimulation of dendritic cell IL-6. J Immunol. 2011;186(12):6746–52. https://doi.org/10.4049/jimmunol.1100117.
Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, et al. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J. 2000;14(13):2065–74. https://doi.org/10.1096/fj.99-0508com.
Hasko G, Szabo C, Nemeth ZH, Kvetan V, Pastores SM, Vizi ES. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol. 1996;157(10):4634–40.
Si QS, Nakamura Y, Kataoka K. Adenosine inhibits superoxide production in rat peritoneal macrophages via elevation of cAMP level. Immunopharmacol. 1997;36(1):1–7. https://doi.org/10.1016/s0162-3109(96)00158-0.
Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation. 2013;36(4):921–31. https://doi.org/10.1007/s10753-013-9621-3.
Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell. 2007;18(1):14–23. https://doi.org/10.1091/mbc.e06-07-0596.
Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P, et al. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front Immunol. 2019;10:925. https://doi.org/10.3389/fimmu.2019.00925.
Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018;78(4):1003–16. https://doi.org/10.1158/0008-5472.CAN-17-2826.
Eltzschig HK, Macmanus CF, Colgan SP. Neutrophils as sources of extracellular nucleotides: functional consequences at the vascular interface. Trends Cardiovasc Med. 2008;18(3):103–7. https://doi.org/10.1016/j.tcm.2008.01.006.
Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19(6):355–67. https://doi.org/10.1016/j.molmed.2013.03.005.
de Leve S, Wirsdorfer, F, Jendrossek, V. The CD73/Ado System-A New Player in RT Induced Adverse Late Effects. Cancers (Basel). 2019;11(10). https://doi.org/10.3390/cancers11101578
Allard D, Allard, B, Stagg, J. On the mechanism of anti-CD39 immune checkpoint therapy. J Immunother Cancer. 2020;8(1). https://doi.org/10.1136/jitc-2019-000186
Levesque SA, Kukulski F, Enjyoji K, Robson SC, Sevigny J. NTPDase1 governs P2X7-dependent functions in murine macrophages. Eur J Immunol. 2010;40(5):1473–85. https://doi.org/10.1002/eji.200939741.
Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;33(5):231–7. https://doi.org/10.1016/j.it.2012.02.009.
de Leve S, Wirsdorfer F, Jendrossek V. Targeting the Immunomodulatory CD73/Adenosine System to Improve the Therapeutic Gain of Radiotherapy. Front Immunol. 2019;10:698. https://doi.org/10.3389/fimmu.2019.00698.
Katsuta E, Tanaka S, Mogushi K, Shimada S, Akiyama Y, Aihara A, et al. CD73 as a therapeutic target for pancreatic neuroendocrine tumor stem cells. Int J Oncol. 2016;48(2):657–69. https://doi.org/10.3892/ijo.2015.3299.
Salimu J, Webber J, Gurney M, Al-Taei S, Clayton A, Tabi Z. Dominant immunosuppression of dendritic cell function by prostate-cancer-derived exosomes. J Extracell Vesicles. 2017;6(1):1368823. https://doi.org/10.1080/20013078.2017.1368823.
Zhang B, Wang, Z, Wu, L, Zhang, M, Li, W, Ding, J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS One. 2013;8(2):e57114. https://doi.org/10.1371/journal.pone.0057114.
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–75. https://doi.org/10.1038/nrc3380.
Golden EB, Apetoh, L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25(1):11-17. https://doi.org/10.1016/j.semradonc.2014.07.005.
Dubyak GR. Luciferase-assisted detection of extracellular ATP and ATP metabolites during immunogenic death of cancer cells. Methods Enzymol. 2019;629:81–102. https://doi.org/10.1016/bs.mie.2019.10.006.
Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014;21(1):79–91. https://doi.org/10.1038/cdd.2013.75.
Bill MA, Srivastava K, Breen C, Butterworth KT, McMahon SJ, Prise KM, et al. Dual effects of radiation bystander signaling in urothelial cancer: purinergic-activation of apoptosis attenuates survival of urothelial cancer and normal urothelial cells. Oncotarget. 2017;8(57):97331–43. https://doi.org/10.18632/oncotarget.21995.
Zanoni M, Sarti AC, Zamagni A, Cortesi M, Pignatta S, Arienti C, et al. Irradiation causes senescence, ATP release, and P2X7 receptor isoform switch in glioblastoma. Cell Death Dis. 2022;13(1):80. https://doi.org/10.1038/s41419-022-04526-0.
Ocadlikova D, Lecciso M, Isidori A, Loscocco F, Visani G, Amadori S, et al. Chemotherapy-Induced Tumor Cell Death at the Crossroads Between Immunogenicity and Immunotolerance: Focus on Acute Myeloid Leukemia. Front Oncol. 2019;9:1004. https://doi.org/10.3389/fonc.2019.01004.
Kim EJ, Lee M, Kim DY, Kim KI, Yi JY. Mechanisms of Energy Metabolism in Skeletal Muscle Mitochondria Following Radiation Exposure. Cells. 2019;8(9):950. https://doi.org/10.3390/cells8090950.
Tsukimoto M. Purinergic Signaling Is a Novel Mechanism of the Cellular Response to Ionizing Radiation. Biol Pharm Bull. 2015;38(7):951–9. https://doi.org/10.1248/bpb.b15-00062.
Zhu M, Yang M, Zhang J, Yin Y, Fan X, Zhang Y, et al. Immunogenic Cell Death Induction by Ionizing Radiation. Front Immunol. 2021;12: 705361. https://doi.org/10.3389/fimmu.2021.705361.
Ide S, Nishimaki N, Tsukimoto M, Kojima S. Purine receptor P2Y6 mediates cellular response to gamma-ray-induced DNA damage. J Toxicol Sci. 2014;39(1):15–23. https://doi.org/10.2131/jts.39.15.
Cho J, Yusuf R, Kook S, Attar E, Lee D, Park B, et al. Purinergic P2Y(1)(4) receptor modulates stress-induced hematopoietic stem/progenitor cell senescence. J Clin Invest. 2014;124(7):3159–71. https://doi.org/10.1172/JCI61636.
Wang YJ, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018;5(3):194–203. https://doi.org/10.1016/j.gendis.2018.05.003.
Ferlazzo G, Morandi B. Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front Immunol. 2014;5:159. https://doi.org/10.3389/fimmu.2014.00159.
Korbecki J, Siminska D, Kojder K, Grochans S, Gutowska I, Chlubek D, et al. Fractalkine/CX3CL1 in Neoplastic Processes. Int J Mol Sci. 2020;21(10):3723. https://doi.org/10.3390/ijms21103723.
Jin H, Ko YS, Kim HJ. P2Y2R-mediated inflammasome activation is involved in tumor progression in breast cancer cells and in radiotherapy-resistant breast cancer. Int J Oncol. 2018;53(5):1953–66. https://doi.org/10.3892/ijo.2018.4552.
Tanamachi K, Nishino K, Mori N, Suzuki T, Tanuma SI, Abe R, et al. Radiosensitizing Effect of P2X7 Receptor Antagonist on Melanoma in Vitro and in Vivo. Biol Pharm Bull. 2017;40(6):878–87. https://doi.org/10.1248/bpb.b17-00083.
Madhav A, Andres A, Duong F, Mishra R, Haldar S, Liu Z, et al. Antagonizing CD105 enhances radiation sensitivity in prostate cancer. Oncogene. 2018;37(32):4385–97. https://doi.org/10.1038/s41388-018-0278-0.
Kachikwu EL, Iwamoto KS, Liao YP, DeMarco JJ, Agazaryan N, Economou JS, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys. 2011;81(4):1128–35. https://doi.org/10.1016/j.ijrobp.2010.09.034.
Reinhardt J, Landsberg J, Schmid-Burgk JL, Ramis BB, Bald T, Glodde N, et al. MAPK Signaling and Inflammation Link Melanoma Phenotype Switching to Induction of CD73 during Immunotherapy. Cancer Res. 2017;77(17):4697–709. https://doi.org/10.1158/0008-5472.CAN-17-0395.
Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunol. 2017;6(6): e1320011. https://doi.org/10.1080/2162402X.2017.1320011.
Allard D, Chrobak P, Allard B, Messaoudi N, Stagg J. Targeting the CD73-adenosine axis in immuno-oncology. Immunol Lett. 2019;205:31–9. https://doi.org/10.1016/j.imlet.2018.05.001.
Chatterjee S, Thyagarajan K, Kesarwani P, Song JH, Soloshchenko M, Fu J, et al. Reducing CD73 expression by IL1beta-Programmed Th17 cells improves immunotherapeutic control of tumors. Cancer Res. 2014;74(21):6048–59. https://doi.org/10.1158/0008-5472.CAN-14-1450.
Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-beta signaling in generation of CD39+CD73+ myeloid cells in tumors. J Immunol. 2014;193(6):3155–64. https://doi.org/10.4049/jimmunol.1400578.
Whiteside TL. Targeting adenosine in cancer immunotherapy: a review of recent progress. Expert Rev Anticancer Ther. 2017;17(6):527–35. https://doi.org/10.1080/14737140.2017.1316197.
Augustin RC, Leone, RD, Naing, A, Fong, L, Bao, R, Luke, JJ. Next steps for clinical translation of adenosine pathway inhibition in cancer immunotherapy. J Immunother Cancer. 2022;10(2). https://doi.org/10.1136/jitc-2021-004089
Kitabatake K, Yoshida E, Kaji T, Tsukimoto M. Involvement of adenosine A2B receptor in radiation-induced translocation of epidermal growth factor receptor and DNA damage response leading to radioresistance in human lung cancer cells. Biochim Biophys Acta Gen Subj. 2020;1864(1): 129457. https://doi.org/10.1016/j.bbagen.2019.129457.
Tanaka Y, Kitabatake K, Abe R, Tsukimoto M. Involvement of A2B Receptor in DNA Damage Response and Radiosensitizing Effect of A2B Receptor Antagonists on Mouse B16 Melanoma. Biol Pharm Bull. 2020;43(3):516–25. https://doi.org/10.1248/bpb.b19-00976.
Tsukui H, Horie H, Koinuma K, Ohzawa H, Sakuma Y, Hosoya Y, et al. CD73 blockade enhances the local and abscopal effects of radiotherapy in a murine rectal cancer model. BMC Cancer. 2020;20(1):411. https://doi.org/10.1186/s12885-020-06893-3.
Wennerberg E, Spada S, Rudqvist NP, Lhuillier C, Gruber S, Chen Q, et al. CD73 Blockade Promotes Dendritic Cell Infiltration of Irradiated Tumors and Tumor Rejection. Cancer Immunol Res. 2020;8(4):465–78. https://doi.org/10.1158/2326-6066.CIR-19-0449.
Kitabatake K, Kaji T, Tsukimoto M. Involvement of CD73 and A2B Receptor in Radiation-Induced DNA Damage Response and Cell Migration in Human Glioblastoma A172 Cells. Biol Pharm Bull. 2021;44(2):197–210. https://doi.org/10.1248/bpb.b20-00654.
Nguyen AM, Zhou J, Sicairos B, Sonney S, Du Y. Upregulation of CD73 Confers Acquired Radioresistance and is Required for Maintaining Irradiation-selected Pancreatic Cancer Cells in a Mesenchymal State. Mol Cell Proteomics. 2020;19(2):375–89. https://doi.org/10.1074/mcp.RA119.001779.
Seitz L, Jin L, Leleti M, Ashok D, Jeffrey J, Rieger A, et al. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Invest New Drugs. 2019;37(4):711–21. https://doi.org/10.1007/s10637-018-0706-6.
Powderly JD, Souza PL, Gutierrez R, Horvath L, Seitz L, Ashok D, et al. AB928, a novel dual adenosine receptor antagonist, combined with chemotherapy or AB122 (anti-PD-1) in patients (pts) with advanced tumors: Preliminary results from ongoing phase I studies. J Clin Oncol. 2019;37(15_suppl):2604–2604. https://doi.org/10.1200/JCO.2019.37.15_suppl.2604.
Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget. 2017;8(5):8738–51. https://doi.org/10.18632/oncotarget.14434.
Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015;75(21):4494–503. https://doi.org/10.1158/0008-5472.CAN-14-3569.
Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunol. 2016;5(8): e1208875. https://doi.org/10.1080/2162402X.2016.1208875.
Bendell JC, LoRusso P, Overman MJ, Noonan AM, Kim D-W, Strickler J, et al. Safety and efficacy of the anti-CD73 monoclonal antibody (mAb) oleclumab ± durvalumab in patients (pts) with advanced colorectal cancer (CRC), pancreatic ductal adenocarcinoma (PDAC), or EGFR-mutant non-small cell lung cancer (EGFRm NSCLC). J Clin Oncol. 2021;39(15_suppl):9047–9047. https://doi.org/10.1200/JCO.2021.39.15_suppl.9047.
Herbst RS, Majem, M, Barlesi, F, Carcereny, E, Chu, Q, Monnet, I, et al. COAST: An Open-Label, Phase II, Multidrug Platform Study of Durvalumab Alone or in Combination With Oleclumab or Monalizumab in Patients With Unresectable, Stage III Non-Small-Cell Lung Cancer. J Clin Oncol. 2022:JCO2200227. https://doi.org/10.1200/JCO.22.00227
Acknowledgements
Not applicable.
Funding
The work was supported by funding from Fudan University Shanghai Cancer Center, Shanghai, China (YJMS201905 to LX).
Author information
Authors and Affiliations
Contributions
XB and LX designed, wrote, edited, and reviewed this manuscript. Both authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bao, X., Xie, L. Targeting purinergic pathway to enhance radiotherapy-induced immunogenic cancer cell death. J Exp Clin Cancer Res 41, 222 (2022). https://doi.org/10.1186/s13046-022-02430-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02430-1