
Abstract
The role of the microbiota in human health and disease is well established, including its effects on several cancer types. However, the role of microbial dysbiosis in prostate cancer development, progression, and response to treatment is less well understood. This knowledge gap could perhaps be implicated in the lack of better risk stratification and prognostic tools that incorporate risk factors such as bacterial infections and inflammatory signatures. With over a decade’s research investigating associations between microbiome and prostate carcinogenesis, we are ever closer to finding the crucial biological link between the two. Yet, definitive answers remain elusive, calling for continued research into this field. In this review, we outline the three frequently used NGS based analysis methodologies that are used for microbiome profiling, thereby serving as a quick guide for future microbiome research. We next provide a detailed overview of the current knowledge of the role of the human microbiome in prostate cancer development, progression, and treatment response. Finally, we describe proposed mechanisms of host-microbe interactions that could lead to prostate cancer development, progression or treatment response.
Similar content being viewed by others


Background
The human body comprises of trillions of microorganisms with the estimated bacterial population in the order of 1013 cells, resulting in an approximate 1:1 ratio between bacterial and human cells in an average human [1]. It is thus fathomable that the human microbial ecosystem (microbiota) can influence aspects of human health and disease through direct or indirect effects [2], for example by manipulating nutrient uptake and drug metabolism or by inducing systemic inflammatory responses [2,3,4,5]. While the resident microbiota is typically associated with beneficial effects to its host, changes to the microbial composition, known as microbial dysbiosis, could be associated with diseases such as inflammatory bowel disease, diabetes mellitus, and obesity (reviewed by [6]). Since inflammation is a signature of many pre-neoplastic and malignant lesions, chronic inflammation has also been implicated in carcinogenesis likely mediated by bacterial toxins as in the case of Helicobacter pylori and gastric carcinoma [7]. In fact, a growing body of evidence now suggests a crucial role for microbial dysbiosis in cancer development and progression, including significant associations with both bacterial and viral species [8,9,10,11,12].
Prostate cancer (PCa) is the second most frequent male malignancy worldwide with over 358,000 estimated deaths in 2018 [13]. PCa has a highly heterogeneous clinical course. Most organ confined (localized) PCa have an indolent course with a 5 year overall survival of ~100% even without any treatment. In such cases, active surveillance is recommended. In aggressive forms of PCa where the tumor is still organ confined, complete removal of the prostate through radical prostatectomy and/or radiation therapy is necessary to prevent further spread of the disease. However, the decision whether or not to treat localized PCa is a major clinical challenge as currently there are no accurate tests to distinguish between indolent and aggressive PCa at the localized (early) stages. This leads to an over-treatment of indolent cases and an under-treatment of aggressive PCa, resulting in patient morbidity and mortality. Thus, there is an urgent need for better risk stratification tools, that incorporate other risk factors such as bacterial infections or inflammatory markers [14].
The advent of next generation sequencing (NGS) technologies has opened a new area of PCa research enabling unparalleled access to the genomic and transcriptomic underpinnings in PCa. Utilizing the potential for these technologies, several molecular markers have been proposed to stratify PCa [15], although complete success in this regard is yet to be achieved. A major reason could be the interaction between the neoplastic cells and the tumor microenvironment, which remains dynamic. Prostate microbiota could also be hypothesized to be a major driver enabling differential clinical course in localized PCa. In fact, among the well known risk factors for PCa such as age and ethnicity, factors such as microbial composition has also found a potentially essential place in recent years due to the increased scientific scrutiny of its role in mediating inflammation and thereby driving prostate carcinogenesis and progression [16]. While earlier studies relied on culturing bacteria from the prostate, NGS based methods have enabled genotyping the microbial ecosystem within a prostate for hundreds to thousands of patients in parallel, providing a better overview of the landscape of the PCa associated microbiome.
The aim of this review is twofold, 1) provide the researcher with the necessary technical know-how to perform microbiome analysis, and 2) inform the reader of the advances that have been made in the field of prostate cancer microbiome research. We start out by describing in general how to analyse microbiome data and note several automated pipelines that are available to the researcher. For the sake of simplicity, we restricted our review to the three most frequently used NGS based analysis methodologies (amplicon sequencing, shotgun DNA and total RNA sequencing) that have been widely adopted by the microbiome research community. We next provide a detailed overview of the current knowledge of the role of the human microbiome in PCa development, progression and treatment response that was made possible by some of the aforementioned methodologies. Finally, we describe the proposed mechanisms of host-microbe interactions that could lead to PCa development, progression, or treatment resistance. A clear distinction between microbial association with PCa development (carcinogenesis) and its association with PCa progression (e.g. metastatic dissemination) is difficult to make due to the lack of healthy non-cancer control samples in most studies, and consequently this remains an outstanding question in the field.

Methodologies for analyzing the microbiome
Most of the current research enumerating the microbial species present in the prostate and various other body sites utilize NGS based methodologies as opposed to the culture based techniques employed during the last century, which could detect only species that could be cultured. Three main methodologies are most commonly used now (Fig. 1A). These include amplicon sequencing, shotgun DNA sequencing, and RNA sequencing based methodologies. An in depth explanation of the analysis methodologies and best practises for microbiome research is beyond the scope of this review, but we direct readers to other published reviews [17, 18]. A graphical summary of the general steps involved in microbiome sequence analysis is shown in Fig. 1B.

A) Prostate tumor microenvironment is shown harboring bacterial, viral, fungal, and archaeal species. 16s amplicon sequencing is useful for profiling e.g. the bacterial taxa, whereas shotgun metagenome (DNA) and metatranscriptome (total RNA) sequencing enables profiling of the entire tumor associated microbiota. B) A general workflow for analysing microbiome data outlining the major steps involved for the three main NGS based methods discussed in this review. QC, quality control. OTU, operational taxonomic unit. ASV, amplicon sequence variants. Figure 1A is inspired by [19]. Image created partly using Biorender.com
Amplicon sequencing
Perhaps the most straight forward method to profile the prokaryotic taxa is to sequence the DNA encoding for the 16s rRNA gene, which is highly conserved among all prokaryotes. Typically, one or more variable regions (V1-V9) of the 16s DNA is amplified and sequenced. Next, for multiple samples sequenced in parallel, the raw sequencing reads are de-multiplexed (e.g. using sabre: https://github.com/najoshi/sabre) and quality checked (e.g. using FastQC [20]) to remove low quality sequences and adapter/primer sequences (e.g. using Trimmomatic [21]). If using paired-end sequencing data, the read pairs are merged, chimeras removed and either an operational taxonomic unit (OTU) or an amplicon sequence variant (ASV) table is generated, which records the number of times each OTU/ASV was observed. While OTUs have been traditionally used as a unit for clustering similar sequences, finer resolution can be achieved using ASVs, that are accurate down to the level of single-nucleotide differences [22]. Finally, taxonomy is assigned to the OTU/ASVs using reference microbial databases such as the SILVA 16s rRNA gene database [23]. These steps are typically performed within dedicated pipelines, such as DADA2 [24], mothur [25], or qiime2 [26]. The data is now ready for further exploratory analyses as well as statistical testing of species compositional differences.
Phyloseq [27] is a powerful R [28] package that is widely used for microbiome analysis of taxonomy-assigned OTU/ASV count data. Here it is possible to analyze the alpha diversity (e.g. total number of species and their relative proportions within a patient or sample group) and beta diversity (compositional difference between populations) using R packages such as vegan [29]. Additionally, differential abundance can be estimated using R packages such as DESeq2 [30] or a Conda formula such as LEfSe [31].
A limitation to amplicon sequencing is that only a particular gene region is amplified, which can bias diversity estimates depending on which variable region is selected. Further, different organisms can have different 16s gene copy numbers [32] which can bias microbial abundance estimates even though tools such as PICRUSt [33] have been developed in an attempt to correct for such biases [34].
Shotgun DNA and RNA sequencing
Using whole genome sequencing or total RNA sequencing as a way to detect non-host DNA/RNA has become a popular alternative to amplicon sequencing, as it enables species-level identification of organisms and generates a complete genome as well as a transcriptome for all the species, meaning that we can obtain information regarding the functional significance of the microbiome. Further, bacterial, viral, fungal and other archaeal reads can all be obtained using these methodologies, making them ideal for microbiome analyses. RNA sequencing based metatranscriptomic analysis can also shed light into which species are contributing actively to the expression profile of the tissue, whereas DNA sequencing based metagenome analysis captures all species, even though they might not be actively contributing to the tissue phenotype. Often, a combination of these two methodologies is required to understand the underlying tumor biology in the context of microbiome interactions with the tumor microenvironment.
The bioinformatics workflow for metagenome or metatranscriptome analyses are similar to that for amplicon sequence based analyses, however with some key differences. Assuming that one has sequenced the entire microbial DNA/RNA pool from e.g. a human tissue sample, to an acceptable coverage, and has performed the pre-processing steps that are generic to raw sequence read analysis, including ribosomal RNA removal and paired-end read merging, then the first step is to align the reads to the reference human genome, e.g. hg38 [35] using tools such as bwa [36]. Reads that do not map to the reference could be considered to be of non-host origin. These reads can then be mapped to reference genomes in the bacterial, viral, fungal, and archaeal sequence databases (e.g. SILVA [23], NCBI RefSeq [37]) and annotated with tools such as DIAMOND [38]. Finally the annotated reads are aggregated to generate the read/taxa count tables which can subsequently be analysed similar to the amplicon sequence generated OTU/ASV count tables.
Many automated bioinformatics pipelines such as Sunbeam [39], MetaWRAP [40] and SqueezeMETA [41] are available for metagenomic analysis, whereas workflows such as IMP [42], SAMSA2 [43], and MetaTrans [44] have been made for analysing the metatranscriptome. An overview of some of these pipelines, including their capabilities and shortcomings have been reviewed by others previously [45]. The steps described above are generic, and variations to these methodologies do exist but are beyond the scope of the current review.
Using these aforementioned methodologies, several studies have investigated the PCa associated microbiome as described in the following sections.
Prostate cancer and the human microbiome
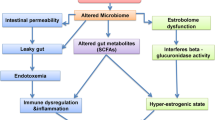
Since 2015 there has been a steady rise in the number of publications looking at the association between the human microbiome and prostate cancer development, progression, and treatment outcome. While most research has focussed on the so called direct effect on PCa of the microbiome in the prostate tissue, others have also investigated associations between PCa and the core microbiota from different body sites, the so called indirect effects [2], as depicted in Fig. 2. These have mainly focussed on the effect of the gastrointestinal microbiota and the urinary microbiome on neoplastic transformation of prostatic epithelia [46, 47], but also include studies evaluating associations between PCa and prostatic and seminal fluid microbiomes [48, 49].

Human microbiome from different body sites have been investigated for its association with prostate cancer (PCa). GI, gastrointestinal. Image created using Biorender.com
Table 1 provides a list of NGS based studies since 2015 investigating microbial dysbiosis associated with PCa. These studies were selected based on a PubMed search for ‘prostate cancer microbiome’ resulting in 159 records since 2015 (as of July 2021). Of these, review articles (n=58) were excluded, and original studies (n=20) profiling the prostate tissue, gut, urinary, seminal fluid and prostatic fluid microbiomes in relation to prostate cancer were selected. A recent study from our group is also discussed in this context.
Prostate microbiome and PCa
With over a decade of research into understanding the role played by the prostatic microbiota in PCa pathophysiology, we are yet to find any causative organisms directly linked to prostate carcinogenesis, despite several studies indicating an association between certain species and the risk of PCa.
We recently investigated the association between the prostate microbiome and PCa using a metatranscriptomic approach based on total RNA sequencing data from 94 PCa patients who underwent curatively intended radical prostatectomy for localized PCa (Salachan et al., 2022; in press). In order to investigate potential dysbiosis associated with PCa, we systematically compared the microbiomes between benign (adjacent normal (AN)) and malignant prostate tissue samples, between less vs. more-aggressive PCa, and between patients who suffered a biochemical disease recurrence compared to those who did not.
We revealed considerable dysbiosis associated with PCa. Notably, species such as Bacteroides fragilis, Saimiriine betaherpesvirus, Staphylococcus saprophyticus, and Vibrio parahaemolyticus had a significantly reduced abundance in the malignant as compared to the benign prostate tissue samples. Similarly, we observed a significant increase in the abundance of Shewanella in the malignant as compared to the benign prostate tissue samples, suggesting a perhaps important biological link between the prostate microbiota and PCa development. Within malignant tissue samples, those that had a higher abundance of Shewanella were associated with dysregulated host immune response, likely mediated by a decrease in enrichment of dendritic cells. We also observed a significant increase in the abundance of Microbacterium species in the T3 tumor samples as compared to the T2 samples, suggesting an association between advanced pathological stage and dysbiosis. While, the lack of true normal comparisons is a limitation to this study, obtaining tissue from healthy individuals is hard and ethically challenging.
Other NGS-based studies have also correlated the abundance of microbial species with known risk factors for PCa. Using large scale whole transcriptome RNA sequencing data obtained from The Cancer Genome Atlas (TCGA) for 242 prostate adenocarcinoma (PRAD) patients from the United States and Germany, a recent study [50] found specific microbes such as Listeria monocytogenes, Methylobacterium radiotolerans JCM 2831, and Xanthomonas albilineans GPE PC73 to be negatively correlated with Gleason score, Tumor-Node-Metastasis (TNM) stage, and prostate-specific antigen (PSA) level, respectively. Microbes such as L. monocytogenes are known to play anti-tumor roles in PCa, for example by stimulating the innate and adaptive immune response [51]. The authors suggest that an over-representation of L. monocytogenes in the tumor as compared to AN samples, indicates a strategy wherein the microbes outcompete the tumor cells in the tumor microenvironment, enable recruitment of immune cells and thereby mitigate tumor growth.
Furthermore, in this study [50], Nevskia ramose was found to have a positive correlation with Gleason score although its significance in PCa is not understood. Staphylococcus aureus was also found to be positively correlated with genomic alterations including amplifications in chromosome 19 and deletions in chromosome 15 and was associated with dysregulated immune-associated genes in this study [50], indicating its pro-tumor roles by inducing inflammatory responses. Strikingly, 234 microbes were significantly associated with elevated levels of PSA [52], the highest number reported from any of the studies included in this review. Such an extent of microbial dysbiosis adds to our knowledge of the crucial role played by microbes in maintaining homeostasis. The lack of true normal comparison is a major limitation to this study [50], preventing us from understanding whether the AN samples truly reflect the normal prostate tissue from men without PCa.
Using both a metagenomic and a metatranscriptomic approach, a study investigating microbial dysbiosis in a Chinese cohort of 65 PCa patients [53] could not differentiate the microbiomes between matched tumor (n=65) and benign (AN) samples (n=65) or between low (n=29) and high (n=36) Gleason score samples. However, both the metagenome and metatranscriptome identified a set of abundant species comprising the core microbiome of the prostate. These included Escherichia, Propionibacterium, Acinetobacter and Pseudomonas. Further investigation of their expression profiles revealed strong correlation between ten Pseudomonas genes and eight host small RNA genes. The authors noted that three of the host small RNA genes may be negatively associated with metastasis as they observed a high expression of these genes in a subset of patients with low rates of metastasis [53]. However, this was not validated in a larger independent cohort.
In another metagenomic study comparing PCa microbiome of patients from different geographic/ethnic origins [54], significant increase in the richness of bacterial species was observed for prostate tumor samples from African men (n=6) when compared to samples from Australian men (n=16), with the former enriched for genera such as Escherichia and Acidovorax. This is perhaps not surprising considering the presence of geographic and ethnic variation in the composition of the human microbiome [55]. The small number of samples is a major challenge to this study [54].
A major stride towards establishing a microbiome signature for PCa was made using an array-based metagenomics and capture sequencing method [56]. The study identified microbial signatures from bacteria, viruses, fungi, and parasites within formalin fixed paraffin embedded prostate tissue samples from American men with PCa. Compared to the benign prostatic hyperplasia control samples (n=15), the malignant samples (n=50) were significantly associated with bacterial phyla such as Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes, fungal phyla such as Ascomycota, and Zygomycota, parasitic phyla such as Nematoda, and Sarcomastogophora, and group I and group IV viruses [56]. Using hierarchical clustering, the authors identified three distinct PCa-specific microbiome signatures that were correlated with disease aggressiveness, suggesting diagnostic and prognostic potential for these signatures [56].
Gastrointestinal microbiome and PCa
Of the indirect effects of the microbiome on prostate carcinogenesis, the association between gut microbiota and PCa has been studied the most. The human gastrointestinal tract harbors the majority of the bacterial population in humans surpassing that of any other bodily site by several orders of magnitude [1]. Coevolution of the gut microbiota enabled symbiotic relationships to exist between humans and the gut microbes, wherein the host provides a conducive environment within the intestine for microbial growth and in turn the microbes aid in digestion [57]. Dysbiosis of the gut microbiota has been implicated in various gastric carcinomas such as colorectal cancer [58], whereas its indirect effect on PCa is less well understood.
In a prospective case-control metagenomic study from 2018, stool samples from 20 Caucasian men with either benign prostatic conditions (n=8) or localized PCa (n=12) were analysed to evaluate their gut microbiome [59]. The study identified biologically significant differences in the composition of the gut microbes between men with PCa compared to men with other benign conditions in the prostate. This included a higher relative abundance of Bacteriodes massiliensis as well as decreased relative abundances of Faecalibacterium prausnitzii and Eubacterium rectalie in the stool from men with PCa compared to the controls. A major limitation to this study [59] is the small sample size and the lack of validation in an independent cohort.
Similarly, enrichment of Bacteroides and Streptococcus species in rectal swabs from patients with PCa (n=64) compared to non-cancer controls (n=41) were found in another study utilizing 16s rRNA amplicon sequencing [47]. The cohort consisted of a mix of races including White, African-American and Latino. This study further generated a microbiome score based on the microbial metabolic profiles, which held predictive potential for PCa risk, indicating the usefulness of the fecal microbial data as a minimally invasive diagnostic tool. However, a recent study [60] utilizing 16s rRNA amplicon sequencing was unable to separate fecal samples obtained from White, non-Hispanic men with benign (n=16) vs. malignant biopsy (n=14) based on their microbial profiles. Nevertheless, a higher abundance of Bacteroides species was observed in patients with PCa compared to control men without PCa in this latter study [60], corroborating the findings from the other studies [47, 59].
While many studies have focused on the role of microbes in PCa development and progression, few have also investigated the changes to the microbiota following treatment against PCa. Using 16s rDNA amplicon sequencing, one such study [61] evaluated the fecal microbiota from 30 PCa patients undergoing gonadotropin releasing hormone agonist/antagonist (GNRH, n=5) or androgen receptor axis-targeted therapy (ATT, n=9) or no treatment (n=16). The study reported altered GI microbiota in men undergoing oral ATT, which could perhaps influence the clinical response to ATT. Specifically, Akkermansia muciniphila and Ruminococcaceae spp. were over-abundant in the fecal specimens of patients undergoing oral ATT compared to the other groups [61].
Similar findings were also observed in two other recent studies employing 16s rRNA amplicon sequencing methodology [62, 63]. Using a Canadian cohort of 68 castration resistant PCa (CRPC) patients undergoing androgen deprivation therapy (ADT) alone (n=21), ADT along with oral abiraterone acetate (AA, n=14) or no treatment controls (n=33), the first study demonstrated depletion of Corynebacterium spp. in patients undergoing ADT, and an enrichment of Akkermansia muciniphila in patients taking oral AA, both compared to patients not receiving any form of treatment [62]. The second study [63] compared the fecal microbiome before (n=21) and after ADT (n=21) in a castration resistant setting using a Chinese cohort of 21 CRPC patients. The authors observed a significant increase in the abundance of Phascolarctobacterium and Ruminococcus as well as for several other bacterial species in patients who received ADT. Whether these insights could be exploited to enhance patient response to ADT needs to be investigated further.
Urinary microbiome and PCa
Few studies have investigated the association between the urinary microbiota and PCa. Urine samples are readily available, non-invasive, and have a higher patient compliance. Associations between the urinary microbiota and PCa can potentially serve as a biomarker that can be incorporated into pre-biopsy models to better predict PCa risk [60].
One of the earliest studies evaluating the urinary microbiome and PCa using 16s rDNA PCR-denaturing gradient gel electrophoresis (DGGE) found significantly decreased abundance of E. coli and Enterococcus in the urine from PCa patients (n=13) compared to men (n=21) with benign prostatic hyperplasia (BPH) [49] in a Chinese cohort of men. In another recent study of 30 White, non-Hispanic men [60], species such as Clostridium XVIII & IV, lachnospira, Acetanaerobacterium, and Faecalibacterium were found to be in significantly higher abundance in the urine from patients with PCa (n=14) compared to benign controls (n=16) as identified using 16s rDNA amplicon sequencing [60]. A prior study analysed urine samples from 129 American men using 16s rDNA sequencing and identified Propionibacterium lymphophilum to have significantly higher abundance in PCa patients (n=61) vs. control men (n=63) who did not have PCa [46]. However, a major limitation to all these studies [46, 49, 60] is the lack of independent validation and the lack of consensus between studies. More streamlined research in this field is required before definitive conclusions can be made about any association (or lack thereof) between the urinary microbiota and PCa.
Prostatic and seminal fluid microbiome and PCa
While it is interesting to profile the prostatic and seminal fluid microbiomes for associations with PCa due to their close proximity to the prostate gland, controlling for contamination from the urinary tract in these samples is often difficult. Few studies have evaluated the prostatic and seminal fluid microbiomes in PCa [48, 49, 64]. Using PCR-DGGE, one study [49] found a significantly increased abundance of Bacteroidetes, Alphaproteobacteria, Firmicutes, Lachnospiraceae, Propionicimonas, Sphingomonas, and Ochrobactrum, and a decreased abundance of Eubacterium and Defluviicoccus in the expressed prostatic secretions (EPS) from Chinese PCa patients (n=13) compared to Chinese men with BPH (n=21). In this study, qPCR detection of E.coli and Enterococcus revealed that E. coli was present in significantly higher number in the EPS and seminal fluid, whereas Enterococcus was present in significantly higher number in the seminal fluid of men with PCa compared to men with BPH [49].
Using 16s rDNA amplicon sequencing, another study found increased proportions of Lactococcus, Carnobacterium, Streptococcus, Geobacillus, and Enterobacter, and decreased proportions of Cronobacter, Alkaliphilus, and Paenibacillus in prostatic fluid samples from patients with PCa (n=32) compared to men without PCa (n=27) in a Chinese cohort [48], whereas Propionibacterium acnes genes were detected in the non-sperm fraction of the seminal fluid from Australian men with PCa (pooled sample from 6 men) but not in men without PCa (pooled sample from 6 men) using small RNA sequencing in a different study [64]. Most of these studies [49, 64] are limited by the small sample sizes and the use of pooled samples. Nevertheless, some interesting observations were made, necessitating further research into this area.
Functional role of specific microbes in PCa
A number of studies have investigated the possible functional role of specific microbes in relation to PCa due to their inflammatory potential and frequent detection in prostatic tissue (Table 2). Cutibacterium acnes (formerly, Propionibacterium acnes) is a skin-associated commensal that has been detected in the prostate of men with PCa in several studies [53, 65] with few studies reporting a higher prevalence in patients with PCa compared to men without [66, 67]. C. acnes has also been associated with chronic inflammation in the prostate of men with PCa [68] and shown to induce acute and chronic inflammation in mice inoculated with human prostatectomy-derived C. acnes isolates [69]. Evidence from cell-based experiments suggests that C. acnes can induce cell proliferation [66] and the secretion of cytokines and chemokines such as IL-6 and IL-8 [70, 71], which are crucial for maintaining active inflammation. However, a later study failed to observe any statistical difference in IL-6 secretion between men with vs. without C. acnes infection [72]. In another study, prostatic epithelial cell lines infected with C. acnes responded via activation of transcription factors such as NF-κB and STAT3 [70], which are associated with cellular proliferation and tumor growth in various cancers, such as PCa and colon cancer [73, 74]. Others have also provided evidence for C. acnes-induced production of reactive oxygen species by keratinocytes in the skin, thereby inducing oxidative stress response and inflammation [75], an event that could perhaps be replicated in the prostate leading to pre-cancerous transformation of the prostatic epithelia [70]. Taken together, all of this evidence points to a possible role for C.acnes-induced inflammation in PCa development or progression.
Many studies have also investigated the role of E. coli in prostate carcinogenesis. Uro-pathogenic strains of E. coli are known to induce prostate tissue damage in rat models of prostatitis [81], mediated by cytotoxic necrotizing factor 1 (CNF1), a virulence factor that has also been shown to promote PCa progression [85]. In a mouse model [52], all mice experimentally infected with E. coli for 12 weeks developed chronic inflammation in the prostate, and with prolonged infection showed cytological changes typical for prostatic intraepithelial neoplasia and high-grade dysplasia. Increased epithelial cell proliferation, and oxidative DNA damage was observed in the prostate glands exhibiting dysplasia, together with decreased androgen receptor and PTEN gene expression, as compared to the control glands [52]. This could indicate a mechanistic link between E. coli-induced inflammation and the onset of PCa or pre-neoplastic lesions.
Others such as Faecalibacterium prausnitzii have also been associated with PCa, with higher fecal abundances of F. prausnitzii observed in benign as compared to malignant patient samples [59]. F. prausnitzii is generally considered to have anti-inflammatory properties with its ability to produce butyrate and induce secretion of anti-inflammatory cytokines such as IL-10, TGF-β2 and IL-1Ra (reviewed by [83]). Furthermore, it has been reported that F. prausnitzii also down-regulated the expression of pro-inflammatory cytokines such as TNF-α, TNF-β and IL-6 in lung cancer cell line [82] and could inhibit the phosphorylation of JAK2/STAT3 in breast cancer cells, potentially leading to growth inhibition of cancer cells [86].
Alterations to host metabolism and immunity mediated by the microbiota
While mechanistic studies investigating microbial regulation of host metabolism in men with PCa are limited, a recent study [84] using PCa mouse models demonstrated that certain species of the intestinal microbiota can modulate the host hormone (e.g. androgen) metabolism and in turn promote cancer growth. Conversely, the study also found that circulating host androgens can alter the composition of the gut microbiota [84]. Notably, the same study showed that Ruminococcus gnavus and Bacteroides acidifaciens were enriched in the fecal microbiota of castrated (as compared to non-castrated) male mice, and that these species can metabolise androgen precursors, pregnenolone and hydroxypregnenolone, into downstream metabolites of the androgen biosynthesis pathway, dehydroepiandrosterone and testosterone. As prostate tumors are reliant on androgens for continued growth, such an alternate source of androgen could lead to endocrine resistance in PCa patients undergoing castration treatment as shown in PCa mouse models [84].
Disentangling host immune responses against tumor cells from those induced by microorganisms is often difficult. We have recently shown enrichment of several immune cell types within malignant prostate tissue samples having low vs. high abundances of V. parahaemolyticus indicating altered host immunity associated with the presence of V. parahaemolyticus (Salachan et al., 2022; in press). Moreover, malignant prostate tissue samples having high (vs. low) abundance of Shewanella showed decreased enrichment of dendritic cells and down-regulation of several toll-like receptors crucial for an active immune system (Salachan et al., 2022; in press), perhaps enabling tumor-immune evasion. Similarly, another recent study [50] found strong correlation between microbe (e.g. Delftia acidovorans, Gardnerella vaginalis) abundance in PCa tissue and regulatory T-cells, as well as with down-regulated immune-associated genes including LPCAT2, TL3, and TGFB2, indicating an immuno-suppressive tumor environment associated with the PCa microbiota.
Future directions
Future research should focus on mapping host microbial species to their functional profiles and delineating specific mechanisms by which microbes enable cancer development and progression or affect treatment response. A better understanding of host-microbiome cross-talk and the associated molecular mechanisms could pave the way for development of novel prevention and/or treatment strategies. Thus, future studies should investigate the clinical utility of targeting the microbiome as a novel mode of anti-cancer treatment. As an example, the genera Ruminococcus could be a top candidate for further investigations of its possible driver role in development of treatment resistant CRPC. Conceivably, future prospective clinical trials could test if a novel treatment strategy aimed at eliminating Ruminococcus sp. from the gut of men with PCa, is able to prevent/delay treatment resistance and hence improve survival. Future research should also combine metagenomic or metatranscriptomic investigations with metabolomics to provide a more complete overview of host-microbe interactions within the context of PCa. Research should also focus on delineating novel blood and/or urine based microbial signatures that could be of diagnostic, prognostic and/or predictive potential.
Conclusions
Through this review we aimed to explore the extent of microbial dysbiosis that is associated with PCa by providing an overview of the current knowledge in the field. Moving away from the notion of cancer as solely being a disease of the genome, we believe a more holistic approach towards cancer treatment, informed by genetic, epigenetic, and host-microbiome interactions could benefit treatment decisions in the future. PCa, like other cancers, is a dynamic and heterogeneous disease that has several layers of molecular and cellular complexity associated with it. Microbiome analyses have revealed a bacteria rich environment in the prostate that might be altered during disease onset, progression or treatment. Several species of the microbial community have been associated with PCa aggressiveness and response to therapy, a finding that has also been observed in many other cancer types. A better understanding of the role microbes play in these processes will help us develop novel treatment strategies as well as better risk stratification tools. For instance, it is possible that removal of certain gut microbial species prior to androgen deprivation/hormonal therapy could delay disease progression to CRPC. Several clinical trials are underway looking at e.g. the efficacy of fecal microbiome transplant in combination with established treatment strategies as a means to control tumor progression in different cancers, including in PCa. If successful, these could give us an upper hand in the battle against cancer.
Availability of data and materials
Not applicable
Abbreviations
- ADT:
-
Androgen deprivation therapy
- AN:
-
Adjacent normal
- ASV:
-
Amplicon sequence variants
- BPH:
-
Benign prostatic hyperplasia
- CNF1:
-
Cytotoxic necrotizing factor 1
- CRPC:
-
Castration resistant prostate cancer
- DGGE:
-
Denaturing gradient gel electrophoresis
- EPS:
-
Expressed prostatic secretions
- GI:
-
Gastrointestinal
- NGS:
-
Next generation sequencing
- OTU:
-
Operational taxonomic unit
- PCa:
-
Prostate cancer
- PRAD:
-
Prostate adenocarcinoma
- PSA:
-
Prostate-specific antigen
- QC:
-
Quality control
- TCGA:
-
The Cancer Genome Atlas
- TNM:
-
Tumor-Node-Metastasis
References
Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016;14(8):e1002533.
Xavier JB, Young VB, Skufca J, Ginty F, Testerman T, Pearson AT, et al. The Cancer Microbiome: Distinguishing Direct and Indirect Effects Requires a Systemic View. Trends Cancer. 2020;6(3):192–204.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31.
Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. BMJ. 2018;360:j5145.
Bjorkholm B, Bok CM, Lundin A, Rafter J, Hibberd ML, Pettersson S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One. 2009;4(9):e6958.
DeGruttola AK, Low D, Mizoguchi A, Mizoguchi E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm Bowel Dis. 2016;22(5):1137–50.
Francescone R, Hou V, Grivennikov SI. Microbiome, inflammation, and cancer. Cancer J. 2014;20(3):181–9.
Hieken TJ, Chen J, Hoskin TL, Walther-Antonio M, Johnson S, Ramaker S, et al. The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci Rep. 2016;6:30751.
Thomas AM, Jesus EC, Lopes A, Aguiar S Jr, Begnami MD, Rocha RM, et al. Tissue-Associated Bacterial Alterations in Rectal Carcinoma Patients Revealed by 16S rRNA Community Profiling. Front Cell Infect Microbiol. 2016;6:179.
Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2017;66(4):633–43.
Liu F, Li J, Guan Y, Lou Y, Chen H, Xu M, et al. Dysbiosis of the Gut Microbiome is associated with Tumor Biomarkers in Lung Cancer. Int J Biol Sci. 2019;15(11):2381–92.
Nené NR, Reisel D, Leimbach A, Franchi D, Jones A, Evans I, et al. Association between the cervicovaginal microbiome, BRCA1 mutation status, and risk of ovarian cancer: a case-control study. The Lancet Oncology. 2019;20(8):1171–82.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Sha S, Ni L, Stefil M, Dixon M, Mouraviev V. The human gastrointestinal microbiota and prostate cancer development and treatment. Investig Clin Urol. 2020;61(Suppl 1):S43–50.
Zhao SG, Chang SL, Erho N, Yu M, Lehrer J, Alshalalfa M, et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017;3(12):1663–72.
de Bono JS, Guo C, Gurel B, De Marzo AM, Sfanos KS, Mani RS, et al. Prostate carcinogenesis: inflammatory storms. Nat Rev Cancer. 2020;20(8):455–69.
Di Bella JM, Bao Y, Gloor GB, Burton JP, Reid G. High throughput sequencing methods and analysis for microbiome research. J Microbiol Methods. 2013;95(3):401–14.
Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform. 2021;22(1):178–93.
Zhang X, Li L, Butcher J, Stintzi A, Figeys D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome. 2019;7(1):154.
Andrew S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11(12):2639–43.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue):D590–6.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–7.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2017.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: Community Ecology Package. R package version 2.5-6. 2019.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60.
Klappenbach JA, Saxman PR, Cole JR, Schmidt TM. rrndb: the Ribosomal RNA Operon Copy Number Database. Nucleic Acids Research. 2001;29(1):181–4.
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–21.
Louca S, Doebeli M, Parfrey LW. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome. 2018;6(1):41.
Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017;27(5):849–64.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95.
Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42(Database issue):D553–9.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60.
Clarke EL, Taylor LJ, Zhao C, Connell A, Lee JJ, Fett B, et al. Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome. 2019;7(1):46.
Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6(1):158.
Tamames J, Puente-Sanchez F. SqueezeMeta, A Highly Portable, Fully Automatic Metagenomic Analysis Pipeline. Front Microbiol. 2018;9:3349.
Narayanasamy S, Jarosz Y, Muller EE, Heintz-Buschart A, Herold M, Kaysen A, et al. IMP: a pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol. 2016;17(1):260.
Westreich ST, Treiber ML, Mills DA, Korf I, Lemay DG. SAMSA2: a standalone metatranscriptome analysis pipeline. BMC Bioinformatics. 2018;19(1):175.
Martinez X, Pozuelo M, Pascal V, Campos D, Gut I, Gut M, et al. MetaTrans: an open-source pipeline for metatranscriptomics. Sci Rep. 2016;6:26447.
Shakya M, Lo CC, Chain PSG. Advances and Challenges in Metatranscriptomic Analysis. Front Genet. 2019;10:904.
Shrestha E, White JR, Yu SH, Kulac I, Ertunc O, De Marzo AM, et al. Profiling the Urinary Microbiome in Men with Positive versus Negative Biopsies for Prostate Cancer. J Urol. 2018;199(1):161–71.
Liss MA, White JR, Goros M, Gelfond J, Leach R, Johnson-Pais T, et al. Metabolic Biosynthesis Pathways Identified from Fecal Microbiome Associated with Prostate Cancer. Eur Urol. 2018;74(5):575–82.
Ma X, Chi C, Fan L, Dong B, Shao X, Xie S, et al. The Microbiome of Prostate Fluid Is Associated With Prostate Cancer. Front Microbiol. 2019;10:1664.
Yu H, Meng H, Zhou F, Ni X, Shen S, Das UN. Urinary microbiota in patients with prostate cancer and benign prostatic hyperplasia. Arch Med Sci. 2015;11(2):385–94.
Ma J, Gnanasekar A, Lee A, Li WT, Haas M, Wang-Rodriguez J, et al. Influence of Intratumor Microbiome on Clinical Outcome and Immune Processes in Prostate Cancer. Cancers (Basel). 2020;12(9).
Wood LM, Guirnalda PD, Seavey MM, Paterson Y. Cancer immunotherapy using Listeria monocytogenes and listerial virulence factors. Immunol Res. 2008;42(1-3):233–45.
Elkahwaji JE, Hauke RJ, Brawner CM. Chronic bacterial inflammation induces prostatic intraepithelial neoplasia in mouse prostate. Br J Cancer. 2009;101(10):1740–8.
Feng Y, Ramnarine VR, Bell R, Volik S, Davicioni E, Hayes VM, et al. Metagenomic and metatranscriptomic analysis of human prostate microbiota from patients with prostate cancer. BMC Genomics. 2019;20(1):146.
Feng Y, Jaratlerdsiri W, Patrick SM, Lyons RJ, Haynes AM, Collins CC, et al. Metagenomic analysis reveals a rich bacterial content in high-risk prostate tumors from African men. Prostate. 2019;79(15):1731–8.
Gupta VK, Paul S, Dutta C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front Microbiol. 2017;8:1162.
Banerjee S, Alwine JC, Wei Z, Tian T, Shih N, Sperling C, et al. Microbiome signatures in prostate cancer. Carcinogenesis. 2019;40(6):749–64.
Zitvogel L, Galluzzi L, Viaud S, Vetizou M, Daillere R, Merad M, et al. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2015;7(271):271ps1.
Mima K, Ogino S, Nakagawa S, Sawayama H, Kinoshita K, Krashima R, et al. The role of intestinal bacteria in the development and progression of gastrointestinal tract neoplasms. Surg Oncol. 2017;26(4):368–76.
Golombos DM, Ayangbesan A, O'Malley P, Lewicki P, Barlow L, Barbieri CE, et al. The Role of Gut Microbiome in the Pathogenesis of Prostate Cancer: A Prospective. Pilot Study. Urology. 2018;111:122–8.
Alanee S, El-Zawahry A, Dynda D, Dabaja A, McVary K, Karr M, et al. A prospective study to examine the association of the urinary and fecal microbiota with prostate cancer diagnosis after transrectal biopsy of the prostate using 16sRNA gene analysis. Prostate. 2019;79(1):81–7.
Sfanos KS, Markowski MC, Peiffer LB, Ernst SE, White JR, Pienta KJ, et al. Compositional differences in gastrointestinal microbiota in prostate cancer patients treated with androgen axis-targeted therapies. Prostate Cancer and Prostatic Diseases. 2018;21(4):539–48.
Daisley BA, Chanyi RM, Abdur-Rashid K, Al KF, Gibbons S, Chmiel JA, et al. Abiraterone acetate preferentially enriches for the gut commensal Akkermansia muciniphila in castrate-resistant prostate cancer patients. Nat Commun. 2020;11(1):4822.
Liu Y, Jiang H. Compositional differences of gut microbiome in matched hormone-sensitive and castration-resistant prostate cancer. Transl Androl Urol. 2020;9(5):1937–44.
Chen Y, Wei J. Identification of Pathogen Signatures in Prostate Cancer Using RNA-seq. PLoS One. 2015;10(6):e0128955.
Yow MA, Tabrizi SN, Severi G, Bolton DM, Pedersen J, Australian Prostate Cancer B, et al. Characterisation of microbial communities within aggressive prostate cancer tissues. Infect Agent Cancer. 2017;12:4.
Davidsson S, Molling P, Rider JR, Unemo M, Karlsson MG, Carlsson J, et al. Frequency and typing of Propionibacterium acnes in prostate tissue obtained from men with and without prostate cancer. Infect Agent Cancer. 2016;11:26.
Kakegawa T, Bae Y, Ito T, Uchida K, Sekine M, Nakajima Y, et al. Frequency of Propionibacterium acnes Infection in Prostate Glands with Negative Biopsy Results Is an Independent Risk Factor for Prostate Cancer in Patients with Increased Serum PSA Titers. PLoS One. 2017;12(1):e0169984.
Cohen RJ, Shannon BA, McNeal JE, Shannon T, Garrett KL. Propionibacterium acnes associated with inflammation in radical prostatectomy specimens: a possible link to cancer evolution? J Urol. 2005;173(6):1969–74.
Shinohara DB, Vaghasia AM, Yu SH, Mak TN, Bruggemann H, Nelson WG, et al. A mouse model of chronic prostatic inflammation using a human prostate cancer-derived isolate of Propionibacterium acnes. Prostate. 2013;73(9):1007–15.
Fassi Fehri L, Mak TN, Laube B, Brinkmann V, Ogilvie LA, Mollenkopf H, et al. Prevalence of Propionibacterium acnes in diseased prostates and its inflammatory and transforming activity on prostate epithelial cells. Int J Med Microbiol. 2011;301(1):69–78.
Drott JB, Alexeyev O, Bergström P, Elgh F, Olsson J. Propionibacterium acnes infection induces upregulation of inflammatory genes and cytokine secretion in prostate epithelial cells. BMC Microbiology. 2010;10(126).
Ugge H, Carlsson J, Soderquist B, Fall K, Anden O, Davidsson S. The influence of prostatic Cutibacterium acnes infection on serum levels of IL6 and CXCL8 in prostate cancer patients. Infect Agent Cancer. 2018;13:34.
Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, et al. Constitutive Activation of Stat3 in Human Prostate Tumors and Cell Lines: Direct Inhibition of Stat3 Signaling Induces Apoptosis of Prostate Cancer Cells. Cancer Research. 2002;62:6659–66.
Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7(6):545–55.
Grange PA, Chereau C, Raingeaud J, Nicco C, Weill B, Dupin N, et al. Production of superoxide anions by keratinocytes initiates P. acnes-induced inflammation of the skin. PLoS Pathog. 2009;5(7):e1000527.
Miyake M, Ohnishi K, Hori S, Nakano A, Nakano R, Yano H, et al. Mycoplasma genitalium Infection and Chronic Inflammation in Human Prostate Cancer: Detection Using Prostatectomy and Needle Biopsy Specimens. Cells. 2019;8(3).
Cavarretta I, Ferrarese R, Cazzaniga W, Saita D, Luciano R, Ceresola ER, et al. The Microbiome of the Prostate Tumor Microenvironment. Eur Urol. 2017;72(4):625–31.
Matsushita M, Fujita K, Motooka D, Hatano K, Fukae S, Kawamura N, et al. The gut microbiota associated with high-Gleason prostate cancer. Cancer Sci. 2021.
Li JKM, Wang LL, Wong CYP, Chiu PKF, Teoh JYC, Kwok HSW, et al. A cross-sectional study on gut microbiota in prostate cancer patients with prostatectomy or androgen deprivation therapy. Prostate Cancer and Prostatic Disease. 2021.
Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends in Microbiology. 2019;27(2):105–17.
Rippere-Lampe KE, Lang M, Ceri H, Olson M, Lockman HA, O'Brien AD. Cytotoxic necrotizing factor type 1-positive Escherichia coli causes increased inflammation and tissue damage to the prostate in a rat prostatitis model. Infect Immun. 2001;69(10):6515–9.
Jafari B, Khavari Nejad RA, Vaziri F, Siadat SD. Evaluation of the effects of extracellular vesicles derived from Faecalibacterium prausnitzii on lung cancer cell line. Biologia. 2019;74(7):889–98.
Lopez-Siles M, Duncan SH, Garcia-Gil LJ, Martinez-Medina M. Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. The International Society for Microbial Ecology Journal. 2017;11(4):841–52.
Pernigoni N, Zagato E, Calcinotto A, Troiani M, Mestre RP, Cali B, et al. Commensal bacteria promote endocrine resistance inprostate cancer through androgen biosynthesis. Science. 2021;374(6564):216–24.
Guo Y, Zhang Z, Wei H, Wang J, Lv J, Zhang K, et al. Cytotoxic necrotizing factor 1 promotes prostate cancer progression through activating the Cdc42-PAK1 axis. J Pathol. 2017;243(2):208–19.
Ma J, Sun L, Liu Y, Ren H, Shen Y, Bi F, et al. Alter between gut bacteria and blood metabolites and the anti-tumor effects of Faecalibacterium prausnitzii in breast cancer. BMC Microbiology. 2020;20(1):82.
Acknowledgements
We would like to thank Aarhus University and Aarhus University Hospital for facilitating our research.
Funding
This study was supported by grants from the Independent Research Fund Denmark and The Danish Cancer Society.
Author information
Authors and Affiliations
Contributions
KDS conceived the study. PVS drafted the manuscript and visualized the tables and figures. KDS edited the manuscript and supervised the study. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Salachan, P.V., Sørensen, K.D. Dysbiotic microbes and how to find them: a review of microbiome profiling in prostate cancer. J Exp Clin Cancer Res 41, 31 (2022). https://doi.org/10.1186/s13046-021-02196-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02196-y