
Abstract
Glioblastoma (GBM) treatment has remained almost unchanged for more than 20 years. The current standard of care involves surgical resection (if possible) followed by concomitant radiotherapy and chemotherapy. In recent years, immunotherapy strategies have revolutionized the treatment of many cancers, increasing the hope for GBM therapy. However, mostly due to the high, multifactorial immunosuppression occurring in the microenvironment, the poor knowledge of the neuroimmune system and the presence of the blood−brain barrier, the efficacy of immunotherapy in GBM is still low. Recently, new strategies for GBM treatments have employed immunotherapy combinations and have provided encouraging results in both preclinical and clinical studies. The lessons learned from clinical trials highlight the importance of tackling different arms of immunity. In this review, we aim to summarize the preclinical evidence regarding combination immunotherapy in terms of immune and survival benefits for GBM management. The outcomes of recent studies assessing the combination of different classes of immunotherapeutic agents (e.g., immune checkpoint blockade and vaccines) will be discussed. Finally, future strategies to ameliorate the efficacy of immunotherapy and facilitate clinical translation will be provided to address the unmet medical needs of GBM.
Similar content being viewed by others

Background
Glioblastoma (GBM) is the most common and aggressive malignant tumor of the central nervous system (CNS) [1, 2]. GBM is a grade IV diffuse astrocytoma that is thought to arise from neural stem cells or progenitor cells, such as oligodendrocyte precursor cells [2,3,4]. Approximately 90% of GBM cases are considered primary GBM, with fast and de novo expansion and without any sign of less malignant precursor tumors. Primary GBM often develops in elderly patients and shows a much poorer prognosis than secondary GBM, which originates from grade II and III astrocytomas, oligodendrogliomas or oligoastrocytomas and most likely manifesting in younger patients [3, 5].
Standard of care (SOC) therapy aims at increasing patient life expectancy and focuses on maximal and safe surgical resection combined with radiotherapy (RT) and adjuvant chemotherapy in the form of oral delivery of temozolomide (TMZ) [6]. Despite this treatment, the median survival of patients diagnosed with GBM is approximately 15 months, with a 2-year life expectancy of less than 30% [7]. For patients with unresectable GBM (up to 35–40% of patients), the prognosis is even poorer [8,9,10]. Indeed, microscopic infiltration of GBM cells and the tumor location render total resection difficult or even impossible and produce inevitable recurrences [9]. Finding new therapies for GBM is therefore an urgent unmet need, although it is very challenging because of unique characteristics of GBM and its tumor microenvironment (TME). GBM are characterized by intratumoral and intertumoral heterogeneity, highly invasive and infiltrative cell properties and an immunosuppressive TME promoting GBM growth via complex interactions [11].
Immunotherapies, by re-educating and harnessing the patient’s immune response against tumors, hold great promise for cancer treatment. These methods have become increasingly used in the treatment of different kinds of cancers, including brain cancers [12, 13]. Current immunotherapy strategies used to treat cancers are mainly based on immune checkpoint blockade (ICB) agents [14, 15], but also therapeutic vaccines [16, 17], adoptive cell therapy [18, 19], monoclonal antibodies (mAbs) [20] and oncolytic viruses [21].
Treatment with ICBs has shown remarkable success in a population of patients with melanomas and other tumors [22,23,24,25]. Therapeutic vaccines have also emerged as promising cancer treatments, with currently 3 therapeutic cancer vaccines approved by the Food and Drug Administration (FDA) [26]. However, even though immunotherapies have shown survival benefit for some proportions of patients with solid tumors, most patients still do not respond to immunotherapy. Less than 15% of cancer patients currently respond to ICBs [27]. Furthermore, these strategies are not as effective as would be desired for GBM treatment. To date, phase III clinical studies with ICB and vaccine therapies have shown no major benefit of immune modulation for GBM treatment or patient survival [13, 28].
A recent clinical study, however, demonstrated that administration of anti-programmed cell death protein 1 (PD-1) mAbs prior to tumor resection increased local and systemic antitumor immune responses [29]. Additionally, interim results of a phase II clinical study evaluating the combination of an allogeneic/autologous therapeutic GBM vaccine in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF), cyclophosphamide and bevacizumab demonstrated a significant survival benefit [30]. These results raise hope for research on GBM immunotherapy treatments. Immune modulation in combination with other treatments has shown encouraging preclinical results. This review summarizes some of these promising combination strategies for the treatment of GBM. We will particularly focus on combinations including ICBs, as they are the most studied combination strategies including immunotherapy for GBM.
The BBB and immune microenvironment in GBM: implications for the development of new therapies
To ensure proper neuronal function, the brain has to be maintained in a homeostatic state. This implies regulation of the influx/efflux of cells, molecules and ions [31]. Two major barriers contribute to separating the CNS from the variable environment of blood: (i) the blood−cerebrospinal fluid barrier, formed by the choroid plexus epithelium and separating the cerebrospinal fluid from the blood; and (ii) the blood−brain barrier (BBB), formed by endothelial cells of the capillary of brain parenchyma and separating the blood from the brain interstitial fluid [32, 33].
Due to its anatomical structure and vascular organization, the BBB is the most selective barrier [34]. For this reason, the BBB is also an important obstacle for the development of successful GBM treatments. While it has been shown to be disrupted in GBM, an intact BBB is still found peritumorally [35, 36]. This heterogeneous disruption leads to protection of most infiltrative components of GBM and limits the delivery of the majority of therapeutics to the tumor [35,36,37].
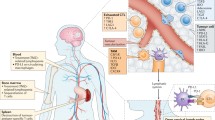
Additionally, due to the presence of the BBB and its tight junctions, the brain has long been considered an immune-privileged site. The identification of an absence of classic lymphatics and tolerance of foreign tissue transplants in the brain also suggested that the brain was immunologically unique [38,39,40]. However, it is now known that the brain is not isolated immunologically [41]. It has been proven that functional lymphatic vessels are present in the CNS, that activated T cells can traffic to the CNS and that CNS antigens can reach the peripheral lymph nodes [38]. This emerging evidence suggests that immunotherapy can be applied to GBM and other brain cancers. However, several characteristics of GBM, e.g., its heterogeneity, BBB, low tumor mutation burden, low infiltration of T cells and microenvironment (which, for example, features a high infiltration of immunosuppressive cells) induce very complex immunosuppression, which is one major hindrance in finding new treatments and translating immunotherapies for GBM (Fig. 1).

Schematic representation of GBM TME. A GBM TME is composed of various cell types. B Factors impeding translation of GBM immunotherapy treatments: (i) the BBB limiting drug delivery efficacy, (ii) the relatively low infiltration of T cells as well as their high exhaustion marker expression, (iii) the high infiltration of immunosuppressive cells (such as Tregs, TAMs and MDSCs) in the TME and (iv) the infiltrative and heterogeneous characteristics of GBM cells. (Abbreviations: GBM = Glioblastoma; LAG-3 = Lymphocyte activation gene 3 protein; MDSC = Myeloid-derived suppressive cell; NK = Natural killer; PD-1 = Programmed cell death-1; TAM = Tumor-associated microglia and macrophage; TIM-3 = T cell immunoglobulin and mucin domain containing-3)
GBM tumors develop in an immunosuppressive microenvironment that stimulates tumor cell growth and aggressiveness. The GBM TME is composed of various cell types: infiltrating tumor cells and cancer stem cells as well as noncancerous cells, such as myeloid cells (including resident microglia and bone marrow-derived macrophages), tissue-resident cells (such as neurons and astrocytes), and lymphocytes, and all these cells can interact together [42, 43].
GBM cells are known to secrete chemokines, growth factors and cytokines into the TME. Liberation of these molecules will attract and stimulate immunosuppressive cells [43,44,45]. In turn, immunosuppressive cells interact with GBM cells through different immunosuppressive receptors, resulting in tumor growth promotion and tumor cell resistance and evasion of immune surveillance [43, 44, 46, 47]. One of the most well-known and described immunosuppressive receptors involved in GBM immune escape is PD-1 [48,49,50]. The PD-1 receptor is mostly expressed on activated T cells, and binding of PD-1 with its ligand PD-L1 leads to inactivation of those T cells [51]. In GBM, PD-L1 has been shown to be overexpressed by GBM and myeloid cells, leading to effective binding of PD-L1 to PD-1 and therefore inhibition of the immune response [48, 52, 53].
The predominant non-neoplastic cells are tumor-associated microglia and macrophages (TAMs), which constitute approximately 30% of GBM TME [54, 55]. Microglia and macrophages are the main innate immune cells in the healthy CNS, where they play a major role in maintaining homeostasis and immune surveillance [56, 57]. Microglia are the resident macrophages of the CNS localized in the parenchyma. They arise from yolk-sac macrophage precursors at early embryonic stage [58,59,60,61]. Three non-parenchymal macrophage populations, or border-associated macrophages, are also found in the CNS under normal conditions: perivascular, meningeal and choroid macrophages [56, 57, 62]. In GBM, the partial disruption of the BBB leads to peripheral bone marrow-derived macrophages infiltration. This macrophage population account for approximately 85% of GBM TAMs [63].
Two TAM phenotypes are commonly described: (i) the classical inflammatory and anti-tumoral M1 phenotype and (ii) the anti-inflammatory and pro-tumoral M2 phenotype [64]. Recent studies also indicate that heterogeneous populations of TAMs expressing both M1- and M2-associated markers are found in human and murine GBM [65, 66]. A high infiltration of M2-like TAMs has been associated with poor prognosis in GBM [55, 67]. Moreover, a higher proportion of M2-like TAMs are found in high grade gliomas, as compared to low grade gliomas [68]. Indeed, GBM cells recruit microglia and macrophages in the TME by means of chemoattractants (e.g., C-C motif chemokine ligand 2 (CCL2), CSF-1) [69, 70] and induce a switch to pro-tumoral subtype [65, 71, 72]. In return, TAMs promote tumor cell proliferation and angiogenesis as well as inhibition of effector T cells proliferation and attraction of T regulatory cells (Tregs) and MDSCs through cytokines secretion [65, 72, 73].
MDSCs constitute a heterogeneous population of cells that play an important role in maintaining an immunosuppressive environment in GBM. They inhibit the immune response by interacting with different cells in the TME, promoting Treg function, limiting antigen presentation and inhibiting effector T cell activity, among other things [74,75,76,77]. In addition, as already mentioned, myeloid cells (including TAMs and MDSCs) have been shown to overexpress the negative checkpoint molecule PD-L1 in GBM patients, therefore promoting negative regulation of the immune response by inducing T cell dysfunction [78].
Compared to other tumors, the number of tumor-infiltrating lymphocytes (TILs) is relatively low in GBM [79]. Additionally, those that are present express high levels of exhaustion markers such as the inhibitory coreceptors T cell immunoglobulin and mucin domain containing-3 (TIM-3), lymphocyte activation gene 3 protein (LAG-3), and PD-1 [80,81,82,83]. Moreover, the fraction of Tregs among TILs is increased in GBM patients [84]. Tregs contribute to the TME immunosuppression by inhibiting effector T cells and antigen-presenting cells (APCs) [83]. Beside a low density of TILs, a low number of infiltrating Natural killer (NK) cells and B cells are found in GBM [85]. Besides, GBM cells have developed mechanisms to escape from NK immune surveillance through inhibitory bindings [86].
In addition to the complex interplay between all the cells present in the TME of GBM, resulting in multifactorial and complex immunosuppression, low tumor mutation burden is an additional likely reason why applying immunotherapy in GBM is difficult. Indeed, tumor-associated antigen production is quite low in GBM, which may lower the efficacy of immunotherapy [87].
Moreover, unique characteristics of GBM also impede the development of new treatments. Indeed, GBM is a very infiltrative tumor that exhibit inter- and intratumoral heterogeneity. Intratumoral heterogeneity refers to molecular diversity within a same tumor, leading to differences in growth rate, cellular morphology, histopathology and differentiation markers expression, among other things [88]. Therefore, different subpopulations of GBM cells coexist and cooperate to promote tumor growth and progression, contributing to divergence in response and resistance to SOC [89, 90]. Intertumoral heterogeneity refers to cellular and genetic differences between GBM tumors from different patients and leads therefore to different molecular subtypes, and heterogeneity in patients’ response to therapy [91].
GBM immunotherapy: disappointing initial clinical results of monotherapies
Immune checkpoints are regulators of the immune system that control immune effector function by maintaining an equilibrium between inhibitory and costimulatory signals. Their role is to protect tissues from damage due to excessive immune response but also to prevent autoimmunity [14, 92]. However, cancer cells have been shown to take advantage of this system to escape immune surveillance by upregulating inhibitory immune checkpoint expression and activating these negative regulators on tumor-specific immune cells [14, 93, 94]. The main example of this mechanism in GBM is the appropriation of the PD-1/PD-L1 pathway [48,49,50].
The main immune checkpoints that have been successfully targeted in cancer treatments are PD-1, its ligand PD-L1, and cytotoxic T-lymphocyte antigen 4 (CTLA-4). Inhibition of these T cell negative regulators with mAbs increases the immune response in many cancers (e.g., melanoma and non-small-cell lung cancer) [25, 95,96,97]. In 2011, the FDA for the first time approved the use of the checkpoint inhibitor ipilimumab – a monoclonal antibody inhibiting CTLA-4 – as a frontline cancer treatment in advanced melanoma [22, 98]. Since 2014, the first mAb directed toward PD-1, nivolumab, has been FDA-approved for the treatment of metastatic melanoma [99]. Nivolumab was further approved for other cancers, such as non-small-lung cancer and squamous cancer of the head and neck [23, 24]. The combination of ipilimumab and nivolumab was also approved for advanced renal cell carcinoma, metastatic melanoma and metastatic colorectal cancer [100,101,102].
By generating and amplifying specific T cell responses against tumors, therapeutic cancer vaccines hold a major place among strategies to fight cancer. In the past decade, research on therapeutic cancer vaccine has extended, thanks to recent progress in delivery technologies and target selection methods as well as continuous advances in understanding tumor immune response mechanisms [103]. Today, there are 3 FDA-approved therapeutic cancer vaccines. The first immunotherapy ever approved by FDA was intravesical BCG (bacillus Calmette-Guérin) in 1990 [26]. This live attenuated vaccine is now part of the standard treatment of early-stage bladder cancer and reduces risks of cancer progression [104, 105]. In 2010, the FDA approved Sipuleucel-T (Provenge®) – an autologous dendritic cell (DC) vaccine that reduces the risk of death in prostate cancer patients [106]. Intralesional vaccination with an oncolytic herpes virus encoding GM-CSF, talimogene laherparepvec (T-VEC; Imlygic®), improved mOS in patients with advanced melanoma [107]. T-VEC was approved by the FDA in 2015 [108].
However, both ICBs and vaccines as monotherapies are still ineffective in multiple cancers, such as GBM. To date, there is no FDA-approved immunotherapy for GBM, even though some are being tested in clinical trials [109]. Moreover, some phase III clinical trials have failed, including trials of immunotherapy treatments such as ICBs or therapeutic vaccines (Table 1).
ICBs, and anti-PD-1 therapy more particularly, have been extensively studied in GBM treatment given their promising results in other solid tumors. Anti-PD-1 agents were the first type of ICB tested in a clinical trial for GBM treatment (CheckMate 143, NCT02017717) [110, 113]. Phase III of this study was conducted on 369 patients with recurrent GBM randomized to receive either nivolumab (anti-PD-1 antibody) or bevacizumab (an antibody targeting and inhibiting vascular endothelial growth factor (VEGF); an antiangiogenic treatment). The results of this study showed no improvement in the median overall survival (mOS) for patients treated with anti-PD-1 antibody compared to those in the other treatment arms [110]. Similarly, the phase III CheckMate 498 trial (NCT02617589) failed to meet its primary endpoint of improving mOS. The study was conducted on 560 newly diagnosed patients and compared nivolumab and TMZ, each of which was given with RT. More recently, Bristol-Myers Squibb (BMS) announced that the phase III trial Checkmate 548 (NCT02667587) did not meet its primary endpoint. The study evaluated the addition of nivolumab to the SOC and was conducted on 693 patients with newly diagnosed GBM.
Likewise, two phase III studies with therapeutic vaccines added to standard treatments failed to produce convincing results [111,112,113]. In the ACT IV phase III study (NCT01480479), the addition of rindopepimut – a vaccine targeting EGFRvIII – to standard chemotherapy did not improve survival of EGFRvIII-positive glioblastoma patients [111]. The NCT00045968 phase III trial evaluating the addition of an autologous tumor lysate-pulsed DC vaccine (DCVax®-L) to SOC for newly diagnosed GBM patients was put on hold by the FDA for unidentified reasons [112, 113].
The main commonality between the agents used in these disappointing phase III trials is that they stimulate only one arm of antitumor immunity: they reduce the immunosuppression exerted on T cells (ICBs), stimulate the immune response against a specific antigen or activate a specific dendritic cell (DC) response against the tumor. Even when combined with SOC, the immune response is not enhanced, mainly because of the immunosuppressive characteristics of systemic chemotherapy and RT (which induce lymphopenia and hypoxia, respectively), as well as corticotherapy [114, 115]. Indeed, the blood of patients treated with dexamethasone has a reduced number of immune cells [116].
Despite these disappointing clinical trial results, the randomized open-label pilot study from Cloughesy and colleagues raises hope for the use of immunotherapy in GBM [29]. In this study, an anti-PD-1 mAb (pembrolizumab) was given as a neoadjuvant drug to patients with recurrent GBM. Administration of pembrolizumab before resection significantly improved overall and progression-free survival with induction of TIL functional activation and production of an interferon (IFN)-γ response within the TME [29]. Besides, a phase II study on the therapeutic GBM vaccine ERC1671 (Gliovac™) showed promising preliminary results. ERC1671 is a vaccine composed of tumor cell lysate from allogeneic and autologous GBM patients mixed with primary irradiated/inactivated whole tumor cells [30]. Combination of ERC1671 with GM-CSF and cyclophosphamide plus bevacizumab resulted in a significant survival benefit with a mOS of 12 months, while placebo plus bevacizumab mOS was 7.5 months. Interim results suggested that the benefit was correlated with the CD4 helper T lymphocytes counts in the peripheral blood [30].
The use of immunotherapy for GBM is therefore not a dead end. According to us, combination strategies targeting different arms of the cancer immunity cycle have great potential to overcome GBM multifactorial immunosuppression and increase antitumor immune response. These strategies are being tested to a large extent in preclinical and clinical studies, giving encouraging results.
Targeting different arms of the GBM immunity cycle to improve immunotherapy efficacy: preclinical aspects
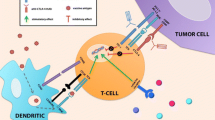
The immune cycle in GBM is a multistep process that can be targeted by treatments at different levels (Fig. 2). Antigens released from dying GBM cells are captured by APCs, processed and displayed on major histocompatibility complex (MHC)-I and -II molecules for presentation to T cells. Effector T cells are primed and activated in response to tumor antigen presentation. Activated T cells kill GBM cells after binding to GBM tumor antigen on MHC-I through the T cell receptor (TCR). However, the immunosuppressive microenvironment can hinder immune control in GBM.

The GBM immunity cycle and associated treatments. The immune response in GBM can be divided into six steps, starting with antigen release from GBM cells and ending with the killing of GBM cells. Potential treatments impacting the immune response steps are written in blue. Step 1 – Antigens are released from dying GBM cells. Step 2 – Tumor antigens are captured by APCs, processed and displayed on MHC-I and -II molecules for presentation to T cells. Step 3 – Effector T cells are primed and activated in response to tumor antigen presentation. Step 4 – Activated T cells traffic through the BBB and infiltrate the tumor site. Step 5 – The immunosuppressive TME must be overcome to allow activated T cells to recognize and bind to GBM cells. Step 6 – Activated T cells kill GBM cells after binding to GBM tumor antigen on MHC-I through the T cell receptor (TCR). The boxes * and ** represent the CTLA-4 and PD-1/PD-L1 pathways. * T cells are activated after the binding of TCR with antigens displayed on MHC and the simultaneous CD28:CD80/86 costimulatory signal. CTLA-4 mediates T cell inhibition by competitively binding to CD80/86. ** T cells are activated after recognizing GBM cells, secreting inflammatory cytokines and inducing GBM cell death. PD-1:PD-L1 binding induces T cell inhibition by reducing T cell proliferation and cytokine production. (Abbreviations: APC = Antigen-presenting cell; Chemo = Chemotherapy; CTLA-4 = Cytotoxic T-lymphocyte antigen 4; CXCR4 = C-X-C chemokine receptor 4; GBM = Glioblastoma; GITR = Glucocorticoid-induced tumor necrosis factor-related protein; IDO = Indoleamine 2,3-dioxygenase; LAG-3 = Lymphocyte activation gene 3 protein; MDSC = Myeloid-derived suppressive cell; MHC = Major histocompatibility complex; PD-1 = Programmed cell death-1; PD-L1 = Programmed death ligand-1; RT = Radiotherapy; TAM = Tumor-associated microglia and macrophage; TCR = T cell receptor; TIGIT = T cell immunoreceptor with Ig and ITIM domains; TIM-3 = T cell immunoglobulin and mucin domain containing-3; VEGF = Vascular endothelial growth factor)
ICBs act by restoring the activity of effector cells so that they can recognize and attack cancer cells again. However, if there is not enough T cell infiltration at the tumor site and/or too many immunosuppressive cells in the TME (e.g., Tregs and TAMs), ICBs may not be enough to promote a strong antitumor response inducing the death of cancer cells. Targeting different arms of the cancer immunity cycle is of great interest, particularly in the context of GBM, in which it could offer better chances to overcome multifactorial immunosuppression [117, 118]. Indeed, it is possible to stimulate the immune system at different times and locations during immune response generation. One possibility to enhance the immune response against GBM (or other cancers) is by stimulating antigen release from dying tumor cells and their presentation to APCs. Immunogenic cell death (ICD) inducers, such as RT and some chemotherapeutics, induce dying cancer cells to release danger signals that stimulate the recruitment of APCs and antigen presentation [117]. Antigen presentation may also be stimulated by using vaccines (e.g., whole-cell tumor vaccines or peptide vaccines) that enhance the recognition of antigens and increase the production of antigen-specific T cells [118]. Another way to improve the immune response against GBM is to decrease the immunosuppression occurring at different stages of the generation of effector T cells. The use of ICBs is helpful to block the inhibition of effector T cells and therefore restore their activity (either early in the course of their activation or in the TME) [119]. Costimulatory agonist mAbs might also be of great interest to potentiate effector T cell function [120] (Fig. 2).
In this chapter, we describe some of the combination strategies that are used in preclinical studies. Most of the described results rely on GL261(−Luc) models, as they are the most common models used nowadays for studying the immunotherapy of GBM. We focus first on combination of chemotherapy with immunotherapy and then on combination of different immunotherapies. ICBs are the most studied immunotherapeutic approach; for this reason, the majority of the combinations presented hereafter includes inhibitory checkpoint molecules. However, we also describe other strategies, including vaccines and agonist costimulatory checkpoints.
Combination of immunotherapy with chemotherapy
The combination of ICBs with chemotherapy, particularly TMZ, is one of the most studied combination therapies for GBM. The combination of chemotherapy and ICBs offers the advantage of enhancing the recognition and elimination of tumor cells (e.g., by increasing tumor antigenicity, inducing ICD and reducing the immunosuppression exerted on effector cells in the TME) [121, 122]. Moreover, as TMZ is part of the SOC, the combination could have potential for inducing a first-line antitumor immune response [13]. However, a standard dose of TMZ causes severe lymphopenia and T cell exhaustion, and anti-PD-1 immunological benefits are almost totally absent. Although the combination of systemic administration of a standard dose of TMZ and anti-PD-1 mAbs has been shown to increase the survival rate in murine GBM, no benefit of immune modulation has been observed, and tumors recur [123,124,125] (Table 2).
One way to enhance ICBs efficacy while using chemotherapy is to modify the dose or the administration route (Table 2). It has in fact been proven that metronomic doses of TMZ do not induce exhaustion of T cells, while standard doses lead to upregulation of exhaustion markers [124]. Moreover, the anti-PD-1 survival benefit was maintained upon the addition of metronomic doses of TMZ, while it was abrogated upon the addition of standard TMZ doses [124]. Comparatively, low doses of TMZ combined with anti-PD-1 therapy also led to survival benefits with an increased number of TILs [126].
Modifying the administration route has also proven to improve the efficacy of combined chemotherapy and immunotherapy, as shown by Mathios and colleagues [125]. In their study, they demonstrated that local administration of chemotherapy, using a wafer impregnated with either TMZ or carmustine (bis-chloroethylnitrosurea (BCNU)), enhanced anti-PD-1 antitumor effects, while systemic chemotherapy abrogated them [125]. One potential explanation for the synergistic effect observed with local chemotherapy is the increased antigen presentation during chemotherapy-induced tumor cell death. This explanation was supported by an immune profile analysis, which showed an increased percentage of DCs in groups treated with local chemotherapy [125].
Similar trends have been observed when TMZ is combined with vaccines in preclinical studies using GBM murine models (Table 2). The preclinical study of Garg and colleagues in 2016 also proved the immune ablative effects of systemic TMZ [127]. The researchers showed a survival benefit after treatment with TMZ combined with an ICD-based DC vaccine. However, the addition of TMZ decreased the levels of infiltrating CD8 T cells [127]. Additionally, a comparison between systemic and intratumoral administration of TMZ in combination with a whole-cell tumor vaccine showed better survival and immune benefits with local administration of chemotherapy [128]. Moreover, while lymphodepletion was observed following systemic administration, there was none after local administration [128]. Finally, the importance of combining immunotherapy with local chemotherapy was again proven in a study using both the GL261 and KR158 glioma models. Therapy with a whole-cell tumor vaccine combined with intratumoral convection-enhanced delivery (CED) of TMZ not only increased the number of long-term survivors and reduced tumor volume but also increased TILs in both models [129].
More recently, studies have also been focusing on immunotherapy targeting innate immune checkpoints given the key role of the innate immune system in the early detection of cancer as well as in the initiation and maintenance of an immune response [131]. CD47 is expressed on the cell surface of solid tumor cells and its ligand, signal regulatory protein alpha (SIRPα), is expressed on macrophages and DCs [132]. CD47 acts like an antiphagocytic signal for phagocytic cells; blockade of CD47 with mAbs induce therefore macrophage phagocytosis of cancer cells [133]. The effects of the innate ICB anti-CD47 in combination with TMZ was evaluated on GL261 and CT-2A mouse models [130]. Combination of TMZ and anti-CD47 mAbs inhibited tumor growth and significantly improved the survival by activating both innate and adaptive immune responses. Indeed, activation of the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway and increased numbers of activated macrophages as well as higher numbers of T cells and IFN-γ-secreting CD8 T cells were observed in mice with GBM [130]. The combination was further improved by adjuvant PD-1 blockade, probably because it helped overcoming adaptive immune resistance [130]. It is worth noting that schedule of TMZ administration was once more of major importance in this study. While concurrent TMZ/anti-CD47 treatment did not induce survival and immune benefits, sequential treatment with metronomic doses of TMZ administered before the concomitant TMZ/anti-CD47 did [130].
Combination of multiple immunotherapies
Combination of different ICBs offers the opportunity to enhance their efficacy and is often studied in many cancers [93]. Indeed, not all immune checkpoints act on effector T cells at the same location or time in the course of their activation (Fig. 2) [134]. Therefore, the use of different ICBs may result in synergistic effects [134, 135]. Moreover, while ICB as a monotherapy induces compensatory upregulation of other immune checkpoints, numerous preclinical studies of different cancers have demonstrated better outcomes and tumor growth decreases with the inhibition of two or more checkpoint receptors [82, 134,135,136]. In preclinical studies of murine GBM, combinations of multiple ICBs or combinations of ICBs with other immunotherapies have been shown to increase the immune response and survival rate (Table 3).
The expression of LAG-3 by CD4 and CD8 T cells was correlated with a significant decrease in their IFN-γ production, corroborating other study results showing that LAG-3 is a marker of T cell exhaustion [137]. LAG-3 is a receptor upregulated on activated NK and T cells, and binding to MHC-II – its main ligand – induces negative regulation of T cells by decreasing proliferation and cytokine production [149,150,151]. The combination of anti-LAG-3 with anti-PD-1 significantly improved long-term survival. The combination was more effective when anti-LAG-3 was given at an early point. However, no difference in the immunological profile was observed when comparing the combination therapy and the other treatment arms [137].
The expression of the checkpoint molecule T cell immunoreceptor with Ig and ITIM domains (TIGIT) was found to be upregulated on CD8 T cells and Tregs in the brains of mice bearing GBM tumors compared to the expression seen in lymph nodes and spleens [138]. TIGIT is a negative checkpoint receptor that is mostly upregulated by NK and T cells, and its ligands are mainly expressed by tumor cells and APCs. TIGIT pathways induce, among other things, negative regulation of T cell-mediated tumor recognition and promotion of NK cell–dependent tumor immunity in different mouse models [152, 153]. Treatment of mice with a combination of anti-TIGIT and anti-PD-1 mAbs significantly improved survival compared to their treatment with monotherapies, with an increase in effector T cell function and downregulation of immunosuppressive cells [138].
The combination of dual ICB therapy with 1-methyltryptophan (1-MT) – an inhibitor of the tryptophan catabolic enzyme indoleamine 2,3-dioxygenase (IDO) – significantly improved the survival of mice bearing intracranial GBM tumors [139]. The inhibition of PD-L1, CTLA-4 and IDO synergistically decreased Treg infiltration. Early blockade of these checkpoints induced an increase in effector CD8 T cell infiltration in the brain and led to 100% long-term survival. Late blockade, however, induced a decrease in TILs and led to cure of 78% of the mice [139].
In addition to inhibitory checkpoint molecules, agonists of costimulatory checkpoint pathways are also promising in the research of new immunotherapy treatments [154]. Treatment of mice with stereotactic radiosurgery (SRS) and a glucocorticoid-induced tumor necrosis factor-related protein (GITR) agonist – which induces stimulation of effector T cells and inhibition of Tregs [155, 156] – induced long-term survival, with an increase in the CD4 T cell/Treg ratio as well as elevated cytokine production by CD4 and CD8 TILs [140]. Combination of costimulatory agonist mAbs with ICB has also shown efficacy in GBM murine models. While using ICB reduces the immunosuppression of effector cells, targeting costimulatory receptors increases effector cell activity, and the combination of both approaches could reinforce the immune response against cancers [135].
The efficacy of combining an anti-PD-1 antibody with an agonist anti-OX40 antibody has been demonstrated, with an increase in the long-term survival rate and the CD8 T cell/Treg ratio in the brain [141]. A triple combination of anti-PD-1 immunotherapy, an anti-OX40 agonist and GVAX – a whole tumor cell vaccine – induced cure of 100% of mice [141]. The synergistic effect was due to complementary actions of the three treatments. Indeed, while GVAX increased the number of activated tumor-specific T cells and infiltrating CD8 T cells, PD-1 blockade further stimulated them, and OX40 induced a vigorous type 1 helper T (Th1) cell response and decreased Treg infiltration [141].
Likewise, neoantigen vaccination has shown synergistic effects when combined with ICB in a CT-2A orthotopic model [142]. Three newly identified neoantigens were used in a polyvalent peptide vaccine and tested in combination with anti-PD-L1 treatment. Following combination therapy, survival was significantly improved compared to that achieved by monotherapies [142].
Other triple therapies targeting different steps of the cancer-immunity cycle have also shown more encouraging results than dual therapies, with a significant improvement in survival and immune profile. The combination of anti-PD-1 mAbs, anti-TIM-3 mAbs and SRS cured 100% of mice. This combination improved the TME immune profile, with an increase in both the CD8 T cell/Treg ratio and the number of IFN-γ-producing CD4 and CD8 TILs [143]. The combination of anti-CTLA-4 mAbs with SRS and an agonist of 4-1BB – a T cell costimulatory checkpoint inducing activation, proliferation and cytokine production [155, 157] – significantly prolonged the survival of mice and increased the number of long-term survivors [144]. The authors demonstrated an increase in TILs and a glioma-specific memory response. The antitumor activity of this triple combination was shown to be CD4 T cell-dependent [144].
Combinations of ICBs with immunotherapies or treatments decreasing GBM TME immunosuppression has also shown synergistic effects. An anti-PD-1 and anti-C-X-C chemokine receptor 4 (CXCR4) combination improved the survival of GBM-bearing mice [145]. CXCR4 overexpression in GBM contributes to treatment resistance through recruitment of immunosuppressive myeloid cells and promotion of tumor aggressiveness [145, 158, 159]. It was demonstrated that targeting myeloid cells with anti-CXCR4 enables anti-PD-1 therapy to induce an antitumor immune response [145].
Modulation of angiogenesis is another way to target the TME. While antiangiogenic therapy and anti-PD-L1 mAbs as monotherapies have both failed in improving the survival of GBM patients, a preclinical study showed that combining both approaches could improve the efficacy of GBM immunotherapy. Anti-VEGF/Ang-2 therapy followed by anti-PD-L1 treatment decreased MDSCs and Tregs in the brain, increased effector CD8 T cell infiltration and improved survival [146].
Targeting the adenosinergic pathway, that has recently been discovered as a major actor in GBM TME immunosuppression, also showed promising preclinical results [160, 161]. Blockade of CD73 – an ectonucleotidase converting ATP to adenosine and involved in chemoresistance and tumor invasion and proliferation [162,163,164] – decreased GBM growth and modulate GBM TME by reducing TAMs and Tregs infiltration [165]. Moreover, silencing CD73 improved survival of mice treated with ICBs [162].
Strategies targeting TAMs in GMB TME in combination with ICBs are also being explored. It was demonstrated that TAMs polarization promoted eradication of GBM tumors following combination of ICBs and immunovirotherapy [147]. G47Δ-mIL12 – an oncolytic herpes simplex virus expressing murine IL-12 – induced M1 polarization of TAMs, plausibly because of IFN-γ expression induced by IL-12. The effect of G47Δ-mIL12 was further increased with the addition of anti-CTLA-4 and anti-PD-1 due to an influx of TAMs. The triple combination also induced an increase in effector CD8 T cells. Altogether, triple combination synergistic effects led to the cure of 89% of 005-GSCs-bearing mice and 50% of CT-2A-bearing mice [147]. What is worthy of note is that combining the oncolytic virus with only one ICB was not sufficient to induce long-lasting effects and overcome GBM immunosuppression [147].
Likewise, it was demonstrated that inhibition of IL-6 reversed TAMs-mediated immunosuppression. However, IL-6 inhibition also induced a reduction in CD40 expression, leading to treatment resistance [148]. CD40 is expressed on APCs (DCs and macrophages) and ligation with CD40L on T cells activates both T cells and APCs, by increasing the latter antigen-presenting and co-stimulatory functions [166, 167]. Anti-CD40 agonist mAbs were able to reprogram the TME and synergized with ICBs in other cancers [168,169,170]. But in GBM preclinical models, agonist CD40 monotherapy did not demonstrate therapeutic improvement [148, 171]. However, combination of IL-6 inhibition and CD40 activation reversed TAMs-mediated GBM immunosuppression and sensitized GBM to anti-PD-1 and anti-CTLA-4 [148].
Limitations of current preclinical models and future outlook to improve combination strategies for the immunotherapy of GBM
Many immunotherapy strategies that have shown successful results in preclinical studies have failed to produce convincing results in clinical trials, revealing limitations and inadequacies of current GBM preclinical models [110, 111, 172]. Translational impact may be improved by developing new relevant preclinical models. In the literature, murine GL261 GBM cell lines are the most commonly used. However, they are highly immunogenic unlike human GBM [172, 173]. Additionally, luciferase-expressing GL261 cells display even more immunogenic features than GL261, with a prolonged median survival time and an elevated inflammatory cytokines production [174]. Transitioning to other models that are less immunogenic and closer to human GBM is highly encouraged [175]. To that purpose, the recently developed SB28 and 005 GSC models are interesting alternatives and are among the best syngeneic models to represent human GBM TME [147, 171, 176,177,178]. Indeed, they are poorly immunogenic, with absence of MHC molecules expression as well as low immune cell infiltration and activation [177,178,179]. These models are moreover resistant to ICBs [147, 178].
An alternative to syngeneic immunocompetent mouse models is the use of genetically engineered mouse models (GEMs). GEMs of GBM reproduce more closely the histology and biology as well as the development of human GBM [172, 180]. Another advantage of these models is that they usually do not require intracranial injections as they are generated through genetic modifications [180]. However, GEMs still do not reflect GBM heterogeneity. Moreover, setting up GEMs requires a lot of expertise and can be expensive [172].
Using patient-derived xenograft models is the best approach to gather human GBM histology and heterogeneity as well as intratumoral heterogeneity [172]. However, these kinds of models generally require immunodeficient mice, which are not suitable for immunotherapy studies [181]. Yet, an immunocompetent mouse model of human GBM was recently developed. By transiently blocking T cell costimulation, the researchers managed to grow human GBM xenograft while keeping intact the mouse immune system [182]. This could be a step forward in the preclinical research for immunotherapy against GBM.
However, the perfect model imitating exactly all characteristics of human GBM (e.g., intratumoral heterogeneity, invasive properties, low immunogenicity, resistance to radio- and chemotherapy) doesn’t exist [172, 175]. It is therefore crucial to design the experiment and select the models according to their unique characteristics. In addition, studies on different models will help to develop a more general therapy against GBM by (i) covering multiple aspects of GBM immunology and (ii) addressing GBM heterogeneity (intratumoral and interpatient) [175].
Nevertheless, to improve immunotherapy for GBM, novel strategies need to be studied. Looking at the development of novel therapeutic targets (e.g., STING, Toll-like receptor (TLR) agonists or old combination therapies applied to new targets) is important to assess the utility of targeting underexplored pathways useful for improving combination immunotherapy [183,184,185,186,187]. In pursuit of this goal, targeting immunosuppressive factors and/or cells within GBM TME is highly valuable [39]. To do so, targeting angiogenesis signaling factors (e.g., VEGF, Ang-2) [146] and adenosinergic pathways components (e.g., adenosine receptors, CD73, CD39) [160, 165, 173, 188] has shown great potential in preclinical studies. Moreover, targeting TAMs are particularly interesting since these cells are the major non-neoplastic cellular components of GBM TME. To this end, many strategies are tested in preclinical studies such as (i) depleting TAMs using CSF-1R inhibitor [189], (ii) activating TAMs by using agonist anti-CD40 mAbs [148, 190], (iii) reprogramming TAMs to induce pro-inflammatory and antitumor immune response by using TLR agonists [186], (iv) promoting TAMs phagocytic activity by using anti-CD47 mAbs [130, 191] and (v) inhibiting TAMs recruitment by targeting the C-C motif chemokine ligand 2/C-C receptor 2 (CCL2/CCR2) axis [192]. CCL2 chemokine production in GBM TME has also been shown to be essential for the recruitment of MDSCs and Tregs [193]. Blockade of CCL2 is therefore a promising approach to overcome GBM TME immunosuppression [194]. Therapies targeting myeloid cells are even more interesting to use in combination with ICBs knowing that they express high levels of PD-L1. This imply furthermore the great potential of targeting both innate and adaptive immunity for GBM treatment.
In addition, development of technologies that improve immunotherapeutic combination against different targets is necessary. As an example, bispecific antibodies targeting two different antigens has shown to be a valuable approach for combination strategies [195]. Antibodies hold a fundamental place in cancer immunotherapy strategies. However, the BBB limits the passage of most of the large molecules such as mAbs [196]. Many physicochemical properties determine the ability of a compound to pass the BBB (e.g., size, lipophilicity, molecular weight, degree of hydrogen bonding) and the delivery of many drugs across the BBB is consequently insufficient [198]. Therefore, improving delivery systems needs to be considered in order to increase delivery at the tumor site. In this regard, novel cyclic peptides modulating the BBB enhanced the brain delivery of mAbs [196]. Similarly, focused ultrasound-mediated BBB disruption improved anti-CD47 mAbs delivery to GBM tumors [199].
To improve local administration, the use of drug delivery systems enhancing brain penetration for intratumoral administration is highly valuable to improve drug distribution and sustained-release. It was demonstrated that densely PEGylated PLGA-based nanoparticles enhanced the penetration of paclitaxel in the brain tissues and therefore improved the treatment efficacy [200, 201]. Similarly, development of novel implantable biomaterials to improve administration in GBM resection cavity and prevent tumor recurrences are encouraged [202, 203]. Among other technologies, a thermoreversible biodegradable chitosan-based hydrogel containing therapeutic T cells showed encouraging results in GBM, offering an interesting platform for local immunotherapy [204]. Furthermore, nanocarriers have to be developed to enhance the local immune response, turning GBM “cold” tumors into “hot” tumors and therefore promoting the infiltration of immune cells. To do so, inducing ICD of GBM cell by using immunostimulant nanocarriers can facilitate the antitumor immune response and improve therapeutic effects [205, 206]. In addition, new administration routes need to be explored. In this regard, intranasal administration is providing promising results [207, 208]. Finally, scalability and clinical translation must be considered when developing such systems.
Conclusions
It was long thought that immunotherapy could not be applied to GBM (e.g., because of the BBB, multifactorial immunosuppressive TME, tumor heterogeneity, etc.). The use of ICBs as immunotherapy during the past decade has revolutionized cancer treatments given their ability to improve patient outcomes. However, ICBs for GBM are still ineffective, as proven by the recent phase III clinical trials that did not reach their primary endpoints when administered in monotherapy. Likewise, vaccines added to the SOC did not improve survival.
In contrast, preclinical studies on combination immunotherapy showed encouraging results. The most successful strategies in terms of survival and immune benefits are the ones that impact the cancer-immunity cycle at different time and/or locations, inducing both stimulation of the immune response and inhibition of immunosuppressive components. Combination of ICBs with chemotherapy, radiotherapy inducing ICD or vaccines have been extensively studied and demonstrated significant improvement over monotherapies. Besides, while immunotherapy for cancer is mainly focusing on factors regulating T cell activation, concomitant targeting of both innate and adaptive immunity holds great promise for GBM treatment. The simultaneous use of immunotherapeutic agents targeting different arms of the immune system is starting to be largely tested in GBM clinical trials, given the success of combination immunotherapy in other cancers and in GBM preclinical studies. There are currently nine ongoing clinical trials (phase I or II) studying the combination of multiple ICBs for GBM treatment [209]. The combination of ICBs with radiotherapy or vaccination is also being tested, with twelve and seven ongoing clinical trials, respectively [209].
We believe that currently ongoing clinical trials and preclinical research on combination strategies will provide key information and better survival without major side effects and that immunotherapies will be added to the SOC for patients facing GBM in the near future.
Availability of data and materials
Not applicable.
Abbreviations
- 1-MT:
-
1-Methyltryptophan
- APC:
-
Antigen-presenting cell
- BBB:
-
Blood−brain barrier
- BCNU:
-
Bis-chloroethyl nitrosurea
- CCL2:
-
C-C motif chemokine ligand 2
- CCR2:
-
C-C receptor 2
- CNS:
-
Central nervous system
- CSC:
-
Cancer stem cell
- CSF:
-
Colony stimulating factor
- CTLA-4:
-
Cytotoxic T-lymphocyte antigen 4
- CXCR4:
-
C-X-C chemokine receptor 4
- FDA:
-
Food and Drug Administration
- GBM:
-
Glioblastoma
- GITR:
-
Glucocorticoid-induced tumor necrosis factor-related protein
- GM:
-
Granulocyte-macrophage
- ICB:
-
Immune checkpoint blockade
- ICD:
-
Immunogenic cell death
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN:
-
Interferon
- LAG-3:
-
Lymphocyte activation gene 3 protein
- mAb:
-
Monoclonal antibody
- MDSC:
-
Myeloid-derived suppressive cell
- MHC:
-
Major histocompatibility complex
- NK:
-
Natural killer
- PD-1:
-
Programmed cell death-1
- PD-L1:
-
Programmed death ligand-1
- RT:
-
Radiotherapy
- SIRPα:
-
Signal regulatory protein alpha
- SOC:
-
Standard of care
- SRS:
-
Stereotactic radiosurgery
- STING:
-
Stimulator of interferon gene
- TAMs:
-
Tumor-associated microglia and macrophages
- TCR:
-
T cell receptor
- Th1:
-
Type 1 helper
- TIGIT:
-
T cell immunoreceptor with Ig and ITIM domains
- TIL:
-
Tumor-infiltrating lymphocyte
- TIM-3:
-
T cell immunoglobulin and mucin domain containing-3
- TLR:
-
Toll-like receptor
- TME:
-
Tumor microenvironment
- TMZ:
-
Temozolomide
- Treg:
-
T regulatory cell
- VEGF:
-
Vascular endothelial growth factor
References
Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol Mech Dis. 2014;9:1–25.
Wesseling P, Capper D. WHO 2016 classification of gliomas. Neuropathol Appl Neurobiol. 2018;44:139–50.
Fan X, Xiong Y, Wang Y. A reignited debate over the cell(s) of origin for glioblastoma and its clinical implications. Front Med. 2019;13:531–9.
Yao M, Li S, Wu X, Diao S, Zhang G, He H, et al. Cellular origin of glioblastoma and its implication in precision therapy. Cell Mol Immunol. 2018;15:737–9.
Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19:764–72.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Stupp R, Brada M, van den Bent MJ, Tonn J-C, Pentheroudakis G. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25:iii93–101.
Fazeny-Dörner B, Wenzel C, Veitl M, Piribauer M, Rössler K, Dieckmann K, et al. Survival and prognostic factors of patients with unresectable glioblastoma multiforme: anticancer. Drugs. 2003;14:305–12.
Lesueur P, Lequesne J, Grellard J-M, Dugué A, Coquan E, Brachet P-E, et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer. 2019;19:198.
Yabroff KR, Harlan L, Zeruto C, Abrams J, Mann B. Patterns of care and survival for patients with glioblastoma multiforme diagnosed during 2006. Neuro Oncol. 2012;14:351–9.
Perrin SL, Samuel MS, Koszyca B, Brown MP, Ebert LM, Oksdath M, et al. Glioblastoma heterogeneity and the tumour microenvironment: implications for preclinical research and development of new treatments. Biochem Soc Trans. 2019;47:625–38.
D’Errico G, Machado HL, Sainz B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Transl Med. 2017;6:3.
Sanders S, Debinski W. Challenges to successful implementation of the immune checkpoint inhibitors for treatment of glioblastoma. Int J Mol Sci. 2020;21:2759.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–5.
Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Abakushina EV, Popova LI, Zamyatnin AA, Werner J, Mikhailovsky NV, Bazhin AV. The advantages and challenges of anticancer dendritic cell vaccines and NK cells in adoptive cell immunotherapy. Vaccines. 2021;9:1363.
Chung H, Jung H, Noh J-Y. Emerging approaches for solid tumor treatment using CAR-T cell therapy. Int J Mol Sci. 2021;22:12126.
Gasser M, Waaga-Gasser AM. Therapeutic antibodies in cancer therapy. In: Böldicke T, editor. Protein targeting compounds. Cham: Springer International Publishing; 2016. p. 95–120.
Rahman MM, McFadden G. Oncolytic viruses: newest frontier for cancer immunotherapy. Cancers. 2021;13:5452.
Cameron F, Whiteside G, Perry C. Ipilimumab: first global approval. Drugs. 2011;71:1093–104.
Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–67.
Kazandjian D, Suzman DL, Blumenthal G, Mushti S, He K, Libeg M, et al. FDA approval summary: nivolumab for the treatment of metastatic non-small cell lung cancer with progression on or after platinum-based chemotherapy. Oncologist. 2016;21:634–42.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.
DeMaria PJ, Bilusic M. Cancer vaccines. Hematol Oncol Clin North Am. 2019;33:199–214.
Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open. 2019;2:e192535.
Heynckes S, Daka K, Franco P, Gaebelein A, Frenking JH, Doria-Medina R, et al. Crosslink between temozolomide and PD-L1 immune-checkpoint inhibition in glioblastoma multiforme. BMC Cancer. 2019;19:117.
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–86.
Bota DA, Chung J, Dandekar M, Carrillo JA, Kong X-T, Fu BD, et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: interim results and correlations with CD4+ T-lymphocyte counts. CNS Oncol. 2018;7:CNS22.
Obermeier B, Verma A, Ransohoff RM. The blood–brain barrier. In: Handbook of clinical neurology: Elsevier; 2016. p. 39–59.
Pardridge WM. CSF, blood-brain barrier, and brain drug delivery. Expert Opin Drug Deliv. 2016;13:963–75.
Serlin Y, Shelef I, Knyazer B, Friedman A. Anatomy and physiology of the blood–brain barrier. Semin Cell Dev Biol. 2015;38:2–6.
Lyon JG, Mokarram N, Saxena T, Carroll SL, Bellamkonda RV. Engineering challenges for brain tumor immunotherapy. Adv Drug Deliv Rev. 2017;114:19–32.
Luo H, Shusta EV. Blood–brain barrier modulation to improve glioma drug delivery. Pharmaceutics. 2020;12:1085.
Mainprize T, Lipsman N, Huang Y, Meng Y, Bethune A, Ironside S, et al. Blood-brain barrier opening in primary brain tumors with non-invasive MR-guided focused ultrasound: a clinical safety and feasibility study. Sci Rep. 2019;9:321.
Karmur BS, Philteos J, Abbasian A, Zacharia BE, Lipsman N, Levin V, et al. Blood-brain barrier disruption in neuro-oncology: strategies, failures, and challenges to overcome. Front Oncol. 2020;10:563840.
Dunn-Pirio AM, Vlahovic G. Immunotherapy approaches in the treatment of malignant brain tumors: immunotherapy for malignant brain tumor. Cancer. 2017;123:734–50.
Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20:12–25.
Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 2015;36:569–77.
Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48–65.
De Vleeschouwer S, Bergers G. Glioblastoma: to target the tumor cell or the microenvironment? In: De Vleeschouwer S, editor. Glioblastoma: Codon Publications; 2017. p. 315–40.
Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol. 2018;14:482–95.
Da Ros M, De Gregorio V, Iorio A, Giunti L, Guidi M, de Martino M, et al. Glioblastoma chemoresistance: the double play by microenvironment and blood-brain barrier. Int J Mol Sci. 2018;19:2879.
Groblewska M, Litman-Zawadzka A, Mroczko B. The role of selected chemokines and their receptors in the development of gliomas. Int J Mol Sci. 2020;21:3704.
Ratnam NM, Gilbert MR, Giles AJ. Immunotherapy in CNS cancers: the role of immune cell trafficking. Neuro Oncol. 2019;21:37–46.
Fanelli GN, Grassini D, Ortenzi V, Pasqualetti F, Montemurro N, Perrini P, et al. Decipher the glioblastoma microenvironment: the first milestone for new groundbreaking therapeutic strategies. Genes. 2021;12:445.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–8.
Wilmotte R, Burkhardt K, Kindler V, Belkouch M-C, Dussex G, de Tribolet N, et al. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. NeuroReport. 2005;16:1081–5.
Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462–7.
Scheffel TB, Grave N, Vargas P, Diz FM, Rockenbach L, Morrone FB. Immunosuppression in gliomas via PD-1/PD-L1 axis and adenosine pathway. Front Oncol. 2021;10:617385.
Litak J, Mazurek M, Grochowski C, Kamieniak P, Roliński J. PD-L1/PD-1 axis in glioblastoma multiforme. Int J Mol Sci. 2019;20:5347.
Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17:1064–75.
Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. 2000;46:957–62.
Sørensen MD, Dahlrot RH, Boldt HB, Hansen S, Kristensen BW. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol Appl Neurobiol. 2018;44:185–206.
Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17:797–805.
Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18:225–42.
Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Dev Brain Res. 1999;117:145–52.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5.
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–80.
Schulz C, Perdiguero EG, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90.
Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14:1227–35.
Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017;77:2266–78.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1(2):e85841.
Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015;10:e0116644.
Prosniak M, Harshyne LA, Andrews DW, Kenyon LC, Bedelbaeva K, Apanasovich TV, et al. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res. 2013;19:3776–86.
Vidyarthi A, Agnihotri T, Khan N, Singh S, Tewari MK, Radotra BD, et al. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol Immunother. 2019;68:1995–2004.
Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18:519–27.
Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol. 2003;54:388–92.
Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19:20–7.
Xu J, Zhang J, Zhang Z, Gao Z, Qi Y, Qiu W, et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis. 2021;12:373.
Andersen RS, Anand A, Harwood DSL, Kristensen BW. Tumor-associated microglia and macrophages in the glioblastoma microenvironment and their implications for therapy. Cancers. 2021;13:4255.
Ugolini A, Tyurin VA, Tyurina YY, Tcyganov EN, Donthireddy L, Kagan VE, et al. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight. 2020;5:e138581.
Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31.
De Leo A, Ugolini A, Veglia F. Myeloid cells in glioblastoma microenvironment. Cells. 2020;10:18.
Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways: T cell suppression by MDSC subpopulations in cancer. Int J Cancer. 2014;134:2853–64.
Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin Cancer Res. 2019;25:3643–57.
Maddison K, Graves MC, Bowden NA, Fay M, Vilain RE, Faulkner S, et al. Low tumour-infiltrating lymphocyte density in primary and recurrent glioblastoma. Oncotarget. 2021;12:2177–87.
Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. 2018;24:4175–86.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
Strepkos D, Markouli M, Klonou A, Piperi C, Papavassiliou AG. Insights in the immunobiology of glioblastoma. J Mol Med. 2020;98:1–10.
Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, et al. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immunother. 2015;64:419–27.
Kmiecik J, Poli A, Brons NHC, Waha A, Eide GE, Enger PØ, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71–83.
Burster T, Gärtner F, Bulach C, Zhanapiya A, Gihring A, Knippschild U. Regulation of MHC I molecules in glioblastoma cells and the sensitizing of NK cells. Pharmaceuticals. 2021;14:236.
Zhang N, Wei L, Ye M, Kang C, You H. Treatment progress of immune checkpoint blockade therapy for glioblastoma. Front Immunol. 2020;11:592612.
Heppner GH, Miller BE. Tumor heterogeneity: biological implications and therapeutic consequences. Cancer Metastasis Rev. 1983;2:5–23.
Bao Z, Wang Y, Wang Q, Fang S, Shan X, Wang J, et al. Intratumor heterogeneity, microenvironment, and mechanisms of drug resistance in glioma recurrence and evolution. Front Med. 2021;15:551–61.
Friedmann-Morvinski D. Glioblastoma heterogeneity and cancer cell plasticity. Crit Rev Oncog. 2014;19:327–36.
Skaga E, Kulesskiy E, Fayzullin A, Sandberg CJ, Potdar S, Kyttälä A, et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer. 2019;19:628.
Zhang Y, Zheng J. Functions of immune checkpoint molecules beyond immune evasion. In: Xu J, editor. Regulation of cancer immune checkpoints. Singapore: Springer Singapore; 2020. p. 201–26.
Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to checkpoint inhibition in cancer immunotherapy. Transl Oncol. 2020;13:100738.
Kim JE, Lim M. The role of checkpoints in the treatment of GBM. J Neuro-Oncol. 2015;123:413–23.
Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65.
Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98–106.
Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with Lambrolizumab (anti–PD-1) in melanoma. N Engl J Med. 2013;369:134–44.
Huang J, Liu F, Liu Z, Tang H, Wu H, Gong Q, et al. Immune checkpoint in glioblastoma: promising and challenging. Front Pharmacol. 2017;8:242.
Hazarika M, Chuk MK, Theoret MR, Mushti S, He K, Weis SL, et al. U.S. FDA approval summary: nivolumab for treatment of unresectable or metastatic melanoma following progression on ipilimumab. Clin Cancer Res. 2017;23:3484–8.
Sheng IY, Ornstein MC. Ipilimumab and nivolumab as first-line treatment of patients with renal cell carcinoma: the evidence to date. Cancer Manag Res. 2020;12:4871–81.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34.
Gourd E. Nivolumab plus ipilimumab in metastatic colorectal cancer. Lancet Oncol. 2018;19:e139.
Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21:360–78.
Guallar-Garrido S, Julián E. Bacillus Calmette-Guérin (BCG) therapy for bladder cancer: an update. ImmunoTargets Ther. 2020;9:1–11.
Sylvester RJ, van der Meijden APM, Lamm DL. Intravesical bacillus Calmette-Guerin reduces the risk of progression in patients with superficial bladder cancer: a meta-analysis of the published results of randomized clinical trials. J Urol. 2002;168:1964–70.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22.
Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–8.
Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol. 2017;18:1–15.
McGranahan T, Therkelsen KE, Ahmad S, Nagpal S. Current state of immunotherapy for treatment of glioblastoma. Curr Treat Options in Oncol. 2019;20:24.
Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6:1003.
Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373–85.
Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16:142.
Medikonda R, Dunn G, Rahman M, Fecci P, Lim M. A review of glioblastoma immunotherapy. J Neuro-Oncol. 2020;151:41–53.
Grossman SA, Ye X, Lesser G, Sloan A, Carraway H, Desideri S, et al. Immunosuppression in patients with high grade gliomas treated with radiation and temozolomide. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17:5473–80.
Cenciarini M, Valentino M, Belia S, Sforna L, Rosa P, Ronchetti S, et al. Dexamethasone in glioblastoma multiforme therapy: mechanisms and controversies. Front Mol Neurosci. 2019;12:65.
Chitadze G, Flüh C, Quabius ES, Freitag-Wolf S, Peters C, Lettau M, et al. In-depth immunophenotyping of patients with glioblastoma multiforme: impact of steroid treatment. OncoImmunology. 2017;6:e1358839.
Ameratunga M, Coleman N, Welsh L, Saran F, Lopez J. CNS cancer immunity cycle and strategies to target this for glioblastoma. Oncotarget. 2018;9:22802–16.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
Pearson JRD, Cuzzubbo S, McArthur S, Durrant LG, Adhikaree J, Tinsley CJ, et al. Immune escape in glioblastoma multiforme and the adaptation of immunotherapies for treatment. Front Immunol. 2020;11:582106.
Mascarelli DE, Rosa RSM, Toscaro JM, Semionatto IF, Ruas LP, Fogagnolo CT, et al. Boosting antitumor response by costimulatory strategies driven to 4-1BB and OX40 T-cell receptors. Front Cell Dev Biol. 2021;9:692982.
Leonetti A, Wever B, Mazzaschi G, Assaraf YG, Rolfo C, Quaini F, et al. Molecular basis and rationale for combining immune checkpoint inhibitors with chemotherapy in non-small cell lung cancer. Drug Resist Updat. 2019;46:100644.
Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. 2018;9:1739.
Park J, Kim CG, Shim J-K, Kim JH, Lee H, Lee JE, et al. Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. OncoImmunology. 2019;8:e1525243.
Karachi A, Yang C, Dastmalchi F, Sayour EJ, Huang J, Azari H, et al. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro Oncol. 2019;21:730–41.
Mathios D, Kim JE, Mangraviti A, Phallen J, Park C-K, Jackson CM, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. 2016;8:370ra180.
Dai B, Qi N, Li J, Zhang G. Temozolomide combined with PD-1 antibody therapy for mouse orthotopic glioma model. Biochem Biophys Res Commun. 2018;501:871–6.
Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell–driven rejection of high-grade glioma. Sci Transl Med. 2016;8:328ra27.
Fritzell S, Sandén E, Eberstål S, Visse E, Darabi A, Siesjö P. Intratumoral temozolomide synergizes with immunotherapy in a T cell-dependent fashion. Cancer Immunol Immunother. 2013;62:1463–74.
Enríquez Pérez J, Kopecky J, Visse E, Darabi A, Siesjö P. Convection-enhanced delivery of temozolomide and whole cell tumor immunizations in GL261 and KR158 experimental mouse gliomas. BMC Cancer. 2020;20:7.
von Roemeling CA, Wang Y, Qie Y, Yuan H, Zhao H, Liu X, et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat Commun. 2020;11:1508.
Rameshbabu S, Labadie BW, Argulian A, Patnaik A. Targeting innate immunity in cancer therapy. Vaccines. 2021;9:138.
Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci. 2012;109:6662–7.
Tseng D, Volkmer J-P, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci. 2013;110:11103–8.
Khair DO, Bax HJ, Mele S, Crescioli S, Pellizzari G, Khiabany A, et al. Combining immune checkpoint inhibitors: established and emerging targets and strategies to improve outcomes in melanoma. Front Immunol. 2019;10:453.
Barbari C, Fontaine T, Parajuli P, Lamichhane N, Jakubski S, Lamichhane P, et al. Immunotherapies and combination strategies for immuno-oncology. Int J Mol Sci. 2020;21:5009.
Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? In: Am. Soc. Clin. Oncol. Educ. Book; 2019. p. 147–64.
Harris-Bookman S, Mathios D, Martin AM, Xia Y, Kim E, Xu H, et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma: LAG-3 expression and therapeutic effect in glioblastoma. Int J Cancer. 2018;143:3201–8.
Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7(8):e1466769.
Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290–301.
Patel MA, Kim JE, Theodros D, Tam A, Velarde E, Kochel CM, et al. Agonist anti-GITR monoclonal antibody and stereotactic radiation induce immune-mediated survival advantage in murine intracranial glioma. J Immunother Cancer. 2016;4:28.
Jahan N, Talat H, Alonso A, Saha D, Curry WT. Triple combination immunotherapy with GVAX, anti-PD-1 monoclonal antibody, and agonist anti-OX40 monoclonal antibody is highly effective against murine intracranial glioma. OncoImmunology. 2019;8:e1577108.
Liu CJ, Schaettler M, Blaha DT, Bowman-Kirigin JA, Kobayashi DK, Livingstone AJ, et al. Treatment of an aggressive orthotopic murine glioblastoma model with combination checkpoint blockade and a multivalent neoantigen vaccine. Neuro Oncol. 2020;22:1276–88.
Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23:124–36.
Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS One. 2014;9:e101764.
Wu A, Maxwell R, Xia Y, Cardarelli P, Oyasu M, Belcaid Z, et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J Neuro Oncol. 2019;143:241–9.
Di Tacchio M, Macas J, Weissenberger J, Sommer K, Bähr O, Steinbach JP, et al. Tumor vessel normalization, immunostimulatory reprogramming, and improved survival in glioblastoma with combined inhibition of PD-1, angiopoietin-2, and VEGF. Cancer Immunol Res. 2019;7:1910–27.
Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32:253–267.e5.
Yang F, He Z, Duan H, Zhang D, Li J, Yang H, et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat Commun. 2021;12:3424.
Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 — potential mechanisms of action. Nat Rev Immunol. 2015;15:45–56.
Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171:1393–405.
Huard B, Tournier M, Hercend T, Triebel F, Faure F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur J Immunol. 1994;24:3216–21.
Chauvin J-M, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. 2020;8:e000957.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19:723–32.
Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11:39.
Donini C, D’Ambrosio L, Grignani G, Aglietta M, Sangiolo D. Next generation immune-checkpoints for cancer therapy. J Thorac Dis. 2018;10:S1581–601.
Ronchetti S, Zollo O, Bruscoli S, Agostini M, Bianchini R, Nocentini G, et al. Frontline: GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol. 2004;34:613–22.
Alderson MR, Smith CA, Tough TW, Davis-Smith T, Armitage RJ, Falk B, et al. Molecular and biological characterization of human 4-1BB and its ligands. Eur J Immunol. 1994;24:2219–27.
Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;24:31–82.
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705.
Jin K, Mao C, Chen L, Wang L, Liu Y, Yuan J. Adenosinergic pathway: a hope in the immunotherapy of glioblastoma. Cancers. 2021;13:229.
Ott M, Tomaszowski K-H, Marisetty A, Kong L-Y, Wei J, Duna M, et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight. 2020;5:e134386.
Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26:39–46.
Yan A, Joachims ML, Thompson LF, Miller AD, Canoll PD, Bynoe MS. CD73 promotes glioblastoma pathogenesis and enhances its chemoresistance via A2B adenosine receptor signaling. J Neurosci. 2019;39:4387–402.
Quezada C, Garrido W, Oyarzún C, Fernández K, Segura R, Melo R, et al. 5′-ectonucleotidase mediates multiple-drug resistance in glioblastoma multiforme cells. J Cell Physiol. 2013;228:602–8.
Azambuja JH, Schuh RS, Michels LR, Iser IC, Beckenkamp LR, Roliano GG, et al. Blockade of CD73 delays glioblastoma growth by modulating the immune environment. Cancer Immunol Immunother. 2020;69:1801–12.
Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393:480–3.
Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. CD40 ligation activates murine macrophages via an IFN-γ-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol. 2005;174:6013–22.
Leblond MM, Tillé L, Nassiri S, Gilfillan CB, Imbratta C, Schmittnaegel M, et al. CD40 agonist restores the antitumor efficacy of anti-PD1 therapy in muscle-invasive bladder cancer in an IFN I/II-mediated manner. Cancer Immunol Res. 2020;8:1180–92.
Ma HS, Poudel B, Torres ER, Sidhom J-W, Robinson TM, Christmas B, et al. A CD40 agonist and PD-1 antagonist antibody reprogram the microenvironment of nonimmunogenic tumors to allow T-cell–mediated anticancer activity. Cancer Immunol Res. 2019;7:428–42.
Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc Natl Acad Sci. 2020;117:8022–31.
Kosaka A, Ohkuri T, Okada H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol Immunother. 2014;63:847–57.
Haddad AF, Young JS, Amara D, Berger MS, Raleigh DR, Aghi MK, et al. Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neuro Oncol Adv. 2021;3:vdab100.
Xu M, Yao Y, Hua W, Wu Z, Zhong P, Mao Y, et al. Mouse glioma immunotherapy mediated by A2B5+ GL261 cell lysate-pulsed dendritic cells. J Neuro-Oncol. 2014;116:497–504.
Sanchez VE, Lynes JP, Walbridge S, Wang X, Edwards NA, Nwankwo AK, et al. GL261 luciferase-expressing cells elicit an anti-tumor immune response: an evaluation of murine glioma models. Sci Rep. 2020;10:11003.
Wouters R, Bevers S, Riva M, De Smet F, Coosemans A. Immunocompetent mouse models in the search for effective immunotherapy in glioblastoma. Cancers. 2020;13:19.
Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110–6.
Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci. 2013;110:12006–11.
Genoud V, Marinari E, Nikolaev SI, Castle JC, Bukur V, Dietrich P-Y, et al. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. OncoImmunology. 2018;7:e1501137.
Khalsa JK, Cheng N, Keegan J, Chaudry A, Driver J, Bi WL, et al. Immune phenotyping of diverse syngeneic murine brain tumors identifies immunologically distinct types. Nat Commun. 2020;11:3912.
Gopinathan A, Tuveson DA. The use of GEM models for experimental cancer therapeutics. Dis Model Mech. 2008;1:83–6.
Kijima N, Kanemura Y. Mouse models of glioblastoma. In: De Vleeschouwer S, editor. Glioblastoma: Codon Publications; 2017. p. 131–9.
Semenkow S, Li S, Kahlert UD, Raabe EH, Xu J, Arnold A, et al. An immunocompetent mouse model of human glioblastoma. Oncotarget. 2017;8:61072–82.
Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, Luke JJ. STING agonists as cancer therapeutics. Cancers. 2021;13:2695.
Nakamura T, Sato T, Endo R, Sasaki S, Takahashi N, Sato Y, et al. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J Immunother Cancer. 2021;9:e002852.
Shi J, Liu C, Luo S, Cao T, Lin B, Zhou M, et al. STING agonist and IDO inhibitor combination therapy inhibits tumor progression in murine models of colorectal cancer. Cell Immunol. 2021;366:104384.
Abarca-Merlin DM, Maldonado-Bernal C, Alvarez-Arellano L. Toll-like receptors as therapeutic targets in central nervous system tumors. Biomed Res Int. 2019;2019:1–9.
Wu D, Zhao Z, Kim J, Razmi A, Wang LL, Kapate N, et al. Gemcitabine and doxorubicin in immunostimulatory monophosphoryl lipid A liposomes for treating breast cancer. Bioeng Transl Med. 2021;6:e10188.
Daniele S, Zappelli E, Natali L, Martini C, Trincavelli ML. Modulation of A1 and A2B adenosine receptor activity: a new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis. 2014;5:e1539.
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–72.
Walker PR, Migliorini D. The CD40/CD40L axis in glioma progression and therapy. Neuro Oncol. 2015;17:1428–30.
Hu J, Xiao Q, Dong M, Guo D, Wu X, Wang B. Glioblastoma immunotherapy targeting the innate immune checkpoint CD47-SIRPα axis. Front Immunol. 2020;11:593219.
Grégoire H, Roncali L, Rousseau A, Chérel M, Delneste Y, Jeannin P, et al. Targeting tumor associated macrophages to overcome conventional treatment resistance in glioblastoma. Front Pharmacol. 2020;11:368.
Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76:5671–82.
Shono K, Yamaguchi I, Mizobuchi Y, Kagusa H, Sumi A, Fujihara T, et al. Downregulation of the CCL2/CCR2 and CXCL10/CXCR3 axes contributes to antitumor effects in a mouse model of malignant glioma. Sci Rep. 2020;10:15286.
Luo H, Hernandez R, Hong H, Graves SA, Yang Y, England CG, et al. Noninvasive brain cancer imaging with a bispecific antibody fragment, generated via click chemistry. Proc Natl Acad Sci. 2015;112:12806–11.
Ulapane, Kopec, Siahaan. Improving in vivo brain delivery of monoclonal antibody using novel cyclic peptides. Pharmaceutics. 2019;11:568.
Stanimirovic D, Kemmerich K, Haqqani AS, Farrington GK. Engineering and pharmacology of blood–brain barrierpermeable bispecific antibodies. Adv Pharm. 2014;71:301–35.
Warren KE. Beyond the blood:brain barrier: the importance of central nervous system (CNS) pharmacokinetics for the treatment of CNS tumors, including diffuse intrinsic pontine glioma. Front Oncol. 2018;8:239.
Sheybani ND, Breza VR, Paul S, McCauley KS, Berr SS, Miller GW, et al. ImmunoPET-informed sequence for focused ultrasound-targeted mCD47 blockade controls glioma. J Control Release. 2021;331:19–29.
Nance EA, Woodworth GF, Sailor KA, Shih T-Y, Xu Q, Swaminathan G, et al. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med. 2012;4:149ra119.
Nance E, Zhang C, Shih T-Y, Xu Q, Schuster BS, Hanes J. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano. 2014;8:10655–64.
Erthal LCS, Gobbo OL, Ruiz-Hernandez E. Biocompatible copolymer formulations to treat glioblastoma multiforme. Acta Biomater. 2021;121:89–102.
Bastiancich C, Malfanti A, Préat V, Rahman R. Rationally designed drug delivery systems for the local treatment of resected glioblastoma. Adv Drug Deliv Rev. 2021;177:113951.
Tsao C-T, Kievit FM, Ravanpay A, Erickson AE, Jensen MC, Ellenbogen RG, et al. Thermoreversible poly(ethylene glycol)-g-chitosan hydrogel as a therapeutic T lymphocyte depot for localized glioblastoma immunotherapy. Biomacromolecules. 2014;15:2656–62.
He C, Ding H, Chen J, Ding Y, Yang R, Hu C, et al. Immunogenic cell death induced by chemoradiotherapy of novel pH-sensitive cargo-loaded polymersomes in glioblastoma. Int J Nanomedicine. 2021;16:7123–35.
Zhang J, Chen C, Li A, Jing W, Sun P, Huang X, et al. Immunostimulant hydrogel for the inhibition of malignant glioma relapse post-resection. Nat Nanotechnol. 2021;16:538–48.
Bruinsmann FA, Richter Vaz G, de Cristo Soares Alves A, Aguirre T, Raffin Pohlmann A, Stanisçuaski Guterres S, et al. Nasal drug delivery of anticancer drugs for the treatment of glioblastoma: preclinical and clinical trials. Molecules. 2019;24:4312.
Wang W, Swenson S, Cho H-Y, Hofman FM, Schönthal AH, Chen TC. Efficient brain targeting and therapeutic intracranial activity of bortezomib through intranasal co-delivery with NEO100 in rodent glioblastoma models. J Neurosurg. 2020;132:959–67.
Chan HY, Choi J, Jackson C, Lim M. Combination immunotherapy strategies for glioblastoma. J Neuro-Oncol. 2021;151:375–91.
Acknowledgments
Not applicable.
Funding
MB is funded by the Télévie (Belgium). This research has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie fellowship (grant agreement no. 887609) and by Fonds National de la Recherche Scientifique – FNRS (grant agreement no. 40000747) (AM). The authors acknowledge the FNRS (Belgium) for financial support (CDR).
Author information
Authors and Affiliations
Contributions
MB wrote the manuscript draft and designed the figures. AM and VP edited the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bausart, M., Préat, V. & Malfanti, A. Immunotherapy for glioblastoma: the promise of combination strategies. J Exp Clin Cancer Res 41, 35 (2022). https://doi.org/10.1186/s13046-022-02251-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02251-2