
Opinion statement
At this time, there are no FDA-approved immune therapies for glioblastoma (GBM) despite many unique therapies currently in clinical trials. GBM is a highly immunosuppressive tumor and there are limitations to a safe immune response in the central nervous system. To date, there have been several failures of phase 3 immune therapy clinical trials in GBM. These trials have targeted single components of an antitumor immune response. Learning from these failures, the future of immunotherapy for GBM appears most hopeful for combination of immune therapies to overcome the profound immunosuppression of this disease. Understanding biomarkers for appropriate patient selection as well as tumor progression are necessary for implementation of immunotherapy for GBM
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is the most common and most aggressive primary malignant brain tumor in adults. Clinical trials in GBM have led to incremental improved median overall survival (mOS) that have improved survival in the general GBM population [1•]. While the most recent positive phase 3 clinical trial of tumor treating fields (TTF) had a mOS of 20.5 months [2••], it is the tail of long-term survivors that continues to provide hope to patients and neuro-oncologists. TTF gained acceptance not just because of a significant improvement in survival, but an increase in 5-year survival to 13% compared to 5% in the standard of care (SOC) arm [2••]. Durable responses to immunotherapy, seen in many cancers, have re-invigorated the study of immunotherapy in GBM.
The brain is not as immuno-privileged as once thought, yet obstacles remain for immunotherapy in treating GBM. Malignant gliomas are one of the most immunosuppressive solid tumors due in part to lymphopenia driven by bone marrow suppression [3]. GBM is also an immunologically quiet tumor, with low tumor mutational burden (TMB), few tumor infiltrating T cells (TILS), and low PD-1/PD-L1 expression, especially when compared to other cancers where immunotherapy has been the most successful [4, 5]. GBM is also a profoundly heterogeneous tumor which facilitates immune evasion [6]. The SOC for newly diagnosed GBM, the combination of radiation therapy (RT) and alkylating chemotherapy, confounds this immunosuppression. Often steroids are necessary for management of peritumoral edema but they decrease the efficacy of immunotherapies [7, 8]. In addition to hurdles for stimulating an effective immune response against GBM, robust immune activation within the intracranial space poses clinical safety risks including complications of cytokine release syndrome and autoimmune encephalitis [9,10,11]. Despite these challenges, long-term responders to immunotherapy have been reported in clinical trials of GBM, but no predictive biomarkers exist at this time [12•].
Immunotherapy in GBM has a long history, including immune stimulation, antibody-mediated immunotherapies, adoptive cellular immunotherapies, and vaccines [13]. At this time, phase 3 clinical trials have not demonstrated efficacy for immunotherapy in GBM and no FDA-approved immunotherapy for GBM exists. Given the challenges of immunotherapy for GBM, a combination approach will likely be required. Additionally, identification of biomarkers for patient selection and disease surveillance are essential. Given the risks of recurrent tumor sampling, there is a search for serum, CSF, and imaging biomarkers for GBM.
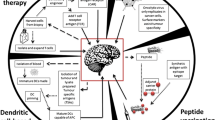
Here, we discuss recent findings and ongoing clinical research into immunotherapy for GBM, including checkpoint inhibitors, vaccines, CAR-T therapy, and viral therapy. We will also discuss our current understanding of biomarkers in GBM that influence candidacy for clinical trials and may explain the response and failures to different immune therapies.
Checkpoints
Checkpoint inhibition has revolutionized treatment of several advanced malignancies providing hope for cancer treatment and resulting in a well-deserved Nobel Prize. Inhibitors of immune checkpoints such as PD-1, PD-L1, and CTLA-4 promote a shift from the normal balance of the adaptive immune system to increased immune activation [14]. In preclinical data, checkpoint inhibitors showed promise for treatment of GBM; however, translation of this preclinical work to patients was complicated by CNS toxicity when used to treat other malignancies [11, 15].
An early phase 1 study of nivolumab (nivo) alone or in combination with ipilimumab (ipi) for treatment of recurrent GBM (rGBM) found higher toxicity with the combination PD-1 and CTLA-4 inhibition, but comparable mOS (12-month mOS 40% v. 30%) [16]. Based on the tolerance of nivo monotherapy and promising mOS compared to historical controls, a large phase 3 trial was developed. CheckMate-143 randomized patients with rGBM to treatment with nivo or bevacizumab (BEV). The genotoxic stress of radiation and chemotherapy used in SOC treatment for newly diagnosed GBM were predicted to increase the TMB in rGBM and favor response to checkpoint inhibition. Unfortunately, at interim analysis of 369 patients, nivo did not demonstrate a mOS benefit over BEV (9.8 m nivo v. 10 m BEV) [17]. It should be noted however that for patients who did respond, those responses were more durable in the nivo arm (11.1 m nivo v 5.3 BEV), again raising enthusiasm for a tail of long term survivors [16]. A single center retrospective of the use of pembrolizumab (pembro) in rGBM also found no benefit [18]. Randomized controlled clinical trials examining the use of nivo in the setting of newly diagnosed GBM (CheckMate 498 and 548) are ongoing. Checkpoint inhibition for GBM was recently reviewed in this journal [19].
Promising data from NSCLC, head and neck cancer, and bladder cancer has stimulated interest in neoadjuvant checkpoint inhibition in GBM [20, 21]. Reported at Society for NeuroOnclogy 2018, patients receiving two doses of pembro prior to re-resection of rGBM had improved mOS (13.7 m neoadjuvant v. 7.5 m adjuvant only) compared to those starting pembro after re-resection [22]. While a small study, this work raises hope that the combination of pembro with surgery may help increase antigen exposure as well as TIL infiltration into rGBM.
Several other phase 1 and 2 clinical trials are currently examining the role of checkpoint inhibitors in combination with other therapies (Table 1). There is great interest in the combination of radiation with checkpoint inhibition, based partly on the idea that radiation may increase antigen presentation as well as promote the abscopal effect. These clinical trials are examining use in both newly diagnosed GBM with standard fractionated chemoradiation, as well as in the rGBM with radiosurgery and hypo-fractioned radiation. There is also a phase 1 study examining use of pembro with MRI-guided laser ablation for rGBM. Addition of pembro to the SOC including TTF is also being studied in the 2-THE-TOP phase 2 clinical trial (NCT03405792). Checkpoint inhibition with combination vaccines and oncolytic viruses will be discussed below.
Preclinical research suggested that low-dose VEGF inhibition promotes the switch in the tumor microenvironment from immunosuppressive (M2-like) to a more immune supportive (M1-like) tumor microenvironment [23]. In melanoma, the combination of CTLA-4 inhibition (ipi) with VEGF inhibition of BEV found disease control rate of 67.4% in 46 patients treated [24]. Aside from the change in the tumor microenvironment, BEV may play another important role in GBM patients as a steroid substitute [25, 26]. A recent retrospective study of non-small cell lung cancer (NSCLC) patients demonstrated a greater than 10% reduction in overall response rate (complete and partial responses) in patients who were on any dose of steroid greater than 10 mg of prednisone a day (= 1.6 mg of dexamethasone) prior to starting treatment with a checkpoint inhibitor. This baseline steroid use was significantly associated with decreased PFS and mOS [8]. This raises concern that necessary management of peritumoral edema in GBM patients, even with the minimal effective dose of dexamethasone needed to control symptoms, may be sufficient to dampen response to checkpoint inhibition and possibly other immunotherapy. In GBM, BEV can be used as a steroid substitute and the safety of the combination of BEV with checkpoint inhibition may provide an opportunity to treat peritumoral edema without the immunosuppressive effects of steroids. For rGBM, there are several ongoing clinical trials examining PD-1 and PD-L1 inhibitors in combination with BEV. Preliminary results from these studies support the safety of this combination; however, among rGBM patients, the combination of pembro with BEV does not improve survival [27].
At this point, there is no role for checkpoint inhibition monotherapy in the treatment of most patients with GBM; however, the combination of checkpoint inhibition with other immune stimulating therapies may be considered. Serious, and even fatal, CNS immune adverse events have been reported with checkpoint inhibition [11]. Given this risk with checkpoint inhibitor monotherapy, as therapies seek to increase immune activation against GBM, there remains concern for complications for over activation of the immune system within the brain.
Checkpoint biomarkers
In parallel with these therapeutic trials, there are several ongoing studies to help better understand biomarkers to predict response to checkpoint inhibition. A number of biomarkers are thought to predict response to PD-1 and PD-L1 inhibitors in other malignancies. Specifically in NSCLC, it has become increasingly clear that response to PD-1 inhibitors correlates with the level of PD-L1 expression in tumor. In Keynote-042, a study of pembro compared to platinum-based chemotherapy in first-line metastatic NSCLC, patients with high expression (> 50%) receiving pembro had a 20.0-month mOS compared to 12.2 months in the chemotherapy group (HR 0.69). In contrast, patients with expression between 1 and 49% receiving pembro had a mOS 13.4 months versus 12.1 months with chemotherapy (HR 0.92) [28]. The CheckMate-057 study of nivo monotherapy versus docetaxel demonstrated no benefit for checkpoint inhibition in tumors with < 1% PD-L1 expression [29]. A study of 94 patients with GBM found median PD-L1 expressional 2.77% and that PD-L1 expression correlated with worse outcome [30] while an earlier study did not find PD-L1 to be a negative prognostic factor [31]. The role of PD-L1 expression on GBM tumor cell in response to checkpoint inhibition is unclear.
To better understand changes in the tumor microenvironment with PD-1 inhibition, pembro was given prior to re-resection in patients with GBM (NCT02337686). Analysis of the resected tumor demonstrated low T cell infiltrate that was not modulated by PD-1 inhibition [32]. Of note, while use of pembro did not improve survival, all patients required steroids after pembro. Studies of neoadjuvant checkpoint inhibition have found a trend toward increased TIL fractions as well as changes in several immune markers [22].
Two tests which reflect the overall genetic stability of tumors, TMB and microsatellite instability (MSI), also play a role in predicting which patients will have meaningful responses to PD-1 axis drugs. In 2017, pembro was approved for patients with MSI or mismatch repair deficiencies for all solid tumors regardless of histology. Higher TMB and MSI correlate with longer mOS [33]. In glioma patients, favorable status across all three of these biomarkers (PD-L1, MSI, TMB) appears to be rare and suggest that only a minority of patients will respond to checkpoint monotherapy [34]. Low frequency of these markers in GBM may be contributing to the disappointing results of PD-1/PD-L1 monotherapies to date [35].
Vaccines
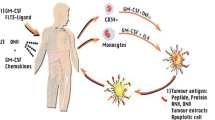
Cancer vaccine therapy in GBM is not preventative, but rather is designed to induce an immune response against the tumor. For GBM, vaccines encompass a range of therapies including direct exposure to antigens (peptide or DNA) in combination with immune-stimulating molecules as well as stimulated patient-derived antigen presenting cells (dendritic cells (DC)). GBM antigen targets are most often tumor-associated antigens given GBM-specific antigens are rare. Some of these antigens are restricted by HLA types, limiting the patient population in which these vaccines may be considered. Use of whole tumor lysate as an antigen was lethal when studied in animal models [36]; however, modifications to GBM tumor lysate, such as heat shock proteins (HSP) and DC vaccines, have been well tolerated with promising early results. Tumor antigen vaccines and customized vaccines will be discussed separately below.
Tumor antigen vaccines
Cancer tumor antigens may be tumor-specific or tumor-associated antigens. At this time, the best studied tumor-specific antigen is a constitutively activated mutation of epidermal growth factor (EGFR), EGFRvIII. The EGFRvIII mutation is reported in 25–30% of GBM and was thought to be an independent negative prognostic factor [37]. Several forms of EGFRvIII vaccines were studied in phase 1 and 2 clinical trials with promising results. This leads to the development of rindopepimut, a conjugated EGFRvIII-specific peptide (also known as CDX-110 and PEPvIII), by Celldex therapeutics. Phase 1 and 2 clinical trials demonstrated promising mOS compared to historical controls leading to an international phase 3, ACT IV, clinical trial in newly diagnosed GBM patients with EGFRvIII mutation. This randomized study of rindopepimut versus control (KLH), added to the SOC adjuvant temozolomide, showed an impressive mOS of 20.1 months in the vaccine arm. However, the control of KLH alone had 20-month mOS, far exceeding the historical controls of 15.2 months. This called into question EGFRvIII as a negative prognostic biomarker. Interestingly, loss of antigen was seen in both the treatment and control arms [38••]. A phase 2 study of rindopepimut in rGBM in combination with BEV does favor treatment with mOS of 12 months in the vaccine group compared to 8.8 months in the KLH group. This finding suggests that combination of vaccine with anti-VEGF therapy may be necessary for a single antigen vaccine to demonstrate survival benefit. At this point, the future of studies of rindopepimut is unclear; however, there are ongoing CAR-T studies targeting this tumor-specific antigen (discussed below).
A number of tumor-associated antigens are being studied in GBM, and while not specific to tumor cells, limited expression elsewhere makes these safe targets for study [39]. Another single antigen vaccine with early promise is SurVaxM, a peptide mimic of survivin conjugated to KLH. While survivin is expressed in the majority of GBM, the vaccine is HLA-restricted, limiting patient inclusion [40]. Phase 2 results of this vaccine added to the SOC adjuvant temozolomide in newly diagnosed GBM were presented at SNO 2018. While immature, mOS in this single arm phase 2 study was a promising 26 months [41]. A randomized phase 2 for new diagnosis GBM of the combination of SurVaxM with PD1 blockade is planned.
There are several other peptide vaccines targeting multiple antigens. Another recently presented vaccine, SL701, consists of short synthetic peptides targeting IL-13Ra2, ephrin A2, and survivin. This is also an HLA-A2-restricted vaccine that has been studied in the first recurrence of GBM, and was found to have a mOS of 12 months in the second phase of this study. Target-specific CD8 response seen in 8/28 patients was associated with longer survival [42].
A six synthetic peptide stimulated DC vaccine with initial promise was ICT-107. In a phase 1 trial, it was found to be safe with a suggestion of benefit to patients who were HLA-A2 positive. STING (NCT 02546102) was a phase 3 clinical trial that opened in 2016, but later suspended in 2017 due to funding. There are many other ongoing studies of vaccines targeting tumor-associated antigens in GBM using peptide, DNA with immune stimulants as well as stimulated DC cells. A review of completed vaccine trials in GBM was recently published this year by Michael Lim [39].
Customized vaccines
For patients who have surgically accessible disease, custom vaccines are a promising area of clinical research. These vaccines require a minimum volume of resectable tumor to generate a custom vaccine, which limits the population of eligible patients.
DC-Vax-L uses whole tumor lysate to pulse patient-derived DCs. Currently over 10 years from diagnosis, some of the patients enrolled in the original phase 1 study of this vaccine are still alive [43]. The phase 3 of DC-Vax-L in newly diagnosed GBM results is still blinded; however, recent reports described a mOS of 23.1 months for all participants (90% of whom received the DC-Vax-L treatment due to crossover design). While the data remains blinded, there are concerns that this may only be interpreted as a single-arm study of 331 patients due to cross over as a result of pseudo-progression and not true progression [44•]. Again promising are reports of durable responders in the phase 3 with survival exceeding 7 years. This vaccine is also being studied in a phase 2 clinical trial in combination with the PD-1 inhibitor, nivo (NCT03014804).
Another promising custom vaccine being studied for new diagnosis GBM is HSPPC-96 (Prophage). In a single-arm phase 2 trial, mOS was 23.8 months; however, when patients were separated by PD-L1 expression on myeloid cells, mOS for those with low expression was an impressive 44.7 months [45]. HSPPC-96 is currently being studied in combination with pembro for newly diagnosed GBM. HSPPC-96 was also studied in 41 patients with rGBM with a mOS of 42.6 weeks [46]. A summary of key vaccine clinical trials in GBM is provided in Table 2.
Vaccine biomarkers
At this time, tumor-associated and tumor-specific antigen vaccines require confirmation that the tumor expresses the targeted antigen. Additionally, many tumor antigens are restricted to specific HLA types (class I restricted cytotoxic T cell or class II restricted helper T cell epitopes), for example HER-2, IL13Ra2, MAGE-1, and survivin. For these tumor-associated antigen vaccines, effective vaccines require tumor antigens that are presented on these restricted HLA alleles to generate an immune response [47]. This narrows the generalizability of these vaccines and clinical trials restrict enrollment to patients with the specific HLA alleles of interest, or stratify results based on HLA type.
Outside of antigen expression and HLA subtyping, there are not prospective biomarkers for response to GBM vaccines. All vaccine clinical trials are looking for correlates to predict immune response and hopefully survival response. Some of those that have been examined include IL-12 production [48], myeloid PD-L1 expression [45], T-reg CTLA-4 expression [49], circulating exomes [50, 51], and antibody production and CD8 response [52].
Additionally, as the DC-Vax-L clinical trial has demonstrated, our current imaging markers have undergone dramatic revolution in the past 10 years limiting the use of MRI for determining progression-free survival as a clinical trial outcome [53•].
CAR-T
CAR-T cell therapy is a newer therapy in oncology, currently approved in B cell lymphoma and leukemia [54]. In brief, CAR-T cells are autologous or allogeneic T cells modified such that the extracellular domains recognizes a unique tumor-associated antigen and the intracellular domain contains a T cell activation signal [55•]. These modified T cells are then administered into the patient, where they can initiate targeted lysis of cells bearing the associated tumor antigen [56]. CAR-T cell therapy has the advantage of bypassing the need for MHC presentation of antigen and development of adaptive immune response as well as bypassing the need for co-stimulatory signals.
Given the success with B cell lymphomas and leukemias, the potential of CAR-T cell therapy for solid tumors, including GBM, has been undergoing investigation [57]. To date, GBM patients treated with CAR-T cell therapy have not had unmanageable CNS effects, a concern given the side effect of elevated intracranial pressure and associated encephalopathy seen in CAR-T cell therapy in B cell lymphoma [58]. The tumor antigens that have been most investigated for CAR targets in GBM to date are IL-13Ra2, EGFRvIII, and Her2.
Interleukin 13 receptor alpha 2 (IL-13Ra2) modulates activation along the rapamycin pathway, and is typically associated with worsened prognosis in GBM [59, 60]. A safety and efficacy trial of CAR T cell therapy in GBM with IL-13Ra2 as the tumor marker was performed on a group of three patients who had a post-resection intracranial infusion, followed by a trial of post-resection direct intratumoral infusion followed intraventricular infusion [61, 62•]. One patient had a dramatic response with clinical and radiographic response that lasted for 7.5 months; however, his disease ultimately did recur [62•]. This CAR-T target is continuing to be studied in intratumoral, intraventricular, and dual delivery systems.
EGFRvIII, as described above in the vaccine section, has also been used a CAR-T cell therapy target. A study regarding tumor infiltration of CAR-T cell performed on ten patients who were given a single peripheral CAR-T cell infusion [63]. Brain specimens in the two patients who underwent post-infusion resection showed increased intratumoral EGFRvIIICAR-T DNA compared to peripheral blood after 2 weeks, and most patients showed decreased expression of EGFRvIII in tumors resected after infusion, suggesting infiltration of the CAR-T cells into the tumor. Patients did not experience tumor regression, although it is notable that this was a particularly poor prognostic group (MGMT negative and most with multifocal disease).
Human epidermal growth factor receptor 2 (HER-2), a tyrosine kinase receptor with high expression in some forms of GBM, has also been used as a potential target [64, 65]. Peripheral infusion of virus-specific (CMV seropositive) HER-2 CAR-T cells demonstrated relative safety of this method as well as persistence of HER-2 CAR-T cells over time (measured for 1 year) [66•]. Seven of the 17 patients did have a period of 8 weeks to 29 months of stable disease and one patient had partial response [66•].
There are currently several active trials investigating the range of intracavitary, intraventricular, and intravenous modes of administration of CAR-T cells for the antigens above as well as EphA2 (NCT02575261). Current studies suggest encouraging results regarding safety and penetrance of CAR-T cells into GBM, although findings regarding effect on tumor growth and recurrence are less conclusive.
No clear biomarkers for treatment response have been established for CAR-T in GBM. In other malignancies, mechanisms of response and resistance have been studied. It is reasonable to consider that similar biomarkers will be found for CAR-T for GBM [67,68,69]. Given at this time, CAR-T cell therapy is targeting single antigens, antigen escape will likely limit effectiveness of CAR-T monotherapy.
Viral therapy
Viral therapy, while initially designed as a mechanism of gene delivery to provide tumor cells with susceptibility to chemotherapy, is now recognized as a form of immunotherapy. Infection of tumor cells with virus attracts the innate immune system leading to cytokine release and tumor cell lysis. This promotes generation of an adaptive immune response to new tumor antigens and potentially development of a long-term immunotherapy effect [70]. While no proven survival benefit has been shown, the excitement about this therapy is largely driven by the population of long-term survivors which was recently reviewed [12•].
Two of these therapies have made it to phase 3 clinical trials, ASPECT and Toca5. The first, ASPECT, studied a replication defective adenovirus, sitimagene ceradenovec, in newly diagnosed GBM [71]. This clinical trial enrolled during early 2000s when the neuro-oncology community was transitioning to the current SOC of adjuvant TMZ. As a result, not all patients within the trial were treated with what is now considered SOC. While there was prolonged time to death or re-intervention in the patients treated with virus, there was no difference in mOS.
The Toca5 clinical trial is comparing Toca-511 (a non-lytic retrovirus expressing cytosine deaminase) to standard therapies for recurrent high grade gliomas. This trial recently completed enrollment; however, phase 3 data is not yet available. If the patients in the phase 1 study were narrowed to patients who would have been eligible for the phase 3, there were 5 patients out of 23 who had durable responses (defined as greater than 24 weeks) and as of August 2017, all of those patients were still alive, one over 4 years [72].
Several other viral therapies have reported GBM patients with durable responses [12•]. These include replication competent HSV1 (G207), parvovirus (ParvOryx01), adenovirus (DNX-2401), and poliovirus (PVSRIPO). Long-term survivors are also reported in another adenovirus that functions as means of gene delivery to allow local delivery of IL-12 [73].
Ongoing study of most of these viruses is now including safety for the combination of viral delivery with checkpoint inhibition.
Conclusion
The success of immunotherapy in GBM faces several obstacles including the highly immunosuppressive nature of GBM and the limitations of the immune response in the central nervous system. Learning from phase 3 clinical trial failures, the future of immunotherapy for GBM appears most hopeful for combination therapies driven by biomarkers for appropriate patient selection. Given the extreme need for improved survival in GBM, current clinical trials are evaluating checkpoint inhibition in combination with novel therapies including vaccines, CAR-T cell therapy, and viral therapy.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Zhu P, Du XL, Lu G, Zhu J-J. Survival benefit of glioblastoma patients after FDA approval of temozolomide concomitant with radiation and bevacizumab: a population-based study. Oncotarget. 2017;8:44015–31 Use of SEER and Texas Cancer Registry databases to confirm population-based increased survival for patients with GBM since advances in care of adjuvant temozolomide.
•• Stupp R, Taillibert S, Kanner A, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. Jama. 2017;318:2306–16 Positive phase 3 clinical trial with use of TTF resulted in increase in overall survival as well as 13% 5-year survival for patients with new diagnosis GBM.
Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24:1459–68.
Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science (New York, NY). 2018;362.
Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. 2018;48:812–30.e14.
Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (New York, NY). 2014;344:1396–401.
Reardon D, Neuberg D, Keskin D, et al. Effect of dexamethasone in glioblastoma (GBM) patients on systemic and intratumoral T-cell responses induced by personalized neoantigen-targeting vaccine. Chicago: ASCO; 2018.
Arbour KC, Mezquita L, Long N, Rizvi H, Auclin E, Ni A, et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non-small-cell lung cancer. J Clin Oncol. 2018;36:2872–8.
Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7:1404–19.
Schneider S, Potthast S, Komminoth P, Schwegler G, Böhm S. PD-1 checkpoint inhibitor associated autoimmune encephalitis. Case Rep Oncol. 2017;10:473–8.
Leitinger M, Varosanec MV, Pikija S, Wass RE, Bandke D, Weis S, et al. Fatal necrotizing encephalopathy after treatment with nivolumab for squamous non-small cell lung cancer: case report and review of the literature. Front Immunol. 2018;9:108.
Chiocca EA, Nassiri F, Wang J, Peruzzi P, Zadeh G. Viral and other therapies for recurrent GBM: is a 24-month durable response unusual? Neuro Oncol. 2018. Meta-analysis examining long-term survivors in prior GBM clinical trials and viral therapy for GBM.
McGranahan T, Li G, Nagpal S. History and current state of immunotherapy in glioma and brain metastasis. Ther Adva Med Oncol. 2017;9:347–68.
Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol. 2015;11:504–14.
Cuzzubbo S, Javeri F, Tissier M, Roumi A, Barlog C, Doridam J, et al. Neurological adverse events associated with immune checkpoint inhibitors: review of the literature. Eur J Cancer. 2017;73:1–8.
Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncology. 2018;20:674–86.
Reardon D, Omuro A, Brandes A, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Pheonix: Society for Neuro Oncology; 2017.
Reiss SN, Yerram P, Modelevsky L, Grommes C. Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J Immunother Cancer. 2017;5:99.
Maxwell R, Jackson CM, Lim M. Clinical trials investigating immune checkpoint blockade in glioblastoma. Curr Treat Options in Oncol. 2017;18:51.
Necchi A, Anichini A, Raggi D, et al. Pembrolizumab as neoadjuvant therapy before radical cystectomy in patients with muscle-invasive urothelial bladder carcinoma (PURE-01): an open-label, single-arm, phase ii study. J Clin Oncol. 2018; 0:JCO.18.01148.
Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–86.
Prins RMA, Orpilla J, Lee A, Davidson T, Gaffey S, Sanders C, et al. Neoadjuvant anti-PD-1 immunotherapy promotes intratumoral and systemic immune responses in recurrent glioblastoma: An Ivy Consortium trial. New Orleans: Society for NeuroOncology; 2018.
Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73:2943–8.
Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2:632–42.
Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, et al. Corticosteroids compromise survival in glioblastoma. Brain J Neurol. 2016;139:1458–71.
Ajlan A, Thomas P, Albakr A, Nagpal S, Recht L. Optimizing bevacizumab dosing in glioblastoma: less is more. J Neuro-Oncol. 2017;135:99–105.
Reardon DA, Groot JFD, Colman H, et al. Safety of pembrolizumab in combination with bevacizumab in recurrent glioblastoma (rGBM). J Clin Oncol. 2016;34:2010.
Lopes G, Wu Y, Kudaba I, et al. Pembrolizumab (pembro) versus platinum-based chemotherapy (chemo) as first-line therapy for advanced/metastatic NSCLC with a PD-L1 tumor proportion score (TPS) ≥ 1%: open-label, phase 3 KEYNOTE-042 study. Chicago: ASCO; 2018.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–39.
Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology. 2016;18:195–205.
Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology. 2015;17:1064–75.
De Groot JF, Penas-Prado M, Mandel JJ, et al. Window-of-opportunity clinical trial of a PD-1 inhibitor in patients with recurrent glioblastoma. Chicago: ASCO; 2017.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–608.
Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro-Oncology. 2017;19:1047–57.
Martinez R, Schackert HK, Appelt H, Plaschke J, Baretton G, Schackert G. Low-level microsatellite instability phenotype in sporadic glioblastoma multiforme. J Cancer Res Clin Oncol. 2005;131:87–93.
Bigner DD, Pitts CM, Wikstrand CJ. Induction of lethal experimental allergic encephalomyelitis in nonhuman primates and guinea pigs with human glioblastoma multiforme tissue. J Neurosurg. 1981;55:32–42.
Weller M, Kaulich K, Hentschel B, Felsberg J, Gramatzki D, Pietsch T, et al. Assessment and prognostic significance of the epidermal growth factor receptor VIII mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy. Int J Cancer. 2014;134:2437–47.
•• Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373–85 Phase III trial of EGFRvIII vaccine, terminated early due to lack of difference in overall survival, despite earlier phase II trial showing a robust anti-EGFRvIII antibody response (suggesting that a humoral response may not be an adequate biomarker of outcome).
Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–42.
Ellis JM, Henson V, Slack R, Ng J, Hartzman RJ, Hurley CK. Frequencies of HLA-A2 Alleles in Five U.S. Population Groups. Predominance of A*02011 and Identification of HLA-A*0231.
Ahluwalia MRD, Abad A, Curry W, Wong E, Peereboom D, Belal A, et al. Fenstermaker R. Phase II trial of a survivin vaccine (SurVaxM) for newly diagnosed glioblastoma. New Orleans: Society for Neuro Oncology; 2018.
Peereboom DNL, Kumthekar P, Badruddoja M, Fink K, Lieberman F, Phuphanich S, et al. Phase 2 trial of SL-701 + bevacizumab in patients with previously treated glioblastoma (GBM) meets primary endpoint of OS-12, with preliminary correlation between long-term survival and target-specific CD8+ T cell immune response. New Orleans: Society for Neuro Oncology; 2018.
Liau LM. Brain tumor immunotherapy: lessons learned and future directions. New Orleans: Society for Neuro Oncology; 2018.
• Liau LM, Ashkan K, Tran DD, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16:142 Unblinded survival data from phase 3 clinical trial of DC-VAX-L tumor lysate vaccine.
Bloch O, Lim M, Sughrue ME, Komotar RJ, Abrahams JM, O'Rourke DM, et al. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD-L1 expression on response to therapy. Clin Cancer Res. 2017;23:3575–84.
Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro-Oncology. 2014;16:274–9.
Zhang JG, Eguchi J, Kruse CA, Gomez GG, Fakhrai H, Schroter S, et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin Cancer Res. 2007;13:566–75.
Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29:330–6.
Fong B, Jin R, Wang X, Safaee M, Lisiero DN, Yang I, et al. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS One. 2012;7:e32614.
Galbo PM Jr, Ciesielski MJ, Figel S, et al. Circulating CD9+/GFAP+/survivin+ exosomes in malignant glioma patients following survivin vaccination. Oncotarget. 2017;8:114722–35.
Manterola L, Guruceaga E, Gallego Perez-Larraya J, et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro-Oncology. 2014;16:520–7.
Schaller TH, Sampson JH. Advances and challenges: dendritic cell vaccination strategies for glioblastoma. Expert Rev Vaccines. 2017;16:27–36.
• Okada H, Weller M, Huang R, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 2015;16:e534–e42 RANO criteria for evaluate response to immunotherapy.
Commissioner Oot. Press announcements - FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma.
Bagley SJ, Desai AS, Linette GP, June CH, O’Rourke DM. CAR T Cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro-Oncology. 2018. An in-depth overview regarding CAR-T cell perceived benefits of CAR T cell therapy for GBM specifically with a comprehensive analysis of published study outcomes as well as discussion of active studies and future considerations.
Chen D, Yang J. Development of novel antigen receptors for CAR T-cell therapy directed toward solid malignancies. Transl Res. 2017;187:11–21.
• Rodriguez A, Brown C, Badie B. Chimeric antigen receptor T-cell therapy for glioblastoma. Transl Res. 2017;187:93–102 A prior review of CAR-T cell therapy in GBM studies which includes a comprehensive and concise discussion of CAR T cell engineering.
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47–62.
Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro-Oncology. 2014;16:1304–12.
Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, et al. Stem-like tumor-initiating cells isolated from IL13Rα2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T cells. Clin Cancer Res. 2012;18:2199–209.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21:4062–72.
• Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375:2561–9 Case report of a patient with multifocal GBM with a dramatic radiographic response to CAR-T cell therapy.
O'Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9.
Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–85.
Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016;108.
• Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3:1094–101 This was a phase I trial of 17 patients who were administered peripheral HER-2 virus specific CAR-T cell therapy that demonstrated evidence for relative safety, persistence of HER-2 CAR-T cells in peripheral blood for up to one year, and suggestion of a period of disease stability.
Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563–71.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–44.
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6.
Peruzzi P, Chiocca EA. Viruses in cancer therapy - from benchwarmers to quarterbacks. Nat Rev Clin Oncol. 2018;15:657–8.
Westphal M, Ylä-Herttuala S, Martin J, Warnke P, Menei P, Eckland D, et al. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14:823–33.
Cloughesy TF, Landolfi J, Vogelbaum MA, Ostertag D, Elder JB, Bloomfield S, et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro-Oncology. 2018;20:1383–92.
Barrett JA, Cai H, Miao J, Khare PD, Gonzalez P, Dalsing-Hernandez J, et al. Regulated intratumoral expression of IL-12 using a RheoSwitch therapeutic system((R)) (RTS((R))) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther. 2018;25:106–16.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Tresa McGranahan declares that she has no conflict of interest. Kate Elizabeth Therkelsen declares that she has no conflict of interest. Sarah Ahmad declares that she has no conflict of interest. Seema Nagpal has received clinical trial funding from Inovio/Regeneron and Tocagen, has received research support from Nektar Therapeutics, and has received compensation from Nektar Therapeutics for service as a consultant.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neuro-oncology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
McGranahan, T., Therkelsen, K.E., Ahmad, S. et al. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options in Oncol. 20, 24 (2019). https://doi.org/10.1007/s11864-019-0619-4
Published:
DOI: https://doi.org/10.1007/s11864-019-0619-4