
Abstract
The advent of chimeric antigen receptor T cells (CAR-T) has been a paradigm shift in cancer immunotherapeutics, with remarkable outcomes reported for a growing catalog of malignancies. While CAR-T are highly effective in multiple diseases, salvaging patients who were considered incurable, they have unique toxicities which can be life-threatening. Understanding the biology and risk factors for these toxicities has led to targeted treatment approaches which can mitigate them successfully. The three toxicities of particular interest are cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and immune effector cell-associated hemophagocytic lymphohistiocytosis (HLH)-like syndrome (IEC-HS). Each of these is characterized by cytokine storm and hyperinflammation; however, they differ mechanistically with regard to the cytokines and immune cells that drive the pathophysiology. We summarize the current state of the field of CAR-T-associated toxicities, focusing on underlying biology and how this informs toxicity management and prevention. We also highlight several emerging agents showing promise in preclinical models and the clinic. Many of these established and emerging agents do not appear to impact the anti-tumor function of CAR-T, opening the door to additional and wider CAR-T applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The earliest clinical reports of chimeric antigen receptor T cells (CAR-T) described a syndrome of fevers, sometimes accompanied by hypoxia, hypotension, and cytopenias, termed cytokine release syndrome (CRS). The first patient who had sustained remission after CAR-T experienced a syndrome of fever, chills, and fatigue beginning two days after infusion and peaking over the subsequent 5 days, conincident with CAR-T cell expansion [1]. A subsequent report described 10 patients with relapsed/refractory B cell leukemias infused with CD19-directed CAR-T, some of the first patients to be treated with this novel therapy: 7 of 9 patients had fever, 3 developed hypotension, and one patient died of a sepsis-like picture with hypotension and renal failure [2]. The first pediatric patient to receive CAR-T received a CD19-directed product for treatment of B cell acute lymphoblastic lymphoma (B-ALL), and developed a syndrome of high-spiking fevers with progression to respiratory and cardiovascular failure, requiring mechanical ventilation and multiple vasopressors [3]. This patient had numerous cytokine elevations, including interleukin-6 (IL-6), interferon-gamma (IFNγ), and IL-10, and clinically did not respond significantly to steroids or etanercept but had a dramatic improvement after she received tocilizumab and remains disease-free now 10 years after infusion. Despite these significant toxicities, CAR-T products have achieved high complete response rates in multiple types of cancer including several pediatric and adult hematologic malignancies [4].
Although the advent of CAR-T therapy has marked a revolution in the treatment of certain cancers, it has also brought unique and serious toxicity profiles, distinct from toxicities seen with traditional cytotoxic therapies. CRS is now recognized as the most common potentially life-threatening CAR-T toxicity and is broadly characterized by inflammation resulting in fever and capillary leak leading to hypotension and hypoxia. The second most common potentially life-threatening toxicity, immune effector cell-associated neurotoxicity syndrome (ICANS), is marked by confusion, ataxia, cranial nerve palsies, and seizure and is hypothesized to be related to cytokine dysregulation in the CNS [5]. An emergent CAR-T toxicity, termed immune effector cell-associated hemophagocytic lymphohistiocytosis (HLH)-like syndrome (IEC-HS), is typified by late onset hyperinflammation with high ferritin, coagulopathy, cytopenias, and multiorgan failure [6]. IEC-HS has only recently been recognized as a distinct entity to underscore its independence from previously described syndromes, requiring discrete diagnostic and management guidelines [6]. These toxicities are due in large part to cytokine dysregulation occurring following CAR-T. They can all occur to varying degrees in other immune effector cell (IEC) therapies, including bispecific T-cell engagers (BiTE) which target endogenous T cells to cancer cells. There is also a growing catalog of adoptive cell therapies utilizing non-T cell effectors, for example NK cells, which may ultimately be found to create their own patterns of toxicities based on their different cellular biology.
The aim of this manuscript is to summarize the current understanding of the biology of these toxicities, the most up to date and evidence-based consensus guidelines for their management, highlight emerging management options, and identify gaps in our understanding of CAR-T mediated toxicities. A few of these emerging toxicity-mitigating therapies have the potential to revolutionize outcomes with CAR-T therapy, paving the way for further CAR-T development and application. This will also lay the groundwork for readers to understand the current major unanswered questions in the field (Box 1).
Cytokine release syndrome
The American Society for Transplantation and Cellular Therapy (ASTCT) recently convened a panel of experts from a variety of disciplines to create a unifying definition and grading system for CRS and ICANS [7]. Prior to this meeting, there were a handful of disparate definitions and grading systems that were developed as the field of CAR-T-related toxicity has matured, and the lack of uniformity created problems in comparing outcomes and toxicities between studies. CRS is currently defined as “a supraphysiologic response following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells. Symptoms can be progressive, must include fever at the onset, and may include hypotension, capillary leak (hypoxia) and end organ dysfunction” [7].
CRS grading
The grading scheme devised in the ASTCT consensus statement inclues fever as a necessary feature of all grades of CRS, and the maximum severity of hypotension or hypoxia defines the grade [7, 8].
-
Grade 1: fever without hypotension or hypoxia, can be associated with constitutional symptoms such as myalgia and malaise.
-
Grade 2: fever with hypotension and/or hypoxia requiring minimal support, such as fluids and low-flow nasal cannula, respectively.
-
Grade 3: hypotension requiring one vasopressor and/or respiratory distress requiring high-flow nasal cannula or facemask.
-
Grade 4: hypotention requiring more than one vasopressor (excluding vasopressin) and/or hypoxia requiring positive pressure ventilation including intubation.
-
Grade 5: death.
CRS incidence
The incidence of CRS is impacted by a number of important variables, including the co-stimulatory domain used in the CAR construct, the target antigen of the CAR, ex vivo processing/culturing parameters, ex vivo T cell selection processes, and the biology of the target tumor itself including tumor burden. Currently, the most widely used CAR-T products are tisagenlecleucel (tisa-cel), axicabtagene ciloleucel (axi-cel), and brexucabtagene autleucel (KTE-X19), all CD19-targeted products that differ in their costimulatory domain with tisa-cel using 4-1BB and axi-cel and KTE-X19 using CD28 [9, 10]. The two landmark trials of CAR-T for relapsed/refractory large B cell lymphoma, ZUMA-1 and JULIET, treated patients with axi-cel and tisa-cel, respectively, and found different frequencies of CRS: on ZUMA-1 93% of patients had any grade of CRS with 13% of patients experiencing severe (grade >=3) CRS, while on JULIET 58% of patients had any grade of CRS with 22% of patients having severe symptoms [11, 12]. The numbers of patients treated for CRS also differed, with 43% of patients receiving IL-6 blockade and 27% receiving steroids on ZUMA-1 compared to 14% and 10%, respectively, on JULIET. The trials ZUMA-2 and ZUMA-3 tested KTE-X19 for relapsed/refractory (r/r) mantle cell lymphoma and r/r adult B-ALL, respectively, and found similar rates of CRS (91% and 89% with any grade CRS, 15% and 24% of patients with severe CRS, respectively) [13, 14]. In a more recent large adult cohort comparing axi-cel and tisa-cel for the treatment of diffuse large B cell lymphoma (DLBCL), CRS occurred in 86% of patients treated with axi-cel (6% of those being severe) versus 76% of patients treated with tisa-cel (12% of those being severe) [15]. Conversely, for non-Hodgkins lymphoma treated with axi-cel on the ZUMA-5 trial, rates were much lower with only 9% of patients experiencing grade >=3 CRS [16]. Practices for what interventions to take for CAR-T related toxicities, when to act, and ways to modulate disease-specific characteristics (e.g. reduction in tumor burden) have changed over time and have likely influenced toxicity incidence.
CRS biology and markers
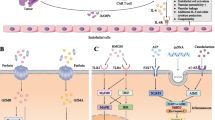
The current understanding of CRS is that CAR-T engagement leads to proliferation of T cells and secretion of pro-inflammatory cytokines that futher activate CAR-T as well as the host immune system, causing symptoms such as fever and off-target tissue damage. Extensive studies have measured the cytokine profiles of CRS with CAR-T and have identified the signaling pathways that are dysregulated in this clinical syndrome (Fig. 1). The importance of IL-6 in CRS is well established [17,18,19,20,21], and IFNγ is increasingly being recognized as a key cytokine in not just CRS but other CAR-T mediated toxicities [20, 22,23,24,25,26]. In general, cytokines released by activated T cells are elevated in CRS, specifically IL-6, IFNγ, soluble IL-2 receptor (sIL-2R), sIL-2Ra, and granulocyte-monocyte colony stimulating factor (GM-CSF). Activated monocytes/macrophages secrete high levels of cytokines in turn, including IL-1Ra, IL-10, IL-6, CXC motif chemokine ligand 9/monokine induced by gamma (CXCL9; MIG), CXCL10 (IP-10), IFNa, macrophage-inflammatory protein-1 alpha (MIP1a), MIP1b, and sIL-6R [5, 27].

Schematic detailing the current understanding of the pathophysiology of CRS and ICANS caused by cross-talk between CAR-T cells and macrophages. Several of the current and emerging therapies for CRS and ICANS are shown in their mechanism of action. Created with biorender.com
In comparing patients with mild (grade 0-3) CRS to those with severe (grade 4-5) CRS in patients treated with a 4-1BB CD19-directed CAR-T product, Teachey et al. found IFNγ, IL-6, IL-8, sIL-2R, sgp130, sIL-6R, MCP1, MIP1α, MIP1β, and GM-CSF to be significantly associated with the more severe grades [20]. Similarly, ferritin and C-reactive protein (CRP) were significantly higher in severe CRS, and notably ferritin was >10,000 mg/dL in every patient with severe CRS. Hypofibrinogenemia was also found in patients with severe CRS. This laboratory profile is nearly identical to that seen in patients with macrophage activation syndrome (MAS)/HLH, for which a value of ferritin >10,000 is considered sensitive and specific, drawing a striking parallel between the two syndromes [20].
An important concept of CAR-T therapy is that many of the pathways that lead to toxicities are not the same as those involved in target cell killing. This is apparent in the finding that the development of toxicity does not correlate with clinical response; while both are outcomes of CAR-T cell activity, they are not mutually necessary. For example, a retrospective analysis of hundreds of adults treated with axi-cel or tisa-cel compared those who developed CRS with those who did not and found no difference in progression free survival (P=0.99) or overall survival (P=0.16) [28].
CRS predictors
Several factors have been found to be predictive of CRS severity, and can broadly be categorized into disease characteristics, therapy characteristics, and laboratory parameters. Disease burden has long been identified as being correlated with severity of toxicity, with higher tumor burden and bulky disease being associated with worse symptoms [26, 29,30,31,32]. In one study of pediatric patients with B-ALL receiving CAR-T, low disease burden had a strong negative predictive value (1 of 15 patients with <5% blasts in bone marrow developed severe CRS), however, disease burden alone was not sufficient to predict which patients will develop severe CRS (10 of 23 patients with >25% disease in bone marrow developed severe CRS) [20]. Highly proliferative malignancies such as B-ALL and DLBCL are associated with more severe CRS when compared to less active diseases including follicular lymphoma and myeloma. However, calculating the growth rate of lymphomas prior to CAR-T infusion was found to be not predictive of toxicity [33], implying that high proliferation is not necessarily associated with a higher risk of CRS [34]. Age within adulthood has not been found to be associated with toxicity rates [35,36,37].
Higher CAR-T doses are predictive of worse symptoms [38], and specific costimulatory domains, such as CD28 (eg. axi-cel), have higher rates of CRS and neurotoxicity than those with 4-1BB (eg. tisa-cel) [15]. The CAR itself can play a role in the development of CRS based on its molecular characteristics. For example, manipulating the structure of CD8α domain of the CAR, while keeping the 4-1BB/CD28 and CD3ζ signaling domains unchanged was found to preserve anti-tumor efficacy in patients with B cell lymphoma while significantly reducing the incidence CRS and neurotoxicity [39, 40]. Separately, lowering the affinity of the antibody-derived domain of the CAR resulted in significantly lower rates of CRS and neurotoxicity with preserved clinical efficacy in a small cohort of pediatric patients with B-ALL [41]. The CAR-T target also plays a role in toxicity profiles based on on-target off-tumor effects. For example, neurotoxicity may be seen more frequently with CD19-directed agents due to the presence of CD19 on mural cells in the brain [42], whereas the unique movement-related toxicities seen with BCMA-directed CAR-T products and may be attributable to BCMA expression in separate areas of the CNS [43].
While cytokine levels have not been found to correlate with CRS severity [44], a number of schema for predicting which patients will develop severe or non-severe CRS have been developed based on retrospective analyses [17, 45, 46]. A recent proteomic analysis identified elevations in MILR1, a negative regulator of hypersensitivity reactions, to be predictive of CRS with high sensitivity and sepecificity (97, 88%) [20]. Pennisi et al. modified the Endothelial Activation and Stress Index (EASIX) score, used to predict outcomes in HSCT, by replacing creatinine with CRP to create the formula lactate dehydrogenase (LDH) x CRP/platelet count and found good association between development of severe CRS and ICANS and this modified score [47]. In a unique approach, Jalota et al explored pretreatment metabolic parameters and found that higher glucose, lower glutamine, and lower cholesterol were associated with earlier onset CRS [48]. While several of these boast remarkable specificities/sensitivities, their utility in prospective risk stratification is yet to be determined.
CRS therapies
While it is clearly established that cytokines play a role in CRS, cytokines are also necessary for T cell function and proliferation, and so the effect on the anti-cancer action of T cells by cytokine blockade has been a major concern. Fortunately, several agents have been found to have mitigating effects on cytokine-related toxicity (Table 1) without dampening cancer cell killing. The IL-6 receptor antagonist tocilizumab is a highly effective and the longest-used treatment for CRS [49], and has been shown to have no effect on CAR-T cell expansion [51]. Notably, tocilizumab administration has not been associated with any major adverse reactions of its own [49]. Tocilizumab is therefore firmly established as the first-line treatment for CRS [8] and can be redosed at 8-hour intervals. Siltuximab, a monoclonal antibody specific to IL-6, has been shown to have similar efficacy to tocilizumab; however, they have not been compared directly in any published trials [60, 73].
As tocilizumab has been found to both be a potent treatment for CRS and not impair the function of CAR-T cells, it follows that tocilizumab could be used to prevent CRS rather than treat it. A few trials have investigated using tocilizumab as a preemptive measure, initiating treatment at low-grade toxicity, and found that this strategy was effective in prevening the onset of severe CRS [51, 69, 70]. Separately, in one single-arm study 20 patients were given tocilizumab prior to CAR-T infusion and reported that there were no cases of severe CRS (grade 3 or higher), and while 5 patients had ICANS, 4 of these were grade 1 [74].
Corticosteroids (CS) are currently recommended as second-line therapy for CRS that persists or progresses after tocilizumab [8]. There has long been concern for CS having deleterious effect on CAR-T function and persistence. Some studies have suggested that CS are disadvantageous for CAR-T cell function [21, 75], however others have not identified any short-term deleterious impact from using CS [51, 52, 76]. Ultimately, it is likely that the effect of CS is a dose-dependent, and short-term use of steroids, even at high dose, were found in retrospective analysis to not affect outcomes of CAR-T therapy while cumulative doses were associated with shorter survival [75]. Earlier administration of CS is associated with shorter courses of CS being used [53], and early CS administration was found to prevent progression to severe CRS on the ZUMA-1 trial [76].
The agent with the most published clinical experience for refractory CRS is likely the IL-1 receptor antagonist anakinra. Preclinical models of CAR-T-derived CRS have found that monocyte/macrophage-derived IL-1 and IL-6 are both important in the pathophysiology of CRS and ICANS and that blockade of IL-1 with anakinra alone is effective in mitigating CRS and ICANS [54, 77]. Clinical data supporting the use of anakinra for severe CRS is growing, and prospective studies are underway for use of anakinra for treatment and prevention of CRS and ICANS [55]. Importantly anakinra does not appear to impact CAR-T efficacy [77, 78].
Several emerging therapies have shown promise for the treatment of CRS (Table 1), to date only reported in small numbers of patients. Based on the finding that IFNγ is elevated in CRS, and is a major avenue for activation of macrophages by T cells, the IFNγ-blocking antibody emapalumab has been utilized successfully for severe CRS [58, 79], and promisingly IFNγ blockade has not been found to compromise CAR-T function in vitro and in vivo in xenograft models of hematologic malignancies [59], although this has found to not hold true for solid tumors [46]. Ruxolitinib has been used in some patients [66], operating on the concept that many inflammatory cytokines signal through JAK/STAT pathways [80]. In one report of 4 patients, ruxolitinib resulted in rapid resolution of CRS symptoms and decrease in serum cytokines associated with CRS [67]. Both emapalumab and ruxolitinib are relatively well tolerated. Ruxolitinib can cause cytopenias [81, 82], and both agents increased the risk of viral reactivations [83, 84]. Another drawback of emapalumab is that it is quite expensive, however 1-2 doses have been sufficient in amelioriating severe CRS in published cases [24, 58].
TNFα blockade with etanercept was reported in three patients treated with BCMA-directed CAR-T [68]. The patients had good therapeutic response, and the authors note no apparent impact on CAR-T efficacy. Etanercept has been previously used with unclear efficacy in CRS related to other CAR-T [3]. GM-CSF levels were found to correlate with severe CRS and neurotoxicity on ZUMA-7 [85], which has sparked interest in the monoclonal antibody against GM-CSF, lenzilumab, for CRS and ICANS. In a preclinical model lenzilumab was found to suppress CAR-T toxicity without decreasing efficacy [86]. In a preventative approach, the single-arm phase 1 ZUMA-19 trial administered lenzilumab prior to axi-cel infusion for 6 adults with (r/r) large B cell lymphoma (LBCL) and no patient developed severe CRS and one developed grade>=3 neurotoxicity with no dose-limiting toxicities attributable to lenzilumab [72].
Also of interest are tyrosine kinase inhibitors which interrupt signal transduction through activating receptors including the CAR. For example, dasatinib has been shown to inhibit phosphorylation of CD3ζ and ZAP70, which blocks signaling through the CAR. This can be used to reversibly tune CAR signaling, and while it does reduce the anti-tumor effect of CAR-T its overall activity has been found to mitigate toxicity and improve CAR-T persistence [61,62,63]. Separately, the Bruton’s tyrosine kinase inhibitor ibrutinib, which inhibits IL-2 signaling, was administered concurrently with CD19-directed CAR-T in 19 patients and resulted in low CRS severity and high rates of disease response [64].
Certain CAR-T are being designed with so-called safety switches, or molecular mechanisms by which the CAR-T can be “turned off,” halting unacceptable toxicities. One approach incorporates an inducible caspase 9 fusion protein into CAR-T cells that dimerizes on exposure to the synthetic drug rimiducid, inducing apoptosis [87]. Numerous novel CAR-T products have been created more recently that incorporate different off switches, some of which are designed to be reversible; others have incorporated on switches, allowing for conditional activation only in the presensce of a specific agent [57]. Another approach that has been considered is T cell-depleting techniques such as antithymocyte globulin or alemtuzumab [88], or with cyclophosphamide [89, 90]. These agents are associated with increased infection risk and would be expected to eliminate CAR-T cells, and should only be considered in extreme circumstances.
Finally, extracorporeal adsorption of cytokines has been reported using Cytosorb®, which is a device designed for the treatment of cytokine storm and granted emergency approval for overwhelming systemic inflammation seen with COVID-19 infection. Cytosorb® has been reported in a single case in the setting of tocilizumab-refractory CRS with rapid response in clinical stability [91]. This has been used for severe refractory neurotoxicity as well [92]. A clinical trial is currently underway investigating the role of Cytosorb® for severe CRS (NCT04048434).
Immune effector cell-associated neurotoxicity
The second most common potentially life-threatening toxicity seen with CAR-T cells is ICANS, a spectrum of neurotoxicy symptoms which encompasses encephalopathy, delirium, motor dysfunction including tremor, ataxia, dysphagia, and seizure. The ASTCT consensus group has defined this broadly as “a disorder characterized by a pathologic process involving the central nervous system following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells. Symptoms or signs can be progressive and may include aphasia, altered level of consciousness, impairment of cognitive skills, motor weakness, seizures, and cerebral edema” [7].
ICANS was initially scored using CTCAE v4, but over time new grading systems have evolved to more specifically capture symptoms attributed specifically to CAR-T. The main challenge with grading IEC-related neurotoxicity is subjectivity regarding neurologic symptoms, especially encephalopathy, and so the CARTOX assessment was devised, then modified with incorporation of parts of the Mini-Mental State Examination into the CARTOX-10 encephalopathy assessment tool [27]. The CARTOX criteria include assessment of elevated ICP into grading, which can be difficult to measure as well as altered by multiple clinical variables, and so an ASTCT consensus group devised a further refined encephalopathy-scoring tool called the Immune Effector Cell-Associated Encephalopathy (ICE) score. ICE scores assess patients based on orientation, naming, command following, writing, and attention with points for each domain adding up to a maximum of 10 [7]. The ICE score is then added to assessments of additional domains of neurologic status: depressed consciousness, seizure, motor dysfunction, and cerebral edema. ICANS grading is summarized as follows [7, 8]:
-
Grade 1: ICE score >6 with preserved alertness
-
Grade 2: ICE score 3-6, mild somnolence but awakens to voice
-
Grade 3: ICE score 0-2, somnolence responsive to tactile stimulation, brief seizure responsive to intervention, and/or limited cerebral edema on imaging
-
Grade 4: ICE score 0, profound somnolence, life-threatening prolonged seizure or status epilepticus, diffuse cerebral edema, and/or symptomatic intracranial hypertension
-
Grade 5: death
In patients youngter than 12 years or with developmental delay the ICE score is replaced by the Cornell Assessment of Pediatric Delerium [8, 93].
ICANS incidence varies with the specific CAR-T product and disease type. The ZUMA-1 trial using axi-cel for relapsed LBCL found ICANS to occur in 64% of patients, with severe in 44% of those cases. The JULIET trial investigating tisa-cel for LBCL had lower rates of ICANS with 26% of patients having any grade of ICANS and 58% of those being severe [11, 12]. The ZUMA-2 and -3 trials investigating KTE-X19 found similar rates of ICANS at 63 and 60%, respectively, with approximately half of each being grade >=3 [13, 14]. An important feature of ICANS is that it has been found to occur at higher rates when using a CAR-T products containing the CD28 co-stimulatory domain, which is apparent when compairing tisa-cel to axi-cel and KTE-X19.
ICANS tends to occur later after infusion than CRS. For example, on the JULIET trial the median time of onset for CRS was 3 days, compared to 6 days for ICANS [12]. Similar timing is observed with axi-cel, where median onset of CRS on the ZUMA-1 trial was 2 days compared to 5 days for ICANS, and on the ZUMA-5 trial using axi-cel for non-Hodgkins lymphoma median onset of CRS was 4 days compared to 8 days for ICANS [11, 16]. Important risk factors for severe ICANS include high pre-infusion disease burden, history of neurologic disease, and development of severe CRS [25, 94, 95].
The biology of ICANS is not fully understood but does appear to be distinct from that of CRS despite both occurring following CAR-T administration. Investigations into the biological correlates of ICANS have identified serum levels of IL-6, IL-18, IFNγ, IL-8, IL-10, and TGFβ to be associated with severe toxicity [96], with increased IL-18 recently identified as a correlate [22]. In the CSF, it was found that concentrations of WBC and CAR-T cells alone did not correlate with neurotoxicity, however levels of IL-6, IL-8, MCP1, and IP10 did correspond [25]. Separately, elevations in the terminal complement complex soluble C5b-9 was found to be significantly associated with ICANS [22], which supports a role for vascular endothelial dysfunction causing blood brain barrier (BBB) disruption as suggested by others [25, 26]. Additionally, excitatory neurotransmitters glutamate and quinolinic acid were elevated in the CSF of patients with ICANS suggesting a mechanistic link to symptoms such as seizures and tremor [25].
Treatment regimens for ICANS (Table 2) are less well established than those for CRS. Tocilizumab is not effective for the treatment of ICANS [11, 25, 104], possibly because tocilizumab, similar to other monoclonal antibodies, does not readily cross the BBB [105]. CS are the primary therapy for ICANS, with expert opinion supporting the use of dexamethasone in particular due to CNS penetration, or methylprednisolone [8, 50, 90]. The ZUMA-1 and -3 trials found reduced incidence of severe ICANS with early CS initiation [14, 76]. For persistent neurotoxicity, dexamethasone is often switched to high-dose methylprednisolone. Increasing CS dosages raises the concern for causing CAR-T dysfunction, as well as significant side effects including metabolic derangements, hypertension, and infection.
Recent practice in the treatment of ICANS reflects the finding that IL-1 plays a significant role in CRS and ICANS [77]. Anakinra is increasingly being used successfully, particularly for ICANS refractory to first-line CS in both adult and pediatric populations [55, 97, 106], and when started early may allow for a more rapid taper of CS [98]. Given these experiences, anakinra is recommended as a second-line agent for ICANS [107], and is being investigated as a prophylactic agent to reduce or prevent toxicity with interim analyses reporting promising reductions in ICANS frequency and severity [71].
Numerous therapies for subsequent-line treatment of refractory ICANS have been proposed. Considering the challenge posed by the BBB, a few patients have been treated with intrathecal hydrocortisone with or without chemotherapy for systemic steroid-refractory disease. Shah et al report rapid and sustained resolution of severe ICANS with IT hydrocortisone and methotrexate with or without cytarabine in two patients [99], and others have demonstrated success with only IT hydrocortisone with or without chemotherapy, finding an overall reduction in total CS dose [100]. Wang et al. describe a patient who developed severe grade 4 cerebral edema after KTE-X19 who was treated with antithymocyte globulin (ATG) and found a rapid decline in CAR-T cell numbers in peripheral blood as well as IL-2 and IFNγ serum concentrations followed by clinical resolution of neurotoxicity within a week [102]. Surprisingly, the authors note that there was lower level CAR-T persistence followed by complete remission over the 24 months reported. Considering the potential role of endothelial dysregulation in ICANS, defibrotide has been investigated as an agent for stabilizing the endothelium. While initial results projected a significant reduction in ICANS rate, a phase 2 trial using defibrotide in a prophylactic role found more modest reductions in ICANS rate and duration, and the trial was terminated early for lack of efficacy [103].
There have now been several cases of a distinct syndrome of bradykinesia associated with limb rigidity, tremor, and other symptoms seen in Parkinson’s disease [108]. To date, all of these cases have occurred after treatment with CAR-T targeting BCMA, and have occurred weeks to months following infusion [32]. Concerningly, many of these cases have proven to be irreversible, although more recently two patients had recovery of symptoms after treatment with CS, anakinra, and high-dose cyclophosphamide [109]. Symptoms appear to correlate with CAR-T infiltration of the CNS, specifically the basal ganglia, suggesting this may be an on-target off-toxicity effect specific to BCMA [43].
Immune effector cell-associated hlh-like syndrome
Some patients with severe CRS develop a state of hyperinflammation that is accompanied by hyperferritinemia, cytopenias, hypofibrinogenemia, and multiorgan dysfunction. It was recognized early on that this was analogous clinically and biochemically with HLH [3, 20], and over time it was observed that some patients developed a similar HLH-like state either after initial improvement from CRS or after complete resolution of CRS. This state does not typically respond to tocilizumab in the same way as severe CRS and is associated with worse outcomes [110,111,112]. As greater numbers of patients with CAR-T-related toxicities were observed, this syndrome has been defined as an entity distinct from severe CRS based on its time course as it suggests a separate underlying biology.
HLH is defined as a disorder characterized fever, hepatosplenomegaly, organ failure, neurologic toxicities, coagulopathy, cytopenias, and hyperferritinemia, and is caused by immune activation due to dysregulated interactions between cytotoxic T cells, macrophages, and natural killer cells resulting in a cascade of cell- and cytokine-mediated tissue damage [113, 114]. HLH is separated into primary HLH (pHLH), caused by germline mutations in immune regulation, classically involving dysfunction in CD8 T cell or NK cells resulting in unchecked activation of macrophages, and secondary HLH (sHLH) characterized by HLH after an initial dysregulating disease such as an autoimmune disorder, EBV infection, malignancy, metabolic disorders, or overwhelming infection [115, 116]. A multidisciplinary team of experts recently defined the syndrome of HLH-like disease following immunotherapy as “Immune effector cell associated HLH-like syndrome,” or IEC-HS [6]. The incidence of IEC-HS has been reported to be ~3.5% across different CAR-T constructs and targets in adults, but varies with CAR-T construct, just as the persistence and propensity to cause CRS/ICANS varies between construct. As an example, a CD22-targeting CAR-T product using a 4-1BB costimuatory domain resulted in a HLH-like syndrome in 21 of 59 patients treated on one trial, much higher rates than previously reported following CART19 [117]. Unlike CRS, IEC-HS is associated with a poor prognosis [79]. Additionally, while the severity of IEC-HS has not been found to correlate with CRS severity, to date all cases of IEC-HS have occurred after or with resolving CRS [6].
The grading of IEC-HS has not been formally established, however the ASTCT consensus group outlines a grading scheme that will likely continue to evolve prior to being universally adopted. The general outlines of the IEC-HS grading system are as follows and adapted from expert panels [6, 90]:
•Grade 1: mild symptoms including fever, but clinical stability not requiring intervention
•Grade 2: mild to moderate symptoms such as hypotension responsive to fluids alone and/or hypoxia requiring low-flow nasal cannula, asymptomatic hypofibrinogenemia
•Grade 3: More severe symptoms including hypotension responsive to a vasopressor, respiratory distress requiring non-invasive support, coagulopathy with bleeding symptoms
•Grade 4: Severe, life-threatening toxicities including respiratory distress requiring intubation, hypotension requiring multiple vasopressors, and/or dialysis
•Grade 5: death
A distinctive feature of both severe CRS and IEC-HS is a very high ferritin, with values frequently greater than 10,000 ng/mL, a value that is considered sensitive and specific for MAS/HLH [20]. While some have proposed using a ferritin value of 10,000 ng/mL as diagnostic criteria [90], the ASTCT definition is more inclusive and delineates a level of twice the upper limit of normal, twice the ferritin for that patient at time of infusion, or rapidly rising ferritin, and note that while experience is insufficient to define a threshold level, a normal ferritin has a high negative predicative value. Additional diagnostic elements from the ASTCT are “features of macrophage activation/HLH, attributable to IEC therapy, and associated with progression or new onset of cytopenias, hyperferritinemia, coagulopathy with hypofibrinogenemia, and/or transaminitis.” There is also an emphasis placed on delayed onset, as this often manifests as CRS is resolving or has completely resolved [Fig. 2a]. Unlike CRS, fever is not required for the diagnosis of IEC-HS [6].

IEC-HS timing is disctinct from that of CRS in that it occurs as CRS is resolving or later, after CRS has resolved (A). Schematic detailing the current understanding of the pathophysiology of IEC-HS, inferred from lines of evidence in sHLH as well. The general concept is that CAR-T activation creates feed-forward loops with macrophages and endogenous T cells, activated by and resulting in cytokine secretion and tissue damage. The mechanisms of action of several proposed therapies for IEC-HS are incorporated into the schematic to illustrate how they may work to ameliorate this pathology (B). Created with biorender.com
While specific cytokine levels were not included in the recent ASTCT diagnostic criteria, cytokines likely play a substantial role in IEC-HS. IL-1b has been shown to be upregulated in patients with HLH-like disese after CAR-T [117] and anakinra has been shown to be an effective treatment for HLH/MAS [118, 119]. IFNγ is elevated in primary and secondary HLH [120,121,122], and essential for disease development in an animal model [123]. Emapalumab is approved for the treatment of pHLH [124, 125]. A model of the biology of IEC-HS can be proposed in which macrophages are activated by CAR-T via IFNγ signaling, as well as by damage signals from target cells being killed by CAR-T. Activated macrophages in turn recruit effector cells and more macrophages/monocytes and activate effectors (including endogenous CD8 T cells) via stimulatory cytokines such as IL-12, IL-18, and IL-6. This creates a feed forward loop resulting in widescale macrophage activation and proliferation causing collateral tissue damage, accompanied by high levels of ferritin and secretion of high levels of IL-1b and TNFα which further mediate tissue damage and immune cell activation (Fig. 2b). IFNγ blockade with emapalumab is approved for pHLH [126] and is being studied in MAS based on favorable outcomes in a pilot study of a small number of patients with refractory disease [127]. IFNγ elevation in IEC-HS has been reported [59, 79, 117]. The efficacy of emapalumab was demonstrated by Rainone et al. who described a patient with relapsed/refractory Ph-like B-ALL treated with tisa-cel who had CRS that improved within the first week after infusion, then developed persistent fevers, transaminitis, and ferritin levels that increased from 3,000 to 15,000 ng/mL over 24h before rising above the detectable range along with development of hemodynamic instability, coagulopathy, worsening cytopenias, hypertriglyceridemia, and hyperbilirubinemia, as well as a high IFNγ serum concentration despite repeated dosing of tocilizumab. The patient was initially treated with CS and anakinra with some clnical improvement, then within a few hours of receiving emapalumab the patient defervesced and stabilized, and anakinra and steroids were able to be weaned off over the subsequent 6 days [24]. CXCL9 is a useful biomarker for monitoring response to emapalumab [126, 128].
Historically, CS were first line for sHLH, however with the advent of newer targeted therapies it is attractive to limit CS use due to their side effects. Suggested therapies for IEC-HS include anakinra, often with CS, based on the established roles of both agents in sHLH. Given more recent description of this entity development of a consensus definition treatment guidelines for IEC-HS are evolving and several options have been used or proposed (Table 3). Growing evidence and experience supports the use of ruxolitinib, the Jak1/Jak2 inhibitor, capitalizing on the JAK/STAT signaling pathway being a common route for pro-inflammatory signals to exert their effects [136]. Ruxolitinib is used increasingly for frontline or salvage therapy in HLH [137, 138], and has been successfully used recently for refractory IEC-HS [112]. The ASTCT consensus group has ultimately advised consideration of ruxolitinib as second line for IEC-HS.
Additional options have been suggested for IEC-HS. IL-6-directed therapy with tocilizubmab or siltuxumab may be beneficial, although this could primarily apply only to IEC-HS with concomitant CRS. Targeted destruction of T cells with ATG and alemtuzumab is used in HLH [130, 132, 139], however such a stragety would be reserved for only the most refractory cases of IEC-HS. More targeted approaches of interest, largely theoretical at the time of this publication, include IL-18 blockade with tadakinig alfa which has limited experience in a subtype of pHLH [134], and is the subject of a trial for MAS therapy (NCT03512314), and TNFα blockade owing to the observation that this cytokine is frequently elevated in HLH and IEC-HS as well [135, 141, 140].
Based on experience in pHLH, the topoisomerase inhibitor etoposide has been used in IEC-HS, and is considered by some to be second-line [8, 90]. Etoposide abrogates effector cell activity through induction of apoptosis rather than more inflammatory cell death mechanisms like pyroptosis [142, 143]. However, avoidance of effector cell destruction is of course beneficial if possible, and etoposide causes increased risk of secondary malignancies. We do not recommend the use of etoposide outside of exceptional circumstances.
Ultimately, both early severe CRS and the later IEC-HS have clinical, biologic and pathophysiologic features highly overlapping with HLH, including high ferritin, low fibrinogen, multisystem organ failure, cytopenias, and hypercytokinemia with elevated IFNγ, IL6, and IL10. IEC-HS portends a worse prognosis, and is less likely to respond to therapies including tocilizumab. Future work is needed to unravel the mechanistic differences between these two entities to inform treatment, especially in refractory cases.
Conclusions
The field of CAR-T cell therapy has grown rapidly in the last 10 years, and our understanding of CAR-T associated toxicities has evolved with it. The definition of CRS has been refined, and now management recommendations can be definitively made through three lines of therapies, with several additional agents showing promise in refractory cases and preemptive strategies emerging as well. ICANS remains pooly understood biologically, although some progress has been made. While a better understanding of ICANS would be helpful for developing treatment strategies, advances in treatment have been made recently and IL-1 blockade with anakinra has emerged as a potent therapy. IEC-HS is a newly-recognized, and named, entity describing a later onset cytokine storm with organ dysfunction following CAR-T. A key feature of IEC-HS is timing: IEC-HS occurs as CRS is resolving or after CRS has resolved. However, it is not clear that this is a biological imperative and CRS and IEC-HS may not be mutually exclusive. Further elucidation of the biological mechanism of each process may help define criteria to better distinguish the two.
Future work should focus on comparative trials with patients randomized to alternative toxicity management strategies to more directly evaluate options and guide clinical decisionmaking. Determination of definitive recommendations is certain to be challenged by the expanding application of CAR-T cell products to more diseases. As new CAR-T products are developed with different features and targets specific immune toxicities may differ. A deeper understaning of the pathophysiology of CRS, ICANS, and IEC-HS will be crucial to achieve continued progress in toxicity amelioration and prevention.
Data availability
No datasets were generated or analyzed in the writing of this manuscript.
References
Porter DL, Levine BL, Kalos M, Bagg A, June CH (2011) Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365(8):725–733
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E et al (2011) Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118(18):4817–4828
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368(16):1509–1518
Cappell KM, Kochenderfer JN (2023) Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol 20(6):359–371
Morris EC, Neelapu SS, Giavridis T, Sadelain M (2022) Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol 22(2):85–96
Hines MR, Knight TE, McNerney KO, Leick MB, Jain T, Ahmed S, Frigault MJ, Hill JA, Jain MD, Johnson WT, Lin Y, Mahadeo KM, Maron GM, Marsh RA, Neelapu SS, Nikiforow S, Ombrello AK, Shah NN, Talleur AC et al (2023) Immune Effector Cell-Associated Hemophagocytic Lymphohistiocytosis-Like Syndrome. Transplant Cell Ther 29(7):438 e1–438 e16
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, Maus MV, Park JH, Mead E, Pavletic S, Go WY, Eldjerou L, Gardner RA, Frey N, Curran KJ, Peggs K, Pasquini M, DiPersio JF, van den Brink MRM et al (2019) ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant 25(4):625–638
Santomasso BD, Nastoupil LJ, Adkins S, Lacchetti C, Schneider BJ, Anadkat M, Atkins MB, Brassil KJ, Caterino JM, Chau I, Davies MJ, Ernstoff MS, Fecher L, Funchain P, Jaiyesimi I, Mammen JS, Naidoo J, Naing A, Phillips T et al (2021) Management of Immune-Related Adverse Events in Patients Treated With Chimeric Antigen Receptor T-Cell Therapy: ASCO Guideline. J Clin Oncol 39(35):3978–3992
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH (2009) Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 17(8):1453–1464
Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, Wilson WH, Rosenberg SA (2009) Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother 32(7):689–702
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U et al (2017) Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377(26):2531–2544
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jager U, Jaglowski S, Andreadis C, Westin JR, Fleury I, Bachanova V, Foley SR, Ho PJ, Mielke S, Magenau JM, Holte H, Pantano S, Pacaud LB et al (2019) Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 380(1):45–56
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, Timmerman JM, Holmes H, Jaglowski S, Flinn IW, McSweeney PA, Miklos DB, Pagel JM, Kersten MJ, Milpied N, Fung H, Topp MS, Houot R, Beitinjaneh A et al (2020) KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med 382(14):1331–1342
Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, Leguay T, Bishop MR, Topp MS, Tzachanis D, O'Dwyer KM, Arellano ML, Lin Y, Baer MR, Schiller GJ, Park JH, Subklewe M, Abedi M, Minnema MC et al (2021) KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 398(10299):491–502
Bachy E, Le Gouill S, Di Blasi R, Sesques P, Manson G, Cartron G, Beauvais D, Roulin L, Gros FX, Rubio MT, Bories P, Bay JO, Llorente CC, Choquet S, Casasnovas RO, Mohty M, Guidez S, Joris M, Loschi M et al (2022) A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nat Med 28(10):2145–2154
Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, Munshi PN, Casulo C, Maloney DG, de Vos S, Reshef R, Leslie LA, Yakoub-Agha I, Oluwole OO, Fung HCH, Rosenblatt J, Rossi JM, Goyal L, Plaks V et al (2022) Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 23(1):91–103
Wang Z, Han W (2018) Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res 6:4
Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, Fitzgerald JC, Aplenc R, Gore L, Grupp SA (2013) Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 121(26):5154–5157
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371(16):1507–1517
Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J, Barrett DM, Weiss SL, Fitzgerald JC, Berg RA, Aplenc R, Callahan C, Rheingold SR, Zheng Z, Rose-John S et al (2016) Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov 6(6):664–679
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J et al (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6(224):224ra25
Diorio C, Shraim R, Myers R, Behrens EM, Canna S, Bassiri H, Aplenc R, Burudpakdee C, Chen F, DiNofia AM, Gill S, Gonzalez V, Lambert MP, Leahy AB, Levine BL, Lindell RB, Maude SL, Melenhorst JJ, Newman H et al (2022) Comprehensive Serum Proteome Profiling of Cytokine Release Syndrome and Immune Effector Cell-Associated Neurotoxicity Syndrome Patients with B-Cell ALL Receiving CAR T19. Clin Cancer Res 28(17):3804–3813
Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, Shaw P, Berg RA, June CH, Porter DL, Frey NV, Grupp SA, Teachey DT (2017) Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit Care Med 45(2):e124–e131
Rainone M, Ngo D, Baird JH, Budde LE, Htut M, Aldoss I, Pullarkat V (2023) Interferon-gamma blockade in CAR T-cell therapy-associated macrophage activation syndrome/hemophagocytic lymphohistiocytosis. Blood Adv 7(4):533–536
Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, Halton E, Wang X, Senechal B, Purdon T, Cross JR, Liu H, Vachha B, Chen X, DeAngelis LM, Li D, Bernal Y, Gonen M, Wendel HG et al (2018) Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov 8(8):958–971
Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, Chen J, Chung D, Harju-Baker S, Ozpolat T, Fink KR, Riddell SR, Maloney DG, Turtle CJ (2017) Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov 7(12):1404–1419
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, Westin J, Gulbis AM, Loghin ME, de Groot JF, Adkins S, Davis SE, Rezvani K, Hwu P, Shpall EJ (2018) Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol 15(1):47–62
Bhaskar ST, Patel VG, Porter DL, Schuster SJ, Nastoupil LJ, Perales MA, Tomas AA, Bishop MR, McGuirk JP, Maziarz RT, Chen AI, Bachanova V, Maakaron JE, Riedell PA, Oluwole OO (2023) Chimeric antigen receptor T-cell therapy yields similar outcomes in patients with and without cytokine release syndrome. Blood Adv 7(17):4765–4772
Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, Roshal M, Maslak P, Davila M, Brentjens RJ, Sadelain M (2018) Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 378(5):449–459
Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, Oluwole OO, Reagan PM, Lekakis LJ, Lin Y, Sherman M, Better M, Go WY, Wiezorek JS, Xue A, Bot A (2020) Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv 4(19):4898–4911
Jain MD, Zhao H, Wang X, Atkins R, Menges M, Reid K, Spitler K, Faramand R, Bachmeier C, Dean EA, Cao B, Chavez JC, Shah B, Lazaryan A, Nishihori T, Hussaini M, Gonzalez RJ, Mullinax JE, Rodriguez PC et al (2021) Tumor interferon signaling and suppressive myeloid cells are associated with CAR T-cell failure in large B-cell lymphoma. Blood 137(19):2621–2633
Cohen AD, Parekh S, Santomasso BD, Gallego Perez-Larraya J, van de Donk N, Arnulf B, Mateos MV, Lendvai N, Jackson CC, De Braganca KC, Schecter JM, Marquez L, Lee E, Cornax I, Zudaire E, Li C, Olyslager Y, Madduri D, Varsos H et al (2022) Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J 12(2):32
Winkelmann M, Blumenberg V, Rejeski K, Quell C, Bucklein VL, Ingenerf M, Unterrainer M, Schmidt C, Dekorsy FJ, Bartenstein P, Ricke J, von Bergwelt-Baildon M, Subklewe M, Kunz WG (2024) Predictive value of pre-infusion tumor growth rate for the occurrence and severity of CRS and ICANS in lymphoma under CAR T-cell therapy. Ann Hematol 103(1):259–268
Lemoine J, Vic S, Houot R (2022) Disease-specific outcomes after chimeric antigen receptor T-cell therapy. Eur J Cancer 160:235–242
Neelapu SS, Jacobson CA, Oluwole OO, Munoz J, Deol A, Miklos DB, Bartlett NL, Braunschweig I, Jiang Y, Kim JJ, Zheng L, Rossi JM, Locke FL (2020) Outcomes of older patients in ZUMA-1, a pivotal study of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood 135(23):2106–2109
Lin RJ, Lobaugh SM, Pennisi M, Chan HT, Batlevi Y, Ruiz JD, Elko TA, Maloy MA, Batlevi CL, Dahi PB, Giralt SA, Hamlin PA, Mead E, Noy A, Palomba ML, Santomasso BD, Sauter CS, Scordo M, Shah GL et al (2021) Impact and safety of chimeric antigen receptor T-cell therapy in older, vulnerable patients with relapsed/refractory large B-cell lymphoma. Haematologica 106(1):255–258
Ram R, Grisariu S, Shargian-Alon L, Amit O, Bar-On Y, Stepensky P, Yeshurun M, Avni B, Hagin D, Perry C, Gurion R, Sarid N, Herishanu Y, Gold R, Glait-Santar C, Kay S, Avivi I (2022) Toxicity and efficacy of chimeric antigen receptor T-cell therapy in patients with diffuse large B-cell lymphoma above the age of 70 years compared to younger patients - a matched control multicenter cohort study. Haematologica 107(5):1111–1118
Jin Z, Xiang R, Qing K, Li X, Zhang Y, Wang L, Zhu H, Mao Y, Xu Z, Li J (2018) The severe cytokine release syndrome in phase I trials of CD19-CAR-T cell therapy: a systematic review. Ann Hematol 97(8):1327–1335
Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, Guo X, Liu H, Ding N, Zhang T, Duan P, Lin Y, Zheng W, Wang X, Lin N, Tu M, Xie Y, Zhang C, Liu W et al (2019) A safe and potent anti-CD19 CAR T cell therapy. Nat Med 25(6):947–953
Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, Xue A, Bot A, Scholler N, Mikkilineni L, Roschewski M, Dean R, Cachau R, Youkharibache P, Patel R, Hansen B, Stroncek DF, Rosenberg SA, Gress RE, Kochenderfer JN (2020) Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med 26(2):270–280
Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, Albon SJ, Casanovas-Company J, Castro F, Popova B, Villanueva K, Yeung J, Vetharoy W, Guvenel A, Wawrzyniecka PA, Mekkaoui L, Cheung GW, Pinner D, Chu J et al (2019) Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med 25(9):1408–1414
Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, Haris M, Wilson NE, Liu F, Gabunia K, Scholler J, Montine TJ, Bhoj VG, Reddy R, Mohan S, Maillard I, Kriegstein AR, June CH, Chang HY et al (2020) Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 183(1):126–142 e17
Van Oekelen O, Aleman A, Upadhyaya B, Schnakenberg S, Madduri D, Gavane S, Teruya-Feldstein J, Crary JF, Fowkes ME, Stacy CB, Kim-Schulze S, Rahman A, Lagana A, Brody JD, Merad M, Jagannath S, Parekh S (2021) Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat Med 27(12):2099–2103
Yang C, Nguyen J, Yen Y (2023) Complete spectrum of adverse events associated with chimeric antigen receptor (CAR)-T cell therapies. J Biomed Sci 30(1):89
Tedesco VET, Mohan C (2021) Biomarkers for Predicting Cytokine Release Syndrome following CD19-Targeted CAR T Cell Therapy. J Immunol 206(7):1561–1568
Wei Z, Xu J, Zhao C, Zhang M, Xu N, Kang L, Lou X, Yu L, Feng W (2023) Prediction of severe CRS and determination of biomarkers in B cell-acute lymphoblastic leukemia treated with CAR-T cells. Front Immunol 14:1273507
Pennisi M, Sanchez-Escamilla M, Flynn JR, Shouval R, Alarcon Tomas A, Silverberg ML, Batlevi C, Brentjens RJ, Dahi PB, Devlin SM, Diamonte C, Giralt S, Halton EF, Jain T, Maloy M, Mead E, Palomba ML, Ruiz J, Santomasso B et al (2021) Modified EASIX predicts severe cytokine release syndrome and neurotoxicity after chimeric antigen receptor T cells. Blood Adv 5(17):3397–3406
Jalota A, Hershberger CE, Patel MS, Mian A, Faruqi A, Khademi G, Rotroff DM, Hill BT, Gupta N (2023) Host metabolome predicts the severity and onset of acute toxicities induced by CAR T-cell therapy. Blood Adv 7(17):4690–4700
Kotch C, Barrett D, Teachey DT (2019) Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev Clin Immunol 15(8):813–822
Maus MV, Alexander S, Bishop MR, Brudno JN, Callahan C, Davila ML, Diamonte C, Dietrich J, Fitzgerald JC, Frigault MJ, Fry TJ, Holter-Chakrabarty JL, Komanduri KV, Lee DW, Locke FL, Maude SL, McCarthy PL, Mead E, Neelapu SS et al (2020) Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer 8(2):e001511
Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, Gust J, Leger KJ, Tarlock K, Cooper TM, Finney OC, Brakke H, Li DH, Park JR, Jensen MC (2019) Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood 134(24):2149–2158
Liu S, Deng B, Zhichao Y, Pan J, Lin Y, Ling Z, Wu T, Chen D, Chang AH, Gao Z, Song Y, Zhao Y, Tong C (2020) Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J 10(15):4
Topp MS, van Meerten T, Houot R, Minnema MC, Bouabdallah K, Lugtenburg PJ, Thieblemont C, Wermke M, Song KW, Avivi I, Kuruvilla J, Duhrsen U, Zheng Y, Vardhanabhuti S, Dong J, Bot A, Rossi JM, Plaks V, Sherman M et al (2021) Earlier corticosteroid use for adverse event management in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol 195(3):388–398
Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M (2018) CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med 24(6):731–738
Diorio C, Vatsayan A, Talleur AC, Annesley C, Jaroscak JJ, Shalabi H, Ombrello AK, Hudspeth M, Maude SL, Gardner RA, Shah NN (2022) Anakinra utilization in refractory pediatric CAR T-cell associated toxicities. Blood Adv 6(11):3398–3403
Dreyzin A, Jacobsohn D, Angiolillo A, Wistinghausen B, Schore RJ, Perez E, Wells E, Terao J, Bonifant C, Rohatgi R, Dave H, Vatsayan A (2022) Intravenous anakinra for tisagenlecleucel-related toxicities in children and young adults. Pediatr Hematol Oncol 39(4):370–378
Sahillioglu AC, Schumacher TN (2022) Safety switches for adoptive cell therapy. Curr Opin Immunol 74:190–198
Schuelke MR, Bassiri H, Behrens EM, Canna S, Croy C, DiNofia A, Gollomp K, Grupp S, Lambert M, Lambrix A, Maude SL, Myers R, Newman H, Petrosa W, Seif A, Sullivan KE, Teachey DT, Diorio C (2023) Emapalumab for the treatment of refractory cytokine release syndrome in pediatric patients. Blood Adv 7(18):5603–5607
Bailey SR, Vatsa S, Larson RC, Bouffard AA, Scarfo I, Kann MC, Berger TR, Leick MB, Wehrli M, Schmidts A, Silva H, Lindell KA, Demato A, Gallagher KME, Frigault MJ, Maus MV (2022) Blockade or Deletion of IFNgamma Reduces Macrophage Activation without Compromising CAR T-cell Function in Hematologic Malignancies. Blood Cancer Discov 3(2):136–153
Patel S, Cenin D, Corrigan D, Hamilton BK, Kalaycio M, Sobecks RM, Anwer F, Khouri J, Dean RM, Winter A, Jagadeesh D, Pohlman B, Hill BT, Sauter CS, Caimi PF (2022) Siltuximab for First-Line Treatment of Cytokine Release Syndrome: A Response to the National Shortage of Tocilizumab. Blood 140:5073–5074
Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, Mades A, Sadelain M, Einsele H, Hudecek M (2019) The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med 11(499):eaau5907
Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, Mackall CL (2019) Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv 3(5):711–717
Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, Malipatlolla M, Xu P, Bashti M, Heitzeneder S, Labanieh L, Vandris P, Majzner RG, Qi Y, Sandor K et al (2021) Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372(6537):eaba1786
Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, Yeung CCS, Sheih A, Pender BS, Hawkins RM, Vakil A, Phi TD, Steinmetz RN, Shadman M, Riddell SR, Maloney DG, Turtle CJ (2020) Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood 135(19):1650–1660
Ruella M, Kenderian SS, Shestova O, Klichinsky M, Melenhorst JJ, Wasik MA, Lacey SF, June CH, Gill S (2017) Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia 31(1):246–248
Zi FM, Ye LL, Zheng JF, Cheng J, Wang QM (2021) Using JAK inhibitor to treat cytokine release syndrome developed after chimeric antigen receptor T cell therapy for patients with refractory acute lymphoblastic leukemia: A case report. Medicine (Baltimore) 100(19):e25786
Pan J, Deng B, Ling Z, Song W, Xu J, Duan J, Wang Z, Chang AH, Feng X, Tan Y (2021) Ruxolitinib mitigates steroid-refractory CRS during CAR T therapy. J Cell Mol Med 25(2):1089–1099
Zhang L, Wang S, Xu J, Zhang R, Zhu H, Wu Y, Zhu L, Li J, Chen L (2021) Etanercept as a new therapeutic option for cytokine release syndrome following chimeric antigen receptor T cell therapy. Exp Hematol Oncol 10(1):16
Kadauke S, Myers RM, Li Y, Aplenc R, Baniewicz D, Barrett DM, Barz Leahy A, Callahan C, Dolan JG, Fitzgerald JC, Gladney W, Lacey SF, Liu H, Maude SL, McGuire R, Motley LS, Teachey DT, Wertheim GB, Wray L et al (2021) Risk-Adapted Preemptive Tocilizumab to Prevent Severe Cytokine Release Syndrome After CTL019 for Pediatric B-Cell Acute Lymphoblastic Leukemia: A Prospective Clinical Trial. J Clin Oncol 39(8):920–930
Banerjee R, Marsal J, Huang CY, Lo M, Kambhampati S, Kennedy VE, Arora S, Wolf JL, Martin TG, Wong SW, Shah N (2021) Early Time-to-Tocilizumab after B Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy in Myeloma. Transplant Cell Ther 27(6):477 e1–477 e7
Park JH, Nath K, Devlin SM, Sauter CS, Palomba ML, Shah G, Dahi P, Lin RJ, Scordo M, Perales MA, Shouval R, Tomas AA, Cathcart E, Mead E, Santomasso B, Holodny A, Brentjens RJ, Riviere I, Sadelain M (2023) CD19 CAR T-cell therapy and prophylactic anakinra in relapsed or refractory lymphoma: phase 2 trial interim results. Nat Med 29(7):1710–1717
Oluwole OO, Kenderian SS, Shiraz P, Karmali R, Reshef R, McCarthy PL, Ghosh N, Lazaryan A, Filosto S, Poddar S, Mao D, Peng A, Kilcoyne A, Nahas M, Neelapu SS (2022) ZUMA-19: A Phase 1/2 Study of Axicabtagene Ciloleucel Plus Lenzilumab in Patients With Relapsed or Refractory Large B-Cell Lymphoma. Blood 140:10318–10320
Gutierrez C, Brown ART, Herr MM, Kadri SS, Hill B, Rajendram P, Duggal A, Turtle CJ, Patel K, Lin Y, May HP, de Moraes AG, Maus MV, Frigault MJ, Brudno JN, Athale J, Shah NN, Kochenderfer JN, Dharshan A et al (2020) The chimeric antigen receptor-intensive care unit (CAR-ICU) initiative: Surveying intensive care unit practices in the management of CAR T-cell associated toxicities,. J Crit Care 58:58–64
Caimi PF, Pacheco Sanchez G, Sharma A, Otegbeye F, Ahmed N, Rojas P, Patel S, Kleinsorge Block S, Schiavone J, Zamborsky K, Boughan K, Hillian A, Reese-Koc J, Maschan M, Dropulic B, Sekaly RP, de Lima M (2021) Prophylactic Tocilizumab Prior to Anti-CD19 CAR-T Cell Therapy for Non-Hodgkin Lymphoma. Front Immunol 12:745320
Strati P, Ahmed S, Furqan F, Fayad LE, Lee HJ, Iyer SP, Nair R, Nastoupil LJ, Parmar S, Rodriguez MA, Samaniego F, Steiner RE, Wang M, Pinnix CC, Horowitz SB, Feng L, Sun R, Claussen CM, Hawkins MC et al (2021) Prognostic impact of corticosteroids on efficacy of chimeric antigen receptor T-cell therapy in large B-cell lymphoma. Blood 137(23):3272–3276
Oluwole OO, Bouabdallah K, Munoz J, De Guibert S, Vose JM, Bartlett NL, Lin Y, Deol A, McSweeney PA, Goy AH, Kersten MJ, Jacobson CA, Farooq U, Minnema MC, Thieblemont C, Timmerman JM, Stiff P, Avivi I, Tzachanis D et al (2021) Prophylactic corticosteroid use in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol 194(4):690–700
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, Traversari C, Bordignon C, Ciceri F, Ostuni R, Bonini C, Casucci M, Bondanza A (2018) Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 24(6):739–748
Ishii K, Pouzolles M, Chien CD, Erwin-Cohen RA, Kohler ME, Qin H, Lei H, Kuhn S, Ombrello AK, Dulau-Florea A, Eckhaus MA, Shalabi H, Yates B, Lichtenstein DA, Zimmermann VS, Kondo T, Shern JF, Young HA, Taylor N et al (2020) Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J Clin Invest 130(10):5425–5443
McNerney KO, DiNofia AM, Teachey DT, Grupp SA, Maude SL (2022) Potential Role of IFNgamma Inhibition in Refractory Cytokine Release Syndrome Associated with CAR T-cell Therapy. Blood Cancer Discov 3(2):90–94
Hu X, Li J, Fu M, Zhao X, Wang W (2021) The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther 6(1):402
Al-Ali HK, Griesshammer M, Foltz L, Palumbo GA, Martino B, Palandri F, Liberati AM, le Coutre P, Garcia-Hernandez C, Zaritskey A, Tavares R, Gupta V, Raanani P, Giraldo P, Hanel M, Damiani D, Sacha T, Bouard C, Paley C et al (2020) Primary analysis of JUMP, a phase 3b, expanded-access study evaluating the safety and efficacy of ruxolitinib in patients with myelofibrosis, including those with low platelet counts. Br J Haematol 189(5):888–903
Ho PJ, Marlton P, Tam C, Stevenson W, Ritchie D, Bird R, Dunlop LC, Durrant S, Ross DM (2015) Practical management of myelofibrosis with ruxolitinib. Intern Med J 45(12):1221–1230
De Benedetti F, Grom AA, Brogan PA, Bracaglia C, Pardeo M, Marucci G, Eleftheriou D, Papadopoulou C, Schulert GS, Quartier P, Anton J, Laveille C, Frederiksen R, Asnaghi V, Ballabio M, Jacqmin P, de Min C (2023) Efficacy and safety of emapalumab in macrophage activation syndrome. Ann Rheum Dis 82(6):857–865
Jordan MB, Locatelli F (2024) Exposure-safety relationship for patients with primary hemophagocytic lymphohistiocytosis treated with emapalumab. Pediatr Blood Cancer 71(2):e30778
Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, Ghobadi A, Rapoport AP, McGuirk J, Pagel JM, Munoz J, Farooq U, van Meerten T, Reagan PM, Sureda A, Flinn IW, Vandenberghe P, Song KW, Dickinson M et al (2022) Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med 386(7):640–654
Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, Hansen MJ, Jin F, Ayasoufi K, Hefazi M, Schick KJ, Walters DK, Ahmed O, Chappell D, Sahmoud T, Durrant C, Nevala WK, Patnaik MM, Pease LR et al (2019) GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 133(7):697–709
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK (2011) Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 365(18):1673–1683
Jain MD, Smith M, Shah NN (2023) How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood 141(20):2430–2442
Garfall AL, Lancaster E, Stadtmauer EA, Lacey SF, Dengel K, Ambrose DE, Chen F, Gupta M, Kulikovskaya I, Vogl DT, Plesa G, Weiss BM, Ferthio R, Richardson C, Melenhorst JJ, Levine BL, June CH, Milone MC, Cohen AD (2016) Posterior Reversible Encephalopathy Syndrome (PRES) after Infusion of Anti-Bcma CAR T Cells (CART-BCMA) for Multiple Myeloma: Successful Treatment with Cyclophosphamide. Blood 128(22):5702
Ragoonanan D, Khazal SJ, Abdel-Azim H, McCall D, Cuglievan B, Tambaro FP, Ahmad AH, Rowan CM, Gutierrez C, Schadler K, Li S, Di Nardo M, Chi L, Gulbis AM, Shoberu B, Mireles ME, McArthur J, Kapoor N, Miller J et al (2021) Diagnosis, grading and management of toxicities from immunotherapies in children, adolescents and young adults with cancer. Nat Rev Clin Oncol 18(7):435–453
Stahl K, Schmidt BMW, Hoeper MM, Skripuletz T, Mohn N, Beutel G, Eder M, Welte T, Ganser A, Falk CS, Koenecke C, David S (2020) Extracorporeal cytokine removal in severe CAR-T cell associated cytokine release syndrome. J Crit Care 57:124–129
Singbartl K, Rosenthal A, Leis J, Patel B, Sen A (2021) Novel Use of Extracorporeal Blood Purification for Treatment of Severe, Refractory Neurotoxicity After Chimeric Antigen Receptor T-Cell Therapy-A Case Report. Crit Care Explor 3(7):e0472
Traube C, Silver G, Kearney J, Patel A, Atkinson TM, Yoon MJ, Halpert S, Augenstein J, Sickles LE, Li C, Greenwald B (2014) Cornell Assessment of Pediatric Delirium: a valid, rapid, observational tool for screening delirium in the PICU*. Crit Care Med 42(3):656–663
Rubin DB, Al Jarrah A, Li K, LaRose S, Monk AD, Ali AB, Spendley LN, Nikiforow S, Jacobson C, Vaitkevicius H (2020) Clinical Predictors of Neurotoxicity After Chimeric Antigen Receptor T-Cell Therapy. JAMA Neurol 77(12):1536–1542
Gofshteyn JS, Shaw PA, Teachey DT, Grupp SA, Maude S, Banwell B, Chen F, Lacey SF, Melenhorst JJ, Edmonson MJ, Panzer J, Barrett DM, McGuire JL (2018) Neurotoxicity after CTL019 in a pediatric and young adult cohort. Ann Neurol 84(4):537–546
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, Soma L, Wood B, Li D, Heimfeld S, Riddell SR, Maloney DG (2016) Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8(355):355ra116
Wehrli M, Gallagher K, Chen YB, Leick MB, McAfee SL, El-Jawahri AR, DeFilipp Z, Horick N, O'Donnell P, Spitzer T, Dey B, Cook D, Trailor M, Lindell K, Maus MV, Frigault MJ (2022) Single-center experience using anakinra for steroid-refractory immune effector cell-associated neurotoxicity syndrome (ICANS). J Immunother Cancer 10(1):e003847
Park JH, Sauter C, Palomba ML, Shah GL, Dahi PB, Lin RJ, Scordo M, Batlevi C, Perales MA, Kane P, Afuye AO, Mead E, Santomasso B, Halton E, Sadelain M, Brentjens R (2021) A Phase II Study of Prophylactic Anakinra to Prevent CRS and Neurotoxicity in Patients Receiving CD19 CAR T Cell Therapy for Relapsed or Refractory Lymphoma. Blood 138:96
Shah NN, Johnson BD, Fenske TS, Raj RV, Hari P (2020) Intrathecal chemotherapy for management of steroid-refractory CAR T-cell-associated neurotoxicity syndrome. Blood Adv 4(10):2119–2122
Zurko JC, Johnson BD, Aschenbrenner E, Fenske TS, Hamadani M, Hari P, Shah NN (2022) Use of Early Intrathecal Therapy to Manage High-Grade Immune Effector Cell-Associated Neurotoxicity Syndrome. JAMA Oncol 8(5):773–775
Yucebay F, Maakaron J, Grana A, Jaglowski S, Roddy J (2020) Intrathecal chemotherapy: an alternative treatment strategy to prolonged corticosteroids for severe CAR T associated neurotoxicity. Biol Blood Marrow Transplant 26(3):S312
Wang M, Jain P, Chi TL, Chen SE, Heimberger A, Weathers SP, Zheng L, Rao AV, Rossi JM (2020) Management of a patient with mantle cell lymphoma who developed severe neurotoxicity after chimeric antigen receptor T-cell therapy in ZUMA-2. J Immunother Cancer 8(2):e001114
Jacobson CA, Rosenthal AC, Arnason J, Agarwal S, Zhang P, Wu W, Amber V, Yared JA (2023) A phase 2 trial of defibrotide for the prevention of chimeric antigen receptor T-cell-associated neurotoxicity syndrome. Blood Adv 7(21):6790–6799
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C et al (2018) Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378(5):439–448
Nellan A, McCully CML, Cruz Garcia R, Jayaprakash N, Widemann BC, Lee DW, Warren KE (2018) Improved CNS exposure to tocilizumab after cerebrospinal fluid compared to intravenous administration in rhesus macaques. Blood 132(6):662–666
Strati P, Ahmed S, Kebriaei P, Nastoupil LJ, Claussen CM, Watson G, Horowitz SB, Brown ART, Do B, Rodriguez MA, Nair R, Shpall EJ, Green MR, Neelapu SS, Westin JR (2020) Clinical efficacy of anakinra to mitigate CAR T-cell therapy-associated toxicity in large B-cell lymphoma. Blood Adv 4(13):3123–3127
Santomasso BD, Gust J, Perna F (2023) How I treat unique and difficult-to-manage cases of CAR T-cell therapy-associated neurotoxicity. Blood 141(20):2443–2451
Gust J (2023) BCMA-CAR T-cell treatment-associated parkinsonism. Blood 142(14):1181–1183
Karschnia P, Miller KC, Yee AJ, Rejeski K, Johnson PC, Raje N, Frigault MJ, Dietrich J (2023) Neurologic toxicities following adoptive immunotherapy with BCMA-directed CAR T cells. Blood 142(14):1243–1248
Hines MR, Keenan C, Maron Alfaro G, Cheng C, Zhou Y, Sharma A, Hurley C, Nichols KE, Gottschalk S, Triplett BM, Talleur AC (2021) Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br J Haematol 194(4):701–707
Martin-Rojas RM, Gomez-Centurion I, Bailen R, Bastos M, Diaz-Crespo F, Carbonell D, Correa-Rocha R, Pion M, Munoz C, Sancho M, Gomez Fernandez I, Oarbeascoa G, Perez-Corral A, Martinez-Laperche C, Anguita J, Buno I, Menarguez J, Diez-Martin JL, Kwon M (2022) Hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) following treatment with tisagenlecleucel. Clin Case Rep 10(1):e05209
Porter TJ, Lazarevic A, Ziggas JE, Fuchs E, Kim K, Byrnes H, Luznik L, Bolanos-Meade J, Ali SA, Shah NN, Wagner-Johnston N, Jain T (2022) Hyperinflammatory syndrome resembling haemophagocytic lymphohistiocytosis following axicabtagene ciloleucel and brexucabtagene autoleucel. Br J Haematol 199(5):720–727
Canna SW, Marsh RA (2020) Pediatric hemophagocytic lymphohistiocytosis. Blood 135(16):1332–1343
La Rosee P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J, Girschikofsky M, Jordan MB, Kumar A, van Laar JAM, Lachmann G, Nichols KE, Ramanan AV, Wang Y, Wang Z, Janka G, Henter JI (2019) Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 133(23):2465–2477
Sepulveda FE, de Saint Basile G (2017) Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol 49:20–26
Schram AM, Berliner N (2015) How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 125(19):2908–2914
Lichtenstein DA, Schischlik F, Shao L, Steinberg SM, Yates B, Wang HW, Wang Y, Inglefield J, Dulau-Florea A, Ceppi F, Hermida LC, Stringaris K, Dunham K, Homan P, Jailwala P, Mirazee J, Robinson W, Chisholm KM, Yuan C et al (2021) Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood 138(24):2469–2484
Charlesworth JEG, Wilson S, Qureshi A, Blanco E, Mitchell A, Segal S, Kelly D, Weitz J, O'Shea D, Bailey K, Kavirayani A (2021) Continuous intravenous anakinra for treating severe secondary haemophagocytic lymphohistiocytosis/macrophage activation syndrome in critically ill children. Pediatr Blood Cancer 68(9):e29102
Phadke O, Rouster-Stevens K, Giannopoulos H, Chandrakasan S, Prahalad S (2021) Intravenous administration of anakinra in children with macrophage activation syndrome. Pediatr Rheumatol Online J 19(1):98
Bracaglia C, de Graaf K, Pires Marafon D, Guilhot F, Ferlin W, Prencipe G, Caiello I, Davi S, Schulert G, Ravelli A, Grom AA, de Min C, De Benedetti F (2017) Elevated circulating levels of interferon-gamma and interferon-gamma-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis 76(1):166–172
Henter JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U (1991) Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 78(11):2918–2922
Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, Shi SW, Shen HP, Mao JQ, Zhang LY, Pan BH (2012) Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr 160(6):984–90 e1
Jordan MB, Hildeman D, Kappler J, Marrack P (2004) An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 104(3):735–743
Al-Salama ZT (2019) Emapalumab: First Global Approval. Drugs 79(1):99–103
Vallurupalli M, Berliner N (2019) Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood 134(21):1783–1786
Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, Degar B, Garrington TP, Sevilla J, Putti MC, Fagioli F, Ahlmann M, Dapena Diaz JL, Henry M, De Benedetti F, Grom A, Lapeyre G, Jacqmin P, Ballabio M, de Min C (2020) Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N Engl J Med 382(19):1811–1822
De Benedetti F, Grom A, Brogan PA, Bracaglia C, Pardeo M, Marucci G, Eleftheriou D, Papadopoulou C, Quartier P, Anton J, Frederiksen R, Asnaghi V, Ballabio M, de Min C (2021) Macrophage Activation Syndrome (MAS) in Systemic Juvenile Idiopathic Arthritis (sJIA): Treatment with Emapalumab, an Anti-Interferon Gamma (IFNγ) Monoclonal Antibody. Blood 138:2058
Kanda N, Shimizu T, Tada Y, Watanabe S (2007) IL-18 enhances IFN-gamma-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur J Immunol 37(2):338–350
Dufranc E, Del Bello A, Belliere J, Kamar N, Faguer S, TAIDI study group (2020) IL6-R blocking with tocilizumab in critically ill patients with hemophagocytic syndrome. Crit Care 24(1):166
Machaczka M, Vaktnas J, Chiang SC, Bryceson YT (2010) Alemtuzumab treatment for hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol 7(10):1–1
Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, Lee ND, Khan SP, Lawrence J, Mo JQ, Blessing JJ, Filipovich AH, Jordan MB (2013) Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer 60(1):101–109
Marsh RA, Jordan MB, Talano JA, Nichols KE, Kumar A, Naqvi A, Vaiselbuh SR (2017) G. Histiocyte Society Salvage Therapy Working, Salvage therapy for refractory hemophagocytic lymphohistiocytosis: A review of the published experience. Pediatr Blood Cancer 64(4):e26308
Papa R, Natoli V, Caorsi R, Minoia F, Gattorno M, Ravelli A (2020) Successful treatment of refractory hyperferritinemic syndromes with canakinumab: a report of two cases. Pediatr Rheumatol Online J 19(1):56
Geerlinks AV, Dvorak AM, Consortium XDT (2022) A Case of XIAP Deficiency Successfully Managed with Tadekinig Alfa (rhIL-18BP). J Clin Immunol 42(4):901–903
Henzan T, Nagafuji K, Tsukamoto H, Miyamoto T, Gondo H, Imashuku S, Harada M (2006) Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol 81(1):59–61
O'Shea JJ, Plenge R (2012) JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36(4):542–550
Wang J, Wang Y, Wu L, Wang X, Jin Z, Gao Z, Wang Z (2020) Ruxolitinib for refractory/relapsed hemophagocytic lymphohistiocytosis. Haematologica 105(5):e210–e212
Wang J, Zhang R, Wu X, Li F, Yang H, Liu L, Guo H, Zhang X, Mai H, Li H, Wang Z (2021) Ruxolitinib-combined doxorubicin-etoposide-methylprednisolone regimen as a salvage therapy for refractory/relapsed haemophagocytic lymphohistiocytosis: a single-arm, multicentre, phase 2 trial. Br J Haematol 193(4):761–768
Keith MP, Pitchford C, Bernstein WB (2012) Treatment of hemophagocytic lymphohistiocytosis with alemtuzumab in systemic lupus erythematosus. J Clin Rheumatol 18(3):134–137
Kikuchi H, Yamamoto T, Asako K, Takayama M, Shirasaki R, Ono Y (2012) Etanercept for the treatment of intractable hemophagocytic syndrome with systemic lupus erythematosus. Mod Rheumatol 22(2):308–311
Oda Y, Urushidani Y, Ooi S, Endoh A, Nakamura R, Adachi K, Fukushima H (2012) Hemophagocytic lymphohistiocytosis in a rheumatoid arthritis patient treated with infliximab. Intern Med 51(6):655–657
Palmblad K, Schierbeck H, Sundberg E, Horne AC, Erlandsson Harris H, Henter JI, Andersson U (2021) Therapeutic administration of etoposide coincides with reduced systemic HMGB1 levels in macrophage activation syndrome. Mol Med 27(1):48
Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB (2014) Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol 192(1):84–91
Funding
DTT: Gabriella Miller Kids First X01HD100702, R03CA256550, Alex’s Lemonade Stand Foundation, the Leukemia and Lymphoma Society, Hyundai Hope of Wheels, R01CA193776 and R01CA264837. CJD: CIHR Fellowship Award, 5K12CA076931-24.
Author information
Authors and Affiliations
Contributions
All authors contributed intellectually and reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
DTT serves on advisory boards for Sobi, Jazz, Servier, and BEAM Therapeutics. DTT receives research funding from BEAM Therapeutics and Neoimmune Tech. DTT has patents (US11747346) and patents pending on CAR-T. All other authors have no conflicts of interest to declare.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hughes, A.D., Teachey, D.T. & Diorio, C. Riding the storm: managing cytokine-related toxicities in CAR-T cell therapy. Semin Immunopathol 46, 5 (2024). https://doi.org/10.1007/s00281-024-01013-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00281-024-01013-w