
Abstract
Cetuximab and panitumumab are monoclonal antibodies (mAbs) against epidermal growth factor receptor (EGFR) that are effective agents for metastatic colorectal cancer (mCRC). Cetuximab can prolong survival by 8.2 months in RAS wild-type (WT) mCRC patients. Unfortunately, resistance to targeted therapy impairs clinical use and efficiency. The mechanisms of resistance refer to intrinsic and extrinsic alterations of tumours. Multiple therapeutic strategies have been investigated extensively to overcome resistance to anti-EGFR mAbs. The intrinsic mechanisms include EGFR ligand overexpression, EGFR alteration, RAS/RAF/PI3K gene mutations, ERBB2/MET/IGF-1R activation, metabolic remodelling, microsatellite instability and autophagy. For intrinsic mechanisms, therapies mainly cover the following: new EGFR-targeted inhibitors, a combination of multitargeted inhibitors, and metabolic regulators. In addition, new cytotoxic drugs and small molecule compounds increase the efficiency of cetuximab. Extrinsic alterations mainly disrupt the tumour microenvironment, specifically immune cells, cancer-associated fibroblasts (CAFs) and angiogenesis. The directions include the modification or activation of immune cells and suppression of CAFs and anti-VEGFR agents. In this review, we focus on the mechanisms of resistance to anti-EGFR monoclonal antibodies (anti-EGFR mAbs) and discuss diverse approaches to reverse resistance to this therapy in hopes of identifying more mCRC treatment possibilities.
Similar content being viewed by others


Background
Metastatic colorectal cancer (mCRC) accounts for almost half of the newly diagnosed colorectal cancer cases and is associated with poor prognosis [1]. Epidermal growth factor receptor (EGFR) is a key factor in cellular proliferation, differentiation and survival [2], which drives the use of EGFR-targeted therapy in malignancy treatment [3]. The advent of cetuximab and panitumumab, two monoclonal antibodies (mAbs) directly targeting EGFR, can prolong survival for 10–20% of mCRC patients [4]. According to the CRYSTAL trial, the application of cetuximab and FOLFIRI in first-line treatment can reduce the risk of progression by 15% and increase overall survival (OS) by 8.2 months in patients who have KRAS WT mCRC compared with patients taking FOLFIRI alone [5].
Although treatment with anti-EGFR monoclonal antibodies (anti-EGFR mAbs) and chemotherapy has a large effect on mCRC, its clinical application is limited because of drug resistance. The clinical benefit in responders treated with anti-EGFR mAbs has been shown to only last 8–10 months [6, 7]. As treatment progresses, approximately 80% of responders develop drug resistance [8]. The mechanisms of resistance to anti-EGFR mAbs have been elucidated previously. Gene mutations downstream of the EGFR signalling pathway, including RAS/RAF/MEK and PI3K/AKT/mTOR, significantly contribute to drug resistance [9,10,11]. The activation of compensatory feedback loops of EGFR, such as erb-b2 receptor tyrosine kinase 2 (ERBB2), MET and insulin-like growth factor 1 receptor (IGF-1R), has been shown to interfere with EGFR inhibitor treatment [12,13,14]. In recent years, the intrinsic mechanisms of metabolism, autophagy [15], cancer stem cells (CSCs) [16] and epithelial-to-mesenchymal transition (EMT )[17] have also been confirmed to be correlated with poor progression despite anti-EGFR mAb treatment. Extrinsic alterations of tumours may appear during treatment with cetuximab and panitumumab [18]. Currently, it is believed that microenvironment remodelling can reduce the cytotoxicity of anti-EGFR mAbs by impairing antibody-dependent cellular cytotoxicity (ADCC) and secreting growth factors [19, 20].
Consequently, strategies to reverse resistance to anti-EGFR mAbs have been explored in experimental studies and clinical trials. These strategies include different aspects, such as new EGFR-targeted inhibitors, combinations of multitargeted inhibitors, metabolic regulators, immune therapy and new cytotoxic drugs. Here, we review the mechanisms underlying resistance to anti-EGFR mAbs and discuss the current studies on improving the efficiency of targeted therapy, increasing the number of available mCRC therapies.
Intrinsic mechanisms of resistance to targeted therapy and related strategies
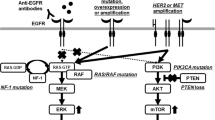
Intrinsic alterations of tumours greatly contribute to resistance to anti-EGFR targeted therapy. Known intrinsic mechanisms are genetic mutations inducing EGFR and compensatory feedback loop signalling activation. Recently, metabolic remodelling, CSCs and EMT have also been confirmed to promote resistance to targeted therapy (Fig. 1). Accordingly, different strategies have been used to reverse the resistance: (i) development of new EGFR targeted inhibitors, (ii) combination of anti-EGFR mAbs with multitargeted inhibitors, (iii) metabolic regulators and (iv) new cytotoxic drugs (Tables 1 and 2).

Intrinsic mechanisms of resistance to anti-EGFR mAbs in metastatic colorectal cancer. The intrinsic mechanisms include abnormal activation of oncogenic signalling pathways, aberrant gene expression, metabolic disorders, increased autophagy function and cancer stem cells. For example, genomic alterations and proteic phosphorylation induce activation of the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR cascades. ERBB2/MET amplification and abnormal IGF-1R activation stimulate compensatory feedback loop signalling of EGFR. The phenotype shift of cancer stem cells (CSCs) into epithelial-to-mesenchymal transition (EMT) contributes to therapy resistance. Glycolysis, lipid synthesis, fatty acid oxidation and vitamin deficiency in cancer cells also reduced the efficiency of EGFR-targeted therapy. The agents for specific points are also shown in the figure. Abbreviations: CSC, cancer stem cell; EMT, epithelial-to-mesenchymal transition; PI3K, phosphoinositide 3-kinase; IGF-1R, insulin-like growth Factor 1 receptor
EGFR ligands and EGFR
EGFR is part of the EGFR tyrosine kinase family [61] and is activated by multiple ligands, such as EGF, TGF-α, HB-EGF, epiregulin (EREG) and amphiregulin (AREG) [62,63,64]. The expression of EGFR ligands in primary tumours is potentially related to anti-EGFR therapy efficiency [65, 66]. KRAS WT mCRC patients with higher expression of AREG and EREG seemed to obtain less survival benefit from cetuximab [64]. EGFR somatic sequence changes, including G465R, G465E, S468R and S492R, located at the extracellular domains (ECDs) of the EGFR-mAb interaction interface, confer resistance to cetuximab and panitumumab by preventing mAb binding [10, 67, 68]. In addition, R198/R200 methylation and mutation in the kinase domain of EGFR (V843I) correlated with disease progression in the presence of cetuximab [69].
Thus, the development of new mAbs that can bind to different or mutated EGFR ECDs is expected to improve the efficiency of anti-EGFR mAbs. MM-151, an oligoclonal antibody that binds multiple regions of the EGFR ECD, was confirmed to inhibit EGFR signalling and cell growth in a preclinical study and decrease mutations in circulating cell-free tumour DNA (ctDNA) of CRC patients [41]. Another FDA-approved EGFR antibody, necitumumab, can bind to S468R, the most common cetuximab-resistant variant of EGFR domain III [70]. Progression-free survival (PFS) and OS of patients taking necitumumab plus mFOLFOX6 were comparable to those of the cetuximab and FOLFOX regimens [21].
Considering the limitations of cetuximab and panitumumab in clinical use, it is necessary to generate more effective anti-EGFR antibodies. Sym004, a novel 1:1 mixture of two nonoverlapping anti-EGFR mAbs, showed significant advantages of abrogating EGFR ligand-induced phosphorylation and suppressing downstream signalling of all individual EGFR mutants both in cetuximab-resistant cell lines and in a tumour xenograft model [23, 71]. A multicentre, phase 2 clinical trial further confirmed that Sym004 improved the OS of anti-EGFR-refractory mCRC by 5.5 months [22]. In addition, GC1118 is a novel, fully humanized anti-EGFR IgG1 antibody that displays inhibitory effects against patient-derived xenografts from CRC tumours with a KRAS mutation [40], especially in those with elevated expression of high-affinity ligands [72].
Compensatory feedback loop signalling
The RAS/RAF/MEK/ERK and PI3K/PTEN/AKT axes are the main downstream signalling pathways of EGFR. Upregulated receptor tyrosine kinases (RTKs), including ERBB2, MET and IGF-1R, activate the PI3K/AKT axis or reactivate the ERK pathway independently of EGFR [73,74,75,76]. Alterations in these pathways, such as gene mutation, gene amplification, gene loss and abnormal phosphorylation, are of great significance in primary and secondary resistance to anti-EGFR mAbs [11, 77]. Combining EGFR-targeted inhibitors with these targeted agents shows potential to reverse resistance to anti-EGFR mAbs.
RAS mutations and RAS regulators
RAS is a master element at the centre of EGFR signalling pathways [78]. Mutations within RAS put the RAS protein in a constitutively active state independent of upstream signals driven by growth factor receptor [79], leading to the failure of EGFR-targeted therapies. Mutations in RAS usually occur in KRAS, NRAS and HRAS, and the KRAS mutation is the most common of these genomic alterations, occurring in 40% of mCRC [9, 80]. Mutations in exons 2, 3 and 4 of KRAS and exons 2, 3, and 4 of NRAS are powerful predictors for cetuximab and panitumumab response in mCRC [81, 82]. However, codon 13 mutations (G13D) in KRAS do not predict nonresponse with complete accuracy [82]. Some missense and nonsense mutations at codons 20, 27, 30, or 31 have also been reported, whereas the function of these mutations on GTPase activity and the outcome of CRC still needs further exploration [83,84,85].
RAS was the first driver gene found, and effective RAS inhibitors have been investigated for over 30 years [86]. For example, sotorasib is a small molecule that selectively and irreversibly targets KRAS (G12C). Nevertheless, drugs again other KRAS mutations in codons 12, 13 and 61 still remain to be developed [87]. Therefore, it is important to find other therapies to improve the therapeutic outcome of these patients. In 2011, Wheeler and colleagues first reported that the addition of dasatinib to cetuximab showed a powerful antiproliferative effect on KRAS mutant cell lines compared to either agent alone in vitro and in vivo [88]. However, the other clinical study did not achieve the expected results. A phase IB/II study of 77 refractory CRC patients treated with dasatinib plus FOLFOX and cetuximab did not demonstrate meaningful clinical activity because the treatment did not fully inhibit the intracellular tyrosine kinase Src [24]. Notably, some untargeted agents displayed positive results in KRAS-mutated CRC cells. The combination of simvastatin and cetuximab suppressed BRAF activity and reduced the proliferation of KRAS-mutant cells [49]. Furthermore, Metformin reversed KRAS-induced resistance to the anti-EGFR antibody by activating AMP-activated protein kinase (AMPK) and inhibiting mTOR [47]. Jung revealed that resistance to cetuximab in CRC cells with KRAS mutations can be bypassed by L-ascorbic acid relying on a sodium-dependent vitamin C transporter 2 [51]. In addition, small chemical compounds such as KY7749 and methylglyoxal scavengers resensitize KRAS-mutated CRC cells to cetuximab in vivo [48, 89]. Despite not specifically targeting the RAS protein, these drugs add alternative methods to reverse resistance induced by RAS.
RAF mutations and RAF inhibitors
BRAF is a serine-threonine kinase just downstream of EGFR/KRAS that activates the MEK/extracellular signal-regulated kinase (ERK) signalling cascade through its phosphorylation and then promotes cancer cell proliferation [90]. BRAF mutation is a powerful biomarker of poor prognosis for mCRC patients receiving anti-EGFR mAbs [80, 91, 92]. The hotspot BRAF V600E mutation at codon 600 of exon 15 increases the activity of BRAF kinase by 130- to 700-fold [26, 93,94,95]. The prevalence of BRAF V600E mutations in mCRC is 8–10%, and they occur mutually exclusively with KRAS mutations [25, 96]. Some BRAF non V600E mutations were also reported, including D594G, G469A, L485F, L525R, Q524L and V600R located in the kinase domain. Non V600E mutations, other than Q524L, may also contribute to primary resistance to anti-EGFR mAbs [25, 96].
BRAF V600E mutations occur in various cancers, such as melanoma, non-small-cell lung cancer, breast cancer and CRC, and inhibitors targeting BRAF have demonstrated clinical benefit for these patients. Vemurafenib, a selective oral inhibitor of the BRAF V600 kinase, achieved an approximately 50% response and improved survival among metastatic melanoma patients with the BRAF V600E mutation [97]. Recently, many clinical trials have been conducted to evaluate EGFR therapeutic resistance with vemurafenib. In 2015, a pilot trial of combined vemurafenib and panitumumab in BRAF-mutant mCRC patients post chemotherapy reported that the treatment limited tumour progression and resulted in modest clinical activity [98]. However, a multicentric clinical study containing 27 CRC patients showed that vemurafenib alone or with cetuximab did not benefit CRC patients [99]. The next year, a phase IB study affirmed the value of vemurafenib again. This study demonstrated that triplets of vemurafenib, irinotecan, and cetuximab were well tolerated and exceeded tumour regression in refractory BRAF-mutated mCRC [100]. Of course, more clinical studies are needed to ensure that vemurafenib is efficacious.
Despite the confusing results of the vemurafenib studies, another BRAF inhibitor, encorafenib, has confirmed the feasibility of dual-targeted EGFR and BRAF treatment to increase the efficiency of anti-EGFR mAbs. The BEACON trial showed promising efficacy results with an objective response rate (ORR) of 48% (95% CI, 29.4–67.5%) among 29 patients in the study [27]. Within the randomized portion of the BEACON trial, the confirmed ORR for the triplet treatment was much better than that for the control (26% vs. 2%). The median OS was 9 months on the triplet regimen compared to 5.4 months in the control group (P < 0.0001, 75]. Based on the randomized, phase III BEACON trial, a combination of encorafenib and binimetinib (a MEK inhibitor) with cetuximab has been recommended as a second-line systemic therapy for BRAF V600E mutation CRC.
MEK activation and MEK inhibitors
MEK/ERK is the most important downstream cascade of the signalling pathways related to anti-EGFR mAbs. However, mutations of RAS/RAF induce constitutive activation of MEK to promote cell proliferation and survival. Encouragingly, combination treatment with MEK and EGFR inhibitors seems to be a possible strategy to overcome the multifaceted clonal heterogeneity in tumours [29, 101]. Additionally, there are already some small molecule MEK inhibitors under research. AS703026 (also known as pimasertib), AZD6244 (also known as selumetinib) and BAY86–9766 have a great ability to hinder the growth of mutant KRAS cells in vitro and in vivo by specifically suppressing the key target kinase ERK, which is downstream of MEK [30, 42]. MEK inhibitors were also confirmed to increase the tumour-suppressive effect of cetuximab. Selumetinib or pimasertib plus cetuximab enhanced antiproliferative and proapoptotic effects in cells resistant to cetuximab in vitro and in vivo [102, 103]. However, Misale found that in vitro and in vivo, the growth of resistant cells could not be hampered by MEK1/2 inhibitors alone; instead, the synergistic pharmacological blockade of EGFR and MEK induced drawn-out ERK inhibition and serious growth impairment of resistant tumour cells [43]. More importantly, the combination of selumetinib and cetuximab failed to achieve positive results in a clinical trial in refractory metastatic CRC patients with KRAS mutations [30, 101] To date, binimetinib is the only MEK inhibitor permitted by the Food and Drug Association for clinical use in mCRC. Clinical verification of the feasibility of MEK inhibitors to reverse EGFR therapeutic resistance is urgently required.
PI3K/AKT activation and PI3K/AKT inhibitors
Phosphor-EGFR is capable of initiating the PI3K/AKT/mTOR pathway, and PI3K mutation and aberrant AKT/mTOR activation promote resistance to anti-EGFR mAbs [104]. The most common mutations in PIK3CA are in exons 9 (68.5%) and 20 (20.4%), and they are detected in 10–18% of mCRC [11, 105]. Mutations in PIK3CA exon 20 were significantly associated with a worse outcome in KRAS WT mCRC patients treated with cetuximab, whereas PIK3CA exon 9 mutations had no effect on outcome in KRAS WT mCRC patients [11]. PTEN is a negative regulator in the PI3K/AKT pathway and was found in 20–40% of mCRC [106, 107]. Loss of PTEN protein results in long-term tumour growth by activating PI3K/AKT. Patients with PTEN-negative status showed a worse response rate and shorter progression-free survival (PFS) than those with PTEN-positive status [106, 108, 109]. PI3K gene mutation and PTEN protein loss are confirmed as novel biomarkers in mCRC patients treated with anti-EGFR mAbs [110].
Combinations of cetuximab and PI3K, AKT or mTOR inhibitors can profoundly control tumour growth in mCRC regardless of driver genotypes [32]. Specifically, PI3K inhibitors have been shown to greatly inhibit the growth of cancer in preclinical and clinical experiments. For example, the PI3K inhibitor XL147 was reported to inhibit the PI3K pathway with a 40–80% reduction in the phosphorylation of AKT and 4EBP1 in tumours and unexpectedly inhibited the MEK/ERK pathway in a phase I trial [111]. Then, in an investigation of the effects of XL147 on proliferation in a panel of tumour cell lines, Shapiro et al. revealed that XL147 was useful for PI3K mutation/amplification cell lines without KRAS/BRAF/PTEN mutation [112]. Another PI3K inhibitor, BKM120, was found to impede KRAS mutation-induced colorectal cancer growth both in vitro and in vivo, regardless of PI3K genotype [45]. Despite the ideal results of preclinical studies, clinical trials on the combination of PI3K inhibitors and EGFR-targeted agents are frustrating. PX-866 is a panisoform inhibitor of PI3K; however, the addition of PX-866 to cetuximab did not improve the PFS and OS of KRAS WT mCRC and caused greater toxicity in a phase II study [32]. Considering the lack of clinical trials on the combination of PI3K inhibitors with cetuximab or panitumumab, the application of PI3K inhibitors in enhancing the response to anti-EGFR mAbs remains unascertainable thus far.
ERBB2 amplification/mutations and ERBB2 inhibitors
A total of 2–7% of unselected CRCs were found to have ERBB2/mutations, and this number was much higher in KRAS wild-type cases (13.6%) and in KRAS/NRAS/BRAF/PIK3CA quadruple wild-type cases (36% )[13, 113, 114]. Bertotti et al. identified ERBB2 as a biomarker of resistance to anti-EGFR mAbs [113]. Importantly, amplification of ERBB2 was also enriched in nonresponsive KRAS WT mCRC [73]. ERBB2 activating mutations of S310F, L755S, V777L, V842I, and L866 increase MAPK phosphorylation and produce resistance to cetuximab and panitumumab [13].
The clinical use of ERBB2-targeted drugs, such as trastuzumab, pertuzumab and lapatinib, improved outcomes for breast cancer and colorectal cancer patients with ERBB2 amplification [115, 116]. Dual-targeted therapy with EGFR and ERBB2 inhibition were found to restore sensitivity to cetuximab in vitro and in vivo. The monoclonal antibody 4D5 is an ERBB2 inhibitory antibody that shows antitumour function in an EGFR-dependent manner. The combination of the mAb 4D5 with cetuximab induced a significant decrease in proliferation in the EGFR-dependent colon cancer cell line and an actual regression of the tumours in xenografted mice [43]. Similarly, trastuzumab, the most common antibody for ERBB2, inhibits the growth of CRC cells when combined with cetuximab [44]. However, the pan ERBB kinase inhibitor neratinib plus cetuximab did not reach an objective response in anti-EGFR treatment refractory mCRC with quadruple-wild-type (KRAS, NRAS, BRAF, PIK3CA) in a phase II clinical trial [31]. In general, it will be important to investigate the efficiency of mAb 4D5 and trastuzumab in the clinic to confirm the value of anti-ERBB2 agents.
IGF-1R activation and anti-IGF-1R mAbs
The IGF-1/IGF-1R pathway plays a crucial role in CRC proliferation, differentiation, apoptosis, migration and angiogenesis. Hyperactivation of IGF-1R results in primary and secondary resistance to EGFR inhibition in RAS wild-type mCRC by upregulating the PI3K/AKT pathway [117, 118]. Analyses from two clinical trials confirmed that the coexpression of pIGF-1R and MMP-7 in RAS wild-type mCRC predicts worse OS after treatment with cetuximab [119].
Therefore, targeting both EGFR and IGF-1R may be a potential therapy for mCRC. Disappointingly, a trial showed that the combination of cetuximab and dalotuzumab or IMC-A12 did not improve the survival of mCRC resistant to cetuximab [35, 36].
MET amplification/activation and MET inhibitors
The MET signalling pathway is another compensatory feedback loop that mostly arises during the treatment of anti-EGFR mAbs. Phosphorylation of MET induces the activation of PI3K/AKT and RAS/RAF/MAPK cascades to rescue tumour cells from EGFR inhibitors [120]. Bardelli et al. highlight that MET amplification is related to acquired resistance to anti-EGFR therapy in tumours without KRAS mutations [12]. Moreover, the EGF ligands HGF and TGF-α can bind to MET and then increase the phosphorylation of MET and its downstream MAPK and AKT [121, 122]. Accordingly, the resistance function of MET was demonstrated by combining MET inhibitors and cetuxima b [123].
Therefore, the application of MET inhibitors has strong therapeutic potential in human cancers. The combination of MET inhibitors with anti-EGFR agents presents encouraging results in both preclinical and clinical studies. Tivantinib (ARQ 197), a selective, non-ATP-competitive inhibitor of c-MET, displayed tolerated toxicity and suggested some activity in previously treated mCRC when combined with cetuximab and chemotherapy [124]. Another phase II clinical study reported a positive outcome in mCRC patients who received tivantinib plus cetuximab. Forty-one patients with tumour progression on cetuximab or panitumumab treatment were enrolled in the study; the ORR was 9.8% (4/41), the median progression-free survival (mPFS) was 2.6 months (95% CI, 1.9–4.2 months), and the mOS was 9.2 months (95% CI, 7.1–15.1 months) [33]. Another small molecule c-MET inhibitor, crizotinib, has been shown to improve the efficiency of radiotherapy in cetuximab-resistant KRAS mutant CRC cell lines [46]. At the American Society of Clinical Oncology (ASCO) 2019, the phase II multicentre, multicohort GEOMETRY mono-1 clinical study showed that the combination of capmatinib (a c-MET inhibitor) with gefitinib (EGFR-TKI) had a good overall response rate in EGFR-TKI-resistant patients, particularly those with MET-amplified disease [34]. Furthermore, capmatinib plus cetuximab suggested that there were preliminary signs of activity in MET-positive mCRC patients who had progressive disease following anti-EGFR mAbs [125]. Suppression of MET will be an important target in overcoming resistance to anti-EGFR therapy.
Microsatellite instability and immune checkpoint inhibitors
Microsatellite instability (MSI) caused by dysfunctional mismatch repair (dMMR) is detected in approximately 15% of all CRC and in nearly all cases with Lynch syndrome [126]. Microsatellite status and cetuximab efficiency is another area of interest. In the CALGB/SWOG 80405 study, patients with microsatellite instability-high (MSI-H) tumours showed worse OS in the cetuximab arm than in the bevacizumab arm [127]. MSI may interact with oncogenic drivers such as BRAF and ERBB2 to promote cetuximab resistance. BRAF V600E occurs in 40% of sporadic MSI-H CRCs and is typically genetically seen subsequent to hMLH1 hypermethylation [128]. In addition, other hotspot mutations in KRAS, PIK3CA and ERBB2 were identified in BRAF WT MSI CRC patients [129]. It has been proven that hMLH1 deficiency plays a role in cetuximab resistance by increasing the expression level of ERBB2 and downstream PI3K/AKT signalling [130]. Although we believe that mismatch repair genes may partly modulate the expression of oncogenic drivers, the mechanism remains largely unclear and a worthwhile focus for further research.
EGFR-targeted treatment increased the infiltration of cytotoxic immune cells and the expression of the PD-L1 immune checkpoint, which may be a potential method to treat cetuximab-resistant CRCs with immunotherapy [19]. In addition, NK cell-mediated ADCC activated by cetuximab triggers immunogenic death of tumour cells, thereby increasing the antitumour activity of immunotherapy [131]. The phase II CAVE mCRC trial demonstrated that rechallenge avelumab (anti-PD-L1) plus cetuximab resulted in a mPFS of 3.6 months and a median OS (mOS) of 11.6 months in a RAS WT mCRC population who developed acquired resistance to anti-EGFR drugs [38]. The ongoing AVETUXIRI trial investigates the efficiency of avelumab combined with cetuximab and irinotecan for refractory mCRC patients with microsatellite stability. The current study data has shown that encouraging results of DCR, PFS and OS were observed in both the RAS MT and RAS WT cohorts [132]. The combination of immune checkpoint inhibitors with anti-EGFR mAbs may bring great breakthroughs to overcome resistance to anti-EGFR drugs and improve the outcome of mCRC regardless of the status of RAS.
Metabolic remodelling and regulators
Alterations in cellular metabolism are essential for rapid tumour proliferation and affect the sensitivity of cancer cells to various drugs [133]. Anti-EGFR treatment causes metabolic rewiring in CRC patients, which makes it possible to increase anti-EGFR mAb efficiency by adding metabolism regulators.
Abnormal glycometabolism reduces the efficiency of anti-EGFR therapy. High glycolytic metabolism regulated by TRAP1 was involved in resistance to EGFR mAbs [15]. Sirt5-positive CRCs develop cetuximab resistance due to an elevated succinate-to-ketoglutarate (αKG) ratio, which inhibits αKG-dependent dioxygenases [134]. Sodium glucose transporter 2 (SGLT2) can ensure glucose entry into cells and is highly expressed in the majority of cancer cells. The SGLT2 inhibitor dapagliflozin combined with cetuximab dramatically reduced carcinoembryonic antigen (CEA) and substantial shrinkage of metastatic tumour lesions [37]. The methylglyoxal scavenger carnosine was confirmed to resensitize KRAS-mutated colorectal tumours to cetuximab in vivo [48]. AMPK activity was consistent with the sensitivity of anti-EGFR mAbs, and metformin overcame KRAS-induced resistance to anti-EGFR antibodies by regulating AMPK/mTOR/Mcl-1 (myeloid cell leukaemia 1) in vivo and in vitro [47]. Fatty acid metabolism displayed strong antiapoptotic effects in cetuximab-nonresponders [135]. Inhibition of lipid synthesis or decomposition with simvastatin or glutaminase 1 inhibitor CB-839 significantly reduced tumour growth of CRC under cetuximab treatment [49, 50]. In addition, vitamin D deficiency has a negative impact on cetuximab-induced ADCC. Supplementation with vitamin D in vitamin-deficient/insufficient CRC cells has been suggested to improve cetuximab-induced ADCC in CRC cell lines [136]. Resistance to cetuximab in mutant KRAS CRC patients can be reversed by L-ascorbic acid by reducing RAF/ERK activity in an SVCT-2-dependent manner [51].
Others
Autophagy and cancer stem cells (CSCs) also contribute to resistance to EGFR target therapy [18, 137]. Treatment with anti-EGFR agents results in dysregulation of autophagy [138]. Increased levels of autophagy-related proteins such as Beclin-1 and LC3 were observed in cetuximab-treated patients [139, 140]. Inhibition of autophagy by chloroquine and 3-methyladenine sensitizes cancer cells to cetuximab [138, 139]. However, blocking general autophagy might greatly affect normal cell growth. Therefore, developing specific autophagy inhibitors that target tumour cells is crucial.
CSCs possess genetic determinants for the EGFR therapeutic response and are primarily supported by a network of pluripotency transcription factors (PTFs). Single-nucleotide polymorphisms of PTFs were significantly associated with PFS of the cetuximab cohort in the FIRE-3 trial [141]. The property of CSCs to EMT is a core transcriptional network to predict the efficacy of EGFR-targeted therapy in KRAS WT CRC [142]. Inhibition of EMT is of great interest for reversing EGFR therapeutic resistance. β-Elemene, a bioactive monomer isolated from the Chinese herb curcumae rhizoma, has been shown to induce ferroptosis and reduce EMT to increase cetuximab activity in RAS-mutated CRC cells [60]. Furthermore, cytotoxic drugs and natural bioactive monomers were confirmed to overcome resistance to EGFR-targeted drugs. TAS-102 is a novel chemotherapeutic agent that contains a thymidine phosphorylase inhibitor, tipiracil hydrochloride, and a cytotoxic thymidine analogue, trifluridine, which has been approved for the treatment of mCRC. Panitumumab/TAS-102 cotreatment showed additive antiproliferative effects in LIM1215 CRC cells in vitro and in vivo [59].
Extrinsic mechanisms of resistance to targeted therapy and related strategies
Microenvironmental plasticity dramatically affected by EGFR inhibition is as powerful of a driver of drug resistance as genetic alterations [18] (Fig. 2). Dysfunction of immune cells, abnormal infiltration of cancer-associated fibroblasts (CAFs) and angiogenesis impair EGFR therapeutic efficiency. Strategies to remodel the tumour microenvironment are part of a larger goal to increase the efficiency of anti-EGFR mAbs. These strategies include (i) modification or activation of NK cells and T cells, (ii) suppression of CAFs, and (iii) inhibition of angiogenesis (Tables 1 and 2).

Extrinsic mechanisms of resistance to anti-EGFR mAbs in metastatic colorectal cancer. Tumour microenvironment plasticity confers resistance to EGFR-targeted therapy. Cetuximab and panitumumab suppress tumours through ADCC mediated by NK cells and macrophages. Dysfunction of NK cells and macrophages with lower ADCC impairs the suppression of EGFR-targeted therapy in cancer. Reduced density of effector T cells and increased PD-L1 expression in cancer cells also promote survival from cancer. CAFs promote resistance to targeted therapy by secreting growth factors that activate the RAS or MET pathway. Abnormal angiogenesis always predicts poor response to anti-EGFR mAbs. Therapies focused on the microenvironment are also shown in the figure. Abbreviations: CAFs, cancer-associated fibroblasts; NK cells, natural killer cells; ADCC, antibody-dependent cellular cytotoxicity; PD-1, programmed death 1; PD-L1, programmed death ligand 1. VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor
Immune cells and agents
Antibody-dependent cellular cytotoxicity (ADCC) mediated through Fc receptors (FcγRs) on immune cells is one of the proposed antitumour mechanisms of anti-EGFR mAbs [20]. Cetuximab-treated patients with FcγRIIa-131R and/or FcγIIIa-158F genotypes had shorter PFS than 131H/H and/or 158 V/V carriers [143]. When exposed to cetuximab in CRC cell lines, human NK cells substantially increase the expression of the costimulatory molecule CD137 (4-1BB )[144]. The combination of cetuximab with anti-CD137 mAb administration was synergistic and resulted in complete tumour resolution and prolonged survival, which were dependent on the participation of NK cells [52]. IL-2 and IL-15 cooperate with cetuximab to stimulate NK cells and improve cytotoxic functionality [145]. Another preclinical study using a mouse model reported that umbilical cord blood stem cell-derived NK (UCB-NK) cells increased antitumour cytotoxicity against CRC regardless of the status of EGFR and RAS [53]. Neither cetuximab nor panitumumab can engage T cells when T cells lack Fcγ receptors, which serve as targets for modifying T cells to enhance the ADCC activity of anti-EGFR agents [54]. T cell-engaging BiTE antibodies targeting the binding domains of cetuximab and panitumumab transiently connect T cells with cancer cells to initiate redirected target cell lysis. Then, they showed that cetuximab-based BiTE antibody mediated potent redirected lysis of KRAS- and BRAF-mutated CRC lines in vitro and prevented the growth of tumours from xenografts [54]. Toll-like receptor 9 (TLR9) is expressed in various immune cells, such as macrophages, NK cells, B lymphocytes and plasmacytoid dendritic cells [146, 147]. Toll-like receptor 9 (TLR9) activation causes antitumour activity by interfering with cancer proliferation and angiogenesis [148]. IMO is a novel second-generation, modified, immunomodulatory TLR9 agonist and was proven to synergistically inhibit tumour growth by improving the ADCC activity of cetuximab in a cetuximab-resistant colorectal cancer line and a mouse model regardless of KRAS genotype [55, 56].
CAFs and inhibitors
Cancer-associated fibroblasts (CAFs) are believed to play a vital role in promoting tumour metastasis and drug resistance by secreting mitogenic growth factors, including FGF1, FGF2, HGF, TGF-β1 and TGF-β 2 [19]. Luraghi et al. reported that HGF can bind to MET receptors and activate MAPK and AKT to induce cetuximab resistance in vitro [75]. The dual inhibition of FGFR and EGFR may be a practical strategy to reverse resistance to anti-EGFR mAbs. The combination of BLU9931,an FGFR4 inhibitor, with cetuximab presented profound antitumour activity compared to cetuximab alone [57]. Regorafenib, a multikinase inhibitor targeting FGFR, VEGF and PDGFR-β, was found to overcome cetuximab resistance in GEO-CR and SW48-CR cells in vitro and in vivo [58].
Angiogenesis and inhibitors
Inhibition of angiogenesis is also one of the mechanisms of cetuximab action. Treatment with cetuximab reduced the expression of vascular endothelial growth factor (VEGF), and a high level of VEGF under cetuximab treatment was associated with a lower response rate and shorter PFS in mCRC [149, 150]. VEGF is one of the most significant angiogenetic factors, and it contributes to cancer prognosis and metastasis. Therefore, it is worth exploring the feasibility of dual-targeted VEGF and EGFR in colorectal cancer. Combination treatment with anti-VEGF and anti-EGFR antibodies demonstrated synergistic activity in vitro, and tumour growth and angiogenesis were strongly suppressed in an in vivo xenograft mouse model [151]. However, in another study, the use of bevacizumab and cetuximab together did not have a greater increase in apoptotic tumour cell death compared to either drug alone [152]. Recently, small molecule inhibitors targeting VEGF have presented the potential to increase the efficiency of anti-EGFR therapy. Pazopanib, a multitargeted tyrosine kinase inhibitor, combined with irinotecan and cetuximab showed manageable safety and feasibility in refractory mCRC [153]. The combination of the anti-EGFR antibody cetuximab and the multikinase VEGF inhibitor regorafenib overcame intrinsic and acquired resistance in mCRC. Eight of 17 mCRC patients, who all were previously receiving anti-VEGF and anti-EGFR therapy, showed clinical benefit from cetuximab and regorafenib, including partial response in 1 patient and stable disease in 7 patients [39]. Dual-targeting of VEGF and EGFR seems to be an effective choice for mCRC patients receiving multiline treatment.
Conclusions and future directions
Heterogeneity and adaptive alterations promote resistance to anti-EGFR targeted therapy and are strongly associated with the clinical outcome of colorectal cancer (Fig. 3). RAS/BRAF/MEK mutations downstream of the EGFR pathway and ERBB2/MET/IGFR/PI3K mutations or amplifications bypassing EGFR are strong biomarkers to predict the efficiency of anti-EGFR mAbs. It is of great importance to ascertain the molecular subtypes in mCRC before treatment. Advances in gene detection methods such as ctDNA, liquid biopsy and exosome DNA sequencing make molecular subtyping feasible. By identifying similarities and differences among tumour subtypes, the use of precision medicine results in greater cancer eradication and better patient care. For subpopulations with driver-gene mutations, combination therapies of different targeted inhibitors make great strides in overcoming resistance to anti-EGFR mAbs. Combining EGFR targeted therapy with inhibitors of BRAF, MET and MEK produces expected results in clinical trials. It is recommended to use encorafenib, binimetinib and cetuximab in the second-line treatment of mCRC. More clinical studies are needed to ensure the effectiveness of MEK inhibitors. In addition, the new generation of anti-EGFR monoclonal antibodies and cytotoxic agents is promising to achieve better outcomes, but further research is needed before clinical application.

Strategies to increase anti-EGFR therapy efficiency in different subtypes of mCRC. Biomarker analysis should be conducted before treatment for mCRC. For patients with disease progression on anti-EGFR therapy, biomarker analysis is still recommended. For mCRC with driver gene alterations, there are some therapies to increase anti-EGFR efficiency. In RAS-mut mCRC, the selected therapies include a combination of RAS inhibitors and anti-EGFR agents, metabolic regulators, immune therapy, cytotoxic drugs and natural bioactive monomers. In RAF-mut mCRC, the main therapy is a BRAF inhibitor. In ERBB2-amp mCRC, ERBB2 inhibitors can be used to promote the antiproliferation of anti-EGFR. In MET-amp mCRC, combined therapy with MET inhibitors and anti-EGFR mAbs was confirmed to be effective. In mCRC with EGFR ECD-mut, new anti-EGFR agents are preferred. In mCRC with no driver gene alteration, multitargeted therapies, metabolic regulators, immune therapy, cytotoxic drugs and antiangiogenic agents can be used with anti-EGFR. Abbreviations: mCRC, metastatic colorectal cancer; EGFR, epidermal growth factor receptor; ERBB2, human epidermal growth factor receptor 2; MET, tyrosine-protein kinase Met; MSI-H, microsatellite instability; dMMR, dysfunctional mismatch repair; PD-1/PD-L1, programmed death-1/programmed death ligand 1; ECD, extracellular domain; WT, wild type; mut, mutation
In this review, we provide new insight into EGFR therapeutic resistance in the tumour microenvironment (TME) and summarize current agents for the TME. The TME, including the immune microenvironment and vascular microenvironment, facilitates tumour growth and metastasis. The ADCC activity of anti-EGFR mAbs mediated by NK cells, T cells and macrophages is one of the antitumour mechanisms targeted by cetuximab and panitumumab. The strong effect of cetuximab on the immune landscape dramatically changes immune infiltrates. Thus, more effective immunotherapies are anticipated to regress the growth and metastasis of tumours. Some antibodies or inhibitors constructed to bind FcγR or TLR9 to stimulate ADCC mediated by NK cells, T cells and macrophages present significant antitumour activity in cell lines and mouse models. Dual-targeted VEGF and EGFR treatments show exciting results in multiline-treated mCRC patients, providing a chance for improved outcomes in refractory patients. Notably, anti-EGFR therapy especially enhances the expression of PD-L1 on tumours and the infiltration of CD8+ T cells. Therefore, this feature may expand indications of immune checkpoint inhibitors in CRC. Treatment with immune checkpoint inhibitors either along with anti-EGFR mAbs or later is a promising therapy for mCRC.
In summary, the recognition of resistance to EGFR-targeted therapy has progressed from driver genes to nongenetic alterations. Different therapies that reverse EGFR therapeutic resistance demonstrate potential in preclinical and clinical trials. These treatments show promise in taking a giant step towards overcoming EGFR therapeutic resistance.
Availability of data and materials
All the data obtained and/or analyzed during the current study were available from the corresponding authors on reasonable request.
Abbreviations
- EGFR:
-
Epidermal growth factor receptor
- WT:
-
Wild type
- mAbs:
-
Monoclonal antibodies
- mCRC:
-
Metastatic colorectal cancer
- CRC:
-
Colorectal cancer
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
- mPFS:
-
median progression-free survival
- mOS:
-
median overall survival
- ORR:
-
Objective response rate
- TKI:
-
Tyrosine kinase inhibitors
- ECD:
-
Extracellular domain
- ERK:
-
Extracellular signal-regulated kinase
- RTKs:
-
Receptor tyrosine kinases
- MSI:
-
Microsatellite instability
- dMMR:
-
Dysfunctional mismatch repair
- CSCs:
-
Cancer stem cells
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- FcγRs:
-
Fc receptors
- CAFs:
-
Cancer-associated fibroblasts
- AMPK:
-
AMP-activated protein kinase
- ASCO:
-
American Society of Clinical Oncology
- SGLT2:
-
Sodium glucose transporter 2
- GLS1:
-
Glutaminase1
- TLR9:
-
Toll-like receptor 9
- FGFR:
-
Fibroblast growth factor receptors
- ctDNA:
-
circulating cell-free tumor DNA
- αKG:
-
a-ketoglutarate
- PTFs:
-
Pluripotency transcription factors
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Wang F, Fu X, Chen P, Wu P, Fan X, Li N, et al. SPSB1-mediated HnRNP A1 ubiquitylation regulates alternative splicing and cell migration in EGF signaling. Cell Res. 2017;27(4):540–58. https://doi.org/10.1038/cr.2017.7.
Mimeault M, Hauke R, Mehta PP, Batra SK. Recent advances in cancer stem/progenitor cell research: therapeutic implications for overcoming resistance to the most aggressive cancers. J Cell Mol Med. 2007;11(5):981–1011. https://doi.org/10.1111/j.1582-4934.2007.00088.x.
Modest DP, Stintzing S, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, et al. Impact of subsequent therapies on outcome of the FIRE-3/AIO KRK0306 trial: first-line therapy with FOLFIRI plus Cetuximab or bevacizumab in patients with KRAS wild-type tumors in metastatic colorectal Cancer. J Clin Oncol. 2015;33(32):3718–26. https://doi.org/10.1200/JCO.2015.61.2887.
Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang CC, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–17. https://doi.org/10.1056/NEJMoa0805019.
Tabernero J, Van Cutsem E, Díaz-Rubio E, Cervantes A, Humblet Y, André T, et al. Phase II trial of cetuximab in combination with fluorouracil, leucovorin, and oxaliplatin in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2007;25(33):5225–32. https://doi.org/10.1200/JCO.2007.13.2183.
Borner M, Koeberle D, Von Moos R, Saletti P, Rauch D, Hess V, et al. Adding cetuximab to capecitabine plus oxaliplatin (XELOX) in first-line treatment of metastatic colorectal cancer: a randomized phase II trial of the Swiss Group for Clinical Cancer Research SAKK. Ann Oncol. 2008;19(7):1288–92. https://doi.org/10.1093/annonc/mdn058.
Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28(7):1254–61. https://doi.org/10.1200/JCO.2009.24.6116.
Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–65. https://doi.org/10.1056/NEJMoa0804385.
Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526(7572):263–7. https://doi.org/10.1038/nature14969.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–62. https://doi.org/10.1016/S1470-2045(10)70130-3.
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–73. https://doi.org/10.1158/2159-8290.CD-12-0558.
Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5(8):832–41. https://doi.org/10.1158/2159-8290.CD-14-1211.
Huang F, Xu LA, Khambata-Ford S. Correlation between gene expression of IGF-1R pathway markers and cetuximab benefit in metastatic colorectal cancer. Clin Cancer Res. 2012;18(4):1156–66. https://doi.org/10.1158/1078-0432.CCR-11-1135.
Maddalena F, Condelli V, Matassa DS, Pacelli C, Scrima R, Lettini G, et al. TRAP1 enhances Warburg metabolism through modulation of PFK1 expression/activity and favors resistance to EGFR inhibitors in human colorectal carcinomas. Mol Oncol. 2020. https://doi.org/10.1002/1878-0261.12814.
Medema JP. Targeting the colorectal Cancer stem cell. N Engl J Med. 2017;377(9):888–90. https://doi.org/10.1056/NEJMcibr1706541.
Gao L, Xu J, He G, Huang J, Xu W, Qin J, et al. CCR7 high expression leads to cetuximab resistance by cross-talking with EGFR pathway in PI3K/AKT signals in colorectal cancer. Am J Cancer Res. 2019;9(11):2531–43.
Sidaway P. Microenvironment plasticity confers cetuximab resistance. Nat Rev Clin Oncol. 2019;16(9):527. https://doi.org/10.1038/s41571-019-0259-4.
Woolston A, Khan K, Spain G, Barber LJ, Griffiths B, Gonzalez-Exposito R, et al. Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti-EGFR treatment in colorectal Cancer. Cancer Cell. 2019;36(1):35–50. https://doi.org/10.1016/j.ccell.2019.05.013.
Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, et al. Impact of fc{gamma}RIIa-fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27(7):1122–9. https://doi.org/10.1200/JCO.2008.18.0463.
Elez E, Hendlisz A, Delaunoit T, Sastre J, Cervantes A, Varea R, et al. Phase II study of necitumumab plus modified FOLFOX6 as first-line treatment in patients with locally advanced or metastatic colorectal cancer. Br J Cancer. 2016;114(4):372–80. https://doi.org/10.1038/bjc.2015.480.
Montagut C, Argilés G, Ciardiello F, Poulsen TT, Dienstmann R, Kragh M, et al. Efficacy of Sym004 in patients with metastatic colorectal Cancer with acquired resistance to anti-EGFR therapy and molecularly selected by circulating tumor DNA analyses. Jama Oncol. 2018;4(4):e175245. https://doi.org/10.1001/jamaoncol.2017.5245.
Sánchez-Martín FJ, Bellosillo B, Gelabert-Baldrich M, Dalmases A, Cañadas I, Vidal J, et al. The first-in-class anti-EGFR antibody mixture Sym004 overcomes Cetuximab resistance mediated by EGFR extracellular domain mutations in colorectal Cancer. Clin Cancer Res. 2016;22(13):3260–7. https://doi.org/10.1158/1078-0432.CCR-15-2400.
Parseghian CM, Parikh NU, Wu JY, Jiang ZQ, Henderson L, Tian F, et al. Dual inhibition of EGFR and c-Src by Cetuximab and Dasatinib combined with FOLFOX chemotherapy in patients with metastatic colorectal Cancer. Clin Cancer Res. 2017;23(15):4146–54. https://doi.org/10.1158/1078-0432.CCR-16-3138.
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3. https://doi.org/10.1038/nature10868.
Shinozaki E, Yoshino T, Yamazaki K, Muro K, Yamaguchi K, Nishina T, et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: the biomarker research for anti-EGFR monoclonal antibodies by comprehensive Cancer genomics (BREAC) study. Br J Cancer. 2017;117(10):1450–8. https://doi.org/10.1038/bjc.2017.308.
Van Cutsem E, Huijberts S, Grothey A, Yaeger R, Cuyle PJ, Elez E, et al. Binimetinib, Encorafenib, and Cetuximab triplet therapy for patients with BRAF V600E-mutant metastatic colorectal Cancer: safety Lead-in results from the phase III BEACON colorectal Cancer study. J Clin Oncol. 2019;37(17):1460–9. https://doi.org/10.1200/JCO.18.02459.
Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-mutated colorectal Cancer. N Engl J Med. 2019;381(17):1632–43. https://doi.org/10.1056/NEJMoa1908075.
Misale S, Bozic I, Tong J, Peraza-Penton A, Lallo A, Baldi F, et al. Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat Commun. 2015;6:8305. https://doi.org/10.1038/ncomms9305.
Troiani T, Napolitano S, Vitagliano D, Morgillo F, Capasso A, Sforza V, et al. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin Cancer Res. 2014;20(14):3775–86. https://doi.org/10.1158/1078-0432.CCR-13-2181.
Jacobs SA, Lee JJ, George TJ, Wade JL, Stella PJ, Wang D, et al. Neratinib plus Cetuximab in quadruple WT (KRAS, NRAS, BRAF, PIK3CA) metastatic colorectal Cancer resistant to Cetuximab or Panitumumab: NSABP FC-7, a phase Ib study. Clin Cancer Res. 2020. https://doi.org/10.1158/1078-0432.CCR-20-1831.
Bowles DW, Kochenderfer M, Cohn A, Sideris L, Nguyen N, Cline-Burkhardt V, et al. A randomized, phase II trial of Cetuximab with or without PX-866, an irreversible Oral phosphatidylinositol 3-kinase inhibitor, in patients with metastatic colorectal carcinoma. Clin Colorectal Cancer. 2016;15(4):337–44. https://doi.org/10.1016/j.clcc.2016.03.004.
Rimassa L, Bozzarelli S, Pietrantonio F, Cordio S, Lonardi S, Toppo L, et al. Phase II study of Tivantinib and Cetuximab in patients with KRAS wild-type metastatic colorectal Cancer with acquired resistance to EGFR inhibitors and emergence of MET overexpression: lesson learned for future trials with EGFR/MET dual inhibition. Clin Colorectal Cancer. 2019;18(2):125–32. https://doi.org/10.1016/j.clcc.2019.02.004.
Wu YL, Zhang L, Kim DW, Liu X, Lee DH, Yang JC, et al. Phase Ib/II study of Capmatinib (INC280) plus Gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung Cancer. J Clin Oncol. 2018;36(31):3101–9. https://doi.org/10.1200/JCO.2018.77.7326.
Reidy DL, Vakiani E, Fakih MG, Saif MW, Hecht JR, Goodman-Davis N, et al. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J Clin Oncol. 2010;28(27):4240–6. https://doi.org/10.1200/JCO.2010.30.4154.
Sclafani F, Kim TY, Cunningham D, Kim TW, Tabernero J, Schmoll HJ, et al. A randomized phase II/III study of Dalotuzumab in combination with Cetuximab and irinotecan in Chemorefractory, KRAS wild-type, metastatic colorectal Cancer. J Natl Cancer Inst. 2015;107(12):v258. https://doi.org/10.1093/jnci/djv258.
Okada J, Matsumoto S, Kaira K, Saito T, Yamada E, Yokoo H, et al. Sodium glucose cotransporter 2 inhibition combined with Cetuximab significantly reduced tumor size and carcinoembryonic antigen level in Colon Cancer metastatic to liver. Clin Colorectal Cancer. 2018;17(1):e45–8. https://doi.org/10.1016/j.clcc.2017.09.005.
ASCO Meeting Library: 2021 ASCO Annual Meeting. https://meetinglibrary.asco.org/record/196464/abstract. Accessed 02 Aug 2021.
Subbiah V, Khawaja MR, Hong DS, Amini B, Yungfang J, Liu H, et al. First-in-human trial of multikinase VEGF inhibitor regorafenib and anti-EGFR antibody cetuximab in advanced cancer patients. JCI Insight. 2017;2(8). https://doi.org/10.1172/jci.insight.90380.
Lee HW, Son E, Lee K, Lee Y, Kim Y, Lee JC, et al. Promising Therapeutic Efficacy of GC1118, an Anti-EGFR Antibody, against KRAS Mutation-Driven Colorectal Cancer Patient-Derived Xenografts. Int J Mol Sci. 2019;20(23). https://doi.org/10.3390/ijms20235894.
Arena S, Siravegna G, Mussolin B, Kearns JD, Wolf BB, Misale S, et al. MM-151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring EGFR extracellular domain mutations. Sci Transl Med. 2016;8(324):314r–24r. https://doi.org/10.1126/scitranslmed.aad5640.
Yoon J, Koo KH, Choi KY. MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer Res. 2011;71(2):445–53. https://doi.org/10.1158/0008-5472.CAN-10-3058.
Kuwada SK, Scaife CL, Kuang J, Li X, Wong RF, Florell SR, et al. Effects of trastuzumab on epidermal growth factor receptor-dependent and -independent human colon cancer cells. Int J Cancer. 2004;109(2):291–301. https://doi.org/10.1002/ijc.11686.
Luca T, Barresi V, Privitera G, Musso N, Caruso M, Condorelli DF, et al. In vitro combined treatment with cetuximab and trastuzumab inhibits growth of colon cancer cells. Cell Prolif. 2014;47(5):435–47. https://doi.org/10.1111/cpr.12125.
Hong S, Kim S, Kim HY, Kang M, Jang HH, Lee WS. Targeting the PI3K signaling pathway in KRAS mutant colon cancer. Cancer Med. 2016;5(2):248–55. https://doi.org/10.1002/cam4.591.
Cuneo KC, Mehta RK, Kurapati H, Thomas DG, Lawrence TS, Nyati MK. Enhancing the radiation response in KRAS mutant colorectal cancers using the c-met inhibitor Crizotinib. Transl Oncol. 2019;12(2):209–16. https://doi.org/10.1016/j.tranon.2018.10.005.
Ye H, Liu Y, Wu K, Luo H, Cui L. AMPK activation overcomes anti-EGFR antibody resistance induced by KRAS mutation in colorectal cancer. Cell Commun Signal. 2020;18(1):115. https://doi.org/10.1186/s12964-020-00584-z.
Bellier J, Nokin MJ, Caprasse M, Tiamiou A, Blomme A, Scheijen JL, et al. Methylglyoxal scavengers Resensitize KRAS-mutated colorectal tumors to Cetuximab. Cell Rep. 2020;30(5):1400–16. https://doi.org/10.1016/j.celrep.2020.01.012.
Lee J, Lee I, Han B, Park JO, Jang J, Park C, et al. Effect of simvastatin on cetuximab resistance in human colorectal cancer with KRAS mutations. J Natl Cancer Inst. 2011;103(8):674–88. https://doi.org/10.1093/jnci/djr070.
Cohen AS, Geng L, Zhao P, Fu A, Schulte ML, Graves-Deal R, et al. Combined blockade of EGFR and glutamine metabolism in preclinical models of colorectal cancer. Transl Oncol. 2020;13(10):100828. https://doi.org/10.1016/j.tranon.2020.100828.
Jung SA, Lee DH, Moon JH, Hong SW, Shin JS, Hwang IY, et al. L-ascorbic acid can abrogate SVCT-2-dependent cetuximab resistance mediated by mutant KRAS in human colon cancer cells. Free Radic Biol Med. 2016;95:200–8. https://doi.org/10.1016/j.freeradbiomed.2016.03.009.
Houot R, Kohrt H. CD137 stimulation enhances the vaccinal effect of anti-tumor antibodies. Oncoimmunology. 2014;3(7):e941740. https://doi.org/10.4161/21624011.2014.941740.
Veluchamy JP, Lopez-Lastra S, Spanholtz J, Bohme F, Kok N, Heideman DA, et al. In vivo efficacy of umbilical cord blood stem cell-derived NK cells in the treatment of metastatic colorectal Cancer. Front Immunol. 2017;8:87. https://doi.org/10.3389/fimmu.2017.00087.
Lutterbuese R, Raum T, Kischel R, Hoffmann P, Mangold S, Rattel B, et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc Natl Acad Sci U S A. 2010;107(28):12605–10. https://doi.org/10.1073/pnas.1000976107.
Damiano V, Caputo R, Bianco R, D'Armiento FP, Leonardi A, De Placido S, et al. Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin Cancer Res. 2006;12(2):577–83. https://doi.org/10.1158/1078-0432.CCR-05-1943.
Rosa R, Melisi D, Damiano V, Bianco R, Garofalo S, Gelardi T, et al. Toll-like receptor 9 agonist IMO cooperates with cetuximab in K-ras mutant colorectal and pancreatic cancers. Clin Cancer Res. 2011;17(20):6531–41. https://doi.org/10.1158/1078-0432.CCR-10-3376.
Hong CS, Sun EG, Choi JN, Kim DH, Kim JH, Ryu KH, et al. Fibroblast growth factor receptor 4 increases epidermal growth factor receptor (EGFR) signaling by inducing amphiregulin expression and attenuates response to EGFR inhibitors in colon cancer. Cancer Sci. 2020;111(9):3268–78. https://doi.org/10.1111/cas.14526.
Napolitano S, Martini G, Rinaldi B, Martinelli E, Donniacuo M, Berrino L, et al. Primary and acquired resistance of colorectal Cancer to anti-EGFR monoclonal antibody can be overcome by combined treatment of Regorafenib with Cetuximab. Clin Cancer Res. 2015;21(13):2975–83. https://doi.org/10.1158/1078-0432.CCR-15-0020.
Baba Y, Tamura T, Satoh Y, Gotou M, Sawada H, Ebara S, et al. Panitumumab interaction with TAS-102 leads to combinational anticancer effects via blocking of EGFR-mediated tumor response to trifluridine. Mol Oncol. 2017;11(8):1065–77. https://doi.org/10.1002/1878-0261.12074.
Chen P, Li X, Zhang R, Liu S, Xiang Y, Zhang M, et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics. 2020;10(11):5107–19. https://doi.org/10.7150/thno.44705.
Jutten B, Keulers TG, Peeters H, Schaaf M, Savelkouls K, Compter I, et al. EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy. 2018;14(2):283–95. https://doi.org/10.1080/15548627.2017.1409926.
Berasain C, Avila MA. Further evidence on the janus-faced nature of the epidermal growth factor receptor: from liver regeneration to hepatocarcinogenesis. Hepatology. 2016;63(2):371–4. https://doi.org/10.1002/hep.28246.
Messa C, Russo F, Caruso MG, Di Leo A. EGF, TGF-alpha, and EGF-R in human colorectal adenocarcinoma. Acta Oncol. 1998;37(3):285–9. https://doi.org/10.1080/028418698429595.
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27(30):5068–74. https://doi.org/10.1200/JCO.2008.21.3744.
Mutsaers AJ, Francia G, Man S, Lee CR, Ebos JM, Wu Y, et al. Dose-dependent increases in circulating TGF-alpha and other EGFR ligands act as pharmacodynamic markers for optimal biological dosing of cetuximab and are tumor independent. Clin Cancer Res. 2009;15(7):2397–405. https://doi.org/10.1158/1078-0432.CCR-08-1627.
Loupakis F, Cremolini C, Fioravanti A, Orlandi P, Salvatore L, Masi G, et al. EGFR ligands as pharmacodynamic biomarkers in metastatic colorectal cancer patients treated with cetuximab and irinotecan. Target Oncol. 2014;9(3):205–14. https://doi.org/10.1007/s11523-013-0284-7.
Van Emburgh BO, Arena S, Siravegna G, Lazzari L, Crisafulli G, Corti G, et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat Commun. 2016;7:13665. https://doi.org/10.1038/ncomms13665.
Price T, Ang A, Boedigheimer M, Kim TW, Li J, Cascinu S, et al. Frequency of S492R mutations in the epidermal growth factor receptor: analysis of plasma DNA from patients with metastatic colorectal cancer treated with panitumumab or cetuximab monotherapy. Cancer Biol Ther. 2020;21(10):891–8. https://doi.org/10.1080/15384047.2020.1798695.
Liao HW, Hsu JM, Xia W, Wang HL, Wang YN, Chang WC, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest. 2015;125(12):4529–43. https://doi.org/10.1172/JCI82826.
Bagchi A, Haidar JN, Eastman SW, Vieth M, Topper M, Iacolina MD, et al. Molecular basis for Necitumumab inhibition of EGFR variants associated with acquired Cetuximab resistance. Mol Cancer Ther. 2018;17(2):521–31. https://doi.org/10.1158/1535-7163.MCT-17-0575.
Dienstmann R, Patnaik A, Garcia-Carbonero R, Cervantes A, Benavent M, Roselló S, et al. Safety and activity of the first-in-class Sym004 anti-EGFR antibody mixture in patients with refractory colorectal Cancer. Cancer Discov. 2015;5(6):598–609. https://doi.org/10.1158/2159-8290.CD-14-1432.
Lim Y, Yoo J, Kim MS, Hur M, Lee EH, Hur HS, et al. GC1118, an anti-EGFR antibody with a distinct binding epitope and superior inhibitory activity against high-affinity EGFR ligands. Mol Cancer Ther. 2016;15(2):251–63. https://doi.org/10.1158/1535-7163.MCT-15-0679.
Ciardiello F, Normanno N. HER2 signaling and resistance to the anti-EGFR monoclonal antibody cetuximab: a further step toward personalized medicine for patients with colorectal cancer. Cancer Discov. 2011;1(6):472–4. https://doi.org/10.1158/2159-8290.CD-11-0261.
Vlacich G, Coffey RJ. Resistance to EGFR-targeted therapy: a family affair. Cancer Cell. 2011;20(4):423–5. https://doi.org/10.1016/j.ccr.2011.10.006.
Luraghi P, Reato G, Cipriano E, Sassi F, Orzan F, Bigatto V, et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 2014;74(6):1857–69. https://doi.org/10.1158/0008-5472.CAN-13-2340-T.
Schirripa M, Zhang W, Heinemann V, Cao S, Okazaki S, Yang D, et al. Single nucleotide polymorphisms in the IGF-IRS pathway are associated with outcome in mCRC patients enrolled in the FIRE-3 trial. Int J Cancer. 2017;141(2):383–92. https://doi.org/10.1002/ijc.30715.
Molinari F, Martin V, Saletti P, De Dosso S, Spitale A, Camponovo A, et al. Differing deregulation of EGFR and downstream proteins in primary colorectal cancer and related metastatic sites may be clinically relevant. Br J Cancer. 2009;100(7):1087–94. https://doi.org/10.1038/sj.bjc.6604848.
Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 2014;4(11):1269–80. https://doi.org/10.1158/2159-8290.CD-14-0462.
Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9(10):962–72. https://doi.org/10.1016/S1470-2045(08)70206-7.
Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011–9. https://doi.org/10.1200/JCO.2010.33.5091.
Bokemeyer C, Köhne CH, Ciardiello F, Lenz HJ, Heinemann V, Klinkhardt U, et al. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur J Cancer. 2015;51(10):1243–52. https://doi.org/10.1016/j.ejca.2015.04.007.
Blons H, Emile JF, Le Malicot K, Julié C, Zaanan A, Tabernero J, et al. Prognostic value of KRAS mutations in stage III colon cancer: post hoc analysis of the PETACC8 phase III trial dataset. Ann Oncol. 2014;25(12):2378–85. https://doi.org/10.1093/annonc/mdu464.
Yen LC, Uen YH, Wu DC, Lu CY, Yu FJ, Wu IC, et al. Activating KRAS mutations and overexpression of epidermal growth factor receptor as independent predictors in metastatic colorectal cancer patients treated with cetuximab. Ann Surg. 2010;251(2):254–60. https://doi.org/10.1097/SLA.0b013e3181bc9d96.
Wang JY, Hsieh JS, Chen FM, Yeh CS, Alexandersen K, Huang TJ, et al. High frequency of activated K-ras codon 15 mutant in colorectal carcinomas from Taiwanese patients. Int J Cancer. 2003;107(3):387–93. https://doi.org/10.1002/ijc.11417.
Duldulao MP, Lee W, Nelson RA, Li W, Chen Z, Kim J, et al. Mutations in specific codons of the KRAS oncogene are associated with variable resistance to neoadjuvant chemoradiation therapy in patients with rectal adenocarcinoma. Ann Surg Oncol. 2013;20(7):2166–71. https://doi.org/10.1245/s10434-013-2910-0.
Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533–52. https://doi.org/10.1038/s41573-020-0068-6.
Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384(25):2371–81. https://doi.org/10.1056/NEJMoa2103695.
Dunn EF, Iida M, Myers RA, Campbell DA, Hintz KA, Armstrong EA, et al. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene. 2011;30(5):561–74. https://doi.org/10.1038/onc.2010.430.
Shin W, Lee SK, Hwang JH, Park JC, Cho YH, Ro EJ, et al. Identification of Ras-degrading small molecules that inhibit the transformation of colorectal cancer cells independent of β-catenin signaling. Exp Mol Med. 2018;50(6):1–10. https://doi.org/10.1038/s12276-018-0102-5.
Sanz-Garcia E, Argiles G, Elez E, Tabernero J. BRAF mutant colorectal cancer: prognosis, treatment, and new perspectives. Ann Oncol. 2017;28(11):2648–57. https://doi.org/10.1093/annonc/mdx401.
Cremolini C, Rossini D, Dell'Aquila E, Lonardi S, Conca E, Del RM, et al. Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal Cancer with acquired resistance to first-line Cetuximab and irinotecan: a phase 2 single-arm clinical trial. Jama Oncol. 2019;5(3):343–50. https://doi.org/10.1001/jamaoncol.2018.5080.
Ciardiello F, Normanno N, Maiello E, Martinelli E, Troiani T, Pisconti S, et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol. 2014;25(9):1756–61. https://doi.org/10.1093/annonc/mdu230.
Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. https://doi.org/10.1038/ncomms3185.
Pakneshan S, Salajegheh A, Smith RA, Lam AK. Clinicopathological relevance of BRAF mutations in human cancer. Pathology. 2013;45(4):346–56. https://doi.org/10.1097/PAT.0b013e328360b61d.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. https://doi.org/10.1038/nature00766.
Molinari F, Felicioni L, Buscarino M, De Dosso S, Buttitta F, Malatesta S, et al. Increased detection sensitivity for KRAS mutations enhances the prediction of anti-EGFR monoclonal antibody resistance in metastatic colorectal cancer. Clin Cancer Res. 2011;17(14):4901–14. https://doi.org/10.1158/1078-0432.CCR-10-3137.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. https://doi.org/10.1056/NEJMoa1103782.
Yaeger R, Cercek A, O'Reilly EM, Reidy DL, Kemeny N, Wolinsky T, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21(6):1313–20. https://doi.org/10.1158/1078-0432.CCR-14-2779.
Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36. https://doi.org/10.1056/NEJMoa1502309.
Hong DS, Morris VK, El OB, Sorokin AV, Janku F, Fu S, et al. Phase IB study of Vemurafenib in combination with irinotecan and Cetuximab in patients with metastatic colorectal Cancer with BRAFV600E mutation. Cancer Discov. 2016;6(12):1352–65. https://doi.org/10.1158/2159-8290.CD-16-0050.
Troiani T, Napolitano S, Martini G, Martinelli E, Cardone C, Normanno N, et al. Maintenance treatment with Cetuximab and BAY86-9766 increases antitumor efficacy of irinotecan plus Cetuximab in human colorectal Cancer xenograft models. Clin Cancer Res. 2015;21(18):4153–64. https://doi.org/10.1158/1078-0432.CCR-15-0211.
Misale S, Arena S, Lamba S, Siravegna G, Lallo A, Hobor S, Russo M, Buscarino M, Lazzari L, Sartore-Bianchi A et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci Transl Med. 2014;6(224):224r-226r. doi:https://doi.org/10.1126/scitranslmed.3007947.
Deming DA, Cavalcante LL, Lubner SJ, Mulkerin DL, LoConte NK, Eickhoff JC, et al. A phase I study of selumetinib (AZD6244/ARRY-142866), a MEK1/2 inhibitor, in combination with cetuximab in refractory solid tumors and KRAS mutant colorectal cancer. Investig New Drugs. 2016;34(2):168–75. https://doi.org/10.1007/s10637-015-0314-7.
Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol. 2009;20(1):84–90. https://doi.org/10.1093/annonc/mdn541.
Mao C, Yang ZY, Hu XF, Chen Q, Tang JL. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2012;23(6):1518–25. https://doi.org/10.1093/annonc/mdr464.
Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol. 2009;27(16):2622–9. https://doi.org/10.1200/JCO.2008.20.2796.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–30. https://doi.org/10.1200/JCO.2008.21.6796.
Negri FV, Bozzetti C, Lagrasta CA, Crafa P, Bonasoni MP, Camisa R, et al. PTEN status in advanced colorectal cancer treated with cetuximab. Br J Cancer. 2010;102(1):162–4. https://doi.org/10.1038/sj.bjc.6605471.
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007;97(8):1139–45. https://doi.org/10.1038/sj.bjc.6604009.
Jhawer M, Goel S, Wilson AJ, Montagna C, Ling YH, Byun DS, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68(6):1953–61. https://doi.org/10.1158/0008-5472.CAN-07-5659.
Shapiro GI, Rodon J, Bedell C, Kwak EL, Baselga J, Brana I, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2014;20(1):233–45. https://doi.org/10.1158/1078-0432.CCR-13-1777.
Foster P, Yamaguchi K, Hsu PP, Qian F, Du X, Wu J, et al. The selective PI3K inhibitor XL147 (SAR245408) inhibits tumor growth and survival and potentiates the activity of chemotherapeutic agents in preclinical tumor models. Mol Cancer Ther. 2015;14(4):931–40. https://doi.org/10.1158/1535-7163.MCT-14-0833.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1(6):508–23. https://doi.org/10.1158/2159-8290.CD-11-0109.
Sclafani F, Roy A, Cunningham D, Wotherspoon A, Peckitt C, Gonzalez DCD, et al. HER2 in high-risk rectal cancer patients treated in EXPERT-C, a randomized phase II trial of neoadjuvant capecitabine and oxaliplatin (CAPOX) and chemoradiotherapy (CRT) with or without cetuximab. Ann Oncol. 2013;24(12):3123–8. https://doi.org/10.1093/annonc/mdt408.
Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(6):738–46. https://doi.org/10.1016/S1470-2045(16)00150-9.
Meric-Bernstam F, Hurwitz H, Raghav K, McWilliams RR, Fakih M, VanderWalde A, et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019;20(4):518–30. https://doi.org/10.1016/S1470-2045(18)30904-5.
Winder T, Zhang W, Yang D, Ning Y, Bohanes P, Gerger A, et al. Germline polymorphisms in genes involved in the IGF1 pathway predict efficacy of cetuximab in wild-type KRAS mCRC patients. Clin Cancer Res. 2010;16(22):5591–602. https://doi.org/10.1158/1078-0432.CCR-10-2092.
Cappuzzo F, Varella-Garcia M, Finocchiaro G, Skokan M, Gajapathy S, Carnaghi C, et al. Primary resistance to cetuximab therapy in EGFR FISH-positive colorectal cancer patients. Br J Cancer. 2008;99(1):83–9. https://doi.org/10.1038/sj.bjc.6604439.
Alonso V, Escudero P, Fernández-Martos C, Salud A, Méndez M, Gallego J, et al. Coexpression of p-IGF-1R and MMP-7 modulates Panitumumab and Cetuximab efficacy in RAS wild-type metastatic colorectal Cancer patients. Neoplasia. 2018;20(7):678–86. https://doi.org/10.1016/j.neo.2018.05.004.
Martinelli E, Ciardiello D, Martini G, Troiani T, Cardone C, Vitiello PP, et al. Implementing anti-epidermal growth factor receptor (EGFR) therapy in metastatic colorectal cancer: challenges and future perspectives. Ann Oncol. 2020;31(1):30–40. https://doi.org/10.1016/j.annonc.2019.10.007.
Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19(24):6751–65. https://doi.org/10.1158/1078-0432.CCR-13-0423.
Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011;17(3):472–82. https://doi.org/10.1158/1078-0432.CCR-10-0568.
Song N, Liu S, Zhang J, Liu J, Xu L, Liu Y, et al. Cetuximab-induced MET activation acts as a novel resistance mechanism in colon cancer cells. Int J Mol Sci. 2014;15(4):5838–51. https://doi.org/10.3390/ijms15045838.
Eng C, Bessudo A, Hart LL, Severtsev A, Gladkov O, Muller L, et al. A randomized, placebo-controlled, phase 1/2 study of tivantinib (ARQ 197) in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with wild-type KRAS who have received first-line systemic therapy. Int J Cancer. 2016;139(1):177–86. https://doi.org/10.1002/ijc.30049.
Delord JP, Argilés G, Fayette J, Wirth L, Kasper S, Siena S, et al. A phase 1b study of the MET inhibitor capmatinib combined with cetuximab in patients with MET-positive colorectal cancer who had progressed following anti-EGFR monoclonal antibody treatment. Investig New Drugs. 2020;38(6):1774–83. https://doi.org/10.1007/s10637-020-00928-z.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073–87. https://doi.org/10.1053/j.gastro.2009.12.064.
Innocenti F, Ou FS, Qu X, Zemla TJ, Niedzwiecki D, Tam R, et al. Mutational analysis of patients with colorectal Cancer in CALGB/SWOG 80405 identifies new roles of microsatellite instability and tumor mutational burden for patient outcome. J Clin Oncol. 2019;37(14):1217–27. https://doi.org/10.1200/JCO.18.01798.
Carethers JM, Jung BH. Genetics and genetic biomarkers in sporadic colorectal Cancer. Gastroenterology. 2015;149(5):1177–90. https://doi.org/10.1053/j.gastro.2015.06.047.
Kloth M, Ruesseler V, Engel C, Koenig K, Peifer M, Mariotti E, et al. Activating ERBB2/HER2 mutations indicate susceptibility to pan-HER inhibitors in lynch and lynch-like colorectal cancer. Gut. 2016;65(8):1296–305. https://doi.org/10.1136/gutjnl-2014-309026.
Han Y, Peng Y, Fu Y, Cai C, Guo C, Liu S, et al. MLH1 deficiency induces Cetuximab resistance in Colon Cancer via her-2/PI3K/AKT signaling. Adv Sci (Weinh). 2020;7(13):2000112. https://doi.org/10.1002/advs.202000112.
Pander J, Heusinkveld M, van der Straaten T, Jordanova ES, Baak-Pablo R, Gelderblom H, et al. Activation of tumor-promoting type 2 macrophages by EGFR-targeting antibody cetuximab. Clin Cancer Res. 2011;17(17):5668–73. https://doi.org/10.1158/1078-0432.CCR-11-0239.
ASCO Meeting Library: 2021 ASCO Annual Meeting. https://meetinglibrary.asco.org/record/194206/abstract. Accessed 02 Aug 2021.
Zaal EA, Berkers CR. The influence of metabolism on drug response in Cancer. Front Oncol. 2018;8:500. https://doi.org/10.3389/fonc.2018.00500.
Du Z, Liu X, Chen T, Gao W, Wu Z, Hu Z, et al. Targeting a Sirt5-positive subpopulation overcomes multidrug resistance in wild-type Kras colorectal carcinomas. Cell Rep. 2018;22(10):2677–89. https://doi.org/10.1016/j.celrep.2018.02.037.
Skvortsov S, Sarg B, Loeffler-Ragg J, Skvortsova I, Lindner H, Werner OH, et al. Different proteome pattern of epidermal growth factor receptor-positive colorectal cancer cell lines that are responsive and nonresponsive to C225 antibody treatment. Mol Cancer Ther. 2004;3(12):1551–8.
Mortara L, Gariboldi MB, Bosi A, Bregni M, Pinotti G, Guasti L, et al. Vitamin D deficiency has a negative impact on Cetuximab-mediated cellular cytotoxicity against human Colon carcinoma cells. Target Oncol. 2018;13(5):657–65. https://doi.org/10.1007/s11523-018-0586-x.
De Angelis ML, Zeuner A, Policicchio E, Russo G, Bruselles A, Signore M, et al. Cancer stem cell-based models of colorectal Cancer reveal molecular determinants of therapy resistance. Stem Cells Transl Med. 2016;5(4):511–23. https://doi.org/10.5966/sctm.2015-0214.
Li X, Fan Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1alpha and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010;70(14):5942–52. https://doi.org/10.1158/0008-5472.CAN-10-0157.
Koustas E, Karamouzis MV, Mihailidou C, Schizas D, Papavassiliou AG. Co-targeting of EGFR and autophagy signaling is an emerging treatment strategy in metastatic colorectal cancer. Cancer Lett. 2017;396:94–102. https://doi.org/10.1016/j.canlet.2017.03.023.
Chen Z, Gao S, Wang D, Song D, Feng Y. Colorectal cancer cells are resistant to anti-EGFR monoclonal antibody through adapted autophagy. Am J Transl Res. 2016;8(2):1190–6.
Arai H, Millstein J, Loupakis F, Stintzing S, Wang J, Battaglin F, et al. Germ line polymorphisms of genes involved in pluripotency transcription factors predict efficacy of cetuximab in metastatic colorectal cancer. Eur J Cancer. 2021;150:133–42. https://doi.org/10.1016/j.ejca.2021.03.048.
Oliveras-Ferraros C, Vazquez-Martin A, Cufi S, Queralt B, Baez L, Guardeno R, et al. Stem cell property epithelial-to-mesenchymal transition is a core transcriptional network for predicting cetuximab (Erbitux) efficacy in KRAS wild-type tumor cells. J Cell Biochem. 2011;112(1):10–29. https://doi.org/10.1002/jcb.22952.
Calemma R, Ottaiano A, Trotta AM, Nasti G, Romano C, Napolitano M, et al. Fc gamma receptor IIIa polymorphisms in advanced colorectal cancer patients correlated with response to anti-EGFR antibodies and clinical outcome. J Transl Med. 2012;10:232. https://doi.org/10.1186/1479-5876-10-232.
Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest. 2014;124(6):2668–82. https://doi.org/10.1172/JCI73014.
Rocca YS, Roberti MP, Juliá EP, Pampena MB, Bruno L, Rivero S, et al. Phenotypic and functional dysregulated blood NK cells in colorectal Cancer patients can be activated by Cetuximab plus IL-2 or IL-15. Front Immunol. 2016;7:413. https://doi.org/10.3389/fimmu.2016.00413.
Akkaya M, Akkaya B, Kim AS, Miozzo P, Sohn H, Pena M, et al. Toll-like receptor 9 antagonizes antibody affinity maturation. Nat Immunol. 2018;19(3):255–66. https://doi.org/10.1038/s41590-018-0052-z.
Ishiguro N, Moriyama M, Furusho K, Furukawa S, Shibata T, Murakami Y, et al. Activated M2 macrophages contribute to the pathogenesis of IgG4-related disease via toll-like receptor 7/Interleukin-33 signaling. Arthritis Rheumatol. 2020;72(1):166–78. https://doi.org/10.1002/art.41052.
Damiano V, Caputo R, Garofalo S, Bianco R, Rosa R, Merola G, et al. TLR9 agonist acts by different mechanisms synergizing with bevacizumab in sensitive and cetuximab-resistant colon cancer xenografts. Proc Natl Acad Sci U S A. 2007;104(30):12468–73. https://doi.org/10.1073/pnas.0705226104.
Vallbohmer D, Zhang W, Gordon M, Yang DY, Yun J, Press OA, et al. Molecular determinants of cetuximab efficacy. J Clin Oncol. 2005;23(15):3536–44. https://doi.org/10.1200/JCO.2005.09.100.
Vincenzi B, Santini D, Russo A, Gavasci M, Battistoni F, Dicuonzo G, et al. Circulating VEGF reduction, response and outcome in advanced colorectal cancer patients treated with cetuximab plus irinotecan. Pharmacogenomics. 2007;8(4):319–27. https://doi.org/10.2217/14622416.8.4.319.
Ding C, Li L, Yang T, Fan X, Wu G. Combined application of anti-VEGF and anti-EGFR attenuates the growth and angiogenesis of colorectal cancer mainly through suppressing AKT and ERK signaling in mice model. BMC Cancer. 2016;16(1):791. https://doi.org/10.1186/s12885-016-2834-8.
Poindessous V, Ouaret D, El OK, Battistella A, Megalophonos VF, Kamsu-Kom N, et al. EGFR- and VEGF(R)-targeted small molecules show synergistic activity in colorectal cancer models refractory to combinations of monoclonal antibodies. Clin Cancer Res. 2011;17(20):6522–30. https://doi.org/10.1158/1078-0432.CCR-11-1607.
Bennouna J, Deslandres M, Senellart H, de Labareyre C, Ruiz-Soto R, Wixon C, et al. A phase I open-label study of the safety, tolerability, and pharmacokinetics of pazopanib in combination with irinotecan and cetuximab for relapsed or refractory metastatic colorectal cancer. Investig New Drugs. 2015;33(1):138–47. https://doi.org/10.1007/s10637-014-0142-1.
Acknowledgements
Not applicable.
Funding
This work was supported by National Science Foundation of China (81830120 to Q.L., 82074225 to Q.J.) and Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR4043 to Y.W.).
Author information
Authors and Affiliations
Contributions
Qi Li generated the ideas and design structure of manuscript. Jing Zhou organized all figures, and wrote the manuscript. Qing Ji and Qi Li revised and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there is no potential competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, J., Ji, Q. & Li, Q. Resistance to anti-EGFR therapies in metastatic colorectal cancer: underlying mechanisms and reversal strategies. J Exp Clin Cancer Res 40, 328 (2021). https://doi.org/10.1186/s13046-021-02130-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02130-2