
Abstract
Many types of human cells self-destruct to maintain biological homeostasis and defend the body against pathogenic substances. This process, called regulated cell death (RCD), is important for various biological activities, including the clearance of aberrant cells. Thus, RCD pathways represented by apoptosis have increased in importance as a target for the development of cancer medications in recent years. However, because tumor cells show avoidance to apoptosis, which causes treatment resistance and recurrence, numerous studies have been devoted to alternative cancer cell mortality processes, namely necroptosis, pyroptosis, ferroptosis, and cuproptosis; these RCD modalities have been extensively studied and shown to be crucial to cancer therapy effectiveness. Furthermore, evidence suggests that tumor cells undergoing regulated death may alter the immunogenicity of the tumor microenvironment (TME) to some extent, rendering it more suitable for inhibiting cancer progression and metastasis. In addition, other types of cells and components in the TME undergo the abovementioned forms of death and induce immune attacks on tumor cells, resulting in enhanced antitumor responses. Hence, this review discusses the molecular processes and features of necroptosis, pyroptosis, ferroptosis, and cuproptosis and the effects of these novel RCD modalities on tumor cell proliferation and cancer metastasis. Importantly, it introduces the complex effects of novel forms of tumor cell death on the TME and the regulated death of other cells in the TME that affect tumor biology. It also summarizes the potential agents and nanoparticles that induce or inhibit novel RCD pathways and their therapeutic effects on cancer based on evidence from in vivo and in vitro studies and reports clinical trials in which RCD inducers have been evaluated as treatments for cancer patients. Lastly, we also summarized the impact of modulating the RCD processes on cancer drug resistance and the advantages of adding RCD modulators to cancer treatment over conventional treatments.
Similar content being viewed by others

Introduction
Cell death (especially cell suicide) plays a fundamental role in maintaining physiological homeostasis by removing damaged cells, and it may also be an aberrant pathological reaction to damaging stimuli [1]. The Nomenclature of Cell Death Committee has developed guidelines to divide cell death modes into accidental cell death and regulated cell death (RCD) according to morphology, biochemistry, and function [2]. Accidental cell death is a biologically uncontrolled process of cell death in response to accidental injury stimuli [3]. However, RCD is characterized by controlled signaling pathways that play key roles in organismal development or tissue renewal [4]. Previously, apoptosis was thought to be the major form of RCD, but with more in-depth study on tumor cell biology and thorough examination of cancer therapy mechanisms, more and more subtypes of RCD are progressively emerging. The novel RCD types we are going to introduce include: necroptosis, pyroptosis, ferroptosis, and cuproptosis, which can occur with or without exogenous environmental or intracellular perturbations [5,6,7]. Malignant cells, on the other hand, continue to evade the RCD routes through evolving a variety of mechanisms [8]. Additionally, RCD pathways have also been reported to be crucial for the prognosis of cancer patients, cancer progression and metastasis, and cancer immune surveillance [9,10,11,12,13,14]. Based on accumulating evidence, distinct forms of RCD might change the tumor microenvironment (TME) by releasing pathogen- or damage-associated molecular patterns (PAMPs or DAMPs), which affect the benefits of anticancer therapy [15,16,17].
Our review outlines the molecular mechanisms and processes of four different types of RCD, necroptosis, pyroptosis, ferroptosis, and the newly discovered cuproptosis, as well as their different roles in the initiation and progression of cancer. We specifically focus on RCD processes that influence the TME and the latest advancements in targeting necroptosis, pyroptosis, ferroptosis, and cuproptosis for cancer therapy. We describe the mechanisms of the various cancer therapies currently available, showing that they mainly depend on different RCD modalities. A reasonable assumption is that these novel RCD modalities constitute a mechanism of defense against tumor progression and migration. Furthermore, the significance and prevalence of RCDs in combating cancer drug resistance have been included, demonstrating that the use of conventional therapy in conjunction with RCD modulators might hold significant potential for cancer treatment. Hopefully, this information will lead to improved guidance for approaches to tumor therapy.
Molecular mechanisms of different cell death pathways
The most extensively studied RCD modality is apoptosis, which leads to immunogenicity or induces no immunogenic response in different contexts [18, 19]. The morphological changes observed during apoptotic cell death include cell shrinkage, externalization of phosphatidylserine on the plasma membrane, and nuclear pyknosis and karyorrhexis; notably, the plasma membrane remains intact [2]. This pathway is believed to function as a natural barrier against malignancy, but the primary hallmark of cancer cells and the emergence of chemotherapy resistance during cancer therapy are limiting or causing cells to resist apoptosis [20, 21]. Therefore, while tackling apoptosis resistance, discover methods that induce nonapoptotic forms of RCD must be discovered as alternative cancer therapies. Excitingly, new forms of RCD have been extensively studied in the past decade; these modalities include necroptosis [22], pyroptosis [23], and ferroptosis [24]. Additionally, a noteworthy finding reported in 2022 is the description of cuproptosis, a previously unknown form of RCD [25].
Necroptosis
Necroptosis is a regulated form of necrosis that depends on the phosphorylation of mixed-lineage kinase-like (MLKL) by receptor interacting kinase-1 (RIPK1) and RIPK3 [26]. The necroptotic process is initiated by the activation of cell surface death receptors (such as FasRs, TNFR1, IFN receptors, and TLRs) and RNA- and DNA-sensing molecules in cells. RIPK3 is required for the necroptotic process, and RIPK3 is activated by three known processes [27]. First, ligation of TNFR1 activates RIPK1, which in turn binds to RIPK3 via shared RIP homology interaction motifs (RHIM) present in both molecules [28, 29]. Similarly, engagement of TLR-3 and TLR-4 recruits the adapter, which contains an RHIM that is capable of binding and activating RIPK3 [27]. Finally, the cytosolic nucleic acid sensor Z-dsDNA/dsRNA-binding protein 1 (ZBP1) also contains a RIPK3-activating RHIM (Fig. 1) [30]. Subsequently, RIPK3 frequently phosphorylates MLKL, which oligomerizes to form an activated “necrosome” complex and is translocated to the plasma membrane. This process eventually leads to cell death characterized by permeabilization of the plasma membrane, cell swelling, and loss of cellular and organelle integrity [31, 32]. The rupture of plasma membrane results in cytokine, chemokine, and potassium efflux, leading to inflammation and immune responses [33]. Necroptosis has been pharmacologically suppressed using chemical compounds, including necrostatin-1 [34]. Recently, the significance of necroptosis in cancer has been increasingly appreciated, and a greater comprehension of necroptotic processes might be helpful in creating novel strategies for controlling cancer [9].

Detailed mechanism of necroptosis. Necroptosis is initiated by the cell surface death receptors (including FasRs, TNFR, IFN receptors, and TLRs) and ZBP1 in cells, and downstream proteins which contain RHIM bind to RIPK3. Subsequently, the necrosome is formed and led to cell lysis
Pyroptosis
Pyroptosis, a proinflammatory RCD pathway used in diverse types of cells, is triggered by human caspase-1, -3, -4, -5 (mouse caspase-11), -6, -8, and -9 and activated by a number of inflammatory bodies, including NLRP3 (Fig. 2) [35,36,37,38,39,40]. The crucial pyroptosis mediators—members of the gasdermin (GSDM) superfamily—are proteolytically activated by these caspases, after which they perforate the plasma membrane [41, 42]. Most members of the GSDM family (A-E) consist of an N-terminal pore-forming domain (PFD) and a C-terminal repressor domain [43, 44]. When a host is stimulated by various stimuli, GSDMs are cleaved by inflammatory caspases at the site of the linker region and liberate the PFD from the repressor domain [44, 45]. Consequently, the N-terminal PFD oligomerizes and forms pores in the cell membrane, leading to cell swelling, chromatin degradation, and expulsion of proinflammatory components (Fig. 2C) [46,47,48].

Summary of pyroptosis mediated by different cellular mechanisms. A Pyroptosis induced by the TNF-α/TRADD pathway and the mechanism of pyroptosis induced by granzymes A and B. B Chemotherapy and CAR-T therapy induce cell death mediated by nonclassical pyroptotic pathways. C Interactions of PAMPs and DAMPs with pattern recognition receptors on the cell surface trigger the classical pyroptotic pathway, leading to the release of HMGB1, IL-1β, and IL-18. (Gzm A: Granzyme A; Gzm B: Granzyme B; PRRs: pattern recognition receptors; PAMPs: pathogen-associated molecular patterns; DAMPs: damage-associated molecular patterns; GSDMD-CT: GSDMD-C-terminus; and GSDME-CT: GSDME-C-terminus.)
GSDMD has been reported to be cleaved by caspase-1, -4, -5, or -11, which is a hallmark of the canonical pyroptosis pathway [41]. PAMPs and DAMPs are detected by pattern recognition receptors, which activate downstream signaling pathways; as a result, ASCs are recruited to establish NLRP3 inflammasomes, which activate pro-caspase-1. Subsequently, activated caspase-1 cleaves GSDMD to free the PFD of GSDMD [49]. In addition, gram-negative bacteria predominantly trigger noncanonical pyroptosis through a mechanism distinct from inflammasomes and caspase-1 by activating human caspase-4/-5 (mouse caspase-11) to cleave GSDMD (Fig. 2B) [47, 50, 51]. Furthermore, the release of granzyme B (Gzm B) from chimeric antigen receptor (CAR)-T cells and chemotherapeutic medicines activates caspase-3, which in turn initiates the caspase-3/GSDME-mediated pyroptotic pathway, resulting in widespread pyroptosis [38, 40, 52]. Moreover, it has been demonstrated that Gzm B directly cleaves GSDME to cause pyroptosis, which subsequently activates the immune system to protect against tumors and slow tumor growth (Fig. 2A) [53]. Researchers have also discovered that natural killer (NK) cells and cytotoxic T lymphocytes kill cells expressing GSDMB via pyroptosis. Cleavage of GSDMB at the Lys229/Lys244 sites by granzyme A leads to lethal effects on target cells. GSDMB has been frequently detected in several tissues, including the epithelium of the digestive system [54]. Chen et al. even discovered that GSDMB binds directly to the caspase recruitment domain in caspase-4, promoting caspase-4 activity, which is necessary for the cleavage of GSDMD in noncanonical pyroptosis [55].
Ferroptosis
Ferroptosis is a unique form of iron-dependent cell death that was originally discovered after tumor cells were exposed to a small-molecule chemical probe named erastin [56]. Morphological characteristics of ferroptosis involve reduced mitochondrial volume, fractured mitochondrial outer membrane, a decreased or absent mitochondrial crest, a normal-sized nucleus with no nuclear concentration, which distinguishes it from other modes of death [57]. Under normal conditions, lipoxygenases such as 12-/15-lipoxygenases often oxidize polyunsaturated fatty acids (PUFAs), but the lipid repair enzyme glutathione peroxidase 4 (GPX4) and its cofactor glutathione (GSH) cause a rapid decrease in the levels of lipoxygenase-oxidized PUFAs [58]. The ferroptosis process is induced by the suppression of the cystine–glutamate antiporter (system Xc−, comprising subunits SLC3A2 and SLC7A11), leading to decreased GSH biosynthesis and inactivation of GPX4 [59]. Subsequently, the cell dies due to overwhelming lipid peroxidation (Fig. 3B) [56, 57]. System XC− inhibitors are categorized as class I ferroptosis-inducing substances, including sorafenib and sulfasalazine [60]. Class II ferroptosis-inducing substances are represented by RSL3, which covalently binds to and directly blocks GPX4, thereby rapidly inducing ferroptotic cell death [59].

The interaction between MΦs and tumor cells in the TME and details of the ferroptosis pathway in tumor cells (by Figdraw). A MΦs engulf red blood cells and digest them into hemoglobin, which is further degraded into heme. Heme is catabolized into Fe(III) and Fe(III), which are released from MΦs or promote ROS production, leading to ferroptosis. B Ferroptotic cell death is induced by the inhibition of system Xc−, resulting in the abrogation of GSH biosynthesis and inactivation of GPX4, which subsequently cause cell death through excess lipid ROS production. PUFAs-OOH and Fe(II) facilitate tumor cell ferroptosis mediated by the Fenton reaction. (ROS: reactive oxygen species; system Xc−: cystine–glutamate antiporter; GSH: glutathione; GPX4: glutathione peroxidase 4; and RBC: red blood cell.)
Another ferroptosis defense system in cells was recently identified, the NAD(P)H-ferroptosis suppressor protein 1-ubiquinone (NAD(P)H-FSP1-CoQ10) pathway, which functions independently of the system XC−-GSH-GPX4 axis [61, 62]. FSP1, ferroptosis suppressor protein 1, is a flavoprotein that has been reported to induce apoptosis. CoQ10 is primarily synthesized in mitochondria, and in addition to its general importance in the mitochondrial electron transport chain, and its reduced form CoQ10H2 is a strong lipophilic antioxidant [63]. Hence, Kirill Bersuker and colleagues observed that FSP1 was recruited to the plasma membrane and then exerted an oxidoreductase function, reducing CoQ10. Subsequently, CoQ10H2 robustly halted the dissemination of lipid peroxides [62].
As the peroxidation of membrane phospholipids possessing PUFAs leads to ferroptosis [60], enzymes mediating the incorporation of PUFAs into phospholipids are important for ferroptotic cell death. For example, acyl-CoA synthetase long-chain family member 4 (ACSL4) leads to the enrichment of long PUFAs in cell membranes and is essential for the execution of ferroptosis [64]. Ferroptosis is also activated by components of the autophagy machinery, such ATG3, ATG5, ATG4B, ATG7, ATG13, and BECN1 [65]. Additionally, knockout or knockdown of the main genes governing autophagy reduces the effects of erastin on ferroptosis because intracellular ferrous iron levels are reduced [66]. Furthermore, Huang et al. documented that ferritinophagy, a proteolytic process through which ferritin is delivered to autophagosomes by NCOA4 [67], generates reactive oxygen species (ROS) and causes ferroptosis [68]. Cancer cells show high sensitivity to ferroptosis, which suggests a unique potential for cancer treatment. In fact, various primary cancers, such as liver cancer, clear cell renal cell carcinoma (RCC), and certain cancer cells with acquired drug resistance exert antitumor effects by inducing ferroptosis [69,70,71].
Cuproptosis
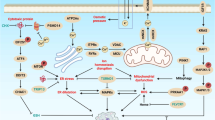
Recently, a novel cell death pathway triggered by copper (Cu), which differs from apoptosis, necroptosis, pyroptosis and ferroptosis, was discovered and coined “cuproptosis” by Peter Tsvetkov and colleagues in 2022 [25]. Cu is a crucial component for various physiological processes, especially tumor growth and metastasis, which have a heightened requirement for Cu [72]. Tsvetkov et al. described Cu-dependent death in 2019 while exploring the anticancer mechanism of elesclomol (a Cu ionophore) [73]. They found that the treatment of a multiple myeloma mouse model with elesclomol reduced the capability of the cancer cells to resist toxicity induced by proteasome inhibitors. Mechanistically, elesclomol-bound Cu(II) interacts with the mitochondrial enzyme ferredoxin 1 (FDX1) and is reduced to produce Cu(I), leading to increased levels of ROS [73, 74]. The lethality of elesclomol was first believed to be caused by lipid peroxidation [75]. Three years later, Tsvetkov and colleagues termed the unique form of Cu-dependent cell death cuproptosis, further supplementing the cell death mechanism induced by elesclomol [25]. The excess Cu(II) within cells can be transported to the mitochondria by ionophores, the FDX1 reduces Cu(II) to Cu(I). Increased amount of Cu(I) directly binds to lipoylated components (like DLAT) of the tricarboxylic acid (TCA) cycle, resulting in the lipoylated proteins aggregation and destabilization of Fe–S cluster proteins, leading to proteotoxic stress and, eventually, cell death (Fig. 4) [25]. Notably, the cell death pathway induced by Cu ionophores was not prevented by treatment with inhibitors of other already known cell death pathways, such as pan-caspase inhibitors (antiapoptotic compounds), ferrostatin-1 (an antiferroptotic compound), necrostatin-1 (an antinecroptotic compound), or N-acetyl cysteine (a suppressor of oxidative stress), suggesting that the cuproptosis mechanism differs from that of previously identified cell death pathways [25].

An excess Cu(II) supply can lead to cell pyroptosis (By BioRender). The uptake of Cu(II) into cells triggers pyroptosis via protein lipoylation, which is an important mechanism for the enzymatic function of proteins in the TCA (tricarboxylic acid) cycle
Cross talk among components of necroptosis, pyroptosis, ferroptosis, and cuproptosis
Accumulating evidence of widespread cross talk between key initiators, effectors and executioners of necroptosis, pyroptosis, ferroptosis, and cuproptosis has been reported. According to recent research, necroptosis activates the NLRP3 inflammasome by releasing potassium from the MLKL pore in macrophages (MΦs) [76]. The ZBP1 protein senses viral/endogenous nucleic acid ligands and triggers innate immune responses [77]. In detail, once ZBP1 is activated, RIPK3 and caspase-8 are recruited to activate the NLRP3 inflammasome, which initiates both necroptosis and pyroptosis [78,79,80]. In addition, the bioinformatics analysis reported by Miao et al. also indicated that the ZBP1, both of the cuproptosis-related and necroptosis-related gene signature, functions as a risk score for prediction of the low-grade glioma patients prognosis [81]. New outcomes from the research of Wei Gao and colleagues also revealed that the administration of elesclomol to CRC cells increased Cu(II) levels in mitochondria and downregulated the expression of the Cu(II) transporter ATP7A, leading to ROS accumulation. The procedure stimulated SLC7A11 degradation, which increased oxidative stress and led to ferroptotic death in CRC cells [75]. The recently constructed glucose oxidase (GOx)-engineered nonporous Cu(I) 1,2,4-triazolate ([Cu(tz)]) coordination polymer nanodrug GOx@[Cu(tz)] efficiently combined cancer starvation and cuproptosis induction [82]. The catalytic function of GOx@[Cu(tz)] can only be “turned on” and depletes glucose upon exposure to high GSH levels in cancer cells. The redox reaction between the released Cu(II) and intracellular GSH will induce GSH depletion and reduce Cu(II) to the Fenton agent Cu(I), which then catalyzes H2O2 to generate •OH via the Fenton reaction [83]. Subsequently, the exhaustion of glucose accompanied by GSH elimination further makes cancer cells more susceptible to cuproptosis [82] and probably ferroptosis.
Diverse cell death modes in cancer biology: cell proliferation and metastasis
Brief overview of cancer cell proliferation and metastasis
Cell death is a physiological regulator of cell proliferation, and both processes exert profound effects on growth and development throughout life [48]. Cancer is characterized by the faulty regulation of cell division and death, which promotes uncontrolled tumor growth and replicative immortality [84]. An elevated cell proliferation rate and cell cycle abnormalities have been reported to be caused by inactivation of tumor suppressor genes such as CDKN2A, PTEN and TP53 [85, 86]. Because the induction of apoptosis alone via traditional anticancer therapeutic strategies does not completely eradicate cancer [84], studying methods to effectively inhibit abnormal cell proliferation and cell growth is important for cancer therapy.
Cancer metastasis is defined as the spread of tumor cells from their original location through the lymphatic system, blood vessels, or body cavities to colonize distant sites, establishing a local surviving cancer cell milieu and continual growth of secondary tumors [87]. Hanahan and Weinberg specified that one hallmark of cancer is “activated invasion and metastasis” [20]. The invasiveness of cancer cells into local tissue and seeding in areas distant from the original tumor to generate metastases are fundamental aspects of cancer malignancy [88], and metastasis remains the leading cause of mortality results from cancer [89]. In addition to cancer cell spread, metastasis facilitates cancer development via the degradation of the extracellular matrix, mediation of the epithelial-to-mesenchymal transition (EMT), promotion of tumor angiogenesis, and other processes [90, 91]. Numerous investigations have revealed that various RCD types are suppressed during tumor metastasis [92], and cell death caused by treatments administered locally or systemically effectively suppresses tumor metastasis [16]. Another widely held belief is that metastasis results from the spread of a malignant lesion through the activation of cellular reprogramming by microenvironmental stressors that promotes cell invasion and migration [93, 94]. Thus, investigating strategies to effectively suppress aberrant cell proliferation and cancer metastasis is essential to cancer treatment.
Necroptosis in proliferation and metastasis
Reports on the relationship between necroptosis and cancer have produced contradictory results, suggesting that necroptosis exerts different effects at different stages of cancer cell proliferation and metastasis. In cancer cells, the expression of major necroptotic pathway regulators is often downregulated, which has been found to be correlated with bad outcomes [95,96,97,98,99]. For example, according to Hockendorf et al., leukemogenesis is significantly accelerated after RIPK3 is knocked out in mice transplanted with bone marrow cells carrying a mutant AML driver gene, and the average lifespan of RIPK3-knockout mice is shorter than that of wild-type mice [100]. Additionally, low RIPK3 expression in colorectal cancer (CRC) patients and the reduced expression of MLKL in pancreatic adenocarcinoma and primary ovarian cancer have been reported to be correlated with reduced DFS and OS [97, 101, 102].
Clinical breast cancer tissues showed noticeably elevated the expression of TNFα, RIPK1, RIPK3, and MLKL at the mRNA and protein levels compared with their paired noncancerous tissues. Moreover, the pharmacological inhibition of necroptosis accelerates the proliferation and metastasis of breast cancer cells [103]. Han et al. discovered that the administration of compounds such as resibufogenin effectively suppresses the occurrence and metastasis of CRC by inducing RIPK3-mediated necroptosis [104]. Additionally, conventional anticancer therapies, such as platinum-based chemotherapy (cisplatin) and proteasome inhibitors (bortezomib), induce tumor cell necroptosis [105]. Furthermore, under certain conditions, necroptosis of cancer cells inhibits metastasis by triggering immunological responses against the tumor through DAMP generation [106].
However, in normal intestinal epithelial cells, the occurrence of MLKL-induced necroptosis disturbs gut homeostasis and results in inflammation [107]. Another study showed that melanoma cell-induced endothelial cell necroptosis significantly promotes the invasion and metastasis of malignant cells. Treating mice with the RIPK1 inhibitor necrostatin-1 or endothelial cell-specific deletion of RIPK3 significantly inhibits endothelial necroptosis and limits the extravasation and metastasis of malignant cells [108]. Similarly, Wang et al. reported that RIPK1 expression is significantly increased in both human lung cancer samples and mouse lung tumor models, suggesting that RIPK1 may exhibit an oncogenic function [109]. According to Liu et al., a higher level of MLKL phosphorylation is correlated with a poorer prognosis and shorter OS of patients with CRC and esophageal cancer [110]. Therefore, necroptosis of tumor cells exerts different effects on cell proliferation and spread and is not always beneficial in the treatment of cancer. In fact, many of the therapies that inhibit necroptosis have also shown good efficacy in the treatment of cancer, which will be introduced in detail at the end of this article.
Pyroptosis in proliferation and metastasis
Pyroptosis has significant therapeutic implications for several malignancies due to its profound effects on the invasion, proliferation, and metastasis of tumor cells. Based on several published investigations, pyroptosis-related modulators show tumor-suppressive activity against CRC [111], liver cancer [49], lung adenocarcinoma [112], and bladder cancer [113]. FL118, a camptothecin analog, inhibits the proliferation, invasion and metastasis of SW480 and HT129 cells by inducing caspase-1-dependent pyroptosis [114]. Additionally, lncRNAs are correlated with the regulation of pyroptosis. LncRNA RP1-85F18.6 is involved in promoting proliferation and invasion and suppressing pyroptosis in CRC cells, and knockdown of RP1-85F18.6 results in GSDMD cleavage to trigger pyroptosis [115]. Moreover, another study revealed that lower GSDMD expression is associated with a poorer CRC prognosis; furthermore, increased GSDMD expression effectively induces cell death [116]. The activation of pyroptosis by upregulating IFN-γ in mouse cancer cells inhibits tumor cell proliferation and enhances antitumor immunity in a mouse colon carcinoma cell line [54].
In recent years, many findings have been reported that pyroptosis suppresses the metastasis of cancer cells. Simvastatin is a statin with anticancer properties that has been implemented in the treatment of non-small cell lung cancer (NSCLC) [117]. The activated NLRP3 inflammasome and caspase-1 by simvastatin induces pyroptosis via the canonical pathway, inhibiting NSCLC cell migration [118]. Analogously, trichosanthin is a single-chain ribosome-inactivating protein extract from the root tuber of a Chinese medicinal herb [119] that inhibits cell growth and metastasis by promoting pyroptosis in NSCLC cells [120]. In addition, berberine is an isoquinoline quaternary alkaloid derived from medicinal plants that causes pyroptosis in HepG2 cells by inducing the expression of caspase-1 and preventing the migration and proliferation of HepG2 cells [121].
However, pyroptosis does not exert an absolutely positive therapeutic anticancer effect. A study revealed that elevated expression of GSDMC is also strongly associated with a worse prognosis for invasive breast carcinoma patients and exhibits a correlation with immune cell infiltration in the tumor [122]. Jianwei Gao and colleagues also found that increased GSDMD expression levels may increase the tumor size, promote more advanced tumor-node-metastasis stages, and affect survival rates. Moreover, GSDMD knockdown significantly restrains NSCLC cell proliferation via intrinsic mitochondrial apoptotic pathways and inhibits EGFR/Akt signaling [123]. Furthermore, as mentioned above, chemotherapy induces pyroptosis in cells which express high level of GSDME, while cells with low or no GSDME expression undergo apoptosis. However, in some tumor cell lines, the expression level of GSDME was lower than that in normal cell lines, leading to the accidental injury of normal tissues during chemotherapy [38, 124]. One explanation for the contradictory effects of pyroptosis is that whereas acute activation of pyroptosis results in necrotic cell death and inhibits tumor formation, persistent stimulation of pyroptosis promotes tumor progression [49]. In another study, abnormally upregulated GSDMB was reported to be critical for promoting the proliferation and invasiveness of bladder cancer cells [125]. Hergueta-Redondo et al. also discovered that a high level of GSDMB in breast cancer patients was related to tumor progression and a low treatment response rate [126]. Elucidating the precise mechanism by which pyroptosis reduces cancer growth and cancer cell proliferation is very important for the development of more-effective anticancer drugs.
Ferroptosis in proliferation and metastasis
Since a common concept related to the development of cancer is based on the specific mutations of oncogenes associated with the redox system [127], cancer cells show a higher level of Fe accumulation, which makes them more susceptible to the modulation of ferroptotic cell death than normal cells. Ferroptosis is a significant force modulating the growth and proliferation of certain types of tumor cells, such as diffuse large B cell lymphoma, RCC, melanoma, and ovarian cancer cells [59, 128, 129]. In 2015, Jiang et al. discovered that cancer cell ferroptotic death is induced by the p53 pathway when ROS are present at high or otherwise ectopic levels. P53 is significant in controlling cell proliferation. Mechanistically, activation of p53 substantially reduces cystine absorption by system XC−, which in turn inhibits intracellular GSH production, hence modulating the proliferation of tumor cells [130, 131]. Additionally, ART is a derivative of artemisinin that inhibits the proliferation of ovarian cancer cells by increasing the generation of ROS and triggering ferroptosis [132]. Furthermore, excessive proliferation of tumor cells is always accompanied by high ROS production, but these cells optimize ROS-driven proliferation via their effective antioxidant activity, enabling them to adapt and thrive under highly oxidative conditions while preventing ROS from reaching that threshold level that triggers ferroptosis [133, 134]. Analogously, tumor cells are also desensitized to ferroptosis by exporting iron through the secretion of ferritin-containing exosomes [135, 136]. Although ferroptosis significantly restricts tumor cell proliferation, effective compounds that induce ferroptosis must be identified while considering the adaptability of tumor cells.
Ferroptosis is also important in inhibiting cancer metastasis. The overexpression of the lncRNA BDNF-AS enhances peritoneal metastasis of gastric cancer by preventing ferroptosis [137]. Another study by Guan et al. also indicated that ferritinophagy-mediated ferroptosis and the KEAP1/NRF2/HO-1 pathway robustly contribute to EMT inhibition in gastric cancer cell lines [138]. Intriguingly, recent findings reported by Ubellacker and colleagues revealed that melanoma cells tend to spreading through the lymphatic system rather than through the bloodstream because lymph fluid contains higher expression levels of GSH and oleic acid and lower levels of free iron than blood plasma. This compositional difference contributes to decreased oxidative stress in lymph and inhibits the ferroptotic death of melanoma cells [139]. Additionally, Li et al. found the main feature of metastatic cells is sustained expression of GPX4, and GPX4 knockdown effectively induces ferroptosis and attenuates the enhanced tumorigenic and metastatic activity of malignant cells [92]. We assume that the induction of ferroptosis by inhibiting GPX4 and GSH might have superb potential for cancer therapy.
Cuproptosis in proliferation and metastasis
Cuproptosis, a Cu-induced cell death pathway, is highly associated with the mitochondrial metabolism [140] and plays a critical role in tumor cell proliferation, metastasis, and drug resistance [141, 142]. The levels of Cu accumulating in both serum and tumor tissues have been discovered to be markedly altered in individuals suffering from various malignancies, such as breast cancer [143], pancreatic cancer [144], thyroid cancer [145], leukemia [146], CRC [147], lung cancer [148], prostate cancer [149], and oral cancer [150]. These changes in Cu homeostasis may enhance tumor development or invasiveness or may confer resistance to treatment [151]. Recent investigations demonstrated that Cu is closely correlated with the expression level of hypoxia-inducible factor 1α [152, 153], which stimulates angiogenesis, and neovascularization in turn induces the production of vascular endothelial growth factor [154]. MEMO1, an oncogenic protein, was identified as an intracellular Cu-dependent protein that is required for breast cancer cell migration and invasion in vitro and spontaneous lung metastasis in vivo [155]. MEMO1 was presumed to bind to Cu(II) and promote ROS production through redox cycling, but Zhang et al. have shown that it preferentially binds to Cu(I) and shields cells from redox activity [155, 156]. Therefore, the identification of an appropriate method to block the binding site of Cu(I) on the MEMO1 protein might be a potential approach for releasing Cu ions and inhibiting the metastasis of tumor cells.
Cuproptosis may function by suppressing cancer cell proliferation and inhibiting metastatic events. For instance, in patients with oral squamous cell carcinoma who take betel nut, the related arecoline stimulation may inhibit cuproptosis, significantly increasing the viability of cancer-associated fibroblasts (CAFs) [157]. CAFs is crucial in cancer progression by contributing to the promotion of the EMT, cancer metastasis and chemotherapy resistance [158, 159]. Cuproptotic tumors also show decreased angiogenesis and are sensitive to sunitinib and sorafenib treatment [160]. To our surprise, cancer cells have evolved to activate a mechanism that prevents Cu-induced death to ensure their survival. Notably, Zhang et al. discovered that the key cuproptosis regulator FDX1 was profoundly downregulated in hepatocellular carcinoma (HCC) patients, leading to HCC cell resistance to cuproptosis [13]. In addition, lower expression of the FDX1 gene was reported to be closely associated with more advanced tumor-node-metastasis stages [161]. Furthermore, lower expression of FDX1 in various cancer types has been correlated with shorter survival times [13, 162, 163]. Despite these findings, cuproptotic events have not been well documented in diverse cancers. Therefore, additional in vitro and in vivo experiments are needed to confirm a role for cuproptosis in the proliferation and metastasis of cancer cells.
Pleiotropic functions of cell death in the TME
Events in TME have been extensively correlated to tumor development, progression, and responses to chemotherapy and antiangiogenic therapy [164,165,166]. The TME includes noncancerous cells, including components that are also present in the tumor, such as immune cells, CAFs, endothelial cells, mesenchymal stroma/stem cells (MSCs), extracellular matrix compounds, and soluble products such as chemokines, cytokines, growth factors, and extracellular vesicles [167, 168]. According to experimental data, tumor immunity within the TME may be influenced by necroptosis, pyroptosis, ferroptosis, and cuproptosis [13, 169]. For example, the novel RCDs increased infiltration of tumor-infiltrating lymphocytes and antitumor immunity have been reported in the tumors of long-term small cell lung cancer survivors [170]. It is widely believed that the treatments with immune checkpoint inhibitors (ICIs) are not effective in “cold tumors,” which are characterized by the lack of T cell infiltration, while “hot tumors” with significant T cell infiltration contribute to better ICI efficacy [171]. In this section, we explore various types of cell death in the TME and show how the interaction of host immune cells with these cells affects tumor progression and cancer treatment.
Cancer cell death influences immune cell infiltration into the TME
Tumor necroptosis influences the TME
The induction of necroptosis in tumor cells has been shown to contribute to an autophagy-mediated increase in DAMPs, which subsequently triggers immunosurveillance [172]. Notably, the necrotic cells transplanted into the TME stimulate the antitumor immune response mediated by BATF3 + cDC1- and CD8 + leukocytes and are accompanied by tumor-associated antigen-presenting cells, which increase the tumor antigen load (Fig. 4) [173]. The enhanced immunogenicity and vaccine efficacy obtained after inducing tumor necroptosis may constitute an approach to the development of cancer vaccines. Furthermore, Aaes et al. documented that vaccinating an experimental mouse model with necroptotic cancer cells induced potent antitumor immunity by promoting the maturation of dendritic cells (DCs), inducing cross-priming of cytotoxic T cells and IFN-γ production in response to tumor antigen stimulation [174]. Moreover, derepression of TRIM28 activity by RIPK3 activation in malignant cells leads to increased production of immunostimulatory cytokines within the TME, contributing to robust cytotoxic antitumor immunity [175]. Hence, de novo necroptotic death creates an inflammatory milieu that modifies tumors that are responsive to ICIs [176].
He et al. discovered 8 differentially expressed necroptosis-related genes in tumors compared with their expression in normal tissues, thereby revealing a prognostic signature they called the NRS score. NRS scores were positively correlated with the number of follicular helper T cells, CD8 + T cells, resting mast cells, M1 MΦs, and M2 MΦs. M1 MΦs typically exert antitumor effects, but M2 MΦs are proposed to be protumorigenic. He et al. found necroptosis might also shield tumors from antitumor immune responses by fostering an immunosuppressive milieu and immune escape mechanisms [177, 178]. Analogously, in pancreatic ductal adenocarcinoma, RIPK3 expression is significantly upregulated compared with that in normal tissues, whereas RIPK3 deletion mitigates the expression of the chemokine CXCL1 in vivo and in vitro. Furthermore, RIPK3 deletion diminishes the infiltration of immunosuppressive myeloid cell subsets (tumor-associated MΦs (TAMs), myeloid-derived suppressor cells (MDSCs) and DCs), and the proportions of T cells and B cells are increased (Fig. 5) [179]. The increases in percentage of T cell and B cell number indicate potent anticancer effects [180].

Summary of novel RCD modalities in tumor cells that influence the TME (By BioRender). We summarize the effects of tumor cell necroptosis, pyroptosis, ferroptosis, and cuproptosis on the number of immune cells and the levels of immune-related factors in the TME
Therefore, the effect of necroptosis on the TME cannot be fully determined, and more evidence obtained from a combination of basic experiments and clinical trials is still required to identify the specific function of necroptosis.
Tumor pyroptosis affects the TME
As mentioned above, compared with necroptosis and ferroptosis, pyroptosis is a more common mechanism of immune defense [181]. Pyroptosis is intimately related to immune cell infiltration into the TME in various cancers. For instance, combinations of BRAF inhibitors and MEK inhibitors (BRAFi + MEKi) modulate the TME by triggering pyroptosis [182]. BRAFi + MEKi have been approved by the FDA to treat BRAF V600E/K-mutant melanoma, but this treatment leads to a certain degree of resistance [183]. Erkes and colleagues revealed that BRAFi + MEKi therapy enhances GSDME cleavage and HMGB1 release. HMGB1 sufficiently induces the infiltration of DCs to expand the proportions of CD4 + and CD8 + T cells, particularly activated (CD44 +) and proliferating (Ki-67 +) T cells, which exert antitumor effects (Fig. 5). Intriguingly, in a BRAFi + MEKi-resistant disease context, intratumoral T cell infiltration is decreased, which is reversed by pyroptosis-inducing chemotherapy [182]. Another study further characterized a distinct prognostic factor for lung adenocarcinoma based on PRGs called the Pyro-score, showing that a low Pyro-score indicates increased immune cell infiltration. And patients with an increased number of infiltrating immune cells exhibit higher sensitivities to anti-PD-1/L1 immunotherapy [184]. Zhang et al. discovered that pyroptosis mediated by GSDME cleavage suppresses tumors by increasing the numbers of tumor-infiltrating NK cells and CD8 + T lymphocytes, as well as by inducing phagocytosis by TAMs (Fig. 5) [53]. However, cancer cells have evolved two strategies to prevent the tumor-suppressing effect of GSDME: (1) epigenetically suppressing GSDME expression [185] and (2) loss-of-function mutations [186]. Indeed, Cai et al. found that the natural product triptolide potently eliminates head and neck cancer cells by inducing GSDME-mediated pyroptosis, which is significantly attenuated after GSDME gene silencing [187]. Therefore, solutions for overcoming these problems will help scientists leverage pyroptosis to improve cancer treatment. Recent findings from a study by Fan and colleagues suggest that harnessing decitabine to demethylate the GSDME gene in malignant cells before inducing pyroptosis effectively overcomes the epigenetic restriction of GSDME [188]. Additionally, strategies have been developed to package recombinant adeno-associated viruses expressing the PFD of GSDM to induce pyroptosis and prolong the life span of cancer models [189].
In spite of the positive experimental findings suggesting the antitumor role of pyroptosis in TME, the study from Tan et al. demonstrated that HMGB1 released from pyroptotic cell death contributes to the tumorigenesis of colitis-associated colorectal cancer through activating ERK1/2 pathway [190]. Activation of ERK1/2 signaling plays a protumorigenic role in modulation of the TME via inducing M2 MΦs polarization [191]. Therefore, it is important to consider the possibility of failure as we continue to research anticancer properties of pyroptosis.
Tumor ferroptosis alters the TME
Ferroptotic cancer cells generate some “find-me” and “eat-me” immunostimulating signals, particularly DAMPs, which robustly recruit DCs, MΦs, and other immune cells properly to the site of dying tumor cells [192, 193]. Another recent study also revealed that ferroptosis induced by a kind of nanomodulators attenuated the self-renewal capability of cancer and downregulated the expression of genes related to angiogenesis (Fig. 5) [194]. Therefore, ferroptosis shows anticancer effect via promoting immunogenicity and inhibiting metastasis-related genomic expression. It was also discovered that early ferroptotic cancer cells can accelerate the phenotypic development of DCs and elicit a vaccination-like response [195]. Additionally, according to research by Géraldine Luis and colleagues, fatty acid-binding protein-4 in TME which increases the production of lipid droplets in cancer cells, and stearoyl-CoA desaturase-1 (SCD1) expressed by cancer cells, cooperatively protects cancer cells from oxidative stress-induced ferroptosis and promotes tumor recurrence [196]. Furthermore, some chemotherapeutic medications, such as cisplatin, targeted medications, such as sorafenib, and radiotherapy strongly induce ferroptosis [197,198,199], which encourages the infiltration of immune cell and raises the immunogenicity of immune-desert tumors, ultimately improving the efficacy of ICI immunotherapy. Tumor cells are significant in constructing immune-stimulating microenvironment, which opens the door for the emergence of innovative solutions to cancer immunotherapy.
Bioinformatics study revealed 7 differentially expressed FRGs in papillary thyroid carcinoma that are positively correlated with a TME enriched with infiltrating immune cells. The three most highly activated subtypes of cells were B cells, CD8 + T cells, and CD4 + T cells (Fig. 5) [200]. Moreover, another risk signature score based on FRGs revealed that lymph node invasion and venous invasion events were more common in high-scoring ovarian cancer samples. Furthermore, infiltrated immune cells and stromal cells were more frequent in the high-scoring group [201]. Xu et al. selected 9 differentially expressed long noncoding RNAs associated with ferroptosis to develop a prognostic signature for patients with HCC, and an increase in the expression of the immunological checkpoint protein B7H3 was identified in the high-risk group [202].
However, cancer cells that undergo ferroptotic death are also associated with the release of PGE2, a significant immunosuppressant that disrupts the anticancer activities of NK cells, DCs, and cytotoxic T cells [59, 203]. Demuynck et al. proposed that cancer cell ferroptosis may substantially increase the levels of oxidized lipids, leading to decreased phagocytosis and antigen cross-presentation by DCs and thus potentially favoring tumor cell evasion of immune surveillance [204]. Additionally, NRF2—nuclear factor erythroid 2-related factor 2, has been proved to inhibit ferroptotic cell death, which can be stimulated by oncoproteins including c-Myc, K-RAS, and B-raf. Intriguingly, downregulating NRF2-targeted genes could increase ferroptosis in the TME and promote cancer progression [205, 206]. Because of persistent uncertainty, we must continue to view the role of tumor cell ferroptosis in the tumor immune microenvironment with skepticism.
Tumor cuproptosis affects the TME
Cuproptosis is associated with immune cell infiltration. As shown in a recent study, melanoma patients with higher expression of CRGs experienced a longer OS. One prominent CRG is LIPT1, whose expression is positively linked to PD-L1 expression and negatively correlated with regulatory T cell infiltration (Fig. 5) [207]. Increased PD-L1 expression suggests that the combination of cuproptosis induction via immune checkpoint blockers may show better efficacy. Additionally, in esophageal carcinoma, higher expression of CRGs was anomalously correlated with an increased number of infiltrating bystander T cells (Fig. 5) [208]. According to Zhang et al., lower CRG expression in HCC patients correlates with an increase in protumor immune components in tumors; however, no change in the percentage of antitumor immune cells was reported [13]. In addition to the role of CRGs in the establishment of TME with antitumor effects, other studies have also shown that cuproptosis-related lncRNAs are also related to the changes of immune cell infiltration. Wang et al. collected a total of 16 cuproptosis-related lncRNAs and constructed high- and low-risk prognostic signatures based on the nomogram and heatmap of cuproptosis-related lncRNAs. They found the high-risk patients have a greater potential for immune escape and less response to cancer immunotherapy of lung adenocarcinoma [209]. Currently, cuproptosis is presumed to play a certain role in shaping an antitumor immune environment, but whether Cu-dependent death exerts an inhibitory effect on cancer immunotherapy remains to be determined. Therefore, clarifying the function of cuproptosis is crucial for the formulation of future combination therapies.
Cell fates in the TME
A 2016 study revealed that alanine released from stroma-associated pancreatic stellate cells by autophagy was a substitute carbon source that fueled the TCA cycle in pancreatic ductal adenocarcinoma. This change in fuel source reduced the tumor cell reliance on glucose and nutrients obtained from serum, which are limited in the pancreatic TME [210]. Based on this finding, we logically suspect that a novel RCD pathway of noncancer cells in the TME may affect cancer cell survival. Therefore, we investigated this hypothesis in detail.
Interestingly, novel forms of RCD for cells that compose the TME profoundly influence the tumor fate. For instance, RIPK3 downregulation in TAMs induces fatty acid oxidation and M2 MΦ polarization in the TME, facilitating HCC tumorigenesis [211]. In addition, Huanrong Lan and colleagues revealed that oxaliplatin resistance in CRC results from the necroptotic evasion of M2 MΦs. Mechanistically, the expression of the methyltransferase METTL3 is increased in oxaliplatin-resistant CRC tissues, and METTL3-mediated N6-adenosine methylation significantly inhibits TRAF5-induced necroptosis both in vitro and in vivo [212]. Thus, the necroptosis of MΦs tends to exert a positive antitumorigenic effect.
The discovery that the serine protease inhibitor Val-boroPro (also called talabostat or PT-100) cleaves the substrate at proline has generated significant interest in this compound as a potential anticancer drug. Val-boroPro achieves its anticancer effects by activating pro-caspase-1, which is subsequently cleaved to activate GSDMD and induces the pyroptosis of monocytes and MΦs [213].
As shown in the study by Hage et al., sorafenib induces pyroptosis in MΦs to stimulate HCC cell killing [214]. Specifically, sorafenib robustly increases the activity of caspase-1, activating GSDM and inducing MΦ pyroptosis. Subsequently, NK cells are activated when cocultured with sorafenib-treated MΦs, and the interplay of MΦs and NK cells induces HCC cell death. Moreover, various cytokines are released from pyroptotic immune cells, including IL-18, which shows established anticancer activity by enhancing the type 1 immune response and can thus be utilized in cancer immunotherapy [215, 216].
MΦs engulf red blood cells and digest them to generate hemoglobin, which is further degraded into heme. Heme is catabolized into iron, which either promotes ROS generation or lipid peroxidation. Through ferroportin, the iron produced by heme is discharged into the environment, increasing the iron level in the TME (Fig. 3A) [217]. Then, iron promotes the Fenton reaction and generates hydroxyl radicals, which cause tumor cells to undergo ferroptosis [218]. Therefore, macrophages increase the content of iron in the TME through their own ferroptosis and promote the subsequent ferroptosis of tumor cells, thus showing a powerful antitumor effect. Ferroptosis of MDSCs was demonstrated to be crucial in fighting malignancies, but Zhu et al. found that N-acyl-sphingosine amidohydrolase (ASAH2) is expressed at high levels in MDSCs in colon carcinoma. ASAH2 reduces MDSC ferroptosis by reducing p53 stability, upregulating Hmox1 expression, and inhibiting lipid ROS production in the TME. The ASAH2 inhibitor NC06 induces ferroptosis in MDSCs by inhibiting ceramidase activity. Animal models confirmed that NC06 inhibits the infiltration of MDSCs into transplanted tumors by promoting MDSC ferroptosis and thus inhibits tumor growth [219]. Furthermore, ferroptosis mediated by tumor-infiltrating lymphocytes significantly enhances the efficacy of ICIs [220, 221].
Since cuproptosis is a novel RCD, determining whether it occurs among noncancerous TME cells is a challenge. However, upon Cu stimulation, exosomes secreted by MΦs increase angiogenesis mediated by endothelial cells in vitro and in vivo [222]. Ryuhei Takemoto and colleagues also found that overexpression of lysyl oxidase, a Cu-containing enzyme, in human leukemic THP-1-cell-derived M2 MΦs promotes tumor metastasis [223]. Therefore, immune cell cuproptosis may have a multifaceted role in TME, and we are awaiting rational animal and cellular investigations to elucidate this role.
Current and future therapeutics targeting different cell death pathways
As we previously discussed, tumor cells and other cells in the TME that undergo necroptosis, pyroptosis, ferroptosis, or cuproptosis possibly contribute to strong antitumor immunity. Additionally, mechanisms for bypassing the apoptosis signaling pathways that cause the death of cancer cells have attracted considerable attention for their use in anticancer therapy [224]. Therefore, we describe small-molecule compounds and other agents targeting novel mechanisms of cell death that might be employed in cancer therapy (Table 1), and we emphasize the therapeutic approaches that have been tested in clinical trials to date (Table 2).
Agents targeting novel cell death pathways
Targeting necroptosis, pyroptosis, and ferroptosis to develop new anticancer medications for therapeutic use has been a long process, and recently, compounds inducing cuproptosis have shown promise as anticancer strategies [225, 226]. In Table 1, we summarize 85 types of therapeutic agents that exert an effect on the mechanisms of newly discovered RCD modalities that have been tested in vivo and/or in vitro.
Agents inducing novel cell death pathways
Approved and investigational drugs inducing novel RCD pathways
According to recent investigations, many clinically approved medications exert potent antitumor effects by inducing (or inhibiting) inflammatory RCD modalities in preclinical studies [169]. CQ has been shown to upregulate endogenous RIPK3 in CRC cell lines, and Hou et al. reported that necroptosis mediates this process, which is not affected by apoptosis inhibitors [235]. Interestingly, shikonin, a naphthoquinone product synthesized from the roots of a Chinese medicinal herb, induces nasopharyngeal carcinoma cell necroptosis in a dose-dependent manner [227]. Mechanistically, shikonin increases ROS production and upregulates the expression levels of RIPK1, RIPK3, and MLKL, which prompts necroptosis in apoptosis-resistant tumor cells [307]. However, the activation of necroptosis can also be mediated by the modulation of the upstream signaling pathways. For instance, the sphingosine analog FTY720, also called fingolimod, induces necroptosis in human lung cancer cells by binding to inhibitor 2 of PP2A (I2PP2A/SET oncoprotein), thus activating the PP2A/RIPK1 pathway [228].
In addition, metformin inhibits cancer cell proliferation by inducing mitochondrial dysfunction to cause pyroptotic cell death [241]. Specifically, metformin is a sensitization agent that enhances AMPK/SIRT1/NF-κB signaling to trigger the activation of caspase-3 and the generation of GSDME-PFD. Lu Wang and colleagues documented that metformin causes pyroptotic death of esophageal squamous cell carcinoma cells by targeting the miR-497/PELP1 axis [242]. Further, chemotherapeutic medications, including actinomycin-D, doxorubicin, topotecan, and bleomycin, stimulate the pyroptotic death of GSDME-expressing cells [38]. Teng et al. also found that the induction of ROS/ NLRP3/GSDMD signal axis via using polyphyllin VI practically leads to pyroptotic death of NSCLC cells [246]. Our Fig. 6 summarizes other methods of action of pyroptosis inducers.

Summary of the modulators of novel RCDs in cancer treatment
Sorafenib is an FDA-approved anticancer drug for the treatment of HCC, RCC, and thyroid cancer [308]. Sorafenib inhibits system XC−, thus promoting ferroptosis by inhibiting GSH production [281]. Additionally, sorafenib and sulfasalazine may synergize to prevent the activation of branched-chain amino acid aminotransferase, a principal enzyme involved in sulfur-based amino acid metabolism. This therapeutic approach induced ferroptosis in HCC cell lines both in vitro and in vivo [309]. Additionally, cisplatin triggers ferroptosis via GSH depletion and inactivation of GPX4 in NSCLC and colon cancer [197]. Etoposide is a phenolic antitumor drug that efficiently removes GSH in myeloperoxidase–rich myelogenous leukemia cells, thus decreasing GPX4 levels and leading to ferroptosis [279]. In the study by Ma et al., combining the lysosome disruptor siramesine with lapatinib, a tyrosine kinase inhibitor, induced ferroptotic death of breast cancer cells by inhibiting iron transportation and induction of lipid peroxidation [282].
Peter Tsvetkov and colleagues identified that the Cu ionophore elesclomol induces cuproptosis by inducing lethal proteotoxic stress in various types of cancer cells (as shown in Table 1). However, as indicated by Gao Wei and colleagues, elesclomol causes CRC cells to undergo Cu-dependent ferroptosis by promoting the degradation of Cu-transporting ATPase 1 and subsequently inducing ROS accumulation, which promotes the degradation of SLC7A11 [75]. Since current experimental study on cuproptosis is still in its infancy, more research is needed to support its potential for cancer treatment.
Nanoparticles (NPs) targeting RCD pathways
Advantages of NPs include easy cell barrier penetration, preferential accumulation in specific organelles and cells, and an increased likelihood of effective fine-tuning, endowing them with great potential as anticancer therapies [310]. As we mentioned above, shikonin shows great potential as an antitumor treatment by inducing necroptosis. However, the clinical application of shikonin has been restricted due to its poor tumor-specific accumulation, low water solubility, short duration in circulating blood, and a high risk for hazardous side effects on normal tissues [311]. Therefore, Feng et al. constructed an FSSN based on the metal-polyphenol coordination of Fe(III) and shikonin, and FSSN showed not only greater water solubility and lower cytotoxicity than shikonin in normal cells but was also integrated with the function of Fe ions. FSSNs effectively reduced the GSH level and induced ferroptosis and necroptosis in mouse breast cancer cell lines [238]. Additionally, the use of graphene oxide NPs in CT26 colon cancer cells successfully induced necroptosis by enhancing the function of RIPK1, RIPK3, and HMGB1 [229]. Similarly, the group of Praveen Sonkusre reported that when treating prostate adenocarcinoma cells with selenium NPs, necroptosis was induced through increased ROS production and TNF and interferon regulatory factor 1 expression [230].
Furthermore, NPs have been used to induce pyroptosis in malignant cells. For example, the biomimetic NP designed by Pengfei Zhao and colleagues consisted of a hydrophobic nucleus composed of indocyanine green and decitabine and a cell membrane shell. Biomimetic NPs induced the accumulation of calcium in the cytoplasm, leading to mitochondrial damage and caspase-3 activation and subsequently inducing GSDME-mediated pyroptosis in 4T1 cell lines [250]. In addition, Kataoka et al. constructed an ROS-responsive nanoreactor based on polyion complex-forming vesicles by introducing thioketal linkers into a covalently cross-linked membrane network. These ROS-responsive NPs shielded glucose oxidase to induce pyroptosis by generating oxidative stress and inducing glucose deprivation [255].
A recent study described the use of an efficient ferroptosis agent, an FePt@MoS2 NP, which induced the release of more than 30% Fe(II) in the TME within 72 h of treatment to accelerate the Fenton reaction and efficiently induce ferroptosis in various cancer cell lines [280]. Analogously, another study showed that zero-valent iron NPs converted Fe(II) to promote the Fenton reaction, which induced mitochondrial lipid peroxidation in oral cancer cells [266]. Furthermore, a positively charged lipophilic nanocarrier (Fe-CO@Mito-PNBE) targeted the negatively charged mitochondrial membrane, and the subsequent release of Fe(III)/Fe(II) ions effectively facilitated the Fenton reaction and ultimately led to cell ferroptosis [267]. More NPs that induce ferroptosis in tumor cells are listed in Table 1.
The administration of NPs reverses cisplatin resistance in cancer cells by inducing cuproptosis. Exogenous platinum is widely presumed to cause drug resistance induced by high concentrations of GSH in cancer cells. According to Lu et al., the diethyldithiocarbamate-Cu complex effectively induces cuproptosis in A549/DDP cell lines by downregulating FDX1 expression. Most of the administered diethyldithiocarbamate-Cu complex maintained a stable chemical structure when mixed with GSH in solvent, suggesting that it potentially combats cisplatin-resistant cancer cells [306]. Accordingly, research into nanomaterials that induce recently discovered RCD pathways is ongoing, and we expect more and better NPs to be clinically used for cancer treatment in the near future.
Small molecules targeting novel RCD pathways
An increasing number of small compounds are being tested to target the necroptotic cell death pathway. For instance, Zhou et al. revealed that emodin, an anthraquinone compound purified from various Chinese medicinal herbs, induces necroptosis in glioma cell lines by enhancing TNF/RIPK1/RIPK3 pathway activation and thus inhibits U251 cell proliferation [231]. Additionally, ophiopogonin D′ induces robust necroptosis in prostate cancer cells through RIPK1 activation [232]. Resibufogenin, a small molecule derived from the bufadienolide family of compounds, significantly inhibits the proliferation of CRC cell lines by upregulating RIPK3 expression [104]. These small-molecule compounds still hold a lot of promise to be applied as clinical medicines because of their remarkable ability to cause tumor cells to undergo necroptosis.
Dobrin et al. found that treatment of triple-negative breast cancer cells with ivermectin induces pyroptosis by activating the P2X4/P2X7-gated pannexin-1 channel [259]. Also, based on accumulating evidence, DHA reduces cancer cell viability and proliferation by modulating different cellular responses [312, 313]. For example, Dumont and colleagues proposed that DHA inhibits NLRP3 inflammasome assembly and the JNK signaling pathway in MDSCs, reducing the 5-fluorouracil-induced generation of IL-1 and increasing the anticancer effectiveness of 5-fluorouracil [314]. Yi-Fan Tan and colleagues also revealed that inhibition of BRD4, either through genetic knockdown or the use of the bromodomain inhibitor JQ1, significantly slows the EMT and the cell proliferation rate and leads to caspase-1/GSDMD-mediated pyroptosis in RCC cells [258]. BRD4 is a member of the BET protein family that is involved in the control of epigenetic modifications [315]. Additionally, the thiopyran derivative L61H10 exhibits great antitumor activity by switching apoptosis to pyroptosis in lung cancer cells [316].
Recently, an increasing number of investigations have demonstrated that small-molecule compounds play essential roles in inducing ferroptosis in tumor cells. Zhang et al. discovered that the benzopyran derivative IMCA significantly downregulates SLC7A11 expression and reduces the contents of cysteine and GSH in cells, resulting in lipid ROS accumulation and ferroptosis in human CRC cell lines [268]. In addition, trigonelline is a plant alkaloid that significantly reduces GSH levels, thus induces ferroptosis in head and neck cancer cells [276]. Furthermore, dihydroartemisinin also exerts a robust effect on inhibiting the proliferation and inducing the ferroptosis of leukemia cells [295]. Similarly, Chang et al. found that a marine terpenoid, heteronemin, induces ferroptosis in HCC cells by initiating lipid peroxidation [317]. As shown in the study by Li et al., the small-molecule MMRi62, which targets MDM2-MDM4, induces ferroptosis by degrading mutant p53 and the heavy chain of ferritin and successfully inhibits the metastasis of pancreatic cancer [298]. As we continue our research, we have gradually discovered much promise in the field of pharmaceuticals that induce ferroptosis in tumor cells, and we are eager to see how these medications will be applied in clinical settings.
Other methods to target novel cell death mechanisms
Wan et al. documented that radiation therapy (RT) causes tumor cells to release microparticles with broad antitumor effects and thus abrogates immunogenicity primarily via ferroptotic cell death [318]. Mechanistically, radiation causes lipid peroxidation and ferroptotic cell death through three parallel mechanisms at least [198, 319, 320]. First, RT causes lipid peroxidation by producing excess ROS. RT-generated ROS remove electrons from PUFAs, resulting in the formation of PUFA radicals (PUFAs-OH). Then, these unstable carbon-centered radicals quickly react with oxygen molecules to generate lipid peroxyl radicals (PUFA-OO·), which remove H· from other molecules via the Fenton reaction and ultimately generate lipid hydroperoxides (PUFAs-OOH). Second, radiation increases the expression of ACSL4 to support PUFA-phospholipid biosynthesis, although the precise mechanism by which RT increases ACSL4 levels is still unknown [198]. Third, RT induces GSH depletion, which impairs GPX4-mediated ferroptosis defenses and subsequently promotes ferroptosis [63, 320]. Furthermore, disulfiram, a medicine approved to treat alcoholism, was shown to cause lysosomal membrane permeabilization via a ROS-dependent process, leading to ferroptosis and increasing cellular susceptibility to radiation [288].
Additionally, human umbilical cord mesenchymal stem cells (hUCMSCs) were recently identified as a viable cancer therapy option. For example, these cells prevent NSCLC and HCC cells from migrating [321]. Additionally, hUCMSCs show some advantages over other MSCs because they exhibit minimal immunogenicity and can be produced in large numbers. Following the overexpression of NLRP1 and caspase-4, hUCMSCs cause pyroptosis of the MCF-7 breast cancer cell line; however, hUCMSC treatment has little to no effect on the cell cycle [322].
The growth of schwannomas is proposed to be inhibited via a unique approach based on both the introduction of an adeno-associated virus (AAV-1) and treatment with the GSDMD PFD. This combination was created using an AAV-1-based vector encoding the mouse GSDMD N-terminus under the control of the promoter P0, which is unique to Schwann cells. This gene did not cause neurotoxicity to surrounding tissues following an intratumor injection and inhibited the development of the NF2 and HEI-193 schwannoma cell lines through GSDMD-mediated pyroptosis [323]. The intratumor delivery of GSDMD PFD via AAV-1 offers a better level of protection for the nearby normal tissue since it is more selective than typical medication therapy.
Agents inhibiting novel cell death pathways
Necroptosis occurs in cancer cells, and the TME is partially protumorigenic because the inflammation underlying necroptosis may trigger tumor development by promoting cell proliferation, genomic instability, angiogenesis, and metastasis [31]. Liu et al. harnessed the MLKL inhibitor NSA to treat a mouse xenograft model, which significantly delayed tumor growth, providing strong evidence of the protumorigenic role of necroptosis [110]. The necroptosis inhibitor necrostatin-1 also helps reduce colitis-associated tumorigenesis in mice [240]. RIPA-56 is a highly potent and metabolically stable inhibitor of RIPK1 that has been employed to treat a mouse model of inflammatory disease and has shown very high selectivity [324]. Another novel RIPK1 inhibitor PK68 which possesses high efficacy and conserved potency among human, mouse, and rat has been reported to effectively inhibit necroptosis and suppress metastasis of both melanoma and lung carcinoma cells in mice [239]. Although the above-mentioned necroptosis inhibitor has not been implemented in cancer patients, the RIPK1 inhibitor, GSK2982772, is currently being tested in phase 2a clinical studies for patients with inflammatory disease [325].
The utilization of pyroptosis inhibitors has significant research promise because of the dual role that pyroptosis plays in cancer. The study in 2019 revealed that delivering the specific anti-GSDMB antibody in biocompatible nanocapsules significantly inhibited the metastasis and drug resistance of HER2 breast cancer cells [264]. In addition, dimethyl fumarate is an inhibitor of pyroptotic cell death that functions by inactivating GSDMD [265]. Recent research by Jun Jacob Hu and colleagues suggests that the use of disulfiram also prevents pyroptosis by preventing the creation of GSDMD pores in a mouse model of inflammation [326]. In addition, Zhang et al. documented that the MLKL inhibitor NSA reverses pyroptosis by suppressing GSDMD oligomerization [327]. The use of these pyroptosis inhibitors in cellular and animal experiments offers great potential for treating patients with certain type of refractory cancers.
Current evidence suggests that ferroptosis induced by doxorubicin (DOX) was proved to contribute to the side effect of cancer therapy, including cardiotoxicity [328]. The DOX cardiomyopathy is caused by the excess free iron released from heme degradation which accumulates on mitochondria. Inhibition of ferroptosis through using ferrostatin-1 and HO-1 antagonist exerts some protective effect against myocardial injury [329]. In addition, the overexpression of ASCL4 also contributes to intestinal injury induced by irradiation therapy. Ji et al. have shown that troglitazone successfully suppresses lipid peroxidation in intestine through inhibiting ASCL4 and inhibited subsequent tissue damage [330]. Further, the novel findings from Soňa Jantová and colleagues demonstrated that the combination of 2,2,6,6, tetramethylpiperidine-N-oxyl (a ferroptosis inhibitor) capped TiO2 nanorods with UV-A light irradiation not only killed MCF-7 cell lines significantly, but also overcame the multidrug resistance [304]. We currently speculate that ferroptosis inhibition might have played a role in this process, but the mechanisms behind it are still blur and need further study. In addition, Dai et al. have found that the DNA damage caused by ferroptosis could facilitate pancreatic tumorigenesis through 8-hydroxy-2′-deoxyguanosine (a major product of oxidative DNA damage)-STING-dependent pathway. And the administration of ferroptosis inhibitor liproxstatin-1 effectively inhibits the pro-tumorigenesis of ferroptosis process [305].
Finally, the use of cuproptosis inhibitors, the most recent kind of cell death, in cancer has yet to be revealed. GSH was found to inhibit cuproptosis in cells, but this can lead to cisplatin resistance in tumor cells [306]. Furthermore, to help visualize the multiple modes of action, we displayed the modulators involved in four RCDs in Fig. 6.
Clinical trials targeting novel RCD modalities
Despite the fact that a variety of reports on novel RCD activators and inhibitors have been published lately, clinical trials evaluating the effects of modulators of novel RCDs are still in their infancy. In this section, we summarize the clinical trials to date in primary outcome measures or interventions that have involved the investigation of relevant biomarkers of novel RCD measurement and list them in Table 2.
One clinical study aimed to investigate whether the immediate necroptosis induced by the nonablative cryosurgical freezing could be beneficial to the subsequent injection of immunotherapeutic drugs (NCT04739618). This study recruited 32 participants with metastatic solid cancer who are first treated by nonablative cryosurgical freezing and then receive multiplex immunotherapy (including pembrolizumab, ipilimumab, and GM-CSF) and evaluate overall response rate of radiographic changes. In addition, another study posted in 2018 sought for the efficacy of RIPK1 inhibitor GSK3145095 alone and in combination with pembrolizumab included 8 participants. The serious adverse event rate of this study is 50% and it was terminated following an internal review of the company (NCT03681951).
Another phase II clinical study aims to evaluate the efficacy and safety of ferroptosis inhibitor MIT-001 for the prevention of oral mucositis in patients with lymphoma or multiple myeloma receiving conditioning chemotherapy with autologous hematopoietic stem cell transplantation (NCT05493800). This research was launched on August 9, 2022, and we shall keep track of its progress and other messages about the relationship between ferroptosis and inflammatory side effect of cancer therapy.
So far, we have found only the modulators of these two forms of RCDs, necroptosis and ferroptosis, in clinical trials and the results of these cancer therapy methods remain to be discovered. As high-quality articles on cell death modalities continue to emerge, more clinical trials will be conducted with the research purpose stated as understanding these four cell death modalities; therefore, we believe that in the near future, better use of necroptosis, pyroptosis, ferroptosis, pyroptosis, and other mechanisms will optimize anticancer treatments.
RCD: is it a potential approach to reverse drug resistance in cancer?
RCD modulation and chemoresistance
The data from Wang et al. have revealed that the epigenetic repression of RIPK3 allows NSCLC cell lines to escape from necroptosis, which subsequently increases resistance to chemotherapy [331]. Xu Zhao and colleagues successfully used trichothecin to induce necroptosis in chemotherapy resistant cancers. Mechanistically, the expression of RIPK3 was significantly upregulated by the natural secondary metabolite, trichothecin, and then RIPK3 enhanced the phosphorylation of MLKL and also activated the mitochondria energy metabolism and ROS production, leading to a novel strategy to sensitize cancer cells to cisplatin therapy [303]. Thus, it is suggested that the necroptosis pathways and lipid peroxidation can act synergistically and both play crucial roles in overcoming chemoresistance. Intriguingly, the combination of DHA with cisplatin can synergistically induce cytotoxicity against pancreatic ductal adenocarcinoma because DHA induces ferroptosis via promoting GPX4 degradation, ROS production, and ferritin degradation mediated by NCOA4 [332]. In addition, Ophiopogonin B, a bioactive component of traditional Chinese medicine, was reported to have significant impact on inducing pyroptotic cell death of A549 cells, which helps to alleviate the cisplatin resistance [260]. Further, Jing Guo and colleagues also revealed that adding GW4064, a synthetic FXR agonist, to oxaliplatin can significantly limit tumor cell proliferation in vitro, and slow tumor growth in xenograft mouse models. GW4064 effectively enhanced caspase-3/GSDME-mediated pyroptosis of HT-29 and SW620 cells, which increased the chemosensitivity of cells to oxaliplatin [261]. Cuproptosis was also demonstrated to fight against platinum-based chemotherapy resistance. Lu et al. revealed that the killing effect of cisplatin was detoxified by GSH in A549 cells, while the nanomedicine based on Cu(II) (CuET) exhibited GSH-resistant cytotoxicity and efficiently reversed cisplatin resistance [306].
RCD modulation and immunotherapy resistance
Nowadays, immunotherapy represented by ICIs has become a major breakthrough in cancer treatment and has achieved considerable success in clinical treatment of some solid tumors [333,334,335]. However, the use of ICIs is restricted by the lack of tumor-associated antigens, which results in more than two thirds of the patients to not react to ICI-based monotherapy [169]. However, due to the intricate role of the novel RCD modes in TME, we may anticipate that manipulating RCDs may affect the efficacy of ICIs in cancer patients. Emerging evidence has demonstrated that CD8 + T cells inhibit tumor cells via induction of necroptosis, pyroptosis, ferroptosis [169, 336, 337], and possibly cuproptosis [338]. Like what we mentioned, the novel RCDs in TME seriously activate proinflammatory cytokines, infiltration of cytotoxic T cells, and other lymphocytes, which are significant for the sensitivity of various tumors to ICIs [176]. In addition, the release of Gzm B from CAR-T cells activates caspase-3/GSDME-dependent pyroptosis of target cells, which enhances the efficacy of CAR-T cell therapy [181]. Thus, similar to chemotherapy, the immunotherapy may partially function as the inducers of the novel RCD mechanisms, which might provide an immune-based underpinnings for some novel combination therapies.
By creating vaccine viruses that loaded MLKL expression, Hoecke et al. directly delivered the necroptosis mediator MLKL to tumor cells, which successfully promoted necroptotic death and enhanced antitumor immunity. The potent antitumor immunity is attributed to the increased immunity directly against neo-epitopes [339]. Additionally, the RNA editing enzyme ADAR1 has long been known to be a major repressor of Z-type dsRNA (a substrate of ZBP1), and this suppression mechanism results in resistance and poor reactivity to ICIs, while the use of small-molecule drug, CBL0137, directly induces Z-type dsDNA formation in cells and results in activation of ZBP1-depenent necroptosis, which significantly reverses ICIs unresponsiveness of melanoma mouse models [236]. Similarly, the RIPK1-dependent necroptosis is inhibited by the cIAPs that can be antagonized by Smac mimetics and activate necroptotic death pathway in Burkitt’s lymphoma cell lines [237]. Also, in melanoma, the use of Smac mimetics enhances the response to ICIs via directly controlling immune cell (including B cells, MDSCs, DCs, and cytotoxic T cells) [340]. The evidence demonstrates that we might harness necroptotic mechanism in modify the TME to be more prepared for immunotherapy.
Pyroptosis is the main host defensive mechanism, and it boosts the tumor-killing activity of immune cells [181]. Wang et al. found that in the presence of pyroptosis, ICI-based therapies were effective in killing cold tumor cells, which is attributed to the fired up TME caused by pyroptosis-induced inflammation [341]. Analogously, the engineering of multienzyme-mimicking covalent organic frameworks induces pyroptosis and remodel the TME to trigger durable antitumor immunity for αPD-1 checkpoint blockade therapy [342]. However, the potent proinflammatory role of pyroptosis may cause undesirable side effect in immunotherapy. We previously mentioned that pyroptotic cell death that mediated via CAR-T cell therapy can positively enhance the efficacy, but it also counteracts the effectiveness of CAR-T therapy via initiating cytokine release syndrome [343]. The cytokine release syndrome is a severe side effect brought on by an amplified inflammatory reaction mediated by pyroptosis. In detail, IL-1β and IL-18 are released through the first activated Gzm B/caspase-3/GSDME pathway in target tumor cells, which later amplifies the inflammatory response by activating the caspase-1/GSDMD axis in MΦs [343].
It has been reported that lipid peroxides caused by ferroptosis can be utilized as a signal to facilitate the recognition and processing of tumor antigens by DCs and present them to CD8 + T cells, activating cytotoxic T lymphocytes to enhance tumor immunotherapy [344]. Thus, the combination of ferroptosis inducers with ICIs might be an excellent choice for sensitizing malignant cells to immunotherapy. Indeed, the research from Weimin Wang and colleagues has shown that the combination of GPX4 inhibitor, cyst(e)inase with PD-L1 blockade, can improve T cell-induced antitumor immunity and ferroptotic death of cancer cells synergistically [345]. Similarly, an innovative NRF2 nanomodulator, ZVI-NP, which both inhibits the antiferroptotic function of NRF2 and generates massive ROS via Fenton reaction, can potently augment antitumor immune response by reprograming the TME [194]. However, similar to the consequence of macrophage pyroptosis, the ferroptotic death of nontumor cells is associated with impaired antitumor ability because of reduced cytotoxic cytokine production. And harnessing ferroptosis inhibitor ferrostatin-1 significantly prevents CD8 + T lymphocytes ferroptosis via suppressing lipid peroxidation; consequently, cytokine production is increased, resulting in tumor clearance. More importantly, ferroptosis inhibition therapy obtains greater antitumor efficacy when in combination with anti-PD-1 antibodies [346].
Since cuproptosis is a novel mode of cell death reported this year, its role in immunotherapy is more focused on research in bioinformatics. The cuproptosis-related modification patterns developed by Zhiyong Cai and colleagues were demonstrated to be employed in prediction of immune cell infiltration in TME and evaluation of an individual’s sensitivity to ICIs [347]. It is highly likely that cuproptosis can also play a role in tumor immunotherapy, so we look forward to more experimental studies on the aspect of cuproptosis. Based on these findings, we assume that the combination therapy strategies might possess great potential to alleviate the challenge of monotherapy resistance, such as 1. combination of RCD modulators with conventional drug therapy; 2. combination of chemotherapy drugs and immunotherapy; and 3. combination of immunotherapy with radiotherapy.
Conclusions
Approaches targeting novel RCD modalities hold promise as novel treatments for cancer, and considerable efforts are devoted to translating novel regulators to the clinic. Thus, we complemented the review of approved drugs that modulate novel RCD pathways with descriptions of some newly developed beneficial small-molecule compounds and nanomaterials and clinical trials which intend to explore the changes in the expression levels of four novel biomarkers of RCD. Finally, RCDs can also make tumors more responsive to immunotherapy by regulating tumor immunogenicity and enhancing lymphocyte infiltration in the TME.
However, in spite of the discovery of many compounds and agents that induce or modulate novel RCD programming and that exert strong antitumor effects, many studies reported opposite outcomes. For instance, Chao-Chieh Lin and colleagues discovered that the expression of the key necroptosis mediator RIPK3 in recurrent tumor cells contributed to clonogenic cell growth, causing p53 destabilization and promoting the activities of the YAP/TAZ pathways [348]. Yee et al. also found that ferroptosis induced by neutrophils played a significant role in promoting the aggressiveness of glioblastoma [349]. Therefore, accurate identification of the role of RCD in different types of cancer allows for better utilization of RCD modulators. Greater knowledge of the role played by the TME in controlling tumor cell death may also facilitate the development of cancer eradication therapy. In conclusion, our review postulates that strategies for the pharmacological modulation of novel tumor cell death pathways may be very helpful in cancer treatments, and we encourage future studies using animal models to identify additional outcomes. Additionally, we hope that more clinical trials investigating the use of novel cell death modulations in cancer patients will be conducted.
Availability of data and materials
The data used to support this study are included within the article.
Abbreviations
- RCD:
-
Regulated cell death
- TME:
-
Tumor microenvironment
- DAMPs:
-
Damage-associated molecular patterns
- PAMPs:
-
Pathogen-associated molecular patterns
- MLKL:
-
Mixed-lineage kinase domain-like protein
- RIPK1:
-
Receptor-interacting protein kinase 1
- RHIM:
-
RIP homology interaction motifs
- ZBP1:
-
Z-dsDNA/dsRNA-binding protein 1
- GSDM:
-
Gasdermin
- PFD:
-
N-terminal pore-forming domain
- Gzm B:
-
Granzyme B
- CAR-T cells:
-
Chimeric antigen receptor T cells
- NK:
-
Natural killer
- PUFAs:
-
Polyunsaturated fatty acids
- GPX4:
-
Glutathione peroxidase 4
- GSH:
-
Glutathione
- System Xc− :
-
Cystine–glutamate antiporter
- ACSL4:
-
Acyl-CoA synthetase long-chain family member 4
- ROS:
-
Reactive oxygen species
- RCC:
-
Renal cell carcinoma
- Cu:
-
Copper
- FDX1:
-
Ferredoxin 1
- TCA cycle:
-
Tricarboxylic acid cycle
- MΦs:
-
Macrophages
- GOx@[Cu(tz)]:
-
Glucose oxidase (GOx)-engineered nonporous Cu(I) 1,2,4-triazolate
- EMT:
-
Epithelial-to-mesenchymal transition
- CRC:
-
Colorectal cancer
- NSCLC:
-
Non-small cell lung cancer
- CAFs:
-
Cancer-associated fibroblasts
- HCC:
-
Hepatocellular carcinoma
- MSCs:
-
Mesenchymal stroma/stem cells
- ICIs:
-
Immune checkpoint inhibitors
- DCs:
-
Dendritic cells
- TAMs:
-
Tumor-associated MΦs
- MDSCs:
-
Myeloid-derived suppressor cells
- BRAFi + MEKi:
-
Combination of BRAF inhibitors and MEK inhibitors
- ASAH2:
-
N-acyl-sphingosine amidohydrolase
- CQ:
-
Chloroquine
- NSA:
-
Necrosulfonamide
- TiO2 NRs:
-
2,2,6,6, Tetramethylpiperidine-N-oxyl capped TiO2 nanorods
- NPs:
-
Nanoparticles
- RT:
-
Radiation therapy
- HUCMSCs:
-
Human umbilical cord mesenchymal stem cells
- DOX:
-
Doxorubicin
- CIAPs:
-
Cellular inhibitor of apoptosis proteins
References
Wang H, Zhou X, Li C, Yan S, Feng C, He J, et al. The emerging role of pyroptosis in pediatric cancers: from mechanism to therapy. J Hematol Oncol. 2022;15(1):140.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541.
Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7(1):286.
Koren E, Fuchs Y. Modes of regulated cell death in cancer. Cancer Discov. 2021;11(2):245–65.
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147(4):742–58.
Conradt B. Genetic control of programmed cell death during animal development. Annu Rev Genet. 2009;43:493–523.
Galluzzi L, Bravo-San Pedro JM, Kepp O, Kroemer G. Regulated cell death and adaptive stress responses. Cell Mol Life Sci. 2016;73(11–12):2405–10.
Cerella C, Teiten MH, Radogna F, Dicato M, Diederich M. From nature to bedside: pro-survival and cell death mechanisms as therapeutic targets in cancer treatment. Biotechnol Adv. 2014;32(6):1111–22.
Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, et al. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18(1):100.
Nie Q, Hu Y, Yu X, Li X, Fang X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022;22(1):12.
Du J, Wan Z, Wang C, Lu F, Wei M, Wang D, et al. Designer exosomes for targeted and efficient ferroptosis induction in cancer via chemo-photodynamic therapy. Theranostics. 2021;11(17):8185–96.
Wang YY, Liu XL, Zhao R. Induction of pyroptosis and its implications in cancer management. Front Oncol. 2019;9:971.
Zhang Z, Zeng X, Wu Y, Liu Y, Zhang X, Song Z. Cuproptosis-related risk score predicts prognosis and characterizes the tumor microenvironment in hepatocellular carcinoma. Front Immunol. 2022;13: 925618.
Shan J, Geng R, Zhang Y, Wei J, Liu J, Bai J. Identification of cuproptosis-related subtypes, establishment of a prognostic model and tumor immune landscape in endometrial carcinoma. Comput Biol Med. 2022;149: 105988.
Wang X, Wu S, Liu F, Ke D, Wang X, Pan D, et al. An immunogenic cell death-related classification predicts prognosis and response to immunotherapy in head and neck squamous cell carcinoma. Front Immunol. 2021;12: 781466.
Chen X, Zeh HJ, Kang R, Kroemer G, Tang D. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat Rev Gastroenterol Hepatol. 2021;18(11):804–23.
Wang H, Liu M, Zeng X, Zheng Y, Wang Y, Zhou Y. Cell death affecting the progression of gastric cancer. Cell Death Discov. 2022;8(1):377.
Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front Cell Infect Microbiol. 2019;9:406.
Medina CB, Mehrotra P, Arandjelovic S, Perry JSA, Guo Y, Morioka S, et al. Metabolites released from apoptotic cells act as tissue messengers. Nature. 2020;580(7801):130–5.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417.
Zhao Z, Liu H, Zhou X, Fang D, Ou X, Ye J, et al. Necroptosis-related lncRNAs: predicting prognosis and the distinction between the cold and hot tumors in gastric cancer. J Oncol. 2021;2021:6718443.
Tan Y, Chen Q, Li X, Zeng Z, Xiong W, Li G, et al. Pyroptosis: a new paradigm of cell death for fighting against cancer. J Exp Clin Cancer Res. 2021;40(1):153.
Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130–43.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–61.
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322–7.
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–79.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–14.
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–21.
Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2019;26(4):564.
Negroni A, Colantoni E, Cucchiara S, Stronati L. Necroptosis in intestinal inflammation and cancer: new concepts and therapeutic perspectives. Biomolecules. 2020;10(10):1431.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–27.
Miller DR, Cramer SD, Thorburn A. The interplay of autophagy and non-apoptotic cell death pathways. Int Rev Cell Mol Biol. 2020;352:159–87.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9.
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–21.
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341(6151):1246–9.
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–92.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103.
Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362(6418):1064–9.
Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019;38(10): e101638.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–71.
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689.
Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal structures of the full-length murine and human gasdermin d reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity. 2019;51(1):43-9e4.
Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature. 2018;557(7703):62–7.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–8.
Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 2017;38(4):261–71.
Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: a new frontier in cancer. Biomed Pharmacother. 2020;121: 109595.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22(10):1264–75.
Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277(1):61–75.
Ruan J, Wang S, Wang J. Mechanism and regulation of pyroptosis-mediated in cancer cell death. Chem Biol Interact. 2020;323: 109052.
Chen J, Ge L, Shi X, Liu J, Ruan H, Heng D, et al. Lobaplatin induces pyroptosis in cervical cancer cells via the caspase-3/GSDME pathway. Anticancer Agents Med Chem. 2022;22(11):2091–7.
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579(7799):415–20.
Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368:6494.
Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, et al. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J Mol Cell Biol. 2019;11(6):496–508.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85.
Hassannia B, Vandenabeele P, Vanden BT. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–49.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–31.
Lee JY, Kim WK, Bae KH, Lee SC, Lee EW. Lipid metabolism and ferroptosis. Biology (Basel). 2021;10(3):184.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–8.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–92.
Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21(1):47.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8.
Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–8.
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–9.
Huang T, Sun Y, Li Y, Wang T, Fu Y, Li C, et al. Growth inhibition of a novel iron chelator, DpdtC, against hepatoma carcinoma cell lines partly attributed to ferritinophagy-mediated lysosomal ROS generation. Oxid Med Cell Longev. 2018;2018:4928703.
Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356(2):971–7.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–7.
Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617.
Ge EJ, Bush AI, Casini A, Cobine PA, Cross JR, DeNicola GM, et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022;22(2):102–13.
Tsvetkov P, Detappe A, Cai K, Keys HR, Brune Z, Ying W, et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol. 2019;15(7):681–9.
Nagai M, Vo NH, Shin Ogawa L, Chimmanamada D, Inoue T, Chu J, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52(10):2142–50.
Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15(12):3527–44.
Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of gasdermin-D. J Immunol. 2017;198(5):2156–64.
Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev. 2020;297(1):26–38.
Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2):aag2045.
Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021;37(3): 109858.
Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Publisher Correction: Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580(7804):E10.
Miao Y, Liu J, Liu X, Yuan Q, Li H, Zhang Y, et al. Machine learning identification of cuproptosis and necroptosis-associated molecular subtypes to aid in prognosis assessment and immunotherapy response prediction in low-grade glioma. Front Genet. 2022;13: 951239.
Xu Y, Liu SY, Zeng L, Ma H, Zhang Y, Yang H, et al. An enzyme-engineered nonporous copper(I) coordination polymer nanoplatform for cuproptosis-based synergistic cancer therapy. Adv Mater. 2022;34(43): e2204733.
Fu LH, Wan Y, Qi C, He J, Li C, Yang C, et al. Nanocatalytic theranostics with glutathione depletion and enhanced reactive oxygen species generation for efficient cancer therapy. Adv Mater. 2021;33(7): e2006892.
Loftus LV, Amend SR, Pienta KJ. Interplay between cell death and cell proliferation reveals new strategies for cancer therapy. Int J Mol Sci. 2022;23(9):4723.
Chao DL, Sanchez CA, Galipeau PC, Blount PL, Paulson TG, Cowan DS, et al. Cell proliferation, cell cycle abnormalities, and cancer outcome in patients with Barrett’s esophagus: a long-term prospective study. Clin Cancer Res. 2008;14(21):6988–95.
Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548(7667):297–303.
Hoshino A, Lyden D. Metastasis: lymphatic detours for cancer. Nature. 2017;546(7660):609–10.
Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther. 2020;5(1):28.
Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16(4):201–18.
Jin X, Demere Z, Nair K, Ali A, Ferraro GB, Natoli T, et al. A metastasis map of human cancer cell lines. Nature. 2020;588(7837):331–6.
Ye X, Brabletz T, Kang Y, Longmore GD, Nieto MA, Stanger BZ, et al. Upholding a role for EMT in breast cancer metastasis. Nature. 2017;547(7661):E1–3.
Liu W, Chakraborty B, Safi R, Kazmin D, Chang CY, McDonnell DP. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat Commun. 2021;12(1):5103.
Suhail Y, Cain MP, Vanaja K, Kurywchak PA, Levchenko A, Kalluri R, et al. Systems biology of cancer metastasis. Cell Syst. 2019;9(2):109–27.
Spill F, Reynolds DS, Kamm RD, Zaman MH. Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol. 2016;40:41–8.
Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25(6):707–25.
Stoll G, Ma Y, Yang H, Kepp O, Zitvogel L, Kroemer G. Pro-necrotic molecules impact local immunosurveillance in human breast cancer. Oncoimmunology. 2017;6(4): e1299302.
Feng X, Song Q, Yu A, Tang H, Peng Z, Wang X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma. 2015;62(4):592–601.
Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015;6: e1636.
McCormick KD, Ghosh A, Trivedi S, Wang L, Coyne CB, Ferris RL, et al. Innate immune signaling through differential RIPK1 expression promote tumor progression in head and neck squamous cell carcinoma. Carcinogenesis. 2016;37(5):522–9.
Hockendorf U, Yabal M, Herold T, Munkhbaatar E, Rott S, Jilg S, et al. RIPK3 restricts myeloid leukemogenesis by promoting cell death and differentiation of leukemia initiating cells. Cancer Cell. 2016;30(1):75–91.
Colbert LE, Fisher SB, Hardy CW, Hall WA, Saka B, Shelton JW, et al. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer. 2013;119(17):3148–55.
He L, Peng K, Liu Y, Xiong J, Zhu FF. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco Targets Ther. 2013;6:1539–43.
Shen F, Pan X, Li M, Chen Y, Jiang Y, He J. Pharmacological inhibition of necroptosis promotes human breast cancer cell proliferation and metastasis. Onco Targets Ther. 2020;13:3165–76.
Han Q, Ma Y, Wang H, Dai Y, Chen C, Liu Y, et al. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3-mediated necroptosis. J Transl Med. 2018;16(1):201.
Sprooten J, De Wijngaert P, Vanmeerbeerk I, Martin S, Vangheluwe P, Schlenner S, et al. Necroptosis in immuno-oncology and cancer immunotherapy. Cells. 2020;9(8):1823.
Zhu F, Zhang W, Yang T, He SD. Complex roles of necroptosis in cancer. J Zhejiang Univ Sci B. 2019;20(5):399–413.
Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M. FADD and caspase-8 regulate gut homeostasis and inflammation by controlling MLKL- and GSDMD-mediated death of intestinal epithelial cells. Immunity. 2020;52(6):978-93e6.
Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536(7615):215–8.
Wang Q, Chen W, Xu X, Li B, He W, Padilla MT, et al. RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis. 2013;34(9):2119–28.
Liu X, Zhou M, Mei L, Ruan J, Hu Q, Peng J, et al. Key roles of necroptotic factors in promoting tumor growth. Oncotarget. 2016;7(16):22219–33.
Derangere V, Chevriaux A, Courtaut F, Bruchard M, Berger H, Chalmin F, et al. Liver X receptor beta activation induces pyroptosis of human and murine colon cancer cells. Cell Death Differ. 2014;21(12):1914–24.
Lin W, Chen Y, Wu B, Chen Y, Li Z. Identification of the pyroptosisrelated prognostic gene signature and the associated regulation axis in lung adenocarcinoma. Cell Death Discov. 2021;7(1):161.
Zhang Q, Tan Y, Zhang J, Shi Y, Qi J, Zou D, et al. Pyroptosis-related signature predicts prognosis and immunotherapy efficacy in muscle-invasive bladder cancer. Front Immunol. 2022;13: 782982.
Tang Z, Ji L, Han M, Xie J, Zhong F, Zhang X, et al. Pyroptosis is involved in the inhibitory effect of FL118 on growth and metastasis in colorectal cancer. Life Sci. 2020;257: 118065.
Ma Y, Chen Y, Lin C, Hu G. Biological functions and clinical significance of the newly identified long noncoding RNA RP185F18.6 in colorectal cancer. Oncol Rep. 2018;40(5):2648–58.
Wu LS, Liu Y, Wang XW, Xu B, Lin YL, Song Y, et al. LPS enhances the chemosensitivity of oxaliplatin in HT29 cells via GSDMD-mediated pyroptosis. Cancer Manag Res. 2020;12:10397–409.
Cardwell CR, Mc Menamin U, Hughes CM, Murray LJ. Statin use and survival from lung cancer: a population-based cohort study. Cancer Epidemiol Biomark Prev. 2015;24(5):833–41.
Wang F, Liu W, Ning J, Wang J, Lang Y, Jin X, et al. Simvastatin suppresses proliferation and migration in non-small cell lung cancer via pyroptosis. Int J Biol Sci. 2018;14(4):406–17.
Shaw PC, Chan WL, Yeung HW, Ng TB. Minireview: trichosanthin—a protein with multiple pharmacological properties. Life Sci. 1994;55(4):253–62.
Tan Y, Xiang J, Huang Z, Wang L, Huang Y. Trichosanthin inhibits cell growth and metastasis by promoting pyroptosis in non-small cell lung cancer. J Thorac Dis. 2022;14(4):1193–202.
Chu Q, Jiang Y, Zhang W, Xu C, Du W, Tuguzbaeva G, et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget. 2016;7(51):84658–65.
Sun K, Chen RX, Li JZ, Luo ZX. LINC00511/hsa-miR-573 axis-mediated high expression of Gasdermin C associates with dismal prognosis and tumor immune infiltration of breast cancer. Sci Rep. 2022;12(1):14788.
Gao J, Qiu X, Xi G, Liu H, Zhang F, Lv T, et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol Rep. 2018;40(4):1971–84.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
He H, Yi L, Zhang B, Yan B, Xiao M, Ren J, et al. USP24-GSDMB complex promotes bladder cancer proliferation via activation of the STAT3 pathway. Int J Biol Sci. 2021;17(10):2417–29.
Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Megias D, Mota A, Rojo-Sebastian A, et al. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS ONE. 2014;9(3): e90099.
Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–47.
Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21(4):648–57.
Lee JY, Nam M, Son HY, Hyun K, Jang SY, Kim JW, et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc Natl Acad Sci U S A. 2020;117(51):32433–42.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62.
Chen LL, Wang WJ. p53 regulates lipid metabolism in cancer. Int J Biol Macromol. 2021;192:45–54.
Greenshields AL, Shepherd TG, Hoskin DW. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol Carcinog. 2017;56(1):75–93.
Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50–64.
Cheung EC, Vousden KH. The role of ROS in tumour development and progression. Nat Rev Cancer. 2022;22(5):280–97.
Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51(5):575-86e4.
Zhang X, Xu Y, Ma L, Yu K, Niu Y, Xu X, et al. Essential roles of exosome and circRNA_101093 on ferroptosis desensitization in lung adenocarcinoma. Cancer Commun (Lond). 2022;42(4):287–313.
Huang G, Xiang Z, Wu H, He Q, Dou R, Lin Z, et al. The lncRNA BDNF-AS/WDR5/FBXW7 axis mediates ferroptosis in gastric cancer peritoneal metastasis by regulating VDAC3 ubiquitination. Int J Biol Sci. 2022;18(4):1415–33.
Guan D, Zhou W, Wei H, Wang T, Zheng K, Yang C, et al. Ferritinophagy-mediated ferroptosis and activation of keap1/Nrf2/HO-1 pathway were conducive to EMT inhibition of gastric cancer cells in action of 2,2’-di-pyridineketone hydrazone dithiocarbamate butyric acid ester. Oxid Med Cell Longev. 2022;2022:3920664.
Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 2020;585(7823):113–8.
Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32(5):417–8.
Ghosh P, Vidal C, Dey S, Zhang L. Mitochondria targeting as an effective strategy for cancer therapy. Int J Mol Sci. 2020;21(9):3363.
Yu D, Liu C, Guo L. Mitochondrial metabolism and cancer metastasis. Ann Transl Med. 2020;8(14):904.
Pavithra V, Sathisha TG, Kasturi K, Mallika DS, Amos SJ, Ragunatha S. Serum levels of metal ions in female patients with breast cancer. J Clin Diagn Res. 2015;9(1):BC25-c7.
Lener MR, Scott RJ, Wiechowska-Kozlowska A, Serrano-Fernandez P, Baszuk P, Jaworska-Bieniek K, et al. Serum concentrations of selenium and copper in patients diagnosed with pancreatic cancer. Cancer Res Treat. 2016;48(3):1056–64.
Baltaci AK, Dundar TK, Aksoy F, Mogulkoc R. Changes in the serum levels of trace elements before and after the operation in thyroid cancer patients. Biol Trace Elem Res. 2017;175(1):57–64.
Zuo XL, Chen JM, Zhou X, Li XZ, Mei GY. Levels of selenium, zinc, copper, and antioxidant enzyme activity in patients with leukemia. Biol Trace Elem Res. 2006;114(1–3):41–53.
Aubert L, Nandagopal N, Steinhart Z, Lavoie G, Nourreddine S, Berman J, et al. Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer. Nat Commun. 2020;11(1):3701.
Jin Y, Zhang C, Xu H, Xue S, Wang Y, Hou Y, et al. Combined effects of serum trace metals and polymorphisms of CYP1A1 or GSTM1 on non-small cell lung cancer: a hospital based case-control study in China. Cancer Epidemiol. 2011;35(2):182–7.
Saleh SAK, Adly HM, Abdelkhaliq AA, Nassir AM. Serum levels of selenium, zinc, copper, manganese, and iron in prostate cancer patients. Curr Urol. 2020;14(1):44–9.
Baharvand M, Manifar S, Akkafan R, Mortazavi H, Sabour S. Serum levels of ferritin, copper, and zinc in patients with oral cancer. Biomed J. 2014;37(5):331–6.
Lelievre P, Sancey L, Coll JL, Deniaud A, Busser B. The multifaceted roles of copper in cancer: a trace metal element with dysregulated metabolism, but also a target or a bullet for therapy. Cancers (Basel). 2020;12(12):3594.
Wu Z, Zhang W, Kang YJ. Copper affects the binding of HIF-1alpha to the critical motifs of its target genes. Metallomics. 2019;11(2):429–38.
Feng W, Ye F, Xue W, Zhou Z, Kang YJ. Copper regulation of hypoxia-inducible factor-1 activity. Mol Pharmacol. 2009;75(1):174–82.
Zimna A, Kurpisz M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: applications and therapies. Biomed Res Int. 2015;2015: 549412.
MacDonald G, Nalvarte I, Smirnova T, Vecchi M, Aceto N, Dolemeyer A, et al. Memo is a copper-dependent redox protein with an essential role in migration and metastasis. Sci Signal. 2014;7(329):ra56.
Zhang X, Walke GR, Horvath I, Kumar R, Blockhuys S, Holgersson S, et al. Memo1 binds reduced copper ions, interacts with copper chaperone Atox1, and protects against copper-mediated redox activity in vitro. Proc Natl Acad Sci U S A. 2022;119(37): e2206905119.
Li J, Chen S, Liao Y, Wang H, Zhou D, Zhang B. Arecoline is associated with inhibition of cuproptosis and proliferation of cancer-associated fibroblasts in oral squamous cell carcinoma: a potential mechanism for tumor metastasis. Front Oncol. 2022;12: 925743.
Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR, et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer. 2019;18(1):91.
Asif PJ, Longobardi C, Hahne M, Medema JP. The role of cancer-associated fibroblasts in cancer invasion and metastasis. Cancers (Basel). 2021;13(18):4720.
Li K, Tan L, Li Y, Lyu Y, Zheng X, Jiang H, et al. Cuproptosis identifies respiratory subtype of renal cancer that confers favorable prognosis. Apoptosis. 2022;27(11–12):1004–14.
Wang T, Liu Y, Li Q, Luo Y, Liu D, Li B. Cuproptosis-related gene FDX1 expression correlates with the prognosis and tumor immune microenvironment in clear cell renal cell carcinoma. Front Immunol. 2022;13: 999823.
Xiao C, Yang L, Jin L, Lin W, Zhang F, Huang S, et al. Prognostic and immunological role of cuproptosis-related protein FDX1 in pan-cancer. Front Genet. 2022;13: 962028.
Zhang C, Zeng Y, Guo X, Shen H, Zhang J, Wang K, et al. Pan-cancer analyses confirmed the cuproptosis-related gene FDX1 as an immunotherapy predictor and prognostic biomarker. Front Genet. 2022;13: 923737.
Runa F, Hamalian S, Meade K, Shisgal P, Gray PC, Kelber JA. Tumor microenvironment heterogeneity: challenges and opportunities. Curr Mol Biol Rep. 2017;3(4):218–29.
De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17(8):457–74.
Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20(4):840.
Song W, Ren J, Xiang R, Kong C, Fu T. Identification of pyroptosis-related subtypes, the development of a prognosis model, and characterization of tumor microenvironment infiltration in colorectal cancer. Oncoimmunology. 2021;10(1):1987636.
Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221: 107753.
Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. 2020;13(1):110.
Niu X, Chen L, Li Y, Hu Z, He F. Ferroptosis, necroptosis, and pyroptosis in the tumor microenvironment: perspectives for immunotherapy of SCLC. Semin Cancer Biol. 2022;86(Pt 3):273–85.
Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. 2021;11(11):5365–86.
Lin SY, Hsieh SY, Fan YT, Wei WC, Hsiao PW, Tsai DH, et al. Necroptosis promotes autophagy-dependent upregulation of DAMP and results in immunosurveillance. Autophagy. 2018;14(5):778–95.
Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol. 2019;4(36):eaaw2004.
Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 2016;15(2):274–87.
Park HH, Kim HR, Park SY, Hwang SM, Hong SM, Park S, et al. RIPK3 activation induces TRIM28 derepression in cancer cells and enhances the anti-tumor microenvironment. Mol Cancer. 2021;20(1):107.
Workenhe ST, Nguyen A, Bakhshinyan D, Wei J, Hare DN, MacNeill KL, et al. De novo necroptosis creates an inflammatory environment mediating tumor susceptibility to immune checkpoint inhibitors. Commun Biol. 2020;3(1):645.
Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. 2020;11: 583084.
He R, Zhang M, He L, Huang J, Man C, Wang X, et al. Integrated analysis of necroptosis-related genes for prognosis, immune microenvironment infiltration, and drug sensitivity in colon cancer. Front Med (Lausanne). 2022;9: 845271.
Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532(7598):245–9.
Liu ZY, Zheng M, Li YM, Fan XY, Wang JC, Li ZC, et al. RIP3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and CXCL1-induced immune suppression. Theranostics. 2019;9(12):3659–73.
Hsu SK, Li CY, Lin IL, Syue WJ, Chen YF, Cheng KC, et al. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics. 2021;11(18):8813–35.
Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020;10(2):254–69.
Hartsough E, Shao Y, Aplin AE. Resistance to RAF inhibitors revisited. J Invest Dermatol. 2014;134(2):319–25.
Wang X, Lin W, Liu T, Xu Z, Wang Z, Cao Z, et al. Cross-talk of pyroptosis and tumor immune landscape in lung adenocarcinoma. Transl Lung Cancer Res. 2021;10(12):4423–44.
Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, et al. The role of pyroptosis in cancer: pro-cancer or pro-"host"? Cell Death Dis. 2019;10(9):650.
Wang Y, Peng J, Xie X, Zhang Z, Li M, Yang M. Gasdermin E-mediated programmed cell death: an unpaved path to tumor suppression. J Cancer. 2021;12(17):5241–8.
Cai J, Yi M, Tan Y, Li X, Li G, Zeng Z, et al. Natural product triptolide induces GSDME-mediated pyroptosis in head and neck cancer through suppressing mitochondrial hexokinase-IotaIota. J Exp Clin Cancer Res. 2021;40(1):190.
Fan JX, Deng RH, Wang H, Liu XH, Wang XN, Qin R, et al. Epigenetics-based tumor cells pyroptosis for enhancing the immunological effect of chemotherapeutic nanocarriers. Nano Lett. 2019;19(11):8049–58.
Lu Y, He W, Huang X, He Y, Gou X, Liu X, et al. Strategies to package recombinant adeno-associated virus expressing the N-terminal gasdermin domain for tumor treatment. Nat Commun. 2021;12(1):7155.
Tan G, Huang C, Chen J, Zhi F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J Hematol Oncol. 2020;13(1):149.
Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. 2018;17(4):428–38.
Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19(7):405–14.
Efimova I, Catanzaro E, Van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer. 2020;8(2): e001369.
Hsieh CH, Hsieh HC, Shih FS, Wang PW, Yang LX, Shieh DB, et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics. 2021;11(14):7072–91.
Xu H, Ye D, Ren M, Zhang H, Bi F. Ferroptosis in the tumor microenvironment: perspectives for immunotherapy. Trends Mol Med. 2021;27(9):856–67.
Luis G, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, et al. Tumor resistance to ferroptosis driven by stearoyl-CoA desaturase-1 (SCD1) in cancer cells and fatty acid biding protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 2021;43: 102006.
Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50(2):445–60.
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30(2):146–62.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280–96.
Lin R, Fogarty CE, Ma B, Li H, Ni G, Liu X, et al. Identification of ferroptosis genes in immune infiltration and prognosis in thyroid papillary carcinoma using network analysis. BMC Genom. 2021;22(1):576.
You Y, Fan Q, Huang J, Wu Y, Lin H, Zhang Q. Ferroptosis-related gene signature promotes ovarian cancer by influencing immune infiltration and invasion. J Oncol. 2021;2021:9915312.
Xu Z, Peng B, Liang Q, Chen X, Cai Y, Zeng S, et al. Construction of a ferroptosis-related nine-lncRNA signature for predicting prognosis and immune response in hepatocellular carcinoma. Front Immunol. 2021;12: 719175.
Johnson AM, Kleczko EK, Nemenoff RA. Eicosanoids in cancer: new roles in immunoregulation. Front Pharmacol. 2020;11: 595498.
Demuynck R, Efimova I, Naessens F, Krysko DV. Immunogenic ferroptosis and where to find it? J Immunother Cancer. 2021;9(12): e003430.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–9.
Jiang M, Qiao M, Zhao C, Deng J, Li X, Zhou C. Targeting ferroptosis for cancer therapy: exploring novel strategies from its mechanisms and role in cancers. Transl Lung Cancer Res. 2020;9(4):1569–84.
Lv H, Liu X, Zeng X, Liu Y, Zhang C, Zhang Q, et al. Comprehensive analysis of cuproptosis-related genes in immune infiltration and prognosis in melanoma. Front Pharmacol. 2022;13: 930041.
Jiang R, Huan Y, Li Y, Gao X, Sun Q, Zhang F, et al. Transcriptional and genetic alterations of cuproptosis-related genes correlated to malignancy and immune-infiltrate of esophageal carcinoma. Cell Death Discov. 2022;8(1):370.
Wang F, Lin H, Su Q, Li C. Cuproptosis-related lncRNA predict prognosis and immune response of lung adenocarcinoma. World J Surg Oncol. 2022;20(1):275.
Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479–83.
Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. 2020;8(5):710–21.
Lan H, Liu Y, Liu J, Wang X, Guan Z, Du J, et al. Tumor-associated macrophages promote oxaliplatin resistance via METTL3-mediated m(6)A of TRAF5 and necroptosis in colorectal cancer. Mol Pharm. 2021;18(3):1026–37.
Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol. 2017;13(1):46–53.
Hage C, Hoves S, Strauss L, Bissinger S, Prinz Y, Poschinger T, et al. Sorafenib induces pyroptosis in macrophages and triggers natural killer cell-mediated cytotoxicity against hepatocellular carcinoma. Hepatology. 2019;70(4):1280–97.
Hou J, Hsu JM, Hung MC. Molecular mechanisms and functions of pyroptosis in inflammation and antitumor immunity. Mol Cell. 2021;81(22):4579–90.
Saetang J, Chonpathompikunlert P, Sretrirutchai S, Roongsawang N, Kayasut K, Voravuthikunchai SP, et al. Anti-cancer effect of engineered recombinant interleukin 18. Adv Clin Exp Med. 2020;29(10):1135–43.
Yang Y, Wang Y, Guo L, Gao W, Tang TL, Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis. 2022;13(4):355.
Zhang F, Li F, Lu GH, Nie W, Zhang L, Lv Y, et al. Engineering magnetosomes for ferroptosis/immunomodulation synergism in cancer. ACS Nano. 2019;13(5):5662–73.
Zhu H, Klement JD, Lu C, Redd PS, Yang D, Smith AD, et al. Asah2 represses the p53-Hmox1 axis to protect myeloid-derived suppressor cells from ferroptosis. J Immunol. 2021;206(6):1395–404.
Zhang W, Wang F, Hu C, Zhou Y, Gao H, Hu J. The progress and perspective of nanoparticle-enabled tumor metastasis treatment. Acta Pharm Sin B. 2020;10(11):2037–53.
Alu A, Han X, Ma X, Wu M, Wei Y, Wei X. The role of lysosome in regulated necrosis. Acta Pharm Sin B. 2020;10(10):1880–903.
Wang Z, Zhao Y, Zhao Y, Zhang Y, Yao X, Hang R. Exosomes secreted by macrophages upon copper ion stimulation can promote angiogenesis. Mater Sci Eng C Mater Biol Appl. 2021;123: 111981.
Takemoto R, Kamiya T, Atobe T, Hara H, Adachi T. Regulation of lysyl oxidase expression in THP-1 cell-derived M2-like macrophages. J Cell Biochem. 2021;122(8):777–86.
Sen T. Identifying and targeting the Achilles heel of a recalcitrant cancer. Sci Transl Med. 2021;13(605):6946.
Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther. 2022;7(1):196.
Li L, Li L, Sun Q. High expression of cuproptosis-related SLC31A1 gene in relation to unfavorable outcome and deregulated immune cell infiltration in breast cancer: an analysis based on public databases. BMC Bioinform. 2022;23(1):350.
Chen C, Xiao W, Huang L, Yu G, Ni J, Yang L, et al. Shikonin induces apoptosis and necroptosis in pancreatic cancer via regulating the expression of RIP1/RIP3 and synergizes the activity of gemcitabine. Am J Transl Res. 2017;9(12):5507–17.
Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med. 2013;5(1):105–21.
Chen GY, Meng CL, Lin KC, Tuan HY, Yang HJ, Chen CL, et al. Graphene oxide as a chemosensitizer: diverted autophagic flux, enhanced nuclear import, elevated necrosis and improved antitumor effects. Biomaterials. 2015;40:12–22.
Sonkusre P, Cameotra SS. Biogenic selenium nanoparticles induce ROS-mediated necroptosis in PC-3 cancer cells through TNF activation. J Nanobiotechnol. 2017;15(1):43.
Zhou J, Li G, Han G, Feng S, Liu Y, Chen J, et al. Emodin induced necroptosis in the glioma cell line U251 via the TNF-alpha/RIP1/RIP3 pathway. Invest New Drugs. 2020;38(1):50–9.
Lu Z, Wu C, Zhu M, Song W, Wang H, Wang J, et al. Ophiopogonin D’ induces RIPK1dependent necroptosis in androgendependent LNCaP prostate cancer cells. Int J Oncol. 2020;56(2):439–47.
Li Y, Tian X, Liu X, Gong P. Bufalin inhibits human breast cancer tumorigenesis by inducing cell death through the ROS-mediated RIP1/RIP3/PARP-1 pathways. Carcinogenesis. 2018;39(5):700–7.
Liu X, Zhang Y, Gao H, Hou Y, Lu JJ, Feng Y, et al. Induction of an MLKL mediated non-canonical necroptosis through reactive oxygen species by tanshinol A in lung cancer cells. Biochem Pharmacol. 2020;171: 113684.
Hou X, Yang C, Zhang L, Hu T, Sun D, Cao H, et al. Killing colon cancer cells through PCD pathways by a novel hyaluronic acid-modified shell-core nanoparticle loaded with RIP3 in combination with chloroquine. Biomaterials. 2017;124:195–210.
Zhang T, Yin C, Fedorov A, Qiao L, Bao H, Beknazarov N, et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature. 2022;606(7914):594–602.
Koch A, Jeiler B, Roedig J, van Wijk SJL, Dolgikh N, Fulda S. Smac mimetics and TRAIL cooperate to induce MLKL-dependent necroptosis in Burkitt’s lymphoma cell lines. Neoplasia. 2021;23(5):539–50.
Feng W, Shi W, Liu S, Liu H, Liu Y, Ge P, et al. Fe(III)-shikonin supramolecular nanomedicine for combined therapy of tumor via ferroptosis and necroptosis. Adv Healthc Mater. 2022;11(2): e2101926.
Hou J, Ju J, Zhang Z, Zhao C, Li Z, Zheng J, et al. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 2019;10(7):493.
Liu ZY, Wu B, Guo YS, Zhou YH, Fu ZG, Xu BQ, et al. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am J Cancer Res. 2015;5(10):3174–85.
Zheng Z, Bian Y, Zhang Y, Ren G, Li G. Metformin activates AMPK/SIRT1/NF-kappaB pathway and induces mitochondrial dysfunction to drive caspase3/GSDME-mediated cancer cell pyroptosis. Cell Cycle. 2020;19(10):1089–104.
Wang L, Li K, Lin X, Yao Z, Wang S, Xiong X, et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 2019;450:22–31.
An H, Heo JS, Kim P, Lian Z, Lee S, Park J, et al. Tetraarsenic hexoxide enhances generation of mitochondrial ROS to promote pyroptosis by inducing the activation of caspase-3/GSDME in triple-negative breast cancer cells. Cell Death Dis. 2021;12(2):159.
Pizato N, Luzete BC, Kiffer L, Correa LH, de Oliveira SI, Assumpcao JAF, et al. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci Rep. 2018;8(1):1952.
Teng JF, Qin DL, Mei QB, Qiu WQ, Pan R, Xiong R, et al. Polyphyllin VI, a saponin from Trillium tschonoskii Maxim. induces apoptotic and autophagic cell death via the ROS triggered mTOR signaling pathway in non-small cell lung cancer. Pharmacol Res. 2019;147:104396.
Teng JF, Mei QB, Zhou XG, Tang Y, Xiong R, Qiu WQ, et al. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-kappaB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers (Basel). 2020;12(1):193.
Kong Y, Feng Z, Chen A, Qi Q, Han M, Wang S, et al. The natural flavonoid galangin elicits apoptosis, pyroptosis, and autophagy in glioblastoma. Front Oncol. 2019;9:942.
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10(3):193.
Zhang X, Zhang P, An L, Sun N, Peng L, Tang W, et al. Miltirone induces cell death in hepatocellular carcinoma cell through GSDME-dependent pyroptosis. Acta Pharm Sin B. 2020;10(8):1397–413.
Zhao P, Wang M, Chen M, Chen Z, Peng X, Zhou F, et al. Programming cell pyroptosis with biomimetic nanoparticles for solid tumor immunotherapy. Biomaterials. 2020;254: 120142.
Elion DL, Jacobson ME, Hicks DJ, Rahman B, Sanchez V, Gonzales-Ericsson PI, et al. Therapeutically active RIG-I agonist induces immunogenic tumor cell killing in breast cancers. Cancer Res. 2018;78(21):6183–95.
Serna N, Alamo P, Ramesh P, Vinokurova D, Sanchez-Garcia L, Unzueta U, et al. Nanostructured toxins for the selective destruction of drug-resistant human CXCR4(+) colorectal cancer stem cells. J Control Release. 2020;320:96–104.
Ploetz E, Zimpel A, Cauda V, Bauer D, Lamb DC, Haisch C, et al. Metal-organic framework nanoparticles induce pyroptosis in cells controlled by the extracellular pH. Adv Mater. 2020;32(19): e1907267.
Hu J, Dong Y, Ding L, Dong Y, Wu Z, Wang W, et al. Local delivery of arsenic trioxide nanoparticles for hepatocellular carcinoma treatment. Signal Transduct Target Ther. 2019;4:28.
Li J, Anraku Y, Kataoka K. Self-boosting catalytic nanoreactors integrated with triggerable crosslinking membrane networks for initiation of immunogenic cell death by pyroptosis. Angew Chem Int Ed Engl. 2020;59(32):13526–30.
Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24(3–4):312–25.
Yue E, Tuguzbaeva G, Chen X, Qin Y, Li A, Sun X, et al. Anthocyanin is involved in the activation of pyroptosis in oral squamous cell carcinoma. Phytomedicine. 2019;56:286–94.
Tan YF, Wang M, Chen ZY, Wang L, Liu XH. Inhibition of BRD4 prevents proliferation and epithelial-mesenchymal transition in renal cell carcinoma via NLRP3 inflammasome-induced pyroptosis. Cell Death Dis. 2020;11(4):239.
Draganov D, Gopalakrishna-Pillai S, Chen YR, Zuckerman N, Moeller S, Wang C, et al. Modulation of P2X4/P2X7/pannexin-1 sensitivity to extracellular ATP via ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci Rep. 2015;5:16222.
Cheng Z, Li Z, Gu L, Li L, Gao Q, Zhang X, et al. Ophiopogonin B alleviates cisplatin resistance of lung cancer cells by inducing caspase-1/GSDMD dependent pyroptosis. J Cancer. 2022;13(2):715–27.
Guo J, Zheng J, Mu M, Chen Z, Xu Z, Zhao C, et al. GW4064 enhances the chemosensitivity of colorectal cancer to oxaliplatin by inducing pyroptosis. Biochem Biophys Res Commun. 2021;548:60–6.
Yu P, Wang HY, Tian M, Li AX, Chen XS, Wang XL, et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol Sin. 2019;40(9):1237–44.
Nam J, Son S, Ochyl LJ, Kuai R, Schwendeman A, Moon JJ. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat Commun. 2018;9(1):1074.
Molina-Crespo A, Cadete A, Sarrio D, Gamez-Chiachio M, Martinez L, Chao K, et al. Intracellular delivery of an antibody targeting gasdermin-B reduces HER2 breast cancer aggressiveness. Clin Cancer Res. 2019;25(15):4846–58.
Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369(6511):1633–7.
Huang KJ, Wei YH, Chiu YC, Wu SR, Shieh DB. Assessment of zero-valent iron-based nanotherapeutics for ferroptosis induction and resensitization strategy in cancer cells. Biomater Sci. 2019;7(4):1311–22.
Gao F, Wang F, Nie X, Zhang Z, Chen G, Xia L, et al. Mitochondria-targeted delivery and light controlled release of iron prodrug and CO to enhance cancer therapy by ferroptosis. New J Chem. 2020;44(8):3478–86.
Zhang L, Liu W, Liu F, Wang Q, Song M, Yu Q, et al. IMCA induces ferroptosis mediated by SLC7A11 through the AMPK/mTOR pathway in colorectal cancer. Oxid Med Cell Longev. 2020;2020:1675613.
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26(5):623-33e9.
Taylor WR, Fedorka SR, Gad I, Shah R, Alqahtani HD, Koranne R, et al. Small-molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci Rep. 2019;9(1):5926.
Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16(5):497–506.
Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14(1):19.
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497–503.
Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14(5):507–15.
Wei G, Sun J, Hou Z, Luan W, Wang S, Cui S, et al. Novel antitumor compound optimized from natural saponin Albiziabioside A induced caspase-dependent apoptosis and ferroptosis as a p53 activator through the mitochondrial pathway. Eur J Med Chem. 2018;157:759–72.
Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–62.
Zhang G, Li N, Qi Y, Zhao Q, Zhan J, Yu D. Synergistic ferroptosis-gemcitabine chemotherapy of the gemcitabine loaded carbonaceous nanozymes to enhance the treatment and magnetic resonance imaging monitoring of pancreatic cancer. Acta Biomater. 2022;142:284–97.
Chen P, Wu Q, Feng J, Yan L, Sun Y, Liu S, et al. Erianin, a novel dibenzyl compound in dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Target Ther. 2020;5(1):51.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90.
Zhang D, Cui P, Dai Z, Yang B, Yao X, Liu Q, et al. Tumor microenvironment responsive FePt/MoS2 nanocomposites with chemotherapy and photothermal therapy for enhancing cancer immunotherapy. Nanoscale. 2019;11(42):19912–22.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3: e02523.
Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7: e2307.
Lv Q, Niu H, Yue L, Liu J, Yang L, Liu C, et al. Abnormal ferroptosis in myelodysplastic syndrome. Front Oncol. 2020;10:1656.
Yu M, Gai C, Li Z, Ding D, Zheng J, Zhang W, et al. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. 2019;110(10):3173–82.
Zhao L, Peng Y, He S, Li R, Wang Z, Huang J, et al. Apatinib induced ferroptosis by lipid peroxidation in gastric cancer. Gastric Cancer. 2021;24(3):642–54.
Tian X, Li S, Ge G. Apatinib promotes ferroptosis in colorectal cancer cells by targeting ELOVL6/ACSL4 signaling. Cancer Manag Res. 2021;13:1333–42.
Xia L, Gong M, Zou Y, Wang Z, Wu B, Zhang S, et al. Apatinib induces ferroptosis of glioma cells through modulation of the VEGFR2/Nrf2 pathway. Oxid Med Cell Longev. 2022;2022:9925919.
Ye L, Jin F, Kumar SK, Dai Y. The mechanisms and therapeutic targets of ferroptosis in cancer. Expert Opin Ther Targets. 2021;25(11):965–86.
Nagpal A, Redvers RP, Ling X, Ayton S, Fuentes M, Tavancheh E, et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Res. 2019;21(1):94.
Birsen R, Larrue C, Decroocq J, Johnson N, Guiraud N, Gotanegre M, et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica. 2022;107(2):403–16.
Zhou X, Zou L, Chen W, Yang T, Luo J, Wu K, et al. Flubendazole, FDA-approved anthelmintic, elicits valid antitumor effects by targeting P53 and promoting ferroptosis in castration-resistant prostate cancer. Pharmacol Res. 2021;164: 105305.
Lee J, You JH, Shin D, Roh JL. Inhibition of glutaredoxin 5 predisposes cisplatin-resistant head and neck cancer cells to ferroptosis. Theranostics. 2020;10(17):7775–86.
Chen JJ, Galluzzi L. Fighting resilient cancers with iron. Trends Cell Biol. 2018;28(2):77–8.
Mao W, Cai Y, Chen D, Jiang G, Xu Y, Chen R, et al. Statin shapes inflamed tumor microenvironment and enhances immune checkpoint blockade in non-small cell lung cancer. JCI Insight. 2022;7(18): e161940.
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–69.
Yi R, Wang H, Deng C, Wang X, Yao L, Niu W, et al. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci Rep. 2020;40(6):BSR20193314.
Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol. 2018;9:1371.
Li J, Lama R, Galster SL, Inigo JR, Wu J, Chandra D, et al. Small-molecule MMRi62 induces ferroptosis and inhibits metastasis in pancreatic cancer via degradation of ferritin heavy chain and mutant p53. Mol Cancer Ther. 2022;21(4):535–45.
Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517–32.
Lippmann J, Petri K, Fulda S, Liese J. Redox modulation and induction of ferroptosis as a new therapeutic strategy in hepatocellular carcinoma. Transl Oncol. 2020;13(8): 100785.
Shishido Y, Amisaki M, Matsumi Y, Yakura H, Nakayama Y, Miyauchi W, et al. Antitumor effect of 5-aminolevulinic acid through ferroptosis in esophageal squamous cell carcinoma. Ann Surg Oncol. 2021;28(7):3996–4006.
Du J, Wang L, Huang X, Zhang N, Long Z, Yang Y, et al. Shuganning injection, a traditional Chinese patent medicine, induces ferroptosis and suppresses tumor growth in triple-negative breast cancer cells. Phytomedicine. 2021;85: 153551.
Zhao X, Quan J, Tan Y, Liu Y, Liao C, Li Z, et al. RIP3 mediates TCN-induced necroptosis through activating mitochondrial metabolism and ROS production in chemotherapy-resistant cancers. Am J Cancer Res. 2021;11(3):729–45.
Fakhar EAM, Aqrab Ul A, Atif M, Alimgeer KS, Suleman Rana M, Yaqub N, et al. Synergistic effect of TEMPO-coated TiO2 nanorods for PDT applications in MCF-7 cell line model. Saudi J Biol Sci. 2020;27(12):3199–207.
Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339.
Lu Y, Pan Q, Gao W, Pu Y, He B. Reversal of cisplatin chemotherapy resistance by glutathione-resistant copper-based nanomedicine via cuproptosis. J Mater Chem B. 2022;10(33):6296–306.
Liu T, Sun X, Cao Z. Shikonin-induced necroptosis in nasopharyngeal carcinoma cells via ROS overproduction and upregulation of RIPK1/RIPK3/MLKL expression. Onco Targets Ther. 2019;12:2605–14.
Sun J, Wei Q, Zhou Y, Wang J, Liu Q, Xu H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst Biol. 2017;11(Suppl 5):87.
Wang K, Zhang Z, Tsai HI, Liu Y, Gao J, Wang M, et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. 2021;28(4):1222–36.
Mohammadinejad R, Moosavi MA, Tavakol S, Vardar DO, Hosseini A, Rahmati M, et al. Necrotic, apoptotic and autophagic cell fates triggered by nanoparticles. Autophagy. 2019;15(1):4–33.
Boulos JC, Rahama M, Hegazy MF, Efferth T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019;459:248–67.
Newell M, Baker K, Postovit LM, Field CJ. A critical review on the effect of docosahexaenoic acid (DHA) on cancer cell cycle progression. Int J Mol Sci. 2017;18(8):1784.
Dekoj T, Lee S, Desai S, Trevino J, Babcock TA, Helton WS, et al. G2/M cell-cycle arrest and apoptosis by n-3 fatty acids in a pancreatic cancer model. J Surg Res. 2007;139(1):106–12.
Dumont A, de Rosny C, Kieu TL, Perrey S, Berger H, Fluckiger A, et al. Docosahexaenoic acid inhibits both NLRP3 inflammasome assembly and JNK-mediated mature IL-1beta secretion in 5-fluorouracil-treated MDSC: implication in cancer treatment. Cell Death Dis. 2019;10(7):485.
Jin X, Yan Y, Wang D, Ding D, Ma T, Ye Z, et al. DUB3 promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol Cell. 2018;71(4):592-605e4.
Chen L, Weng B, Li H, Wang H, Li Q, Wei X, et al. A thiopyran derivative with low murine toxicity with therapeutic potential on lung cancer acting through a NF-kappaB mediated apoptosis-to-pyroptosis switch. Apoptosis. 2019;24(1–2):74–82.
Chang WT, Bow YD, Fu PJ, Li CY, Wu CY, Chang YH, et al. A marine terpenoid, heteronemin, induces both the apoptosis and ferroptosis of hepatocellular carcinoma cells and involves the ROS and MAPK pathways. Oxid Med Cell Longev. 2021;2021:7689045.
Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated tumor cell-derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv. 2020;6(13):eaay9789.
Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9(12):1673–85.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15(2):469–84.
Yuan Y, Zhou C, Chen X, Tao C, Cheng H, Lu X. Suppression of tumor cell proliferation and migration by human umbilical cord mesenchymal stem cells: a possible role for apoptosis and Wnt signaling. Oncol Lett. 2018;15(6):8536–44.
Jiao Y, Zhao H, Chen G, Sang X, Yang L, Hou Z, et al. Pyroptosis of MCF7 cells induced by the secreted factors of hUCMSCs. Stem Cells Int. 2018;2018:5912194.
Ahmed SG, Abdelanabi A, Doha M, Brenner GJ. Schwannoma gene therapy by adeno-associated virus delivery of the pore-forming protein Gasdermin-D. Cancer Gene Ther. 2019;26(9–10):259–67.
Ren Y, Su Y, Sun L, He S, Meng L, Liao D, et al. Discovery of a highly potent, selective, and metabolically stable inhibitor of receptor-interacting protein 1 (RIP1) for the treatment of systemic inflammatory response syndrome. J Med Chem. 2017;60(3):972–86.
Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, et al. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem. 2017;60(4):1247–61.
Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21(7):736–45.
Zhang J, Wei K. Necrosulfonamide reverses pyroptosis-induced inhibition of proliferation and differentiation of osteoblasts through the NLRP3/caspase-1/GSDMD pathway. Exp Cell Res. 2021;405(2): 112648.
Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5(9): e132747.
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–80.
Ji Q, Fu S, Zuo H, Huang Y, Chu L, Zhu Y, et al. ACSL4 is essential for radiation-induced intestinal injury by initiating ferroptosis. Cell Death Discov. 2022;8(1):332.
Wang Q, Wang P, Zhang L, Tessema M, Bai L, Xu X, et al. Epigenetic regulation of RIP3 suppresses necroptosis and increases resistance to chemotherapy in nonsmall cell lung cancer. Transl Oncol. 2020;13(2):372–82.
Du J, Wang X, Li Y, Ren X, Zhou Y, Hu W, et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis. 2021;12(7):705.
Robert C, Ribas A, Schachter J, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019;20(9):1239–51.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–301.
Latif F, Bint Abdul Jabbar H, Malik H, Sadaf H, Sarfraz A, Sarfraz Z, et al. Atezolizumab and pembrolizumab in triple-negative breast cancer: a meta-analysis. Expert Rev Anticancer Ther. 2022;22(2):229–35.
Chen L, Niu X, Qiao X, Liu S, Ma H, Shi X, et al. Characterization of interplay between autophagy and ferroptosis and their synergistical roles on manipulating immunological tumor microenvironment in squamous cell carcinomas. Front Immunol. 2021;12: 739039.
Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40(4):365-78e6.
Wang Z, Yao J, Dong T, Niu X. Definition of a novel cuproptosis-relevant lncRNA signature for uncovering distinct survival, genomic alterations, and treatment implications in lung adenocarcinoma. J Immunol Res. 2022;2022:2756611.
Van Hoecke L, Riederer S, Saelens X, Sutter G, Rojas JJ. Recombinant viruses delivering the necroptosis mediator MLKL induce a potent antitumor immunity in mice. Oncoimmunology. 2020;9(1):1802968.
Michie J, Kearney CJ, Hawkins ED, Silke J, Oliaro J. The immuno-modulatory effects of inhibitor of apoptosis protein antagonists in cancer immunotherapy. Cells. 2020;9(1):207.
Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579(7799):421–6.
Zhang L, Yang QC, Wang S, Xiao Y, Wan SC, Deng H, et al. Engineering multienzyme-mimicking covalent organic frameworks as pyroptosis inducers for boosting antitumor immunity. Adv Mater. 2022;34(13): e2108174.
Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020;5(43):eaax7969.
Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (Lond). 2022;42(2):88–116.
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4.
Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33(5):1001–12.
Cai Z, He Y, Yu Z, Hu J, Xiao Z, Zu X, et al. Cuproptosis-related modification patterns depict the tumor microenvironment, precision immunotherapy, and prognosis of kidney renal clear cell carcinoma. Front Immunol. 2022;13: 933241.
Lin CC, Mabe NW, Lin YT, Yang WH, Tang X, Hong L, et al. RIPK3 upregulation confers robust proliferation and collateral cystine-dependence on breast cancer recurrence. Cell Death Differ. 2020;27(7):2234–47.
Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun. 2020;11(1):5424.
Acknowledgements
Not applicable.
Funding
This study was jointly supported by the National Natural Science Foundation of China (U21A20374 and 82072698), Shanghai Municipal Science and Technology Major Project (21JC1401500), Scientific Innovation Project of the Shanghai Education Committee (2019-01-07-00-07-E00057), Clinical Research Plan of the Shanghai Hospital Development Center (SHDC2020CR1006A), Xuhui District Artificial Intelligence Medical Hospital Cooperation Project (2021-011), and Shanghai Rising-Star Program (no. 20QA1402100).
Author information
Authors and Affiliations
Contributions
SS and XY designed the study. XT, TR, and MX were responsible for writing the paper and generating the figures and tables. WW, JX, BZ, and JL performed detailed analyses. XY and SS supervised the review project. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tong, X., Tang, R., Xiao, M. et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol 15, 174 (2022). https://doi.org/10.1186/s13045-022-01392-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01392-3