
Abstract
In recent years, cancer immunotherapy based on immune checkpoint inhibitors (ICIs) has achieved considerable success in the clinic. However, ICIs are significantly limited by the fact that only one third of patients with most types of cancer respond to these agents. The induction of cell death mechanisms other than apoptosis has gradually emerged as a new cancer treatment strategy because most tumors harbor innate resistance to apoptosis. However, to date, the possibility of combining these two modalities has not been discussed systematically. Recently, a few studies revealed crosstalk between distinct cell death mechanisms and antitumor immunity. The induction of pyroptosis, ferroptosis, and necroptosis combined with ICIs showed synergistically enhanced antitumor activity, even in ICI-resistant tumors. Immunotherapy-activated CD8+ T cells are traditionally believed to induce tumor cell death via the following two main pathways: (i) perforin-granzyme and (ii) Fas-FasL. However, recent studies identified a new mechanism by which CD8+ T cells suppress tumor growth by inducing ferroptosis and pyroptosis, which provoked a review of the relationship between tumor cell death mechanisms and immune system activation. Hence, in this review, we summarize knowledge of the reciprocal interaction between antitumor immunity and distinct cell death mechanisms, particularly necroptosis, ferroptosis, and pyroptosis, which are the three potentially novel mechanisms of immunogenic cell death. Because most evidence is derived from studies using animal and cell models, we also reviewed related bioinformatics data available for human tissues in public databases, which partially confirmed the presence of interactions between tumor cell death and the activation of antitumor immunity.
Similar content being viewed by others

Introduction
The mechanism by which the host immune system recognizes and kills tumor cells has not been well established. Whether dead tumor cells have immunogenic potential to elicit effective antitumor responses remains controversial because only nonself antigens are able to induce an immune response according to the “self/nonself” model that emerged in the 19th century [1]. Between the 1960s and 1980s, many studies reported that some treatment modalities, such as chemotherapy and radiotherapy, endow cancer cells with the ability to promote potent anticancer immunity [2, 3]. However, these findings were not widely acknowledged due to the lack of direct molecular evidence of tumor-associated antigen (TAA) involvement in antitumor immunity. In the 1990s and 2000s, several studies gradually elaborated TAA-directed antitumor immunity, ranging from how TAA-specific T cells survive negative selection in the thymus to how these T cells in the tumor microenvironment kill cancer cells [4, 5].
The “danger model” of immunity, which emerged in the 1990s, partially attributes the activation of the antitumor immune response to nonphysiological cell death and the subsequent release of specific molecules, which are referred to as damage-associated molecular patterns (DAMPs). These DAMPs bind receptors in various immune cells and trigger a series of immune responses, including the activation of innate and adaptive immune cells [6], opsonization or phagocytosis of dying cancer cells [7], and maturation of dendritic cells (DCs) [8]. Over a long period, the cell death mechanisms have been inaccurately classified in a dichotomized manner as follows: (i) apoptosis regulated by intrinsic pathways and extrinsic intervention and (ii) accidental necrosis. However, apoptosis is usually regarded as an immune-tolerogenic process. In 2014, two studies reported a potential mechanism by which apoptotic cells maintain immune silence [9, 10]; the authors postulated that apoptotic caspases play a key role in the immune-tolerogenic process despite the lack of a clear molecular mechanism. Five years later, Jiang et al. elucidated that caspase 3/6/7 activation causes the downregulation of cGAS, MAVS, and IRF3, which are essential proteins for the activation of innate immunity [11]. Similarly, necrosis-induced inflammation facilitates only tissue repair responses (which are largely immunoregulatory) but not effective anticancer immunity [1]. In this context, researchers introduced a novel concept, i.e., immunogenic cell death (ICD), which might be elicited by tumor vaccination, radiotherapy, and some types of chemotherapy [12]. For a long time, ICD was also referred to as immunogenic apoptosis (IA) because most types of ICD occur via apoptosis. In recent years, with the increasing awareness of cell death mechanisms, many nonapoptotic cell deaths have been defined. Necroptosis, pyroptosis, and ferroptosis are three widely studied nonapoptotic cell deaths, all of which harbor unique molecular characteristics. Functionally, their role under physiological conditions in mammals has not been well-defined. According to previous studies, mammalian necroptosis and pyroptosis primarily exist to counteract pathogen infections and trigger an inflammatory anti-microbial response through the release of DAMPs. Interestingly, several lines of evidence suggest that crosstalk exists between necroptosis and pyroptosis [13,14,15]. Activated necroptosis by the induction of receptor-interacting protein kinase 1 (RIPK3) facilitates NLRP3-caspase-1-mediated IL-1β secretion [13]. Subsequent experiments supported that necroptosis signaling could trigger the RIPK3-mixed lineage kinase-like (MLKL)-NLRP3-Caspase-1 axis using MLKL and inflammasome knockout models [14]. In contrast to the deletion of core apoptotic effectors, the genetic deletion of the key necroptotic machinery, i.e., RIPK3 and MLKL, has no important influence on animal development. Ferroptosis can be induced by physiological conditions, such as high extracellular glutamate. Evolutionally, the incorporation of polyunsaturated fatty acids (PUFAs) into cell membranes is significant for the development of complicated neuronal circuits, modulating membrane fluidity, and cell adaptation to environments with different temperatures [16]. The accumulation of PUFAs in membranes creates a vulnerability to lethal lipid peroxidation due to their ability to form stabilized radicals, and many reactive electrophiles targeting nucleophilic sites in vital proteins are generated in this process. Hence, one possible physiological function of ferroptosis is the elimination of cells with the excessive production of electrophilic intermediates [17]. Another thought-provoking question relates to the identification of the factors that switch the cell death pattern from apoptosis to nonapoptotic cell death. Existing evidence suggests that necroptosis is a backup cell death mechanism triggered when apoptosis is hindered, which is highlighted by caspase-8 inhibition of lethal necroptotic signaling [18]. Acyl-CoA synthetase long-chain family member 4 (ACSL4) is a key factor that controls the sensitivity of cells to ferroptosis induction by regulating pro-ferroptotic lipid. Researchers unveiled ACSL4 as a target of caspase cleavage during bortezomib-induced apoptosis [19]. Therefore, it is biologically plausible that the inactivation of ACSL4 during apoptosis may inhibit the insertion of PUFAs into the membrane, thereby limiting the capability of cells to undergo ferroptosis. In contrast, cells that undergo ferroptosis because of cysteine deprivation have approximately 10% the normal level of intracellular GSH [20]. The reducing power of GSH may be required for the processing and activation of caspases 3 and 8 [21, 22]; hence, cells depleted of GSH could be unable to activate caspases. The main signal pathways of these nonapoptotic cell deaths are shown as Fig. 1. Many studies found that these cell death modalities were widely involved in cancer clearance. For example, genetically enhanced tumor-selective ferroptosis sensitivity obviously inhibited the formation and development of pancreatic cancer in genetically engineered mice [23]. This phenomenon was replicated through the administration of cyst(e)inase, which is a drug that depletes cysteine and cystine, suggesting a translatable means to inducing ferroptosis in pancreatic cancer [23]. Similarly, inducing necroptosis could dramatically increase the survival times of mice with orthotopic pancreatic cancer and reduce tumor growth, stroma, and metastasis [24]. In addition, pyroptosis induction eradicates neoplastic cells in multiple cancers [25]. Although these novel cell death modalities show obvious anticancer function based on many laboratory lines of evidence, whether they could affect the response of the immune system to tumors remains unclear. Some studies conducted over the past 5 years revealed that necroptosis, ferroptosis, and pyroptosis are tightly associated with antitumor immunity. Tumor cells undergoing necroptosis, ferroptosis, and pyroptosis could trigger robust anti-tumor immunity in vivo and in vitro, and their efficacy can be synergistically improved by immune checkpoint inhibitors (ICIs), even in ICI-resistant tumors. Coincidentally, the clinical use of ICIs has achieved great success in recent years [26, 27]. Regarding the mechanism, ICIs restrain tumor development by relieving the dysfunction of effector T cells. Immunotherapy-activated CD8+ T cells are traditionally believed to induce tumor cell death via the following two main approaches: (i) perforin-granzyme and (ii) Fas-FasL [28, 29]. However, many studies identified a new mechanism by which CD8+ T cells suppress tumors through the induction of ferroptosis and pyroptosis [30,31,32,33].

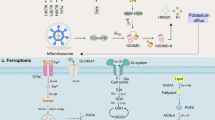
Pathways controlling ferroptosis, necroptosis, and pyroptosis. Xc-complex imports cystine, which is used to synthesize glutathione. Glutathione is used by GPX4 to prevent lipid reactive oxygen species accumulation. In this context, the normal expression and function of Xc-complex and GPX4 are essential for the inhibition of ferroptosis under physiological conditions. Gasdermins form membrane pores to cause pyroptosis. The following three pathways have been confirmed to induce pyroptosis in mammals: (1) NLRP3/ASC/caspase-1/GSDMD axis, (2) caspase-4/5/11/GSDMD axis, and (3) caspase-3/GSDME axis. Membrane-associated MLKL induces necroptosis. When the function of caspase-8 is inhibited, the binding of TNF-α and its receptor could promote the assembly of a RIPK1-RIPK3-MLKL signaling complex. RIPK3-mediated phosphorylation of MLKL leads to MLKL translocation to the plasma membrane to trigger membrane damage. As a result of membrane damage, potassium ion outflow could further activate NLRP3 through NEK7, which may be a crosstalk with pyroptosis pathway
In this context, we compiled this review to summarize knowledge regarding the crosstalk among ferroptosis, pyroptosis, necroptosis, and antitumor immunity and highlight promising modalities for cancer treatment based on these novel findings.
Traditional view of immunogenic cell death
The ability of dying cells to drive adaptive immunity depends on the following two major parameters: antigenicity and adjuvanticity [34, 35]. Antigenicity confers antigens with the ability to be recognized by host naive T cells. However, most naive T cells harboring antigens with high antigenicity are cleared by negative selection in the thymus during childhood [35]. The exception is some naive T cells expressing self-reactive low-affinity T cell receptors (TCRs) that evade thymic selection, implying that these antigens may initiate ICD once peripheral tolerance is disrupted. This finding partially explains why TAAs trigger an adaptive immune response under specific conditions [4, 36]. In addition, many non-self neoantigens are derived as TAAs from proteins harboring point mutations in tumors, which also confers tumor cells with high antigenicity [37]. Adjuvanticity is acquired from the spatiotemporally coordinated release of or exposure to DAMPs, which are necessary for the recruitment and maturation of antigen-presenting cells (APCs). Most therapy-induced antitumor immune responses observed in laboratories have been attributed to ICD [12, 38]. Nonetheless, many years elapsed before researchers established a widely acknowledged protocol for determining whether a specific type of cell death is categorized as ICD. The gold standard approach used to evaluate the capability of dying cells to induce adaptive immunity involves vaccination assays using immunocompetent, syngeneic mice [39]. Specifically, mouse-derived tumor cells are exposed to a potential ICD inducer ex vivo and then administered as a vaccine in the absence of any immunological adjuvant after the removal of exogenous chemical entities, such as the ICD inducer. After 7 to 10 days, mice are simultaneously treated with the vaccine and challenged with live cancer cells (at a minimal dose that is 100% effective in generating progressive lesions in naive mice). After 40 to 60 days, the tumor incidence and growth parameters are measured and compared with the control [39].
ICD features the spatiotemporally defined release of DAMPs (such as ATP and HMGB1), a type I IFN response, and the production of pathogen response-like chemokines that together enhance the immunogenic potential of dying cancer cells [40]. Different DAMPs mediate distinct immunostimulatory responses [41, 42]. For instance, surface-exposed HSPs not only trigger phagocytosis but also facilitate the formation of immunostimulatory Th1 and Th17 cells by inducing the secretion of proinflammatory cytokines from DCs [41]. In other cases, ATP release activates the NLRP3 inflammasome, resulting in the secretion of active IL1β, which is essential for ICD [42]. However, tumor cells with innate or experimentally enforced defects in pathways necessary for cell death-associated DAMP release, such as autophagy and the unfolded protein response, fail to undergo ICD in response to stimuli that would otherwise induce this process [35]. Therefore, extrinsic interventions applied to induce ICD have been widely researched in recent years. A qualified ICD inducer should be capable of promoting reactive oxygen species (ROS)-based ER stress in tumor cells [43, 44]. To date, many drugs approved for anticancer therapy, including doxorubicin, mitoxantrone, oxaliplatin, and bortezomib, have proven to be effective in inducing ICD as evidenced by vaccination experiments in mice [6, 45]. Other cancer therapy modalities, such as fractionated radiotherapy, but not single-dose therapy, exert optimal immunostimulatory effects, at least in mouse models [46].
However, over time, ICD was also termed IA because most ICD occurs via apoptosis. In recent years, many other cell death mechanisms distinct from apoptosis were reported to be associated with antitumor immunity, which is introduced in the following sections.
Necroptosis and antitumor immunity
Necroptosis is a form of programmed cell death that occurs downstream of PRK1 and RIPK3, which assemble into an oligomeric complex termed the necrosome [47]. Necroptotic cells undergo rapid membrane permeabilization through the executioner protein mixed-lineage kinase-like (MLKL), thereby mediating the release of intracellular contents, including immunogenic DAMPs. Although accidental necrosis is widely regarded as a self-sacrifice strategy used by tumors to develop a favorable environment for proliferation and metastasis [48], its genetically programmed counterpart, i.e., necroptosis, has been reported to play a tumor inhibitory role in most cases [14, 49]. The levels of the RIPK3 protein were decreased in two thirds of a cohort of more than 60 cancer cell lines, suggesting that cancer cells prefer to evade necroptosis for survival. In addition, low RIPK3 expression indicates a worse prognosis in patients with multiple cancers [50, 51]. Drug-induced necroptosis directly inhibits tumor proliferation and reduces the incidence of metastasis through the accumulation of high ROS levels [14, 49, 52], which might be a rational explanation for the observed association between the expression level of necroptotic markers and patient survival.
Although tumor cell necroptosis appears to be a favorable factor for tumor clearance, some cells undergoing necroptosis are unable to explain the entire antitumor effect of necroptosis inducers, suggesting a potential connection between necroptosis and antitumor immunity [37, 53]. While cells undergoing necroptosis are involved in the activation of the immune system, particularly antigen presentation and cross-priming of CD8+ T cells [54, 55], in 2016, Aaes et al. were the first to confirm that necroptosis in tumors is ICD [56]. Necroptotic tumor cells were phagocytized, and then, bone marrow-derived DC maturation was induced [56]. Through in vivo and in vitro experiments, necroptotic tumor cells were shown to induce antitumor immunogenicity through the cross-priming and proliferation of CD8+ T cells [56]. Furthermore, the authors showed that necroptotic tumor cells serve as potent immunizers in a prophylactic tumor vaccination model, which is an essential step for confirming that the cell death type is immunogenic [35]. However, these researchers did not clarify the mechanism by which effector immune cells interact with necroptotic cells [56]. In a recent study, tumor control by necroptotic cells was shown to require BATF3+ cDC1 cells and CD8+ leukocytes [57]. In mice deficient in these two types of immune cells, necroptotic cells failed to trigger obvious anticancer effects. In contrast to the previous study by Aaes et al. [56], the authors of this study showed that immune-mediated tumor control by necroptotic fibroblasts requires nuclear factor κB (NF-κB) activation within dying cells but not MLKL-mediated and cell lysis-dependent DAMP release. In addition, in contrast to the traditional view of ICD [38], this study denied the contribution of newly primed CD8+ T cells from the tumor-draining lymph to tumor clearance and further implicated the local effects of necroptotic cells within the tumor microenvironment [57]. Notably, these authors observed a potential synergistic effect between necroptosis induction in the tumor microenvironment and immune checkpoint blockade (ICB) on promoting durable tumor rejection [57]. Interestingly, with advances in nanomedicine, a necroptotic cancer cell-mimicry nanovaccine was recently reported to boost antitumor immunity with a tailored immunostimulatory modality [58]. Vaccine-administered mice showed efficient lymph node trafficking and multiepitope T cell responses. This nanovaccine induced the expansion of IFN-γ-expressing CD8+ T cells and NKG2D+ natural killer (NK) cells. Importantly, the vaccination-induced tumor regression in vivo was optimized in combination with ICIs [58].
The role of NF-κB activation in necroptosis-provoked antitumor immunity is controversial. As shown in the study by Yatim et al., robust cross-priming requires RIPK1 signaling and NF-κB-induced transcription within dying cells [55], and decoupling NF-κB signaling from necroptosis reduces the priming efficiency and the antitumor immune response. Similarly, Snyder and colleagues identified the important role of NF-κB-derived signals in DC activation and subsequent anticancer immunity [57]. However, Aaes et al. failed to correlate the immunogenicity of necroptotic tumor cells with the NF-κB activation status in vitro [56]. A potential explanation for the observed discrepancy may lie in the differences between the dying cells administered as a vaccine (PBS-washed dead tumor cells in the study by Aaes et al. and live fibroblasts in the studies by Yatim et al. and Snyder et al.). Interestingly, in mice with tumor-infiltrating immune cells lacking the expression of pattern recognition receptors capable of recognizing DAMPs, the administration of necroptotic fibroblasts into tumor tissues still controls tumor outgrowth, which reduces the importance of DAMPs in necroptosis-activated tumor immunity [57]. The crosstalk between necroptosis and anticancer immunity is summarized in Fig. 2.

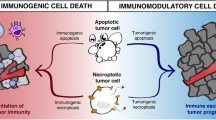
Crosstalk between necroptosis and antitumor immunity. Two strategies have been reported to trigger antitumor immunity through necroptosis. (1) Vaccination with necroptotic tumor cells: DAMPs released from tumor cells undergoing necroptosis promote the maturation of BMDCs, cross-priming of effector T cells, and subsequent cytotoxic effects. Excessive IFN-γ production is observed during this process, likely representing another anticancer approach used by CD8+ T cells. (2) Vaccination with fibroblasts: necroptotic cells release NF-κB-derived signals, further leading to DC activation, increased antigen loading, and robust CD8+ T cell-mediated tumor control. In this context, DAMPs do not appear to be involved in the activation of antitumor immunity. Tumor clearance is increased by the concomitant administration of PD1 inhibitors
Ferroptosis and antitumor immunity
Ferroptosis is a novel form of regulated cell death characterized by the iron-dependent accumulation of lipid ROS to lethal levels [59]. The sensitivity to ferroptosis is determined by many essential molecules. Acyl-CoA synthetase long-chain family member 4 (ACSL4) dictates ferroptosis sensitivity by shaping the cellular lipid composition [60]. Mechanistically, ACSL4 enriches cellular membranes with long polyunsaturated ω6 fatty acids, which are vulnerable to ferroptosis execution [60]. Glutathione peroxidase 4 (GPX4), a selenoprotein harboring a selenocysteine (Sec) in its catalytic center, is required for an efficient reduction in peroxidized phospholipids [61, 62]. Hence, a low GPX4 expression level is associated with an increased sensitivity to ferroptosis [63]. Similarly, the activity of a cystine-glutamate antiporter (system Xc-) is important for ferroptosis execution. Small compounds, such as erastin, exclusively bind system Xc- and hinder the cellular influx of cystine, which further obstructs glutathione synthesis and increases the sensitivity to ferroptosis. Recent studies also defined ferroptosis as a type of autophagy-dependent cell death [64]. Hou et al. reported the contribution of autophagy to ferroptosis by degrading ferritin in fibroblasts and cancer cells [65]. The knockout or knockdown of the key gene controlling autophagy limits erastin-induced ferroptosis by decreasing the intracellular ferrous iron levels [65]. Moreover, lipophagy-mediated lipid droplet degradation promotes lipid peroxidation in ferroptosis, which is reversed by the knockdown of RAB7A (cargo receptor of lipid droplets) and autophagy-associated gene 5 (ATG5) [64].
Many studies have implicated ferroptosis in carcinogenesis [66,67,68]. Strategies manipulating the induction of ferroptosis effectively repress tumor development, even in some chemoresistant tumors [68, 69]. The tumor suppressor p53 is closely associated with the sensitivity to ferroptosis [70, 71]. In p53-intact mice, p53 binds the SLC7A11 promotor region and inhibits its transcription [71], which is essential for ferroptosis induction. However, mice with multiple mutations in acetylation sites within p53 (K98R, K117R, K161R, and K162R) show a marked loss of p53-dependent ferroptotic responses [71]. Based on the widespread p53 mutations in distinct cancers [72], ferroptosis is speculated to be an intrinsic mechanism of resisting tumor initiation.
Previous studies have investigated the role of ferroptosis in cancer under the following two themes: (i) the up/downregulation of specific signaling pathways that sensitize/desensitize tumor cells to ferroptosis induction [73, 74] and (ii) drugs or noncoding RNAs that induce ferroptosis in tumor models [75,76,77]. However, few studies reported the direct crosstalk between ferroptosis and antitumor immunity, although a biologically plausible hypothesis is that dying cells communicate with immune cells through a set of signals, such as the “find me” and “eat me” signals produced during cell death [78]. Cancer cells undergoing ferroptosis release HMGB1 in an autophagy-dependent manner [79, 80]. As a significant DAMP, HMGB1 is a key protein required for the immunogenicity of cancer cells [81]. Nevertheless, direct evidence of the connection between ferroptosis and antitumor immunity was not available until Wang et al. reported that CD8+ T cells induce ferroptosis in tumor cells in vivo [33]. Immunotherapy-activated CD8+ T cells downregulate the expression of SLC7A11, which is a molecule required for ferroptosis induction. CD8+ T cell-derived IFN-γ increases the binding of signal transducer and activator of transcription 1 (STAT1) to the SLC7A11 transcription start site, subsequently inhibiting its transcription. STAT1 deficiency in tumor cells abolishes the IFN-γ-mediated downregulation of SLC7A11 and reverses RSL3-induced lipid peroxidation and cell death [33]. In contrast, ferroptosis-resistant or ferroptosis inhibitor-treated tumor cells are insensitive to a PD-L1 inhibitor treatment. Further in vivo experiments revealed that T cells induce ferroptosis in mice bearing ovarian tumors [33]. Immunohistochemical studies have shown that the level of CD8 is negatively associated with Xc- complex expression, suggesting that the sensitivity to ferroptosis is parallel to anticancer immunity. Subsequently, the same team reported that IFN-γ derived from immunotherapy-activated CD8+ T cells synergizes with radiotherapy-activated ataxia-telangiectasia mutated (ATM) to induce ferroptosis in human fibrosarcoma cells and melanoma cells [32], which strengthened the status of ferroptosis among common anticancer modalities. However, these studies failed to elucidate the mechanism by which tumor cells undergoing ferroptosis enhance antitumor immunity. Because HMGB1 was recently reported to be a ferroptosis-related DAMP [79], the mechanism by which ferroptotic cells trigger potent immune responses may share some similarities with traditional ICD [82]. Unfortunately, due to the lack of evidence in the prophylactic tumor vaccination model, which is the gold standard for ICD detection, the definition of ferroptosis as an ICD is premature, despite its promising potential. While these findings indicate that ferroptosis has a synergistic effect on antitumor immunity, some theoretical discrepancies require additional investigation. Tumor cells undergoing ferroptosis might conceivably function as arachidonic acid (AA) donors for the transcellular biosynthesis of eicosanoids, thereby participating in the generation of biologically active immunomodulatory AA metabolites that affect antitumor immunity [83]. In addition, based on accumulating evidence, the increased intratumor production of prostaglandin E2 (PGE2) facilitates tumor evasion of immune surveillance [84, 85]. The induction of ferroptosis in tumor cells is associated with an increased expression of PTGS2 and the release of PGE2 [62]. Hence, PGE2 production may be an intrinsic obstacle to the induction of a robust immune response by ferroptotic cells. The crosstalk between ferroptosis and anticancer immunity is summarized in Fig. 3.

Crosstalk between ferroptosis and pyroptosis and antitumor immunity. Pyroptosis in less than 15% of tumor cells is sufficient to clear an entire tumor graft, suggesting that robust anticancer immunity plays an important role in pyroptosis-initiated tumor killing. On the one hand, tumor cells undergoing pyroptosis facilitate the recruitment of anticancer immune cells, including CD8+ T cells and NK cells, by releasing danger signals. However, the level of infiltration of tumor-promoting cells, such as MDSCs, is significantly decreased during this process. On the other hand, CD8+ T cells and NK cells induce cancer cell pyroptosis by secreting GzmA and GzmB, which are enzymes capable of cleaving GSDMB and GSDME, respectively. Activated macrophage-derived IL-1β is required for the antitumor immunity induced by tumor cell pyroptosis. Similarly, CD8+ T cells induce tumor cell ferroptosis by secreting IFN-γ, which mediates the downregulation of SLC7A11 and leads to the accumulation of lipid ROS. Notably, PD1/PDL-1 inhibitors exert an obvious synergistic effect with pyroptosis/ferroptosis inducers on tumor inhibition
Pyroptosis and antitumor immunity
As a lytic and inflammatory type of regulated cell death, pyroptosis is characterized by cell swelling, lysis, and the release of many proinflammatory factors, including IL-1β, IL-18, ATP, and HMGB1. Dying cells activate pyroptosis through the following two main approaches: (i) GSDMD (gasdermin D)-dependent activation regulated by caspase 1/4/5/11 and (ii) GSDME-dependent activation regulated by caspase 3 [86,87,88,89,90]. Activated caspases cleave the hinge region between the N- and C-terminal domains of GSDMD or GSDME, releasing the segment with lethal activity and leading to pyroptosis [91, 92]. Few studies investigated the roles of other GSDM family members in pyroptosis, but GSDMA, GSDMB, and GSDMC also harbor a pore-formation domain and can induce pyroptosis.
Because the activation of pyroptosis leads to the release of inflammatory mediators, such as IL-1 and IL-18, which might promote cancer development and progression [93,94,95], some researchers have viewed pyroptosis as another protumorigenic mechanism of cell death similar to accidental necrosis [93, 95]. Gao et al. reported significantly increased levels of the GSDMD protein in non-small cell lung cancer compared to matched adjacent specimens. Higher GSDMD expression has been associated with aggressive traits, including a larger tumor size and more advanced TNM stage in lung cancer [96]. Nonetheless, the exogenous activation of pyroptosis has recently been shown to elicit robust antitumor activity [97, 98]. Cancers of the digestive [99,100,101,102], respiratory [103, 104], reproductive [105, 106], and hemopoietic systems are sensitive to pyroptosis induction [107, 108]. Chemotherapeutic drugs, such as paclitaxel and cisplatin, effectively inhibit tumor proliferation and metastasis by inducing pyroptosis [109,110,111]. Interventions with certain chemotherapeutic drugs evoke a switch from caspase 3-dependent apoptosis to pyroptosis [111, 112]. As tumor cells show innate resistance to apoptosis, the development of new strategies aiming to induce pyroptosis may provide more efficient cancer therapy options and improve patient survival [113, 114]. Interestingly, although many studies have reported the role of pyroptosis in cancer, the association between pyroptosis and anticancer immunity remains unclear. Recently, two simultaneously published studies reported that tumor cells undergoing pyroptosis recruit tumor-suppressed immune cells [30, 31]. Wang et al. constructed a bioorthogonal system to reveal that pyroptosis in less than 15% of tumor cells was sufficient to clear an entire tumor graft operating in live animals. This bioorthogonal system enabled the controlled release of a drug from an antibody-drug conjugate in mice. When combined with nanoparticle-mediated delivery, desilylation catalyzed by Phe-BF3 could release a client protein, including an active gasdermin, from a nanoparticle conjugate selectively into tumor cells in mice. Another study conducted by Zhang et al. showed that in the pyroptosis-activated immune microenvironment, CD8+ T cells and NK cells reciprocally induce pyroptosis in tumor cells via granzyme B (an enzyme capable of cleaving GSDME), thereby establishing a positive feedback loop. However, the tumor suppression was abrogated in perforin-deficient mice or mice depleted of killer lymphocytes. These authors also showed that 20 of the 22 tested cancer-associated GSDME mutations reduce GSDME function, suggesting that GSDME inactivation is a strategy developed by cancer cells to escape immune attack.
Similarly, CD8+ T cells and NK cells were recently shown to trigger tumor clearance through the GSDMB-granzyme A axis, and this process is enhanced by IFN-γ [115]. Researchers confirmed that the expression of GSDMB, but not other GSDMs, induced pyroptosis through a mechanism facilitated by granzyme A. In fact, all findings combined indicate that the mechanism of pyroptosis induction by NK cells may vary among different cell lines, suggesting that tumor cells with different molecular characteristics potentially dictate the activation of the respective GSDM-granzyme axis [30, 31, 115]. Additional experimental evidence is needed to support this hypothesis. Indeed, an in vitro study previously implicated GSDMD as essential for the antitumor function of CD8+ T cells [103]. The colocalization of GSDMD and granzyme B was observed in the proximity of immune synapses, and a GSDMD deficiency reduced the cytolytic capacity of CD8+ T cells. Perforin was postulated to be the only pore-forming protein used by CD8+ T cells [116], but the authors proposed that GSDMD may be a new pore-forming protein that can be harnessed by effector T cells and form pores within mammalian cells, although the mechanism by which GSDMD is delivered into tumor cells from CD8+ T cells remains unclear.
In most cancer types, ICIs are significantly limited by the fact that approximately only one third of patients are responsive [117]. Tumors resistant to ICIs are deemed “cold” [118, 119]. However, Wang et al. found that ICIs efficiently killed cold tumor cells only in the context of the concomitant induction of pyroptosis. Similarly, pyroptosis induction alone failed to trigger efficient tumor inhibition, highlighting the importance of treating cold tumors with a combination of pyroptosis inducers and ICIs. Nonetheless, pyroptosis induction may not benefit all immunotherapy modalities. Chimeric antigen receptor (CAR) T cells were recently shown to rapidly activate extensive caspase 3/GSDME-dependent pyroptosis in targeted cells through the release of granzyme B. Consequently, pyroptosis-released factors activate caspase 1, which cleaves GSDMD in macrophages, leading to cytokine release and subsequent cytokine release syndrome (CRS), which is a severe adverse reaction characterized by fever, hypotension, and respiratory insufficiency [120]. The authors also found that the amount of perforin/granzyme B used by CAR T cells rather than existing CD8+ T cells is critical for the induction of target cell pyroptosis. Hence, the administration of a combination of ICIs and pyroptosis inducers for the treatment of solid tumors may not cause severe side effects because CAR T cells are not involved. The crosstalk between pyroptosis and anticancer immunity is summarized in Fig. 3.
Approved drugs that induce necroptosis, ferroptosis, and pyroptosis
The development of new anticancer drugs targeting these novel mechanisms of cell death for clinical application is a lengthy process. Hence, studies exploring the effects of approved drugs with a known ability to induce ICD in patients with cancer are meaningful (Table 1). For example, artesunate, a widely prescribed antimalarial medicine, was reported to induce necroptosis and ferroptosis in tumor cells. Many clinical trials have reported the benefit of artesunate alone or in combination with other anticancer drugs in the treatment of cancer [75, 149]. Calreticulin exposure, autophagic ATP release, and HMGB1 upregulation are postulated to be the major mechanisms by which traditional chemotherapeutics trigger anticancer immunity. In addition, some chemotherapy drugs, including sorafenib, cisplatin, and paclitaxel, induce ferroptosis and pyroptosis, prompting a careful re-evaluation of the type of cell death induced by chemotherapy.
Although the drugs listed in Table 1 have shown moderate efficacy in treating cancer, given their potential role in inducing ICD, we hypothesize that appropriate combinations of these drugs with ICIs may achieve greater therapeutic benefits. Many studies have reported the increased benefit of combinations of chemotherapy and ICIs in cancer therapy (Table 2) [124, 150,151,152]. Based on the results of the KEYNOTE-189 trial involving patients with metastatic non-squamous non-small cell lung cancer, the addition of an ICI to standard chemotherapy with pemetrexed and a platinum-based drug results in significantly longer overall and progression-free survival than chemotherapy alone [150]. Similarly, in another trial (IMpower150), improved survival was noted among patients treated with atezolizumab plus chemotherapy compared with patients treated with chemotherapy alone regardless of the presence of liver metastasis or epidermal growth factor receptor mutations [151].
In addition to cytotoxic chemotherapy drugs, some non-toxic reagents may induce anticancer immunity. Cancer-derived oxysterols have consistently been shown to generate an immunosuppressive tumor microenvironment by dampening the DC antigen presentation ability, reducing the number of CD8+ T cells, and blocking the recruitment of LOX-1-positive MDSCs, which perform pro-tumor functions. Hence, reagents that block cholesterol synthesis may stimulate the host antitumor immune response and synergistically improve the efficacy of ICIs. Actually, many clinical observations have supported this hypothesis. Patients who have taken statins for more than 5 years exhibit a 47% decreased risk of colorectal cancer than non-statin users after adjusting for confounding factors [153]. Statins reduce patient mortality and prolong the relapse-free survival of patients with various cancers regardless of whether the statins were taken before or after the cancer diagnosis [154,155,156]. Moreover, inhibitors of the mevalonate pathway, including statins, are robust cancer vaccinations and synergize with ICIs in multiple mouse cancer models [157]. Interestingly, statins were recently shown to induce ferroptosis in cancer cells by reducing the production of CoQ10, which desensitizes tumor cells to ferroptosis [127]. Hence, given the crosstalk between ferroptosis in cancer cells and activated anticancer immunity, statins should be highlighted as an important reagent for cancer therapy and a candidate adjuvant to immunotherapy.
The abscopal effect is a phenomenon in which local radiotherapy is associated with the regression of metastatic cancer at a distance from the irradiated site [158, 159]. In 2012, Postow et al. reported a case of the abscopal effect in a patient with melanoma treated with ipilimumab and radiotherapy [160]. Recently, radiotherapy was reported to kill tumor cells by inducing ferroptosis and necroptosis [32, 161]. A plausible hypothesis is that dying cancer cells release “danger signals” to recruit anticancer immune cells and enhance antigen presentation by DCs. Reciprocally, ICI-activated anticancer immunity might restore the structure of vessels in the tumor microenvironment, which relieves the hypoxia state of tumor cells and increases the efficacy of radiotherapy [162]. Hence, the combination of radiotherapy and immunotherapy is a promising modality for cancer treatment and is already supported by a well-designed clinical trial [163].
In addition, many nanoparticles, such as zero-valent iron nanoparticles and arginine-rich manganese silicate nanobubbles, induce ferroptosis in cancer cells depending on the nanoparticle structure and chemical modifications [164,165,166,167]. Similarly, nanoparticles trigger necroptotic tumor cell death by modulating autophagy [168]. Although most nanoparticle-based treatments have not been approved for clinical application to date, nanoparticles alone and as drug transporters and biomimetic nanotechnology have a bright future in next-generation cancer therapy [169, 170].
Summary of the bioinformatic evidence of the roles of necroptosis, ferroptosis, and pyroptosis in anticancer immunity
Although laboratory studies have revealed the crosstalk between distinct cell death mechanisms and anticancer immunity, a substantial lack of evidence from human samples has hindered a better understanding of the potential for clinical translation.
With the increasing number of RNA sequencing-based studies and advances in bioinformatic methods, we estimated the degree of infiltration of distinct immune cells and the enrichment of immunity-related signatures in the human tumor microenvironment. In this review, we systematically analyzed a panel of genes essential for necroptosis, ferroptosis, and pyroptosis to explore their roles in T cell dysfunction and CD8+ T cell infiltration. Specifically, we reviewed the published data in the Tumor Immune Dysfunction and Exclusion (TIDE) (http://tide.dfci.harvard.edu/query/) and Tumor Immune Estimation Resource (TIMER) (https://cistrome.shinyapps.io/timer/) databases [117, 171]. Overall, the relationship between key signatures of cell death and T cell dysfunction depends on the tumor type (Fig. 4a). For example, in an endometrial carcinoma cohort (TCGA-UCEC), a higher percentage of cytotoxic T lymphocytes (CTLs) predicted a longer overall survival only in patients with lower SLC7A11 expression, which parallels the results of increased anticancer immunity with increased ferroptosis sensitivity (Fig. 4b). Hence, the combination of ICIs and ferroptosis inducers may prolong the survival of patients with endometrial carcinoma. However, this relationship was absent in other core cohorts. Three molecules essential for the initiation of necroptosis, i.e., RIP1, RIP3, and MLKL, were associated with obvious T cell dysfunction in more than two core cohorts; however, their overexpression predicted a prolonged survival in many clinical studies of ICIs, suggesting that the detrimental role of necroptosis markers in T cell function is rescued by ICIs.

Bioinformatic evidence of the effects of necroptosis, ferroptosis, and pyroptosis on T cell dysfunction and CD8+ T cell infiltration based on molecular signatures. a The effects of necroptosis, ferroptosis, and pyroptosis on T cell dysfunction and CD8+ T cell infiltration were evaluated based on the molecular signatures of four sets with 37 independent cohorts (core cohorts, immunotherapy datasets, CRISPR screen datasets, and datasets of immunosuppressive cell types). The core cohorts consisted of the five most confident results obtained using gene expression data, and a high z-score (red) suggests that the indicated gene promotes T cell dysfunction. The immunotherapy datasets consisted of 12 datasets of patients who received either ICIs or ACT. In this set, a high z-score (red) represents an unfavorable role of the indicated gene in improving the effects of immunotherapy. CRISPR screening of mouse cancer cells identified genes whose knockout enhanced the efficacy of T cell-mediated tumor cell killing based on 17 cohorts; in these studies, a z-score < 0 (blue) reflects the downregulation of the indicated gene after an increase in either T cell function or the efficacy of immunotherapy, suggesting the negative effects of the indicated gene on immunotherapy outcomes. Immunosuppressive cells restrict the tumor infiltration of T cells, including cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), and the M2 subtype of tumor-associated macrophages (TAMs). This section presents the gene expression levels in these T cell exclusion signatures, and a high z-score (red) indicates that the specified gene is overexpressed in immunosuppressive cells. b An example of the evaluation of T cell dysfunction. A considerable amount of CTL infiltration predicts prolonged survival only in tumor samples from patients with low SLC7A11 expression, suggesting that SLC7A11 potentially promotes T cell dysfunction. c The effects of necroptosis, ferroptosis, and pyroptosis on CD8+ T cell infiltration based on molecular signatures are shown
In addition, we calculated the correlation between CD8+ T cell infiltration and the expression of essential markers of distinct types of cell death (Fig. 4c). Then, we compared these correlations across seven common cancers and found that the pro-necroptosis, pro-pyroptosis, and pro-ferroptosis signatures were broadly associated with greater CD8+ T cell infiltration. In contrast, the anti-ferroptosis signatures were correlated with less CD8+ T cell infiltration. Because a greater enrichment of CD8+ T cells usually reflects better ICI efficacy [172], rational combinations of ICIs and agents that induce cell death may be effective anticancer modalities.
Microsatellite instability (MSI) and the tumor mutation burden (TMB) are important predictive biomarkers for cancer immunotherapy. Therefore, we further explored the correlations among genes involved in necroptosis, ferroptosis, and pyroptosis, MSI, and TMB across 33 cancers, and the results are visualized in Fig. 5. The results revealed that the sensitivity of necroptosis, ferroptosis, and pyroptosis may be positively correlated with higher MSI and TMB only in a portion of cancers. For example, the expression of the GSDM family, which are the executors of pyroptosis, is positively associated with a higher MSI and/or TMB. In addition, increased MSI and TMB are correlated with RIPK1 and MLKL, which are key molecules in necroptotic cell death. Whether the sensitivity of ferroptosis is increased in tumor samples harboring higher MSI and TMB is unclear given that a ferroptosis-insensitivity marker (SLC7A11) is positively associated with MSI and TMB, while a ferroptosis-sensitivity marker (ACSL4) is also positively correlated with MSI and TMB. In contrast to the other members of the GSDM family, the role of GSDMC in anticancer immunity has not been reported to date; hence, we constructed a radar plot to highlight the relationship among GSDMC expression, MSI, and TMB. The results showed that the expression level of GSDMC is positively correlated with MSI or TMB in seven cancers (BRCA, COAD, READ, GBM, THCA, THYM, and LUSC) but negatively correlated with MSI or TMB in seven other cancers (CHOL, ESCA, KIRC, KIRP, LGG, PRAD, and SKCM). The activation of GSDMB to induce pyroptosis in some specific cancers may be a promising modality to improve the efficacy of ICIs given the positive correlation between GSDMB expression and MSI or TMB in these cancers. This hypothesis is expected to be validated by future studies.

Correlations among ferroptosis-, pyroptosis-, and necroptosis-related genes; microsatellite instability (MSI); and the tumor mutation burden (TMB) across 33 cancers. The correlations among ferroptosis-, pyroptosis-, and necroptosis-related genes, MSI, and TMB were visualized as a heatmap. The colors of the up-pointing triangles reflect the correlation strength between the expression levels of ferroptosis-, pyroptosis-, and necroptosis-related genes and MSI. The colors of the down-pointing triangles reflect the correlation strength between the expression levels of ferroptosis-, pyroptosis-, and necroptosis-related genes and TMB. The correlations between GSDMC and MSI or TMB were further visualized as a radar map
Conclusions
In patients with most cancer types, the application of ICIs is significantly limited by the fact that approximately only one third of patients are responsive. In this review, we reveal broad crosstalk between anticancer immunity and nonapoptotic cell death mechanisms based on existing laboratory and bioinformatic evidence. In this context, the concomitant induction of nonapoptotic tumor cell death is a promising strategy for treating cancer. A long-standing challenge is the design of potent drugs that specifically activate the above-mentioned nonapoptotic cell deaths in humans with rigorous safety testing. However, in the near future, medical organizations are encouraged to carry out clinical trials that treat patients with approved drugs that function in activating ferroptosis, pyroptosis, or necroptotic cell death with the concomitant use of ICIs. Although these drugs may not be pathway specific, the efficacy, safety, and side-effects of such combinations could provide valuable suggestions for future investigations.
Availability of data and materials
Not applicable.
Abbreviations
- AA:
-
Arachidonic acid
- ACSL4:
-
Acyl-CoA synthetase long-chain family member 4
- ATG5:
-
Autophagy-associated gene 5
- ATM:
-
Ataxia-telangiectasia mutated
- CAR:
-
Chimeric antigen receptor
- CRS:
-
Cytokine release syndrome
- CTL:
-
Cytotoxic T lymphocytes
- DAMP:
-
Damage-associated molecular patterns
- DC:
-
Dendritic cell
- GPX4:
-
Glutathione peroxidase 4
- GSDM:
-
Gasdermin
- HMGB1:
-
High mobility group box 1
- IA:
-
Immunogenic apoptosis
- ICIs:
-
Immune checkpoint inhibitors
- ICB:
-
Immune checkpoint blockade
- ICD:
-
Immunogenic cell death
- MLKL:
-
Mixed-lineage kinase-like
- MSI:
-
Microsatellite instability
- NF-κB:
-
Nuclear factor κB
- NK:
-
Natural killer
- PGE2:
-
Prostaglandin E2
- PRK:
-
Receptor-interacting protein kinase
- Sec:
-
Selenocysteine
- STAT1:
-
Signal transducer and activator of transcription 1
- System Xc-:
-
Cystine-glutamate antiporter
- TAA:
-
Tumor-associated antigen
- TCR:
-
T cell receptors
- TIDE:
-
Tumor immune dysfunction and exclusion
- TIMER:
-
Tumor immune estimation resource
- TMB:
-
Tumor mutation burden
References
Medzhitov R, Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300.
Liao SK, Carr DH. Comparative immunogenicity of irradiated, neuraminidase treated, and fused cells of a strain-restricted sarcoma. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol. 1974;82:133–42.
Bogden AE, Esber HJ. Influence of surgery, irradiation, chemotherapy, and immunotherapy on growth of a metastasizing rat mammary adenocarcinoma. Natl Cancer Inst Monogr. 1978;49:97–100.
Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25:261–70.
Chappell DB, Restifo NP. T cell-tumor cell: a fatal interaction? Cancer Immunol Immunother. 1998;47:65–71.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701.
Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–77.
White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–62.
Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, Zhang R, Huang X, Feng JM, Jiang Z. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74:19–31.e17.
Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8(1):e000337.
Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA, Ma A. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity. 2015;42:55–67.
Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282.
Conos SA, Chen KW. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A. 2017;114:E961–e969.
Barelli H, Antonny B. Lipid unsaturation and organelle dynamics. Curr Opin Cell Biol. 2016;41:25–32.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Brault M, Oberst A. Controlled detonation: evolution of necroptosis in pathogen defense. Immunol Cell Biol. 2017;95:131–6.
Crawford ED, Seaman JE, Agard N, Hsu GW, Julien O, Mahrus S, Nguyen H, Shimbo K, Yoshihara HA, Zhuang M, et al. The DegraBase: a database of proteolysis in healthy and apoptotic human cells. Mol Cell Proteomics. 2013;12:813–24.
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–6.
Hentze H, Schmitz I, Latta M, Krueger A, Krammer PH, Wendel A. Glutathione dependence of caspase-8 activation at the death-inducing signaling complex. J Biol Chem. 2002;277:5588–95.
Ueda S, Nakamura H, Masutani H, Sasada T, Yonehara S, Takabayashi A, Yamaoka Y, Yodoi J. Redox regulation of caspase-3(-like) protease activity: regulatory roles of thioredoxin and cytochrome c. J Immunol. 1998;161:6689–95.
Badgley MA, Kremer DM. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–9.
Xie Y, Zhu S, Zhong M, Yang M, Sun X, Liu J, Kroemer G, Lotze M, Zeh HJ 3rd, Kang R, Tang D. Inhibition of Aurora kinase A induces necroptosis in pancreatic carcinoma. Gastroenterology. 2017;153:1429–1443.e1425.
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, Wang K, Sun X, Zheng J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodríguez-Abreu D, Moro-Sibilot D, Thomas CA, Barlesi F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288–301.
Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, Narwal R, Steele K, Gu Y, Karakunnel JJ, Rizvi NA. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17:299–308.
Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat Rev Immunol. 2015;15:45–56.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–52.
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–20.
Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, Huang H, Shao F. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579:421–6.
Lang X, Green MD. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9:1673–85.
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4..
Bloy N, Garcia P, Laumont CM, Pitt JM, Sistigu A, Stoll G, Yamazaki T, Bonneil E, Buqué A, Humeau J, et al. Immunogenic stress and death of cancer cells: contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol Rev. 2017;280:165–74.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Immunol Rev. 2017;17:97–111..
Bobisse S, Foukas PG, Coukos G, Harari A. Neoantigen-based cancer immunotherapy. Ann Transl Med. 2016;4:262.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691.
Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Breckpot K, Brough D, Chaurio R, Cirone M, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6:588.
Zappasodi R, Pupa SM, Ghedini GC, Bongarzone I, Magni M, Cabras AD, Colombo MP, Carlo-Stella C, Gianni AM, Di Nicola M. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010;70:9062–72.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8.
Dudek-Perić AM, Ferreira GB, Muchowicz A, Wouters J, Prada N, Martin S, Kiviluoto S, Winiarska M, Boon L, Mathieu C, et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res. 2015;75:1603–14.
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. Embo j. 2012;31:1062–79.
Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013;24:319–33.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379–88.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20.
Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, Santacroce R, Di Corcia MG, Lucchese A, Dini L, et al. Cell death: apoptosis versus necrosis (review). Int J Oncol. 2002;21:165–70.
Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347–53.
Feng X, Song Q, Yu A, Tang H, Peng Z, Wang X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma. 2015;62:592–601.
Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707–25.
Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, Zeng H, Zuo D, Hua Y, Cai Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013;13:580.
Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335–7.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96.
Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O. Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, Albert ML: RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science. 2015;350:328–34.
Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P, et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 2016;15:274–87.
Snyder AG, Hubbard NW. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol. 2019;4(36):eaaw2004.
Kang T, Huang Y, Zhu Q, Cheng H, Pei Y, Feng J, Xu M, Jiang G, Song Q, Jiang T, et al. Necroptotic cancer cells-mimicry nanovaccine boosts anti-tumor immunity with tailored immune-stimulatory modality. Biomaterials. 2018;164:80–97.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–70.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–52.
Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2019;S1044-579X(19)30006–9.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–8.
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, Li B. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34.
Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. J Cell Mol Med. 2019;35:830–49.
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, Wang J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23:4900–12.
Friedmann Angeli JP. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. J Cell Mol Med. 2019;19:405–14.
Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–8.
Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016;17:366–73.
Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18:89–102.
Beckers J, Aichler M, Walch A, Prokisch H, Trümbach D, Mao G, Qu F, Bayir H, Füllekrug J, Scheel CH, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nat Chem Biol. 2019;572:402–6.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Adv Mater. 2017;13:91–8.
Efferth T. From ancient herb to modern drug: artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol. 2017;46:65–83.
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, Stockwell BR. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26:623 633.e629.
Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, O'Connell D, Zhang P, Li Y, Gao T, et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25:1457–72.
Elliott MR, Ravichandran KS. The dynamics of apoptotic cell clearance. Dev Cell. 2016;38:147–60.
Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510:278–83..
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2:e1054549.
Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, Prada N, Adjemian S, Catani JP, Freudenberg M, et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ. 2014;21:69–78.
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75.
Friedmann Angeli JP, Krysko DV. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–14.
Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569:73–8.
Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21–8.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Agard NJ, Maltby D, Wells JA. Inflammatory stimuli regulate caspase substrate profiles. Mol Cell Proteomics. 2010;9:880–93.
Julien O, Wells JA. Caspases and their substrates. Cell Death Differ. 2017;24:1380–9.
Crawford ED, Wells JA. Caspase substrates and cellular remodeling. Annu Rev Biochem. 2011;80:1055–87.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8.
Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A. 2010;107:21635–40.
Janowski AM, Kolb R, Zhang W, Sutterwala FS. Beneficial and detrimental roles of NLRs in carcinogenesis. Front Immunol. 2013;4:370.
Dunn JH, Ellis LZ, Fujita M. Inflammasomes as molecular mediators of inflammation and cancer: potential role in melanoma. Cancer Lett. 2012;314:24–33.
Gao J, Qiu X, Xi G, Liu H, Zhang F, Lv T, Song Y. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in non-small cell lung cancer. Oncol Rep. 2018;40:1971–84.
Wang YY, Liu XL, Zhao R. Induction of pyroptosis and its implications in cancer management. Front Oncol. 2019;9:971.
Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, Hu J. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis. 2019;10:650.
Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, Zhao W, Huai W, Guo P, Han L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest. 2014;94:52–62.
Wei Q, Zhu R, Zhu J, Zhao R, Li M. E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC cells. Oncol Res. 2019;27:827–34.
Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, Heid B, Cecere TE, Allen IC. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol. 2015;194:3369–80.
Qiu S, Liu J, Xing F. ‘Hints’ in the killer protein gasdermin D: unveiling the secrets of gasdermins driving cell death. Cell Death Differ. 2017;24:588–96.
Xi G, Gao J, Wan B, Zhan P, Xu W, Lv T, Song Y. GSDMD is required for effector CD8(+) T cell responses to lung cancer cells. Int Immunopharmacol. 2019;74:105713.
Lu H, Zhang S, Wu J, Chen M, Cai MC, Fu Y, Li W, Wang J, Zhao X, Yu Z, et al. Molecular targeted therapies elicit concurrent apoptotic and GSDME-dependent pyroptotic tumor cell death. Clin Cancer Res. 2018;24:6066–77.
Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233:2116–32.
So D, Shin HW. Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense. Oncogene. 2018;37:5191–204.
Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, Poplawski SE, Wu W, Liu Y, Lai JH, et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol. 2017;13:46–53.
Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med. 2018;24:1151–6.
Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ, Zeng CY, Mai FY, Hu B, Ouyang DY. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24:312–25.
Wang F, Liu W, Ning J, Wang J, Lang Y, Jin X, Zhu K, Wang X, Li X, Yang F, et al. Simvastatin suppresses proliferation and migration in non-small cell lung cancer via pyroptosis. Int J Biol Sci. 2018;14:406–17.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
Dasgupta A, Nomura M, Shuck R, Yustein J. Cancer’s Achilles’ heel: apoptosis and necroptosis to the rescue. Int J Mol Sci. 2016;18(1):23.
Huang X, Xiao F, Li Y, Qian W, Ding W, Ye X. Bypassing drug resistance by triggering necroptosis: recent advances in mechanisms and its therapeutic exploitation in leukemia. J Exp Clin Cancer Res. 2018;37:310.
Zhou Z, He H. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368(6494):eaaz7548.
Young JD, Hengartner H, Podack ER, Cohn ZA. Purification and characterization of a cytolytic pore-forming protein from granules of cloned lymphocytes with natural killer activity. Cell. 1986;44:849–59.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–8.
Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–87.
Abril-Rodriguez G, Ribas A. SnapShot: immune checkpoint inhibitors. Cancer Cell. 2017;31:848–848.e841.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225.
Kim EH, Shin D, Lee J, Jung AR, Roh JL. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018;432:180–90.
Morikawa N, Tachibana M, Ago Y, Goda H, Sakurai F, Mizuguchi H. LY341495, an mGluR2/3 antagonist, regulates the immunosuppressive function of myeloid-derived suppressor cells and inhibits melanoma tumor growth. Biol Pharm Bull. 2018;41:1866–9.
Sun X, Niu X, Chen R, He W, Chen D, Kang R, Tang D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64:488–500.
Kimura T, Kato Y, Ozawa Y, Kodama K, Ito J, Ichikawa K, Yamada K, Hori Y, Tabata K, Takase K, et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018;109:3993–4002.
Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, Dai X, Li Z, Wu G. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50:445–60.
Ursic K, Kos S, Kamensek U, Cemazar M, Scancar J, Bucek S, Kranjc S, Staresinic B, Sersa G. Comparable effectiveness and immunomodulatory actions of oxaliplatin and cisplatin in electrochemotherapy of murine melanoma. Bioelectrochemistry. 2018;119:161–71.
Chen JJ, Galluzzi L. Fighting resilient cancers with iron. Trends Cell Biol. 2018;28:77–8.
Pisanti S, Picardi P, Ciaglia E, D'Alessandro A, Bifulco M. Novel prospects of statins as therapeutic agents in cancer. Pharmacol Res. 2014;88:84–98.
Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–62.
Qian P, Zhang YW, Zhou ZH, Liu JQ, Yue SY, Guo XL, Sun LQ, Lv XT, Chen JQ. Artesunate enhances γδ T-cell-mediated antitumor activity through augmenting γδ T-cell function and reversing immune escape of HepG2 cells. Immunopharmacol Immunotoxicol. 2018;40:107–16.
Cui C, Feng H, Shi X, Wang Y, Feng Z, Liu J, Han Z, Fu J, Fu Z, Tong H. Artesunate down-regulates immunosuppression from colorectal cancer Colon26 and RKO cells in vitro by decreasing transforming growth factor β1 and interleukin-10. Int Immunopharmacol. 2015;27:110–21.
Zhang LX, Liu ZN, Ye J, Sha M, Qian H, Bu XH, Luan ZY, Xu XL, Huang AH, Yuan DL, et al. Artesunate exerts an anti-immunosuppressive effect on cervical cancer by inhibiting PGE2 production and Foxp3 expression. Cell Biol Int. 2014;38:639–46.
Wang H, Tang Y, Fang Y, Zhang M, Wang H, He Z, Wang B, Xu Q, Huang Y. Reprogramming Tumor Immune Microenvironment (TIME) and metabolism via biomimetic targeting codelivery of Shikonin/JQ1. Nano Lett. 2019;19:2935–44.
Han Q, Ma Y, Wang H, Dai Y, Chen C, Liu Y, Jing L, Sun X. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3-mediated necroptosis. J Transl Med. 2018;16:201.
Oliver Metzig M, Fuchs D, Tagscherer KE, Gröne HJ, Schirmacher P, Roth W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-α-dependent necroptosis driven by RIP1 kinase and NF-κB. Oncogene. 2016;35:3399–409.
Zhao D, Zhang H, Tao W, Wei W, Sun J. A rapid albumin-binding 5-fluorouracil prodrug with a prolonged circulation time and enhanced antitumor activity. Biomater Sci. 2017;5:502–10.
Wang L, Li K, Lin X, Yao Z, Wang S, Xiong X, Ning Z, Wang J, Xu X, Jiang Y, et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 2019;450:22–31.
Kheirandish M, Mahboobi H, Yazdanparast M, Kamal W, Kamal MA. Anti-cancer effects of metformin: recent evidences for its role in prevention and treatment of cancer. Curr Drug Metab. 2018;19:793–7.
Yue E, Tuguzbaeva G, Chen X, Qin Y, Li A, Sun X, Dong C, Liu Y, Yu Y, Zahra SM, et al. Anthocyanin is involved in the activation of pyroptosis in oral squamous cell carcinoma. Phytomedicine. 2019;56:286–94.
Zhou L, Wang H, Yi J, Yang B, Li M, He D, Yang W, Zhang Y, Ni H. Anti-tumor properties of anthocyanins from Lonicera caerulea ‘Beilei’ fruit on human hepatocellular carcinoma: in vitro and in vivo study. Biomed Pharmacother. 2018;104:520–9.
Pizato N, Luzete BC, Kiffer L, Corrêa LH, de Oliveira Santos I, JAF A, Ito MK, Magalhães KG. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci Rep. 2018;8:1952.
Paixão E, Oliveira ACM, Pizato N, Muniz-Junqueira MI, Magalhães KG, Nakano EY, Ito MK. The effects of EPA and DHA enriched fish oil on nutritional and immunological markers of treatment naïve breast cancer patients: a randomized double-blind controlled trial. Nutr J. 2017;16:71.
Zhu L, Chen L. Progress in research on paclitaxel and tumor immunotherapy. Cell Mol Biol Lett. 2019;24:40.
Wang S, Luo J, Zhang Z, Dong D, Shen Y, Fang Y, Hu L, Liu M, Dai C, Peng S, et al. Iron and magnetic: new research direction of the ferroptosis-based cancer therapy. Am J Cancer Res. 2018;8:1933–46.
Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, Sun RY, Zhou D. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171–85.
Mora J, Mertens C, Meier JK, Fuhrmann DC, Brüne B. Strategies to interfere with tumor metabolism through the interplay of innate and adaptive immunity. Cells. 2019;8(5):445.
Yu P, Wang HY, Tian M, Li AX, Chen XS, Wang XL, Zhang Y, Cheng Y. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol Sin. 2019;40:1237–44.
Nam J, Son S, Ochyl LJ, Kuai R, Schwendeman A, Moon JJ. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat Commun. 2018;9:1074.
Efferth T. Cancer combination therapy of the sesquiterpenoid artesunate and the selective EGFR-tyrosine kinase inhibitor erlotinib. Phytomedicine. 2017;37:58–61.
Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–92.
Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodríguez-Abreu D, Moro-Sibilot D, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. 2019;7:387–401.
Reck M, Schenker M, Lee KH, Provencio M, Nishio M, Lesniewski-Kmak K, Sangha R, Ahmed S, Raimbourg J, Feeney K, et al. Nivolumab plus ipilimumab versus chemotherapy as first-line treatment in advanced non-small-cell lung cancer with high tumour mutational burden: patient-reported outcomes results from the randomised, open-label, phase III CheckMate 227 trial. Eur J Cancer. 2019;116:137–47.
Rosch PJ, McCully K. Statin use and reduced cancer-related mortality. N Engl J Med. 2013;368:576.
Cardwell CR, Hicks BM, Hughes C, Murray LJ. Statin use after colorectal cancer diagnosis and survival: a population-based cohort study. J Clin Oncol. 2014;32:3177–83.
Shao YY, Hsu CH, Yeh KH, Chen HM, Yeh YC, Lai CL, Lin ZZ, Cheng AL, Lai MS. Statin use is associated with improved prognosis of colorectal cancer in Taiwan. Clin Colorectal Cancer. 2015;14:177–184.e174.
Higashi T, Hayashi H, Kitano Y, Yamamura K, Kaida T, Arima K, Taki K, Nakagawa S, Okabe H, Nitta H, et al. Statin attenuates cell proliferative ability via TAZ (WWTR1) in hepatocellular carcinoma. Med Oncol. 2016;33:123.
Xia Y, Xie Y, Yu Z, Xiao H, Jiang G, Zhou X, Yang Y, Li X, Zhao M, Li L, et al. The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell. 2018;175:1059–1073.e1021.
Liu Y, Dong Y, Kong L, Shi F, Zhu H, Yu J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J Hematol Oncol. 2018;11:104.
Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18:313–22.
Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366:925–31.
Wang HH, Wu ZQ, Qian D, Zaorsky NG, Qiu MH, Cheng JJ, Jiang C, Wang J, Zeng XL, Liu CL, et al. Ablative hypofractionated radiation therapy enhances non-small cell lung cancer cell killing via preferential stimulation of necroptosis in vitro and in vivo. Int J Radiat Oncol Biol Phys. 2018;101:49–62.
Wang Y, Liu ZG, Yuan H, Deng W, Li J, Huang Y. The reciprocity between radiotherapy and cancer immunotherapy. Clin Cancer Res. 2019;25:1709–17.
Theelen W, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts J, Dumoulin DW, Bahce I, Niemeijer AN, de Langen AJ, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol. 2019;5(9):1276–82.
Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977–85.
Szwed M, Sønstevold T, Øverbye A. Small variations in nanoparticle structure dictate differential cellular stress responses and mode of cell death. Nanotoxicology. 2019;13:761–82.
Wang S, Li F, Qiao R, Hu X, Liao H, Chen L, Wu J, Wu H, Zhao M, Liu J, et al. Arginine-rich manganese silicate nanobubbles as a ferroptosis-inducing agent for tumor-targeted theranostics. ACS Nano. 2018;12:12380–92.
Ou W, Mulik RS, Anwar A, McDonald JG, He X, Corbin IR. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Radic Biol Med. 2017;112:597–607.
Arya BD, Mittal S, Joshi P, Pandey AK, Ramirez-Vick JE, Singh SP. Graphene oxide-chloroquine nanoconjugate induce necroptotic death in A549 cancer cells through autophagy modulation. Nanomedicine (Lond). 2018;13:2261–82.
Mahvi DA, Liu R, Grinstaff MW, Colson YL, Raut CP. Local cancer recurrence: the realities, challenges, and opportunities for new therapies. CA Cancer J Clin. 2018;68:488–505.
Wang H, Mooney DJ. Biomaterial-assisted targeted modulation of immune cells in cancer treatment. Nat Mater. 2018;17:761–72.
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, Liu XS. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77:e108–10.
McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019;37:457–95.
Acknowledgements
Not applicable.
Funding
This study was jointly funded by the National Natural Science Foundation of China (no. 81772555, 81802352, and 81902428), the National Science Foundation for Distinguished Young Scholars of China (no. 81625016), the Shanghai Sailing Program (no. 19YF1409400 and 20YF1409000), the Shanghai Rising-Star Program (no. 20QA1402100), the Shanghai Anticancer Association Young Eagle Program (no. SACA-CY19A06), the Clinical and Scientific Innovation Project of Shanghai Hospital Development Center (no. SHDC12018109 and SHDC12019109), and the Scientific Innovation Project of Shanghai Education Committee (no. 2019-01-07-00-07-E00057).
Author information
Authors and Affiliations
Contributions
S.S. and X.Y. designed the study. J.X., C.L., and Q.M. developed the study details. R.T. and J.H. were responsible for writing the paper and performing data mining. B.Z. and J.L. performed the statistical analysis. S.S. and X. Y proofread the final version. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tang, R., Xu, J., Zhang, B. et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol 13, 110 (2020). https://doi.org/10.1186/s13045-020-00946-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00946-7