
Abstract
Chimeric antigen receptor (CAR) T-cell therapy has yielded impressive outcomes and transformed treatment algorithms for hematological malignancies. To date, five CAR T-cell products have been approved by the US Food and Drug Administration (FDA). Nevertheless, some significant toxicities pose great challenges to the development of CAR T-cell therapy, most notably cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). Understanding the mechanisms underlying these toxicities and establishing prevention and treatment strategies are important. In this review, we summarize the mechanisms underlying CRS and ICANS and provide potential treatment and prevention strategies.
Similar content being viewed by others
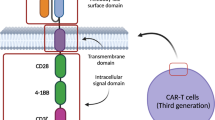

Background
Chimeric antigen receptor (CAR) T-cell therapy, one of the most significant developments in immunotherapy, has yielded impressive outcomes in hematological malignancies. To date, five different CAR T-cell products have been approved by the US Food and Drug Administration (FDA), including four products targeting CD19 for acute lymphocyte leukemia (ALL) or lymphoma [1,2,3,4] and idecabtagene vicleucel (Abecma) targeting B cell maturation antigen (BCMA), which has recently been approved for relapsed or refractory multiple myeloma (R/R MM) [5].
Genetically engineered to express CAR molecules that can specifically recognize tumor antigens, CAR T-cells can be activated, proliferate and exert antitumor effects without major histocompatibility complex (MHC) restriction. With the optimization of products and treatment regimens, the efficacy of CAR T-cell therapy is improving, and its application fields are expanding. Despite these achievements, some severe toxicities associated with CAR T-cells dampen their development. The most common toxicity is cytokine release syndrome (CRS), which is a systemic inflammatory response mediated by the overactivation of effector cells and large amounts of cytokines [6]. Neurotoxicity, another common toxicity related to CAR T-cell therapy, is a toxic encephalopathy state with a broad spectrum of neuropsychiatric symptoms. Such neurotoxicity has been designated “immune effector cell-associated neurotoxicity syndrome (ICANS)” by the American Society for Transplantation and Cellular Therapy (ASTCT) [7]. Clarifying the mechanisms underlying CRS and ICANS could facilitate the prevention and treatment of CAR T-cell-related toxicities. Herein, we review the key pathways involved in the mechanisms of CRS and ICANS based on the current understanding and provide promising prevention and management strategies to improve the safety of this beneficial therapy and expand its application. Since CAR T-cell-related toxicities are investigated mostly in the field of hematological malignances, they are the focus of this review.
Current understanding of the mechanisms of CRS
CRS is the most common toxicity related to CAR T-cell therapy, with an incidence of 42–100%, and 0–46% of patients develop severe CRS after CAR T-cell infusion (Table 1). It is believed that CRS is a systemic disease induced by the overactivation of immune effector cells and supraphysiological levels of various proinflammatory cytokines, including IL-1, IL-6, IFN-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [7]. This toxicity manifests as a constellation of symptoms (Table 2), most of which are reversible; 0–9.1% of patients progressed to fatal cases (Table 1), whereas there were no deaths from CRS in most clinical trials. A meta-analysis of 2592 patients from 84 eligible studies showed that the mortality rate of CRS was less than 1% [8]. A schematic representation of the CRS mechanisms is briefly shown in Fig. 1A. The interplay between CAR T-cells and tumor cells activates host bystander cells, especially macrophages, eliciting a distortion of the cytokine network. Then, massive cytokines induce endothelial cell activation, contributing to constitutional symptoms in relation to CRS.

Mechanisms of CRS. A. Cell interactions involved in CRS. Upon recognizing tumor antigens, CAR T-cells secrete perforin, granzyme and inflammatory cytokines, including IFN-γ and TNF-α, to induce pyroptosis of tumor cells, releasing large amounts of DAMPs that stimulate macrophages for massive cytokine production and CRS. Macrophages can also be activated by cytokines, such as GM-CSF, IFN-γ, TNF-α and catecholamine, or CD40/CD40L interactions with CAR T-cells. Pyroptosis of macrophages and further DAMPs leakage amplify the inflammatory cascade. IL-6 and other cytokines in CRS bind to their receptors on endothelial cells, causing an increase in vascular permeability and leakage and promoting cytokine production to exacerbate the CRS. B. Signaling pathway of pyroptosis in tumor cells. GZMA or GZMB enters tumor cells through perforin-induced pores. GZMB cleaves GSDME or activates caspase-3 to cleave GSDME. GZMA cleaves and activates GSDMB. Subsequently, the released gasdermin-N domain (N-GSDME or N-GSDMB) oligomerizes on the cell membrane to form membrane pores and disrupts the osmotic potential, resulting in cell swelling and lysis. C. Inflammatory signaling pathways in macrophages. Pyroptotic products include HMGB1, ATP, and dsDNA. HMGB1 activates TLR2 and TLR4 and subsequently recruits MyD88 and TRIF to activate MAPKs and IKK, leading to the subsequent production of inflammatory cytokines via AP-1 and NF-κB; ATP binds to the P2X7 receptor and induces NLRP3 activation; dsDNA is phagocytized by macrophages and activates AIM2. Activated AIM2 or NLRP3 combines with ASC and pro-caspase-1 to induce the maturation of caspase-1, which can cleave the N-terminus of GSDMD to form pores on the membrane, triggering pyroptosis and producing bioactive IL-1β. In addition, catecholamine can be recognized by α1-AR and activate the AIM2/ASC-caspase-1 pathway
CAR T-cell activation and pyroptotic target cells: the root factor
After the recognition of tumor antigens, CAR T-cells release massive amounts of perforin/granzymes and cytokines, including TNF-α and IFN-γ, resulting in tumor pyroptosis [9,10,11,12]. Pyroptosis is a type of programmed cell death that differs from apoptosis [13], and is characterized by cellular swelling, lysis and subsequent cell content and proinflammatory factor release. It is believed that pyroptosis of the target cell represents the onset of CRS. Two pathways are likely to be involved, which are mediated by granzyme B (GZMB) and granzyme A (GZMA) (Fig. 1B). GZMA and GZMB can both enter cells through pores formed by perforin [9, 12]. Subsequently, GZMB cleaves gasdermin E (GSDME) directly or by activating caspase-3 [9], while GZMA directly cleaves gasdermin B (GSDMB) for its activated form [12]. Then, the N-domains of gasdermin, which are veiled by the C-terminus, can be released and oligomerize on the cell membranes to form pores, causing decreased cell viability, bubbles blowing from the plasma membrane, cell swelling and finally cell lysis [9, 13, 14]. The different types of gasdermin and their pyroptotic pathways differ among tumor cells. GSDME widely exists in hematologic malignances [9], while GSDMB is found more frequently in bladder cancer, skin cancer and renal clear cell carcinoma, and its expression can be upregulated by cytokines, such as IFN-γ [12].
Cell death through either apoptosis or pyroptosis mainly depends on the amount of gasdermin expression [9, 12, 15]. Low levels of gasdermin induce apoptosis, while high levels of gasdermin switch apoptosis to pyroptosis [9]. Cytotoxic T lymphocytes (CTLs) mediate apoptosis in tumor cells via a low level of perforin/granzyme release, consequently activating little gasdermin and producing a few pores on the cytoplasmic membrane [16, 17]. Notably, cells are capable of repairing perforin pore formation to a certain degree, protecting cells from pyroptosis [18]. In contrast, CAR T-cells release a large amount of perforin/granzymes to induce massive gasdermin release, surpassing their self-repair capability and leading to pyroptosis [9]. Clinically, GSDME is widely expressed in B leukemic cells after CD19 CAR T-cell infusion, and the severity of CRS is positively associated with an increase in GSDME [9].
Activated macrophages: the key mediator
An increasing number of studies indicate that monocyte and macrophage lineages are the key origin of inflammatory cytokines in relation to CRS [19,20,21]. Macrophages can be activated by damage-associated molecular patterns (DAMPs), including high-mobility group box 1 (HMGB1), adenosine 5′-triphosphate (ATP) and double-stranded DNA (dsDNA) [9, 22], which are pyroptotic products of tumor cells (Fig. 1C). HMGB1 can bind Toll-like receptor 2 (TLR2) and TLR4 on the surface of macrophages. Then, the adaptor proteins myeloid differentiation primary-response 88 (MyD88) and TIR-domain-containing adaptor inducing IFNβ (TRIF) recruit and activate mitogen-activated protein kinases (MAPKs) and IκB kinase (IKK). MAPKs and IKK regulate the release of a wide range of cytokines, including IL-6, via transcription factors activator protein 1 (AP-1) and nuclear factor κB (NF-κB) [9, 22]. In addition, TLR2 induces soluble IL-6R (sIL-6R) secretion, which can combine with IL-6 and facilitate the proinflammatory effects of IL-6 [23]. ATP, which is recognized by its receptor P2X7 on macrophages, can induce the activation of NOD-, LRR- and pyrin domain-containing 3 (NLRP3) in the cytoplasm and recruit apoptosis-associated speck-like protein (ASC) and pro-caspase 1 to form the NLRP3 inflammasome, subsequently leading to the maturation of caspase 1 [22]. On the one hand, caspase 1 is responsible for pro-IL-1β cleavage and the secretion of IL-1β; on the other hand, caspase 1 transforms GSDMD into the active form and leads to pyroptosis in macrophages [9, 22]. Pyroptotic macrophages consequently produce more DAMPs and proinflammatory factors, creating a vicious cycle and leading to the further activation of macrophages [22]. Additionally, the dsDNA-mediated absent in melanoma 2 (AIM2) inflammasome pathway could be involved in caspase 1 formation [24]. Macrophages phagocytize dsDNA released by pyroptotic tumor cells and activate the AIM2 inflammasome, which is a dsDNA sensor, in the cytoplasm. Similar to the NLRP3 inflammasome pathway, activated AIM2 forms an AIM2/ASC-pro-caspase 1 complex to trigger caspase 1-dependent IL-1β maturation [24], thereby causing more IL-1β release and aggravation of CRS.
In addition to the “pyroptosis–DAMPs–macrophage” pattern, macrophages can be recruited and activated by cytokines produced by CAR T-cells, such as TNF-α, IL-2, GM-CSF and IFN-γ [25, 26]. Contact-dependent CD40 ligand (CD40L)-CD40 interactions between CAR T-cells and macrophages may also play a role in triggering IL-6 and IL-1 release and higher inducible nitric oxide synthase (iNOS) expression [21]. iNOS stimulates NO production, resulting in potentiated vasodilatation and hemodynamic instability, which are common clinical features of CRS [27]. In addition, catecholamines are produced after the coculture of CAR T-cells and malignant cells, which may be involved in cytokine release [24, 28]. By binding α1-adrenergic receptors (ARs) on the surface of macrophages, catecholamines can enhance the AIM2/ASC-caspase-1 pathway and further promote IL-1β production and macrophage pyroptosis [22, 24]. In addition, catecholamines lead to a self-amplifying feed-forward loop in macrophages, promoting the release of catecholamines, IL-6 and other cytokines, including macrophage inflammatory protein (MIP)-1α, IFN-γ, IL-2 and TNF [28]. Monocytes are also likely to be the key mediators and the main source of IL-6 and IL-1 in CRS, as Norelli et al. reported that monocyte depletion prior to CAR T-cell therapy can protect mice from lethal CRS [20].
IL-6 and endothelial cell activation: the final core pathway
Among the cytokines released by monocytes/macrophages, including IL-6, IL-1, IL-2, TNF-α, GM-CSF and IFN-γ, IL-6 plays a central role [29,30,31]. Notably, in addition to macrophages and monocytes, dendritic cells, endothelial cells, and even CAR T-cells are considered to participate in IL-6 production [21, 32]. IL-6 activates its downstream Janus kinase (JAK) and signal transducer and activator of transcription 3 (STAT3) mainly by binding the membrane-bound IL-6 receptor (mIL-6R) (classic signaling) or sIL-6R (trans-signaling) and another membrane protein, gp130. gp130 is ubiquitously expressed, whereas mIL-6R is mainly expressed on hepatocytes and immune cells. Cells that do not express mIL-6R, such as endothelial cells, are activated by trans-signaling in which IL-6 binds sIL-6R and forms a complex in the serum, triggering the dimerization of gp130 on the cell membrane [33,34,35]. As a result, activated endothelial cells secrete additional IL-6 and other proinflammatory factors, such as vascular endothelial growth factor (VEGF), IL-8, monocyte chemoattractant protein-1 (MCP-1) and coagulation cascade activator plasminogen activator inhibitor-1 (PAI-1) [36,37,38], leading to a positive loop of cytokine release and amplified inflammatory responses. In addition, endothelial cells are injured by cytokines and contribute to increased vascular permeability and leakage, edema, organ hypoperfusion, coagulopathy and organ dysfunction [39]. In CRS-associated coagulopathy, tissue factor (TF), factor VIIa, factor Xa, thrombin and platelets can also play roles as proinflammatory factors, upregulating cytokine synthesis in endothelial cells [40,41,42].
In addition to the most common systemic CRS, it is reported that local CRS (L-CRS) is the earliest toxicity in non-Hodgkin lymphoma (NHL) and mainly manifests as local swelling and redness [43]. Initially, CAR T-cells distribute locally in the tumor mass, triggering L-CRS. Then, with CAR T-cell and cytokine accumulation, they overflow into the circulation and cause systemic CRS [43]. However, the mechanisms of L-CRS are likely to be unique since the inhibition of IL-6 may aggravate L-CRS. More research investigating its specific mechanisms is warranted.
Current understanding of the mechanisms of ICANS
ICANS is the second most common toxicity associated with CAR T-cell therapy, with incidences ranging from 2% to 64% for all-grade ICANS and 0–50% for severe ICANS (Table 1). ICANS is strongly related to CRS and always occurs and reaches a peak several days after CRS [39, 44,45,46,47] despite some rarely independent occurrences [39, 48]. The clinical presentations are summarized in Table 2. The brain blood barrier (BBB) plays an important role in maintaining homeostasis of the central nervous system (CNS). In an inflammatory state, the integrity of the BBB is disrupted, triggering a series of pathophysiological reactions in the brain. It appears that the activation of the cerebral vascular endothelium and disruption of the BBB occur at the onset of ICANS. Exposure of other components of the BBB, especially astrocytes and pericytes, to large amounts of cytokines is likely to cause injury, cytokine secretion and further breakdown of the BBB. Without the barrier function of the BBB, extensive numbers of immune cells and cytokines infiltrate the CNS. Together with activated resident proinflammatory cells, infiltrated immune cells exacerbate the inflammatory cascade in the CNS, accounting for the cerebral edema, thrombosis, hemolysis and other neuropsychiatric symptoms observed in ICANS (Fig. 2).

Mechanisms of ICANS. A. Normal state. BBB is integral, consisting of endothelial cells with tight junctions, EBM, pericytes, PBM and endfeet of astrocytes. Tie2 on endothelial cells binds with Ang I to maintain the quiescent state of endothelium. B. ICANS. Systemically increased cytokines, such as IFN-γ, IL-6, GM-CSF and TNF, can activate brain endothelial cells to release W-P bodies and their contents, Ang II and vWF. Platelets adhere vWF to form the vWF-platelet string. Possibly because of the lack of ADAMTS13, vWF fails to be cleaved and thus causes microvascular thrombosis and consumptive coagulopathy. An increase in the Ang II/Ang I ratio can lead to endothelial activation and BBB disruption through abnormalities of the Ang-Tie2 axis. As a result, cytokines and CAR T-cells infiltrate the peripheral vascular space (PVS). Cytokines have access to pericytes, inducing pericyte stress and consequent VEGF and IL-6 release to further activate endothelial cells. CD19 CAR T-cells trigger CD19-positive pericyte depletion. Astrocytes can also be injured by cytokines, causing cell swelling, abnormal osmotic forces, and consequently cerebral edema. In addition, when stimulated by cytokines, astrocytes produce VEGF-A to aggravate the BBB disruption. The disrupted BBB allows myeloid cells to infiltrate into the brain parenchyma, cooperating with activated resident microglia to trigger the immune response in the CNS. Macrophages and microglia can also produce QA and Glu, activating NMDA receptors on synapses to induce seizures and other excitotoxicity. Cytokines may also play a role in neuronal injury
Brain vascular endothelial activation and BBB disruption
The BBB consists of endothelial cells with tight junctions, the endothelial basement membrane (EBM), pericytes surrounding capillaries, the parenchymal basement membrane (PBM) and the endfeet of astrocytes, also called glia limitans [49, 50]; of these components, the endothelium is the most important component. Some characteristics of brain vascular endothelial cells are distinct from those of peripheral vascular endothelial cells, such as tight junctions and specific transport systems, restricting various molecules and cells from entering the CNS [51, 52]. However, inflammation can lead to endothelial activation, reversible contraction, damage to tight junctions, and even cell death [53, 54], disrupting the integrity of the BBB and activating coagulation. The angiopoietin (Ang)-Tie2 axis plays a significant role in balancing the endothelium between quiescence and activation. Ang I is produced by platelets and pericytes constitutively, while Ang II is primarily reserved in Weibel-Palade (W-P) bodies in endothelial cells. Normally, Ang I binds Tie2 on endothelial cells, promoting cell spreading and accumulating vascular endothelial cadherin (VE cadherin), which is an important component of tight junctions. This characteristic ensures the integrity of the endothelial barrier [55, 56]. When stimulated by inflammatory factors, such as VEGF, thrombin and epinephrine, W-P bodies and Ang II are released from the endothelium via Ca2+-mediated or cAMP-mediated pathways [57]. As a result, concentrated Ang II in serum replaces Ang I, inhibiting the Tie2 pathway and increasing the permeability of the endothelium [58, 59]. Disorder of the Ang-Tie2 axis is likely to be implicated in CAR T-cell-associated ICANS, and severe ICANS patients have been observed to present a higher ratio of Ang II/Ang I [39, 45]. Interestingly, another study showed that a decrease in Ang I triggered a higher Ang II/Ang I ratio rather than increased Ang II, which is associated with the thrombocytopenia observed in severe ICANS patients [45].
Endothelial activation is likely to contribute to ICANS-associated microthrombi and disseminated intravascular coagulation (DIC). Laboratory markers indicate that patients with severe ICANS suffer DIC, and an autopsy study showed intravascular von Willebrand factor (vWF) binding and platelet microthrombi [39, 45]. vWF can be released from W-P bodies in the form of ultralarge vWF and bind endothelial cells [60]. Then, vWF exposes the binding sites for platelet adhesion and vWF-platelet string formation. The blood flow shearing force induces vWF to expose another binding site for a disintegrin and metalloproteinase with thrombospondin type 1 repeats member 13 (ADAMTS13), which is responsible for vWF cleavage [60, 61]. Cytokines influence this process. IFN-γ, TNF, IL-4 and IL-6 can directly inhibit the production of ADAMTS13 or impede its cleavage [61, 62]. It is assumed that thrombocytopenia and consumptive coagulopathy in severe ICANS patients are due to ADAMTS13 deficiency [39], which is similar to the mechanism responsible for thrombotic thrombocytopenic purpura (TTP) [63]. However, since there is no evidence of other systematic manifestations of thrombotic microangiopathy, such as renal failure and morphological changes in red blood cells, thrombotic microangiopathy in ICANS presents a unique pathogenesis requiring further investigation.
Dysfunction of other components of the BBB and inflammation amplification
Detrimental cytokines could be accessible to other components of the BBB due to the high permeability of the endothelial barrier, exacerbating the inflammatory response and further damaging the integrity of the BBB. Astrocytes represent a component with endfeet in the inner layer of the BBB and have direct contact with the brain parenchyma, regulating the osmotic pressure of the brain [64, 65]. Autopsy studies of severe ICANS patients have revealed that astrocytes are activated and injured [39, 66, 67], and the astrocyte markers glial fibrillary acidic protein (GFAP) and calcium-binding protein B (S100b) [68, 69] are increased in the CSF of ICANS patients after treatment with CD19 CAR T-cells [66]. Astrocyte injury is likely to play a role in the development of CAR T-cell-associated cerebral edema [66]. Iron channels and aquaporins on astrocytes can be interrupted by extracellular environmental changes, such as glutamate (Glut), hypoxia and stimulation by cytokines, such as IL-6, IL-1β and TNF, leading to cell swelling and abnormal osmotic forces, followed by cerebral edema and neuronal injury [64, 70, 71]. In addition, astrocytes have proinflammatory potential [64]. Triggered by IL-1β, astrocytes can release VEGF-A and destroy the tight junctions of endothelial cells [72, 73]. Astrocytes can also increase BBB permeability through various mechanisms, including the production of apolipoprotein E (APOE) and inhibition of the cyclophilin A (CYA)–NF-κB–matrix metalloproteinase 9 (MMP9) pathway in pericytes [74].
Pericytes lining along the endothelium play crucial roles in regulating BBB permeability and neuroinflammation [44, 66, 75, 76]. High concentrations of IFN-γ, TNF-α and other cytokines transmitted from serum to the CSF can cause pericyte stress. These cytokines stimulate pericytes to secrete additional inflammatory factors to further activate endothelial cells as positive feedback [39]. The incubation of primary human brain vascular pericytes with IFN-γ and TNF-α can induce large amounts of IL-6 and VEGF secretion [39]. IFN-γ can also inhibit the platelet-derived growth factor receptor β (PDGFRβ) signaling pathway, which is crucial for the proliferation and migration of pericytes, inducing pericyte stress and disrupting their regulatory role in the BBB [77, 78]. Furthermore, a recent study suggested that the on-target, off-tumor effect of CD19 CAR T-cells is also responsible for the breakdown of the BBB. Through single-cell RNA sequencing analysis (scRNA-seq), Parker et al. [79] found that CD19 is expressed on mural cells, including pericytes and vascular smooth muscle cells. These CD19-positive cells can be recognized by CAR T-cells, resulting in pericyte depletion and further BBB disruption. This phenomenon could also explain the higher incidence of ICANS in CD19 CAR T-cell therapy compared with other targets, such as CD22 and CD30 [80,81,82,83].
Inflammatory cellular infiltrates and neuronal dysfunction
Increased BBB permeability allows plasma leakage into the brain parenchyma as confirmed by imaging and histopathologic examinations of patients with CAR T-cell-associated cerebral edema [39, 67, 84,85,86]. In addition, immune cells and cytokines can penetrate the brain parenchyma and trigger inflammatory reactions in the CNS, thereby inducing neuronal injury and dysfunction. Accumulating evidence sheds light on the contribution of myeloid cells to the pathophysiology of ICANS. Monocytes and macrophages are likely to be recruited to the CNS and produce IL-1β and IL-6, which are key factors in ICANS [20, 87]. Coincidentally, Deng et al. [88] used scRNA-seq to identify rare but significant ICANS-associated cells (IACs), possibly belonging to the myeloid lineage. Moreover, it was demonstrated that GM-CSF, which is crucial for myeloid cell proliferation and activation, was the factor most significantly associated with ICANS in the ZUMA-1 clinical trial [89]. GM-CSF was also highly increased in the CSF of nonhuman primate ICANS models [90]. The increase in GM-CSF may be driven by the diffusion of increased serum GM-CSF or be produced by CNS-infiltrating T cells or activated endothelium. Consequently, GM-CSF promotes the production of IFN-γ-inducible protein 10 (IP-10), MCP-1, and CC chemokine ligand 1 (CXCL1) and attracts myeloid cells to infiltrate the brain [25, 45, 91]. GM-CSF also plays a role in activating microglia [92]. Microglia, which are brain-resident macrophages, can polarize into a proinflammatory phenotype after exposure to GM-CSF, IL-6, and IFN-γ and amplify the inflammatory cascade [93, 94]. Clinically, microglial activation has also been observed in ICANS induced by CAR T-cells [39, 45, 87, 95]. It is assumed that the inflammatory cytokines in the brain parenchyma, including both those produced locally and those traveling from the bloodstream, cause neuron dysfunction and a series of neuropsychiatric symptoms. In preclinical studies, IL-1β and TNF could change neuronal excitability, inducing delirium and other neuropsychiatric symptoms [96, 97]. Other cytokines, such as IL-6 and IL-8, may also be implicated in neural injury [98].
Moreover, myeloid cells are likely to mediate excitotoxicity in ICANS by producing endogenous excitatory agonists [45]. Stimulated by IFN-α2, IFN-γ, and TNF-α, microglia and macrophages can produce the N-methyl-d-aspartate (NMDA) receptor agonists quinolinic acid (QA) and excitatory neurotransmitter Glut, causing seizures and other excitatory symptoms [99, 100]. In addition, QA can promote the secretion of Glut [101], activate astrocytes to secrete numerous cytokines, such as TNF-α, IL-6, and MCP-1, and change the cohesion of the BBB, representing a feed-forward mechanism exacerbating brain dysfunction [101, 102].
Other functions of microglia include synaptic pruning and scavenging damaged cellular debris to maintain hemostasis of the brain and normal cognitive function [103,104,105]. Abnormalities in these functions contribute to cognitive disorders, which are the main pathophysiology of neurodegenerative diseases and aging and may be implicated in ICANS. Microglia may be depleted through off-tumor effects since low levels of CD19 were detected in human microglia by scRNA-seq and another single-cell transcriptomics database [79, 106]. Moreover, microglia were lost in a murine model treated with CD19 CAR T-cells, which was possibly associated with CD19 targeting [106]. CD22 is also expressed in microglia in the human brain [107], whereas there is no evidence indicating that the severity of ICANS is increased in CD22 CAR T-cell therapy. In summary, the possible on-target off-tumor mechanism of cognitive disorder induced by microglial injury in the ICANS remains to be clarified in further studies.
In addition to the infiltration of myeloid cells and resident microglial activation, T cells and CAR T-cells can also enter the CNS [30, 39, 45, 66, 88, 90]. It was assumed that the cytokines secreted by infiltrated CAR T-cells in the brain constitute another crucial factor causing ICANS [30, 108]. However, CAR T-cells can also be found in CSF from patients without ICANS [45, 66], suggesting that they may play a less important role in neurotoxicity.
Prevention and management of toxicities
Optimization of the infusion dose
Because the infusion dose of CAR T-cells and the disease burden are strongly associated with CRS and ICANS [44], Turtle et al. [84] proposed low-dose infusions for patients with high tumor burden, which achieved a high complete remission (CR) rate with no CRS and less ICANS. However, there was a high relapse rate in this study, raising the concern that a reduction in CAR T-cells could impair the long-term prognosis [84]. Recently, split dosing, also called fractionated dosing, has been recommended. This approach delivers CAR T-cells several times in the form of dose escalation, and subsequent infusions can be stopped if early clinical CRS is found [109]. Individualized dose modifications are considered superior to single-dose infusion since they can achieve a balance between the efficacy and safety of CAR T-cell therapy. In the trial reported by Frey et al. [109], there were three schemes of CAR T-cell infusion as follows: high-dose single-infusion (HDS; 5 × 108 CAR T-cells; n = 6), low-dose single or fractionated infusion (LD; 5 × 107 CAR T-cells; n = 9) and high-dose fractionated infusion (HDF; 5 × 108 CAR T-cells; n = 20). In the HDF group, CAR T-cells were planned to be infused within 3 days by split dosing (Day 1, 10%; Day 2, 30%; and Day 3, 60%); however, 9 patients had a high tumor burden; among these patients, 3 received Day 1 only, 3 received Day 1 and Day 2, and the other 3 received all 3 doses. Eighteen patients in the HDF group achieved CR (18/20, 90%), and only 1 patient developed grade 4 CRS (1/20, 5%). In contrast, in the HDS group, only 3 patients achieved CR (3/6, 50%), while the other 3 patients died from CRS and concurrent infections (3/6, 50%). In the LD group, 3 patients (3/9; 33%) achieved CR, and 2 patients experienced CRS > grade 4 (2/9, 22%). The 2-year survival rate was also improved in the HDF group (73% in the HDF group, 22% in the LD group, and 17% in the HDS group). The fractionated dosing scheme was also applied in another clinical trial [110]. The rates of severe CRS and ICANS were decreased in the fractionated infusion cohort, while no remarkable differences in the 1-year progression-free survival (PFS) or 1-year overall survival (OS) were found. Collectively, these data preliminarily demonstrate that split dosing and individualized modification represent promising strategies to alleviate CRS and ICANS. It is more practical for clinicians to adapt infusion dosing than explore drugs or novel CAR T-cell products. More studies are required to validate whether this strategy dampens the efficacy and long-term outcome of CAR T-cell therapy and optimize the specific timing and dosing of infusion.
Optimization of the CAR structure
The basic components of CAR usually include an antibody-derived single chain variable fragment (scFv), hinge domain (HD), transmembrane domain (TMD), and intracellular domain, which consists of one or more costimulatory domains and a CD3-zeta (CD3ζ) domain. These components all contribute to the transmission of signals for CAR T-cell activation, and optimizing the structure for a safer CAR is a feasible strategy.
Recent studies have indicated that modifying the affinity of scFv for tumor antigens has an impact on toxicities. Ghorashian et al. [111] designed a novel CD19 CAR (CAT) with a lower affinity but more robust cytotoxicity than FMC63, which showed a high CR rate (12/14, 86%) with a low toxicity rate (no severe CRS; 1 severe ICANS) in the treatment of R/R pediatric B-ALL. In addition, the replacement of murine-derived FMC63 with humanized scFv [112] or the addition of an anti-IL-6 antibody to the scFv sequences to neutralize macrophage-derived IL-6 [113] are regarded as possible strategies to mitigate CRS.
The origin, length and flexibility of the HD and TMD also have effects on CAR function and the occurrence of CRS [114,115,116]. CD8α-HD/TMD and CD28-HD/TMD are most frequently applied in clinical practice, and CD8α-HD/TMD CARs present a lower ICANS incidence [45, 87, 117, 118]. Additionally, including humanized scFv and CD8α-HD/TMD in one CAR molecule may exert synergistic effects on reducing toxicities [119]. In addition to the HD/TMD types, changes in length may impact toxicities. A CD19 CAR T-cell product containing a longer CD8α-HD/TMD was found to be associated with lower cytokine production, inducing no severe CRS or ICANS in 25 patients with refractory B cell lymphoma [116].
Several studies have indicated that CAR T-cells with 4-1BB costimulatory domains are associated with less toxicity than CAR T-cells with CD28 costimulatory domains [45, 120, 121]. A possible explanation for this finding is that CAR T-cells with 4-1BB costimulation have a mild but persistent killing effect mediated by the THEMIS-SHP1 complex, which can counteract the phosphorylation of the CD3ζ domain and consequently downregulate the expression of IFN-γ and other cytokines [122]. In comparison, CD28 CAR T-cells with no THEMIS-SHP1 complex have more rapid and intense phosphoprotein signaling, inducing large amounts of cytokine production and powerful, rapid, but transient killing effects [122]. Based on this mechanism, Sun et al. [122] added the small molecule AP21967 to a modified SHP1 on the CD28 domain to reduce cytokine release and ameliorate CRS without compromising the antitumor effects. However, a retrospective analysis showed that the incidences of CRS and ICANS are likely to be more related to HD and TMD than the costimulatory domain [123]. The role of the costimulatory domain is still unclear and remains to be clarified in further research.
Usually, CD3ζ with 3 immunoreceptor tyrosine-based activation motif (ITAM) domains is contained in the intracellular domain of the CAR and transmits activating signals through ζ-associated protein of 70 kDa (ZAP70) [124]. Reducing the number of ITAMs on CD3ζ or using other CD3 subunits with one ITAM, such as CD3ε, may be effective in inhibiting excessive CAR T-cell activation and decreasing CRS [125,126,127,128]. Other optimization strategies include knocking down genes of key cytokines in CAR T-cells that contribute to toxicities, such as GM-CSF [25, 91] and IL-6 [32, 129].
Elimination switches
Adding switches on CAR T-cells to regulate the inactivation of CAR signaling is being actively explored to eliminate toxicities. Most switches are controlled by small-molecule agents and can be divided into reversible and irreversible switches. The former can reactivate CAR T-cells when the inducers are removed, while irreversible switches induce permanent CAR T-cell damage.
Reversible switches can be achieved through the chemically disruptable heterodimerization of functional chains. Giordano-Attianese et al. [130] designed a STOP CAR (Fig. 3A) that contains the following 2 chains: recognition (R) chain and signaling (S) chain. The extracellular domain of the S chain consists of a c-Myc-tag and DAP10, and DAP10 maintains the expression stability of the S chain. The two chains are spontaneously dimerized through a computationally designed protein pair, apolipoprotein E4 (apoE4) and B cell lymphoma-extra large (Bcl-XL). ApoE4 is expressed at the end of the R chain, and Bcl-XL is located between the CD28 costimulatory domain and the CD3 ζ domain of the S chain. The specific disruption between apoE4 and Bcl-XL is triggered by the Bcl-XL inhibitor A1155463, consequently interfering with CAR signaling and transiently inactivating CAR T-cells. Another off-switch design is combined with proteolysis-targeting chimera (PROTAC) technology and triggered by lenalidomide (Fig. 3B) [131]. The hybrid zinc finger degron tag ZFP91-IKZF3 is added at the C-terminus of the CAR. The presence of lenalidomide can induce CRL4CRBN E3-mediated ubiquitylation and proteasomal degradation of CAR, while CAR signaling can be restored by decreasing the lenalidomide levels. Although the two forms of switches mentioned above have not been used in clinical settings, they have good clinical suitability since these switches originate from nonimmunogenic human sequences, and the small molecule agents are FDA-approved. Additionally, CAR molecules can be incorporated with cleavable degradation moieties (degrons) called switch-off CARs (SWIFF-CARs) (Fig. 3C) [132]. SWIFF-CARs are controlled by hepatitis C virus (HCV) NS3 protease, and the administration of asunaprevir can inhibit the cleavage activity of HCV NS3 protease, preventing the degradation of protease/degron complex from CAR. As a result, the degron induces the rapid degradation of CAR. However, the strategies mentioned above all require the addition of a switch gene, which is challenging for the vector virus loaded with a CAR gene since the viral payload capacity is limited. Therefore, Park et al. generated a conditional scFv based on a camelid antibody, which is capable of recognizing both tumor-associated antigen (TAA) and methotrexate (MTX) (Fig. 3D) [133]. Upon binding MTX, the conformation of MTX-based scFv changes to disrupt TAA binding, resulting in CAR T-cell inactivation [133]; when MTX is removed, CAR T-cells are reactivated.

Elimination switches. Reversible switches A. The STOP CAR is a dimer of two functional chains that can be disrupted by a Bcl-XL inhibitor. B. Lenalidomide induces CRL4CRBN E3 ubiquitin ligase-mediated ubiquitination and degradation of the hybrid zinc finger degron ZFP91-IKZF3-incorporated CAR. C. Asunaprevir binds to HCV NS3 protease in SWIFF-CAR and inhibits the degradation of the protease/degron complex. Therefore, the whole CAR would be degraded by degron. D. The CAR contains a conditional scFv based on the camelid antibody. MTX binds to the scFv and induces scFv conformational changes, therefore inhibiting CAR from recognizing TAA. Irreversible switches E. Upon the administration of monoclonal antibodies, CAR T-cells expressing CD20 or EGFRt can be irreversibly eliminated through the CDC or ADCC effect. F. The administration of AP1903 induces the dimerization of iCasp9, which triggers downstream apoptotic cascades, resulting in CAR T-cell death. G. When ganciclovir is administered, HSV-TK phosphorylates ganciclovir to form the toxic ganciclovir-triphosphate compound, leading to the inhibition of DNA synthesis and CAR T-cell death. H. T-cells transfected with mRNA transiently encode CAR, the expression of which can be downregulated with mRNA degradation
Alternatively, irreversible switches also represent a strategy to eliminate the activities and toxicities of CAR T-cells. It can be achieved through complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC). CAR T-cells engineered with surface antigens can be neutralized by FDA-approved therapeutic antibodies, such as rituximab (targeting CD20) [134] and cetuximab (targeting truncated epidermal growth factor receptor, EGFRt) [135], and cause irreversible clearance (Fig. 3E). However, this strategy takes effect slowly and, therefore, may be unsuitable for patients with severe cytokine-mediated toxicities requiring urgent treatment. Another feasible solution is to express inducible caspase 9 (iCasp9) in CAR T-cells (Fig. 3F). iCasp9 is an apoptosis-triggering fusion protein, and rimiducid (AP1903) can induce the dimerization of iCasp9 and activate downstream caspase, eliminating the engineered CAR T-cells within 30 min [136, 137]. This strategy induced dramatic and immediate CAR T-cell elimination in a 26-year-old female, effectively alleviating the ICANS symptoms and improving the ICANS grades (from 3 to 1) and the Immune Effector Cell-Associated Encephalopathy (ICE) score (from 0 to 7) within 12 h [138]. The efficacy of iCasp9 CAR T-cells in CD19 B cell malignancies is still being evaluated in clinical trials (NCT03016377 and NCT03696784). Herpes simplex virus thymidine kinase (HSV-tk)/ganciclovir is another safety switch that inhibits DNA synthesis through death-inducing signaling cascades and, therefore, induces CAR T-cell death (Fig. 3G). However, its clinical applications are likely to be limited by its immunogenicity and slow induction of cell death [139, 140]. In addition, transfection with mRNA can construct a ‘biodegradable’ product with the transient expression of CAR molecules. CAR is downregulated along with the degradation of mRNA [141], reducing the risk of supercytokine production from the long-term overactivation of CAR T-cells (Fig. 3H). However, to achieve remission, multiple infusions of such CAR T-cells are required, which may increase the risk of phylaxis [142].
In summary, the off-switch is a promising strategy to eliminate toxicities, especially reversible switches, which avoid the permanent elimination of costly and robust CAR T-cells. More studies are expected to explore the on/off kinetics of such switches and confirm their immediate reactivity in vivo before translation into the clinic. Setting a specific timing for clinicians to turn off the switch is also of great importance. Moreover, safety issues associated with the small-molecule controller should be considered, and using approved agents as controllers may be better choices. In the future, clinical validations of the antitumor effects and toxicity-management ability of these novel products are needed.
Anticytokine agents
Targeting IL-6
Tocilizumab is a humanized IgG1 monoclonal antibody that blocks both membrane-bound and soluble IL-6 receptors, inhibiting IL-6, the key mediator in the pathogenesis of CRS. As the only FDA-approved and first-line agent for severe or life-threatening CRS [143] (Table 3), tocilizumab effectively induces the rapid reversal of symptoms and normalizes laboratory markers in most patients [31, 144]. Although it was assumed that inhibiting the downstream pathways of IL-6 may disrupt the cytokine environment necessary for maintaining CAR T-cell potency, clinical experience has shown that tocilizumab does not dampen their expansion, persistence and antitumor efficacy [89, 145].
Tocilizumab is usually applied in cases of severe CRS, while mild CRS can be resolved through supportive care alone [146, 147]. Consensus regarding the specific timing for tocilizumab administration has not been reached and is an active field of investigation. The latest research shows that early intervention (EI) for CRS may be a better option by administering tocilizumab within a short time after CAR T-cell infusion or at the onset of mild CRS symptoms [145, 148]. Gardner et al. [145] found that the EI strategy with tocilizumab and/or corticosteroids could reduce the incidence of severe CRS in ALL patients receiving CD19 CAR T-cells without dampening the antitumor efficacy. Another clinical trial also demonstrated that the EI strategy was effective for high-tumor-burden B-ALL [149]. More aggressively, in some studies, tocilizumab was even administered before the infusion of CAR T-cells and led to a decrease in subsequent CRS [150].
However, tocilizumab has shown limited efficacy in most ICANS cases [39, 45]. Early intervention with tocilizumab may even aggravate ICANS. In a safety expansion trial of ZUMA-1, prophylactic tocilizumab was administered to NHL patients 2 days after the CAR T-cell infusion, and an increase in severe ICANS was observed compared with the original research in which tocilizumab was administered after the occurrence of CRS; in addition, one death occurred due to cerebral edema [89, 148]. The reason for ICANS aggravation is likely to be the increased BBB permeability to IL-6 and poor BBB penetration of tocilizumab. After the extensive blockade of IL-6R in the peripheral circulation, large amounts of IL-6 enter the BBB through passive diffusion and trigger an inflammatory response in the CNS, which cannot be neutralized by tocilizumab [151].
Siltuximab may be an alternative agent to tocilizumab. By binding IL-6, siltuximab removes IL-6 from the circulation [33], thus reducing IL-6 entry into the CNS and alleviating ICANS. Shah et al. successfully resolved CRS and ICANS with either tocilizumab, corticosteroids, and/or siltuximab in 5 patients [152], preliminarily showing the efficacy of siltuximab. However, to date, solid clinical evidence is lacking, and more research is needed to verify the therapeutic effects of siltuximab.
Corticosteroids
Corticosteroids have been shown to effectively resolve the symptoms of CRS and ICANS after CAR T-cell infusion by exerting powerful nonspecific anti-inflammatory effects [31, 153, 154]. Corticosteroids are the first-line therapy for isolated ICANS [155, 156] (Table 3), while for CRS, corticosteroids are usually reserved for life-threatening (grade 3 or 4) or tocilizumab-resistant CRS [151, 155, 157]. Dexamethasone is the most commonly used among corticosteroids because it shows good CNS penetration and can improve the integrity of the BBB [48, 151, 158].
A concern is that corticosteroids may compromise the antitumor efficacy by dampening T-cell function, expansion, and persistence and even inducing T cell apoptosis [153, 159], and it was found that using corticosteroids reduced the OS rate of patients receiving CAR T-cell therapy in a clinical trial [160]. However, increasing evidence indicates that rational dosage regimens of corticosteroids could maintain the intact efficacy of CAR T-cell therapy. It is likely that the short-term use of corticosteroids is practicable [161]. Moreover, the earlier administration of corticosteroids is under investigation [145, 162,163,164]. The ZUMA-3 trial administered corticosteroids at grade 2 ICANS after CD19 CAR T-cell therapy [162]. Compared with administering corticosteroids at grade 3 ICANS, there were significant reductions in the incidence rates of severe ICANS (11% vs 64%) and peak levels of cytokines and other proinflammatory markers in the early intervention group; CAR T-cell expansion was similar between the two groups [162]. A more aggressive measure may also be feasible. A safety management cohort of ZUMA-1 adopted the prophylactic administration of corticosteroids during the first 3 days after the CAR T-cell infusion and early corticosteroid and/or tocilizumab intervention for grade 1 CRS or ICANS, which resulted in no severe CRS and low rates of severe ICANS (13%) without detrimental effects on the efficacy of CAR T-cells [164]. In clinical practice, further explorations are needed to optimize corticosteroid regimens, including their dosage, timing and duration.
Targeting IL-1
In addition to IL-6 blockade and corticosteroids, other anticytokine candidates are being actively explored and are summarized in Table 3. One potential strategy is targeting IL-1. IL-1β, which is released earlier than IL-6 and promotes IL-6 production, is another key cytokine involved in CRS and ICANS [20, 165]. Anakinra, an FDA-approved recombinant IL-1 receptor antagonist for neonatal onset multisystem inflammatory disease (NOMID) and rheumatoid arthritis (RA), has been proven to successfully resolve CRS in mouse models without affecting the expansion of CAR T-cells [21]. In addition, notably, anakinra can penetrate the BBB and target IL-1β in the CNS [166], ameliorating ICANS symptoms and the histopathological signs of meningeal inflammation in a mouse model, which cannot be resolved by tocilizumab [20]. A clinical trial confirmed that anakinra is effective in the treatment of hemophagocytic lymphohistiocytosis (HLH) after CD22 CAR T-cell therapy in ALL patients [81]. Strati et al. applied anakinra in 6 patients with severe ICANS refractory to tocilizumab and dexamethasone, and 4 of the 6 patients showed improvement in symptoms [167]. It is hypothesized that a high disease burden and early progression limit the efficacy of anakinra since it was used as the last remedy for toxicities in this trial, and the preemptive administration of anakinra may result in better outcomes. More relevant clinical trials are ongoing (NCT04148430, NCT04150913, NCT04205838, and NCT04432506).
Targeting GM-CSF
GM-CSF is also an important cytokine associated with CAR T-cell-related adverse events, especially ICANS [89, 91]. Lenzilumab, a monoclonal antibody against GM-CSF, can resolve ICANS and CRS induced by CAR T-cells in mouse models by inhibiting myeloid cells and T cells entering the CNS [91]. Intriguingly, instead of dampening CAR T-cell expansion, lenzilumab even enhanced its antitumor effect [91]. The ZUMA-19 trial (NCT04314843) preliminarily showed the efficacy and safety of lenzilumab and Axi-Cel (an FDA-approved CD19 CAR T-cell product) in treating patients with relapsed or refractory diffuse large B cell lymphoma (DLBCL). It was observed that lenzilumab could induce a lower risk of severe CRS and ICANS and reduce inflammatory markers, such as IL-6, CRP, ferritin, MCP-1, IL-8, and IP-10 [168].
Targeting the JAK signaling pathway
The JAK family consists of JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2), which are bound to the intracellular domains of type I and type II cytokine receptors, including interleukins, IFNs, and colony-stimulating factors, and regulate gene transcription by phosphorylating STAT [169]. Different JAK subunits selectively bind different cytokine receptors, while compared with other JAKs, JAK1 plays a broader role [170]. In terms of IL-6, its receptor binds JAK1, JAK2 and TYK2, and JAK1 is the dominant kinase [171, 172].
Some researchers are exploring JAK inhibitors for CRS treatment. Ruxolitinib, a JAK1/2 inhibitor approved for the treatment of myeloproliferative neoplasms (MPN) and steroid-refractory acute graft-versus-host disease (GVHD) [173, 174], showed feasibility in preventing severe CRS refractory to steroid therapy in four R/R B-ALL patients receiving CD19 or CD22 CAR T-cell therapy [175]. Grade 3 CRS induced by CD7 CAR T-cells was also resolved by ruxolitinib-based therapy in 2 T-ALL patients [176]. In addition, itacitinib, which is a selective JAK1 inhibitor developed for the treatment of graft-versus-host disease, may resolve CRS with a lower risk of immunosuppression [177]. A phase 2 trial evaluating itacitinib for the prevention of CRS is ongoing (NCT04071366).
Tyrosine kinase inhibitor
Dasatinib, an FDA-approved tyrosine kinase inhibitor (TKI), blocks the adenosine triphosphate binding sites of lymphocyte-specific protein tyrosine kinase (LCK) and inhibits the phosphorylation of CD3ζ and ZAP70 [178, 179], effectively depressing the activation of CAR T-cells and T cells. Mestermann et al. demonstrated the on/off switch effect of dasatinib [180]. Dasatinib can transform CAR T-cells into a functional off state both in vitro and in a mouse model, reversibly inhibiting the proliferation and cytotoxicity of CAR T-cells. Upon the removal of dasatinib, CAR T-cell functions can be quickly restored without compromising the overall efficacy. Moreover, during the pause phase, CAR T-cells can undergo epigenetic remodeling to reverse exhaustion, thereby enhancing the cell fitness and antitumor potency [181]. Since dasatinib directly interferes with the CAR T-cell signaling pathway, inhibiting CAR T-cells more rapidly and completely than dexamethasone, which regulates transcription, may be suitable for the emergency treatment of CRS [180].
Ibrutinib is a Bruton’s tyrosine kinase (BTK) inhibitor approved by the FDA for the treatment of chronic lymphocytic leukemia (CLL). Ibrutinib can also inhibit the IL-2-induced tyrosine kinase (ITK) pathway, regulating the cytokine release of various immune cells, such as T cells, monocytes and tumor cells [182,183,184]. Several trials have demonstrated that the concurrent use of ibrutinib and CAR T-cells for CLL can exert synergistic antitumor efficacy [185, 186] while ameliorating CRS [185].
Other rational therapies
With an evolving understanding of the mechanisms of CRS and ICANS, more potential therapeutic targets are emerging. Transforming pyroptosis into apoptosis and disrupting the onset of CRS by inhibiting the production of gasdermins, inflammasomes, caspases or DAMPs or regulating the killing pathway of the CAR appear promising [187,188,189]. Based on the role of catecholamines in CRS, experiments in a mouse model have shown that the early use of metyrosine (MTR) or atrial natriuretic peptide (ANP) can prevent CRS by reducing the production of catecholamines and various cytokines without affecting the efficacy of CAR T-cells [28]. It was previously emphasized that the activation of endothelial cells plays an important role in CAR T-cell toxicities, especially ICANS. Strategies, such as rescuing the disorder of the Ang-Tie2 axis by Ang I or vascular endothelial protein tyrosine phosphatase (VE-PTP)-dependent restoration [59, 190] and using defibrotide to regulate endothelial cell-leukocyte interactions [191], may be promising for reversing hemodynamic disorders and maintaining the stability of the BBB as a result abrogating ICANS.
Blocking other important cytokines in the development of CRS and ICANS to interrupt the inflammatory cascade is also a potential strategy. Some centers have attempted to use etanercept to block TNF-α, and its feasibility has been preliminarily confirmed [29, 192, 193]. Moreover, an in vitro experiment demonstrated that the use of adalimumab, a TNF-α antibody, combined with an anti-IL-1β antibody could exert a synergistic effect on preventing the activation of endothelial cells [194]. Emapalumab, which blocks IFN-γ in the treatment of HLH [195], may also inhibit CAR T-cell-associated toxicities but poses a significant risk to CAR T-cell efficacy [6]. By preventing mRNA maturation, TO-207 can abrogate the monocyte production of multiple cytokines, such as TNF-α, IL-6, IL-1β, MCP-1, IL-18, IL-8, and GM-CSF, without dampening the cytotoxicity of CAR T-cells [196]. Pretreatment with THZ1 to block cyclin-dependent kinase 7 (CDK7) may also be an option. THZ1 can suppress inflammatory genes at the transcriptional level, particularly inhibiting STAT1 and IL-1, therefore, alleviate CRS without impairing the antitumor efficacy [197]. In addition, nonspecific cytokine filtering through extracorporeal cytokine removal, plasma exchange, or hemofiltration is also effective [198,199,200].
Conclusion and future perspectives
The toxicities of CAR T-cell therapy, especially CRS and ICANS, impede the broader adoption of this robust antitumor agent. Studies investigating the mechanisms of CRS have made great breakthroughs. The key involvement of pyroptosis is highlighted, and the role of macrophages as the main source of important cytokines in CRS has been confirmed. Regarding ICANS, some crucial links between the main cellular and molecular mediators are still elusive. It is reasonable to speculate that the mechanisms of common reversible neurocognitive disorders differ from those of lethal events, and further studies are warranted to explain the wide spectrum of clinical manifestations. Animal models play critical roles in both further understanding the detailed mechanisms of toxicities and exploring promising strategies. Rapid progress is underway in the optimization of the modeling, including humanized mice and primate models, in an effort to narrow the gap with clinical settings. In addition, advances in sequencing and omics techniques provide new insight into mechanistic studies. Such techniques can help identify certain cell populations and components that cannot be identified by traditional technologies. The development of multidimensional parameter analyses, combining preclinical and clinical evidence and collaborating phenotypic and functional information is expected to provide more comprehensive insight into the underlying mechanisms.
For the prevention and management of toxicities, strategies have been proposed from different perspectives. Optimizing the dosing schemes is a practical clinical strategy, and laboratory strategies include developing less toxic CAR structures and reversible off-switches, which may radically improve the safety of CAR T-cell therapy. However, more research is still required before these strategies can be broadly available in clinical settings. Currently, the most popular intervention is pharmacotherapy. Tocilizumab, as the staple of CRS treatment, and corticosteroids, another common agent for CRS and ICANS, are being explored for preemptive toxicity mitigation and gradually showing their superiority. More efforts are expected to determine the optimal dosing and timing for prophylactic use. Notably, ideal targeted interventions for ICANS are lacking, and recent studies are shedding light on other specific immune modulators, such as inhibitors of IL-1 and GM-CSF. Since mechanisms are being gradually unveiled, it is hopeful that they will provide more insight for novel intervention strategies. For instance, targeting pyroptosis is a future direction to abolish the onset of CRS. In general, ideal solutions for toxicity are effective without a negative impact on the effectiveness of CAR T-cells and are easy to implement in most medical centers in an effort to promote the widespread use of a safer product in real-world practice.
CAR T-cell therapy is also broadening its field to various cancers and nonmalignant diseases, including infections, autoimmune diseases, transplantation, cardiac fibrosis and senescence-associated pathologies [201,202,203,204]. The manifestations, severities, and outcomes of such toxicities are likely to differ mainly in terms of the primary diseases, distinctive targeting cells and different categories of T cells adopted for CAR T-cell engineering. It is hoped that the mechanisms summarized in this article could facilitate an understanding of the toxicities of different indications. Simultaneously, deeper investigation of their unique features and underlying mechanisms is of great importance, which can aid the accurate recognition of toxicities and the development of rational management strategies, enabling the application of safer CAR T-cell therapy in a wider range of fields and providing greater benefits to more patients.
Availability of data and materials
Not applicable.
Abbreviations
- CAR:
-
Chimeric antigen receptor
- FDA:
-
US Food and Drug Administration
- CRS:
-
Cytokine release syndrome
- ICANS:
-
Immune effector cell associated neurotoxicity syndrome
- ALL:
-
Acute lymphocyte leukemia
- BCMA:
-
B cell maturation antigen
- R/R MM:
-
Relapse or refractory multiple myeloma
- MHC:
-
Major histocompatibility complex
- ASTCT:
-
American Society for Transplantation and Cellular Therapy
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- GZMB:
-
Granzyme B
- GZMA:
-
Granzyme A
- GSDME:
-
Gasdermin E
- GSDMB:
-
Gasdermin B
- CTLs:
-
Cytotoxic T lymphocytes
- DAMPs:
-
Damage-associated molecular patterns
- HMGB1:
-
High-mobility group box 1
- ATP:
-
Adenosine 5′-triphosphate
- dsDNA:
-
Double-stranded DNA
- TLR:
-
Toll-like receptor
- MyD88:
-
Myeloid differentiation primary-response 88
- TRIF:
-
TIR-domain-containing adaptor inducing IFNβ
- MAPKs:
-
Mitogen activated protein kinases
- IKK:
-
IκB kinase
- AP-1:
-
Activator protein 1
- NF-κB:
-
Nuclear factor κB
- sIL-6R:
-
Soluble IL-6R
- NLRP3:
-
NOD-, LRR-, and pyrin domain-containing protein 3
- ASC:
-
Apoptosis-associated speck-like protein
- AIM2:
-
Absent in melanoma 2
- CD40L:
-
CD40 ligand
- iNOS:
-
Inducible nitric oxide synthase
- AR:
-
α1-adrenergic receptors
- MIP-1α:
-
Macrophage inflammatory protein-1α
- JAK:
-
Janus kinase
- STAT3:
-
Signal transducer and activator of transcription 3
- mIL-6R:
-
Membrane-bound IL-6 receptor
- VEGF:
-
Vascular endothelial growth factor
- MCP-1:
-
Monocyte chemoattractant protein-1
- PAI-1:
-
Plasminogen activator inhibitor-1
- TF:
-
Tissue factor
- L-CRS:
-
Local CRS
- NHL:
-
Non-Hodgkin lymphoma
- BBB:
-
Brain blood barrier
- CNS:
-
Central nerve system
- PVS:
-
Perivascular space
- EBM:
-
Endothelial basement membrane
- PBM:
-
Parenchymal basement membrane
- Ang:
-
Angiopoietin
- W-P:
-
Weibel-Palade
- VE cadherin:
-
Vascular endothelial cadherin
- DIC:
-
Disseminated intravascular coagulation
- vWF:
-
Von Willebrand factor
- ADAMTS13:
-
A disintegrin and metalloproteinase with thrombospondin type 1 repeats member 13
- TTP:
-
Thrombotic thrombocytopenic purpura
- GFAP:
-
Glial fibrillary acidic protein
- Glut:
-
Glutamate
- APOE:
-
Apolipoprotein E
- CYA:
-
Cyclophilin A
- MMP9:
-
Matrix metalloproteinase 9
- PDGFRβ:
-
Platelet-derived growth factor receptor β
- scRNA-seq:
-
Single-cell RNA sequencing analysis
- IACs:
-
ICANS associated cells
- IP-10:
-
IFN-γ-inducible protein 10
- CXCL1:
-
CC chemokine ligand 1
- NMDA:
-
N-methyl-d-aspartate
- QA:
-
Quinolinic acid
- CR:
-
Complete remission
- HDS:
-
High-dose single-infusion
- LD:
-
Low-dose single or fractionated infusion
- HDF:
-
High-dose fractionated infusion
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
- scFv:
-
Single chain variable fragment
- HD:
-
Hinge domain
- TMD:
-
Transmembrane domain
- ITAM:
-
Immunoreceptor tyrosine based activation motif
- ZAP70:
-
ζ-associated protein of 70 kDa
- R:
-
Recognition
- S:
-
Signaling
- apoE4:
-
Apolipoprotein E4
- Bcl-XL:
-
B cell lymphoma-extra large
- PROTAC:
-
Proteolysis-targeting chimera
- SWIFF-CARs:
-
Switch-off CARs
- HCV:
-
Hepatitis C virus
- TAA:
-
Tumor-associated antigen
- MTX:
-
Methotrexate
- CDC:
-
Complement dependent cytotoxicity
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- EGFRt:
-
Truncated epidermal growth factor receptor
- iCasp9:
-
Inducible caspase 9
- ICE:
-
Immune Effector Cell Encephalopathy
- HSV-tk:
-
Herpes simplex virus thymidine kinase
- EI:
-
Early intervention
- NOMID:
-
Neonatal onset multisystem inflammatory disease
- RA:
-
Rheumatoid arthritis
- HLH:
-
Hemophagocytic lymphohistiocytosis
- DLBCL:
-
Diffuse large B cell lymphoma
- TYK2:
-
Tyrosine kinase 2
- MPN:
-
Myeloproliferative neoplasms
- GVHD:
-
Graft-versus-host disease
- TKI:
-
Tyrosine kinase inhibitor
- LCK:
-
Lymphocyte-specific protein tyrosine kinase
- BTK:
-
Bruton’s tyrosine kinase
- CLL:
-
Chronic lymphocytic leukemia
- ITK:
-
IL-2-induced tyrosine kinase
- MTR:
-
Metyrosine
- ANP:
-
Atrial natriuretic peptide
- VE-PTP:
-
Vascular endothelial protein tyrosine phosphatase
- CDK7:
-
Cyclin-dependent kinase 7
References
US Food and Drug Administration: KYMRIAH (tisagenlecleucel). (2021). https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel. Accessed 11 Oct 2021.
US Food and Drug Administration: YESCARTA (axicabtagene ciloleucel). (2021). https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel. Accessed 11 Oct 2021.
US Food and Drug Administration: TECARTUS (brexucabtagene autoleucel). (2021). https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/tecartus-brexucabtagene-autoleucel. Accessed 11 Oct 2021.
US Food and Drug Administration: BREYANZI (lisocabtagene maraleucel). (2021). https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/breyanzi-lisocabtagene-maraleucel. Accessed 11 Oct 2021.
US Food and Drug Administration: ABECMA (idecabtagene vicleucel). (2021). https://www.fda.gov/vaccines-blood-biologics/abecma-idecabtagene-vicleucel. Accessed 11 Oct 2021.
Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020;383(23):2255–73.
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–38.
Lei W, Xie M, Jiang Q, Xu N, Li P, Liang A, et al. Treatment-related adverse events of chimeric antigen receptor T-cell (CAR T) in clinical trials: a systematic review and meta-analysis. Cancers (Basel). 2021;13(15):3912.
Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020;5(43):eaax7969.
Michie J, Beavis PA, Freeman AJ, Vervoort SJ, Ramsbottom KM, Narasimhan V, et al. Antagonism of IAPs enhances CAR T-cell efficacy. Cancer Immunol Res. 2019;7(2):183–92.
Textor A, Listopad JJ, Wührmann LL, Perez C, Kruschinski A, Chmielewski M, et al. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNγ. Cancer Res. 2014;74(23):6796–805.
Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368(6494):eaaz7548.
Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20(3):143–57.
Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–54.
Jiang M, Qi L, Li L, Li Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020;6:112.
Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 1994;370(6491):650–2.
Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–70.
Lopez JA, Susanto O, Jenkins MR, Lukoyanova N, Sutton VR, Law RH, et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood. 2013;121(14):2659–68.
Hao Z, Li R, Meng L, Han Z, Hong Z. Macrophage, the potential key mediator in CAR-T related CRS. Exp Hematol Oncol. 2020;9:15.
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–48.
Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–8.
Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20(2):95–112.
Flynn CM, Garbers Y, Lokau J, Wesch D, Schulte DM, Laudes M, et al. Activation of toll-like receptor 2 (TLR2) induces Interleukin-6 trans-signaling. Sci Rep. 2019;9(1):7306.
Liu D, Xu X, Dai Y, Zhao X, Bao S, Ma W, et al. Blockade of AIM2 inflammasome or α1-AR ameliorates IL-1β release and macrophage-mediated immunosuppression induced by CAR-T treatment. J Immunother Cancer. 2021;9(1):e001466.
Sachdeva M, Duchateau P, Depil S, Poirot L, Valton J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J Biol Chem. 2019;294(14):5430–7.
Titov A, Petukhov A, Staliarova A, Motorin D, Bulatov E, Shuvalov O, et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018;9(9):897.
Ohashi Y, Kawashima S, Hirata K, Yamashita T, Ishida T, Inoue N, et al. Hypotension and reduced nitric oxide-elicited vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. J Clin Invest. 1998;102(12):2061–71.
Staedtke V, Bai RY, Kim K, Darvas M, Davila ML, Riggins GJ, et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature. 2018;564(7735):273–7.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17.
Chen LY, Kang LQ, Zhou HX, Gao HQ, Zhu XF, Xu N, et al. Successful application of anti-CD19 CAR-T therapy with IL-6 knocking down to patients with central nervous system B-cell acute lymphocytic leukemia. Transl Oncol. 2020;13(11):100838.
Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov. 2018;17(6):395–412.
Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol. 2018;18(12):773–89.
Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. 2015;34:75–82.
Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016;8(8):959–70.
Zegeye MM, Lindkvist M, Fälker K, Kumawat AK, Paramel G, Grenegård M, et al. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Commun Signal. 2018;16(1):55.
Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A. 2020;117(36):22351–6.
Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404–19.
Jones A, Geczy CL. Thrombin and factor Xa enhance the production of interleukin-1. Immunology. 1990;71(2):236–41.
Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A. 2000;97(10):5255–60.
Freedman JE, Loscalzo J. Platelet-monocyte aggregates: bridging thrombosis and inflammation. Circulation. 2002;105(18):2130–2.
Wei J, Liu Y, Wang C, Zhang Y, Tong C, Dai G, et al. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct Target Ther. 2020;5(1):134.
Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130(21):2295–306.
Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018;8(8):958–71.
Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–30.
Gofshteyn JS, Shaw PA, Teachey DT, Grupp SA, Maude S, Banwell B, et al. Neurotoxicity after CTL019 in a pediatric and young adult cohort. Ann Neurol. 2018;84(4):537–46.
Gust J, Taraseviciute A, Turtle CJ. Neurotoxicity associated with CD19-targeted CAR-T cell therapies. CNS Drugs. 2018;32(12):1091–101.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–96.
Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18(2):123–31.
Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201.
Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. 2007;100(2):174–90.
Huang X, Hussain B, Chang J. Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci Ther. 2021;27(1):36–47.
Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. 2017;60:1–12.
Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov. 2017;16(9):635–61.
Milam KE, Parikh SM. The angiopoietin-Tie2 signaling axis in the vascular leakage of systemic inflammation. Tissue barriers. 2015;3(1–2):e957508.
Schillemans M, Karampini E, Kat M, Bierings R. Exocytosis of Weibel-Palade bodies: how to unpack a vascular emergency kit. J Thromb Haemost. 2019;17(1):6–18.
Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, et al. The tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood. 2004;103(11):4150–6.
Higgins SJ, Purcell LA, Silver KL, Tran V, Crowley V, Hawkes M, et al. Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Sci Transl Med. 2016;8(358):358ra128.
De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von Willebrand factor strings puzzle. Blood. 2013;121(2):270–7.
Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104(1):100–6.
Cao WJ, Niiya M, Zheng XW, Shang DZ, Zheng XL. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J Thromb Haemost. 2008;6(7):1233–5.
Schwameis M, Schörgenhofer C, Assinger A, Steiner MM, Jilma B. VWF excess and ADAMTS13 deficiency: a unifying pathomechanism linking inflammation to thrombosis in DIC, malaria, and TTP. Thromb Haemost. 2015;113(4):708–18.
Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–63.
Min R, van der Knaap MS. Genetic defects disrupting glial ion and water homeostasis in the brain. Brain Pathol. 2018;28(3):372–87.
Gust J, Finney OC, Li D, Brakke HM, Hicks RM, Futrell RB, et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy. Ann Neurol. 2019;86(1):42–54.
Torre M, Solomon IH, Sutherland CL, Nikiforow S, DeAngelo DJ, Stone RM, et al. Neuropathology of a case with fatal CAR T-cell-associated cerebral edema. J Neuropathol Exp Neurol. 2018;77(10):877–82.
Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. 2014;20(2):160–72.
Kim HJ, Tsao JW, Stanfill AG. The current state of biomarkers of mild traumatic brain injury. JCI Insight. 2018;3(1):e97105.
Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A. 2011;108(2):846–51.
Rama Rao KV, Jayakumar AR, Tong X, Alvarez VM, Norenberg MD. Marked potentiation of cell swelling by cytokines in ammonia-sensitized cultured astrocytes. J Neuroinflammation. 2010;7:66.
Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122(7):2454–68.
Potokar M, Jorgačevski J, Zorec R. Astrocyte aquaporin dynamics in health and disease. Int J Mol Sci. 2016;17(7):1121.
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–6.
Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–61.
Bhattacharya A, Kaushik DK, Lozinski BM, Yong VW. Beyond barrier functions: roles of pericytes in homeostasis and regulation of neuroinflammation. J Neurosci Res. 2020;98(12):2390–405.
Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19(6):771–83.
Jansson D, Scotter EL, Rustenhoven J, Coppieters N, Smyth LC, Oldfield RL, et al. Interferon-γ blocks signalling through PDGFRβ in human brain pericytes. J Neuroinflammation. 2016;13(1):249.
Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell. 2020;183(1):126–42.e17.
Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–8.
Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol. 2020;38(17):1938–50.
Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. 2017;127(9):3462–71.
Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin lymphoma: an open-label phase I trial. Clin Cancer Res. 2017;23(5):1156–66.
Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–38.
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8(355):355ra116.
Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified t cells after failure of ibrutinib. J Clin Oncol. 2017;35(26):3010–20.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor t cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–54.
Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med. 2020;26(12):1878–87.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel car t-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Taraseviciute A, Tkachev V, Ponce R, Turtle CJ, Snyder JM, Liggitt HD, et al. Chimeric antigen receptor T cell-mediated neurotoxicity in nonhuman primates. Cancer Discov. 2018;8(6):750–63.
Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697–709.
Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediat Inflamm. 2015;2015:568543.
Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178(1):39–48.
Menèndez Iglesias B, Cerase J, Ceracchini C, Levi G, Aloisi F. Analysis of B7-1 and B7-2 costimulatory ligands in cultured mouse microglia: upregulation by interferon-gamma and lipopolysaccharide and downregulation by interleukin-10, prostaglandin E2 and cyclic AMP-elevating agents. J Neuroimmunol. 1997;72(1):83–93.
Topp MS, Gökbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66.
Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. 2012;33(1):116–25.
McCusker RH, Kelley KW. Immune-neural connections: how the immune system's response to infectious agents influences behavior. J Exp Biol. 2013;216(Pt 1):84–98.
de Rooij SE, van Munster BC, Korevaar JC, Levi M. Cytokines and acute phase response in delirium. J Psychosom Res. 2007;62(5):521–5.
Heyes MP, Saito K, Major EO, Milstien S, Markey SP, Vickers JH. A mechanism of quinolinic acid formation by brain in inflammatory neurological disease. Attenuation of synthesis from L-tryptophan by 6-chlorotryptophan and 4-chloro-3-hydroxyanthranilate. Brain. 1993;116(Pt 6):1425–50.
Pemberton LA, Kerr SJ, Smythe G, Brew BJ. Quinolinic acid production by macrophages stimulated with IFN-gamma, TNF-alpha, and IFN-alpha. J Interf Cytokine Res. 1997;17(10):589–95.
Tavares RG, Tasca CI, Santos CE, Alves LB, Porciúncula LO, Emanuelli T, et al. Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem Int. 2002;40(7):621–7.
Clark IA, Vissel B. Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J Neuroinflammation. 2016;13(1):236.
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–94.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–8.
Peri F, Nüsslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133(5):916–27.
Pennell CA, Barnum JL, McDonald-Hyman CS, Panoskaltsis-Mortari A, Riddle MJ, Xiong Z, et al. Human CD19-targeted mouse t cells induce B cell aplasia and toxicity in human CD19 transgenic mice. Mol Ther. 2018;26(6):1423–34.
Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature. 2019;568(7751):187–92.
Hu Y, Sun J, Wu Z, Yu J, Cui Q, Pu C, et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J Hematol Oncol. 2016;9(1):70.
Frey NV, Shaw PA, Hexner EO, Pequignot E, Gill S, Luger SM, et al. Optimizing chimeric antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol. 2020;38(5):415–22.
Ortíz-Maldonado V, Rives S, Castellà M, Alonso-Saladrigues A, Benítez-Ribas D, Caballero-Baños M, et al. CART19-BE-01: a multicenter trial of ARI-0001 cell therapy in patients with CD19(+) relapsed/refractory malignancies. Mol Ther. 2021;29(2):636–44.
Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019;25(9):1408–14.
Myers RM, Li Y, Barz Leahy A, Barrett DM, Teachey DT, Callahan C, et al. Humanized CD19-targeted chimeric antigen receptor (CAR) T cells in CAR-naive and CAR-exposed children and young adults with relapsed or refractory acute lymphoblastic leukemia. J Clin Oncol. 2021;39(27):3044-55.
Tan AHJ, Vinanica N, Campana D. Chimeric antigen receptor-T cells with cytokine neutralizing capacity. Blood Adv. 2020;4(7):1419–31.
Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452–65.
Muller YD, Nguyen DP, Ferreira LMR, Ho P, Raffin C, Valencia RVB, et al. The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front Immunol. 2021;12:639818.
Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25(6):947–53.
Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–59.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. 2020;26(2):270–80.
Ying Z, He T, Wang X, Zheng W, Lin N, Tu M, et al. Parallel comparison of 4-1BB or CD28 co-stimulated CD19-targeted CAR-T cells for B cell non-Hodgkin's lymphoma. Mol Ther Oncolytics. 2019;15:60–8.
Zhao X, Yang J, Zhang X, Lu XA, Xiong M, Zhang J, et al. Efficacy and safety of CD28- or 4-1BB-based CD19 CAR-T cells in B cell acute lymphoblastic leukemia. Mol Ther Oncolytics. 2020;18:272–81.
Sun C, Shou P, Du H, Hirabayashi K, Chen Y, Herring LE, et al. THEMIS-SHP1 recruitment by 4-1BB tunes LCK-mediated priming of chimeric antigen receptor-redirected T cells. Cancer Cell. 2020;37(2):216–25.e6.
Davey AS, Call ME, Call MJ. The influence of chimeric antigen receptor structural domains on clinical outcomes and associated toxicities. Cancers (Basel). 2020;13(1):38.
Ramello MC, Benzaïd I, Kuenzi BM, Lienlaf-Moreno M, Kandell WM, Santiago DN, et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci Signal. 2019;12(568):eaap9777.
Ng YY, Tay JCK, Li Z, Wang J, Zhu J, Wang S. T cells expressing NKG2D CAR with a DAP12 signaling domain stimulate lower cytokine production while effective in tumor eradication. Mol Ther. 2021;29(1):75–85.
Wu W, Zhou Q, Masubuchi T, Shi X, Li H, Xu X, et al. Multiple signaling roles of CD3ε and its application in CAR-T cell therapy. Cell. 2020;182(4):855–71.e23.
James JR. Tuning ITAM multiplicity on T cell receptors can control potency and selectivity to ligand density. Sci Signal. 2018;11(531):eaan1088.
Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25(1):82–8.
Kang L, Tang X, Zhang J, Li M, Xu N, Qi W, et al. Interleukin-6-knockdown of chimeric antigen receptor-modified T cells significantly reduces IL-6 release from monocytes. Exp Hematol Oncol. 2020;9:11.
Giordano-Attianese G, Gainza P, Gray-Gaillard E, Cribioli E, Shui S, Kim S, et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat Biotechnol. 2020;38(4):426–32.
Jan M, Scarfò I, Larson RC, Walker A, Schmidts A, Guirguis AA, et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci Transl Med. 2021;13(575):eabb6295.
Juillerat A, Tkach D, Busser BW, Temburni S, Valton J, Duclert A, et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 2019;19(1):44.
Park S, Pascua E, Lindquist KC, Kimberlin C, Deng X, Mak YSL, et al. Direct control of CAR T cells through small molecule-regulated antibodies. Nat Commun. 2021;12(1):710.
Serafini M, Manganini M, Borleri G, Bonamino M, Imberti L, Biondi A, et al. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum Gene Ther. 2004;15(1):63–76.
Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. 2016;126(11):4262–72.
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–83.
Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105(11):4247–54.
Foster MC, Savoldo B, Lau W, Rubinos CA, Grover N, Armistead PM, et al. Utility of safety switch to abrogate CD19.CAR T cell-associated neurotoxicity. Blood. 2021;137(23):3306–9.
Tiberghien P, Ferrand C, Lioure B, Milpied N, Angonin R, Deconinck E, et al. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood. 2001;97(1):63–72.
Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107(6):2294–302.
Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–47.
Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1(1):26–31.
US Food and Drug Administration: FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. (2017). https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome. Accessed 11 Oct 2021.
Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017;45(2):e124–e31.
Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood. 2019;134(24):2149–58.
Varadarajan I, Kindwall-Keller TL, Lee DW. In: Lee DW, Shah NN, editors. Chapter 5 - management of cytokine release syndrome. Amsterdam: Elsevier; 2020. p. 45–64.
Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat Rev Clin Oncol. 2019;16(1):45–63.
Locke FL, Neelapu SS, Bartlett NL, Lekakis LJ, Jacobson CA, Braunschweig I, et al. Preliminary results of prophylactic tocilizumab after axicabtageneciloleucel (axi-cel; KTE-C19) treatment for patients with refractory, aggressive Non-Hodgkin Lymphoma (NHL). Blood. 2017;130(Suppl 1):1547.
Kadauke S, Myers RM, Li Y, Aplenc R, Baniewicz D, Barrett DM, et al. Risk-adapted preemptive tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric B-cell acute lymphoblastic leukemia: a prospective clinical trial. J Clin Oncol. 2021;39(8):920–30.
Caimi PF, Ahmed N, Ojas PR, Andrade L, Cytotherapy MLJ. Prophylactic tocilizumab before CD3/4-1bb anti-CD19 car-T cell infusion decreases incidence of severe crs without increased risk of neurotoxicity. Cytotherapy. 2020;22(5):S16–7.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95.
Shah B, Huynh V, Sender LS, Lee DW III, Castro JE, Wierda WG, et al. High rates of minimal residual disease-negative (mrd−) complete responses (cr) in adult and pediatric and patients with relapsed/refractory acute lymphoblastic leukemia (r/r all) treated with KTE-C19 (anti-CD19 chimeric antigen receptor [CAR] T cells): preliminary results of the ZUMA-3 and ZUMA-4 trials. Blood. 2016;128(22):2803.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25.
Karschnia P, Jordan JT, Forst DA, Arrillaga-Romany IC, Batchelor TT, Baehring JM, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood. 2019;133(20):2212–21.
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62.
Gust J, Ceppi F, Turtle CJ. In: Lee DW, Shah NN, editors. Chapter 7-Neurotoxicities after CAR T-cell immunotherapy. Amsterdam: Elsevier; 2020. p. 83–105.
Maus MV, Alexander S, Bishop MR, Brudno JN, Callahan C, Davila ML, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer. 2020;8(2):e001511.
Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T-cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433–44.
Lanza L, Scudeletti M, Puppo F, Bosco O, Peirano L, Filaci G, et al. Prednisone increases apoptosis in in vitro activated human peripheral blood T lymphocytes. Clin Exp Immunol. 1996;103(3):482–90.
Strati P, Ahmed S, Furqan F, Fayad LE, Lee HJ, Iyer SP, et al. Prognostic impact of corticosteroids on efficacy of chimeric antigen receptor T-cell therapy in large B-cell lymphoma. Blood. 2021;137(23):3272–6.
Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. 2020;10(2):15.
Shah BD, Bishop MR, Oluwole OO, Logan AC, Baer MR, Donnellan WB, et al. KTE-X19 anti-CD19 CAR T-cell therapy in adult relapsed/refractory acute lymphoblastic leukemia: ZUMA-3 phase 1 results. Blood. 2021;138(1):11–22.
Topp M, Van Meerten T, Houot R, Minnema MC, Milpied N, Lugtenburg PJ, et al. Earlier steroid use with Axicabtagene Ciloleucel (Axi-Cel) in patients with relapsed/refractory large B cell lymphoma. Blood. 2019;134(Suppl 1):243.
Oluwole OO, Bouabdallah K, Muñoz J, De Guibert S, Vose JM, Bartlett NL, et al. Prophylactic corticosteroid use in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol. 2021;194(4):690–700.
Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448–57.
Fox E, Jayaprakash N, Pham TH, Rowley A, McCully CL, Pucino F, et al. The serum and cerebrospinal fluid pharmacokinetics of anakinra after intravenous administration to non-human primates. J Neuroimmunol. 2010;223(1–2):138–40.
Strati P, Ahmed S, Kebriaei P, Nastoupil LJ, Claussen CM, Watson G, et al. Clinical efficacy of anakinra to mitigate CAR T-cell therapy-associated toxicity in large B-cell lymphoma. Blood Adv. 2020;4(13):3123–7.
Humanigen: Humanigen reports positive data with lenzilumab in the ZUMA-19 CAR-T Phase 1b study in DLBCL and plans to initiate a potential registrational study. (2021). https://ir.humanigen.com/news/news-details/2021/Humanigen-Reports-Positive-Data-With-Lenzilumab-in-the-ZUMA-19-CAR-T-Phase-1b-Study-in-DLBCL-and-Plans-to-Initiate-a-Potential-Registrational-Study/default.aspx. Accessed 22 Aug 2021.
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O'Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16(12):843–62.
O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–50.
Guschin D, Rogers N, Briscoe J, Witthuhn B, Watling D, Horn F, et al. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. EMBO J. 1995;14(7):1421–9.
Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93(3):373–83.
US Food and Drug Administration: FDA approves treatment for patients with rare bone marrow disorder. (2019). https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-patients-rare-bone-marrow-disorder. Accessed 11 Oct 2021.
US Food and Drug Administration: FDA approves ruxolitinib for acute graft-versus-host disease. (2019). https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ruxolitinib-acute-graft-versus-host-disease. Accessed 11 Oct 2021.
Pan J, Deng B, Ling Z, Song W, Xu J, Duan J, et al. Ruxolitinib mitigates steroid-refractory CRS during CAR T therapy. J Cell Mol Med. 2021;25(2):1089–99.
Li S, Wang X, Yuan Z, Liu L, Luo L, Li Y, et al. Eradication of T-ALL cells by CD7-targeted universal CAR-T cells and initial test of Ruxolitinib-based CRS management. Clin Cancer Res. 2021;27(5):1242–6.
Huarte E, O'Connor RS, Peel MT, Nunez-Cruz S, Leferovich J, Juvekar A, et al. Itacitinib (INCB039110), a JAK1 inhibitor, reduces cytokines associated with cytokine release syndrome induced by CAR T-cell therapy. Clin Cancer Res. 2020;26(23):6299–309.
Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M, Kiefer SE, et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006;66(11):5790–7.
Blake S, Hughes TP, Mayrhofer G, Lyons AB. The Src/ABL kinase inhibitor dasatinib (BMS-354825) inhibits function of normal human T-lymphocytes in vitro. Clin Immunol. 2008;127(3):330–9.
Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. 2019;11(499):eaau5907.
Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021;372(6537):eaba1786.
Long M, Beckwith K, Do P, Mundy BL, Gordon A, Lehman AM, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest. 2017;127(8):3052–64.
Ferrarini I, Rigo A, Montresor A, Laudanna C, Vinante F. Monocyte-to-macrophage switch reversibly impaired by Ibrutinib. Oncotarget. 2019;10(20):1943–56.
Ruella M, Kenderian SS, Shestova O, Klichinsky M, Melenhorst JJ, Wasik MA, et al. Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia. 2017;31(1):246–8.
Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood. 2020;135(19):1650–60.
Kenderian SS, Ruella M, Shestova O, Kim M, Klichinsky M, Chen F, et al. Ruxolitinib prevents cytokine release syndrome after CAR T-cell therapy without impairing the anti-tumor effect in a xenograft model. Blood. 2016;128(22):652.
Deng T, Tang C, Zhang G, Wan X. DAMPs released by pyroptotic cells as major contributors and therapeutic targets for CAR-T-related toxicities. Cell Death Dis. 2021;12(1):129.
Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6:128.
Li Y, Huang H, Liu B, Zhang Y, Pan X, Yu XY, et al. Inflammasomes as therapeutic targets in human diseases. Signal Transduct Target Ther. 2021;6(1):247.
Gurnik S, Devraj K, Macas J, Yamaji M, Starke J, Scholz A, et al. Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol. 2016;131(5):753–73.
Pellegatta F, Lu Y, Radaelli A, Zocchi MR, Ferrero E, Chierchia S, et al. Drug-induced in vitro inhibition of neutrophil-endothelial cell adhesion. Br J Pharmacol. 1996;118(3):471–6.
Dong L, Chang L-J, Gao Z, Lu D-P, Zhang J-P, Wang J-B, et al. Chimeric antigen receptor 4SCAR19-modified T cells in acute lymphoid leukemia: a phase II multi-center clinical trial in China. Blood. 2015;126(23):3774.
Zhang L, Wang S, Xu J, Zhang R, Zhu H, Wu Y, et al. Etanercept as a new therapeutic option for cytokine release syndrome following chimeric antigen receptor T cell therapy. Exp Hematol Oncol. 2021;10(1):16.
Chen Y, Li R, Shang S, Yang X, Li L, Wang W, et al. Therapeutic potential of TNFα and IL1β blockade for CRS/ICANS in CAR-T therapy via ameliorating endothelial activation. Front Immunol. 2021;12:623610.
Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. 2020;382(19):1811–22.
Futami M, Suzuki K, Kato S, Ohmae S, Tahara Y, Nojima M, et al. The novel multi-cytokine inhibitor TO-207 specifically inhibits pro-inflammatory cytokine secretion in monocytes without affecting the killing ability of CAR T cells. PLoS One. 2020;15(4):e0231896.
Wei Y, Li C, Bian H, Qian W, Jin K, Xu T, et al. Targeting CDK7 suppresses super enhancer-linked inflammatory genes and alleviates CAR T cell-induced cytokine release syndrome. Mol Cancer. 2021;20(1):5.
Stahl K, Schmidt BMW, Hoeper MM, Skripuletz T, Möhn N, Beutel G, et al. Extracorporeal cytokine removal in severe CAR-T cell associated cytokine release syndrome. J Crit Care. 2020;57:124–9.
Xiao X, He X, Li Q, Zhang H, Meng J, Jiang Y, et al. Plasma exchange can be an alternative therapeutic modality for severe cytokine release syndrome after chimeric antigen receptor-T cell infusion: a case report. Clin Cancer Res. 2019;25(1):29–34.
Liu Y, Chen X, Wang D, Li H, Huang J, Zhang Z, et al. Hemofiltration successfully eliminates severe cytokine release syndrome following CD19 CAR-T-cell therapy. J Immunother. 2018;41(9):406–10.
Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573(7774):430–3.
Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127–32.
Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol. 2018;18(10):605–16.
Orvain C, Boulch M, Bousso P, Allanore Y, Avouac J. Is there a place for CAR-T cells in the treatment of chronic autoimmune rheumatic diseases? Arthritis Rheumatol. 2021. https://doi.org/10.1002/art.41812.
Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–31.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–52.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–42.
Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267–80.
Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141.
Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129(6):2210–21.
Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–37.
Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–16.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139.
Frey NV, Gill S, Hexner EO, Schuster S, Nasta S, Loren A, et al. Long-term outcomes from a randomized dose optimization study of chimeric antigen receptor modified T cells in relapsed chronic lymphocytic leukemia. J Clin Oncol. 2020;38(25):2862–71.
Alvi RM, Frigault MJ, Fradley MG, Jain MD, Mahmood SS, Awadalla M, et al. Cardiovascular events among adults treated with chimeric antigen receptor T-cells (CAR-T). J Am Coll Cardiol. 2019;74(25):3099–108.
Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood. 2018;131(1):121–30.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–9.
Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86–104.
Ruark J, Mullane E, Cleary N, Cordeiro A, Bezerra ED, Wu V, et al. Patient-reported neuropsychiatric outcomes of long-term survivors after chimeric antigen receptor T cell therapy. Biol Blood Marrow Transplant. 2020;26(1):34–43.
Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6(6):664–79.
Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR T cell-associated neurotoxicity. Front Immunol. 2020;11:577027.
Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Müller-Tidow C, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. 2021;32(1):34–48.
Neelapu SS. Managing the toxicities of CAR T-cell therapy. Hematol Oncol. 2019;37(Suppl 1):48–52.
Zhang JP, Zhang R, Tsao ST, Liu YC, Chen X, Lu DP, et al. Sequential allogeneic and autologous CAR-T-cell therapy to treat an immune-compromised leukemic patient. Blood Adv. 2018;2(14):1691–5.
Acknowledgements
XXu, XXiao, SH, SC, YW, QS are the Small Small bird (SSb) group members and acknowledge the strong support from SSb.
Funding
This research was supported by grants from the Clinical Research Startup Program of Southern Medical University by High-level University Construction Funding of Guangdong Provincial Department of Education (No. LC2016ZD027), the National Key Research and Development Program of China (No. 2017YFA0105503), Frontier Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (No. 2018GZR110105014), Basic and Applied Basic Research Fund of Guangdong Province (No. 2019A1515111113), China Postdoctoral Science Foundation (No. 2020 M682626), Special Funds for the Cultivation of Guangdong College Students’ Scientific and Technological Innovation (No. pdjh2021b0101), Guangdong Students’ Platform for Innovation and Entrepreneurship Training Program (No. S202112121059).
Author information
Authors and Affiliations
Contributions
XXu and YL contributed to conception and manuscript design. XXiao, SH, SC and XXu drafted the manuscript. YW, QS, SC, SH and XXiao prepared the tables and figures. SH and SC participated in data collection. XXu, XXiao, SH and SC participated in the revision of the manuscript. XXu and YL were involved in funding acquisition. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xiao, X., Huang, S., Chen, S. et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J Exp Clin Cancer Res 40, 367 (2021). https://doi.org/10.1186/s13046-021-02148-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02148-6