
Abstract
Biomarkers-guided precision therapeutics has revolutionized the clinical development and administration of molecular-targeted anticancer agents. Tailored precision cancer therapy exhibits better response rate compared to unselective treatment. Protein kinases have critical roles in cell signaling, metabolism, proliferation, survival and migration. Aberrant activation of protein kinases is critical for tumor growth and progression. Hence, protein kinases are key targets for molecular targeted cancer therapy. The serine/threonine kinase Akt is frequently activated in various types of cancer. Activation of Akt promotes tumor progression and drug resistance. Since the first Akt inhibitor was reported in 2000, many Akt inhibitors have been developed and evaluated in either early or late stage of clinical trials, which take advantage of liquid biopsy and genomic or molecular profiling to realize personalized cancer therapy. Two inhibitors, capivasertib and ipatasertib, are being tested in phase III clinical trials for cancer therapy. Here, we highlight recent progress of Akt signaling pathway, review the up-to-date data from clinical studies of Akt inhibitors and discuss the potential biomarkers that may help personalized treatment of cancer with Akt inhibitors. In addition, we also discuss how Akt may confer the vulnerability of cancer cells to some kinds of anticancer agents.
Similar content being viewed by others


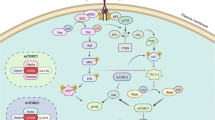
Introduction
Molecular targeted therapy has been widely introduced into clinical practice to treat different types of human cancer [1,2,3,4,5]. One of the most successful molecular targeted cancer therapeutics is the anti-estrogen therapy for patients with estrogen receptor (ER)-positive breast cancer (Fig. 1) [6,7,8]. However, the importance of molecular targeted therapy was not well recognized until the deregulation of protein kinases in cancer was clarified. The development of selective protein kinase inhibitors to treat cancer with aberrant activation of specific signaling pathways has been considered a promising approach since 1990s. BCR-ABL, cyclin-dependent kinases (CDKs), epidermal growth factor receptor (EGFR), epidermal growth factor receptor-2 (HER2/neu/ERBB2) and vascular endothelium growth factor receptor (VEGFR) have been identified as targets for the development of selective inhibitors. The BCR-ABL fusion gene, which is created through chromosomes 9 and 22 translocation (also called Philadelphia chromosome), is a key oncogene in chronic myeloid leukemia (CML) [9,10,11]. The Abl tyrosine kinase inhibitor imatinib (CGP 57148B/STI571) was first introduced in 1996, tested in the clinic from 1998 and approved for treating BCR-ABL-positive CML, Philadelphia chromosome-positive acute lymphoblastic leukemia and gastrointestinal stroma tumor [12,13,14,15].

Timeline of key progresses in ER-, Bcr-Abl-, EGFR-, HER2- and Akt-targeted cancer therapy. Based on the identification of ER, Bcr-Abl, EGFR, HER2 and Akt in promoting tumorigenesis, ER-, Bcr-Abl-, EGFR- and HER2-targeted therapy have been approved for treating various types of cancer, while Akt inhibitors are still being evaluated in phase III clinical trials
While imatinib is a small-molecule inhibitor, monoclonal antibodies has been developed to target HER2, an oncogene that is amplified in some types of cancer [16,17,18,19,20,21,22,23,24]. The series of preclinical studies and clinical trials led to the regulatory approval of Herceptin/trastuzumab, a humanized monoclonal antibody targeting HER2, to treat breast cancer (Fig. 1). HER2 amplification or overexpression is the biomarker to allow precision treatment with Herceptin. EGFR/ERBB1, the first EGFR family member, was cloned just one year before HER2 and then identified as an oncogene [25,26,27,28]. EGFR inhibitors such as gefitinib and erlotinib have been developed to treat cancer [29,30,31]. Based on the encouraging results from the phase II trials in advanced non-small-cell lung cancer (NSCLC) patients [32, 33], gefitinib received accelerate approval by the US Food and Drug Administration (FDA) to treat advanced NSCLC in 2003, followed by the approval of erlotinib (Tarceva) in 2004 [34]. However, the later disclosed phase III trials of gefitinib were disappointing [35], which led to the restriction of gefitinib use by the U.S. FDA [36]. Nevertheless, subsequent studies demonstrate that patients with sensitizing mutations of EGFR gene are more responsive to gefitinib or erlotinib compared with patients without such mutations [37,38,39], which leads to the administration of EGFR inhibitors for precision cancer therapy. In contrast, some mutations (e.g., T790M) in EGFR lead to resistance of NSCLC to gefitinib [40]. So far, the EGFR tyrosine kinase inhibitors gefitinib, erlotinib, afatinib, osimertinib and dacomitinib have been approved as first-line treatment for metastatic EGFR mutant patients with NSCLC [41]. The lessons from development of EGFR inhibitors highlight the importance of predictive biomarkers in guiding precision cancer therapy. Over the last two decades, a number of tyrosine kinase inhibitors with different targets have been developed and administered in precision cancer therapy [29, 42].
The phosphatidylinositol 3'-kinase (PI3K)/Akt pathway is one of the critical signaling outputs of Bcr-Abl, EGFR and HER2. Human homologs of v-Akt oncogene were preliminarily identified in 1987 and fully cloned, sequenced and characterized as a protein kinase in 1991 [43,44,45,46]. Subsequently, Akt was identified as a target of PI3K in 1995 [47]. The third member of Akt family, Akt3, was cloned firstly from a rat brain cDNA library and then cloned from human tissues [48, 49]. Of note, the three isoforms of Akt have different and even opposing functions in tumorigenesis [50, 51]. In general, Akt signaling is aberrantly activated in many cancers. Amplification of AKT1, AKT2 and AKT3 has been reported in gastric, ovarian, pancreatic and breast cancers, glioma and melanoma [44, 52,53,54,55,56,57,58]. In addition, overexpression of individual Akt isoforms in the absence of gene amplification has been detected in breast cancer, colorectal cancer, hepatocellular carcinoma and melanoma [59,60,61,62]. Somatic activating mutations in Akt1 and Akt3 also contribute to the hyperactivation of Akt in some types of tumors [63,64,65,66,67]. Akt activation promotes tumor progression and resistance to many chemotherapeutic agents [68,69,70]. Therefore, Akt is considered as a rational target for cancer treatment. The first Akt inhibitor ML-9 (1-(5-chloronaphthalene-1-sulphonyl)-1H-hexahydro-1,4-diazepine) were reported in 2000 [71]. However, this inhibitor also inhibits myosin light-chain kinase and stromal interaction molecule 1 [72, 73]. On the basis of the protein kinase A (PKA) inhibitor H-89, a new compound NL-71-101 was developed to inhibit Akt 2.4-fold better than PKA in 2002 [74]. The synthesis from l-quebrachitol of a series of 3-deoxygenated ether lipid-type phosphatidylinositol analogues as Akt inhibitors was reported in 2003 [75]. One year later, the results of phase I clinical trial for one of the Akt inhibitors, perifosine, was reported [76]. Currently, some Akt inhibitors, such as capivasertib (AZD5363) and ipatasertib, are being evaluated in phase III clinical trials. Herein, we review our current knowledge about Akt signaling in cancer and update recent progress in the clinical trials of Akt inhibitors. We also discuss how Akt may confer vulnerability to some kinds of anticancer agents and how gene mutations may render cancer cells vulnerable to Akt inhibition. These may help realize the goal of personalized medicine and shed new light on how to treat cancer precisely by exploiting Akt as a biomarker or molecular target [77].
Mechanisms of Akt activation and inactivation
The three members of Akt family are encoded by 3 genes (AKT1, AKT2 and AKT3), which are located at 14q32.33, 19q13.2 and 1q43-q44, respectively (Fig. 2a). Akt consists of three domains, including an N-terminal pleckstrin homology (PH) domain, a central kinase domain containing an activation loop, and an AGC kinase C-terminal domain (Fig. 2b). Structurally, the PH domain interacts with the kinase domain and elicits intramolecular autoinhibition of Akt kinase activation [78]. The PH domain is a docking site for phosphatidylinositol 3,4,5-trisphosphate (PIP3) or phosphatidylinositol 3,4-bisphosphate (PIP2). Whereas PIP3 engagement of PH domain reportedly promotes Akt activation [79], other studies indicate that PIP3 binding to the Akt1 PH domain per se does not induce Akt1 activation [80, 81]. Rather, PIP3 engagement of the PH domain may facilitate the membrane translocation of Akt and subsequent phosphorylation by PDK1, mTORC2 and other kinases [80]. Of note, a basic patch in the linker between PH and kinase domains can interact with phosphorylated C-terminal residue S473, thereby promoting Akt1 activation [81]. While the basic residue arginine in the basic patch is conserved in Akt1, Akt2 and Akt3, it remains to know whether this residue is also critical for activating Akt2 and Akt3.

An overview of AKT1/2/3 genes and proteins. a Human AKT1 gene has 15 exons, while AKT2 and AKT3 gene have 14 exons. b Akt protein has 3 domains, the plecstrin homology (PH) domain, kinase domain (KD) and C-terminal domain (CTD). The three conserved phosphorylation sites in Akt1, Akt2 and Akt3, and the kinases that phosphorylate these residues are shown
Various types of stimuli including growth factors, cytokines, hormones and stresses can activate Akt. The activity of Akt is tightly regulated by post-translational modifications such as phosphorylation, ubiquitination, acetylation and palmitoylation. Dynamic activation of Akt is dictated by post-translational modifications and removal of these modifications. Although the plasma membrane distribution is critical for Akt activation, Akt can be activated at other subcellular compartments such as the endosome, lysosome, endoplasmic reticulum and nucleus. A detailed discussion of the activation of Akt at these different compartments is beyond the scope of this review. A recent review explicitly introduced the compartment-specific activation of Akt [82].
Activation of Akt by posttranslational modification
In general, receptor- or non-receptor tyrosine kinases, PI3K, the 3-phosphoinositide-dependent protein kinase 1 (PDK1) and mTOR complex 2 (mTORC2) can activate Akt by promoting the phosphorylation of Akt1, Akt2 and Akt3 at residues T308/S473, T309/S474 and T305/S472, respectively [83]. As a catalytic product of PI3K, PIP3 is a critical activator of PDK1 and Akt. The binding of PIP3 or PI(3,4)P2 to the PH domains of Akt and PDK1 triggers membrane localization of Akt and PDK1 [84]. PDK1 directly phosphorylates Akt1 at T308, while mTORC2 not only directly phosphorylates Akt1 at S473, but also phosphorylates and activates insulin receptor (InsR)/insulin-like growth factor receptor (IGF-IR) through the tyrosine kinase activity of mTOR, thereby indirectly promoting Akt activation [83, 85]. Except for PDK1 and mTORC2, IkappaB kinase epsilon (IKKE) and TANK-binding kinase 1 (TBK1) may directly phosphorylate Akt at S473 and T308, which is dependent on PI3K but independent of PDK1 and mTORC2 [86,87,88]. Meanwhile, phosphorylation of C-terminal S477/T479 residues by CDK2/cyclin A2 or mTORC2, together with phosphorylation of S473, synergistically promotes the phosphorylation of Akt1 by PDK1 [81, 89]. Mechanistically, C-terminal phosphorylations can abrogate the intramolecular autoinhibition of Akt by the PH domain [81].
In addition, Mer tyrosine kinase (MERTK) can phosphorylate tyrosine 26 (Y26) of Akt1 and facilitate the activation of Akt by PI3K pathway [90]. Of note, it was reported that heat shock could induce Akt1 activation without plasma translocation, phosphorylation at S473 and T308 and active PI3K [91]. However, another study indicates that Akt phosphorylation is induced by heat shock [92]. Hence, it remains to know whether heat shock induces Akt1 phosphorylation on residues other than S473 and T308, and if kinases other than PI3K, PDK1 and mTORC2 mediate this process. Moreover, one study indicates that heat shock-induced activation of monomeric Akt is dependent on PI3K, whereas activation of oligomeric Akt upon heat shock is independent of PI3K [93].
Except for heat shock, oxidative stress may induce Akt activation through the non-receptor tyrosine kinase Src [93]. In addition, cyclic AMP (cAMP), an important second messenger, may promote Akt activation via PKA and exchange protein directly activated by cAMP [94,95,96,97]. Together, these studies indicate that the PDK1- and mTORC2-independent mechanisms of Akt activation are complex. Elucidation of such mechanisms may be important for exploring how cancer cells become resistant to PI3K/mTOR inhibition. Moreover, the Akt C-terminal modulatory protein was initially identified as an Akt inhibitor [98]. However, later studies indicate that it is overexpressed in breast cancer, head and neck cancer and positively regulates Akt phosphorylation [99, 100]. The reasons for discrepancy among these studies are unclear.
Besides Akt phosphorylation, K63-linked ubiquitinylation of Akt may promote Akt activation by facilitating membrane distribution. Tumor necrosis factor receptor associated factor (TRAF) 4, TRAF6 and S-phase kinase-associated protein 2 (Skp2) mediate K63-linked ubiquitination of Akt [101,102,103]. Furthermore, K63-linked polyubiquitination of Akt is promoted by SET domain bifurcated histone lysine methyltransferase 1-dependent Akt1 methylation at lysine 64, which facilitates lysine demethylase 4A-mediated recruitment of TRAF6 or Skp2 [104]. On the other hand, the deubiquitinating enzyme CYLD and ubiquitin specific peptidase 1 can remove K63-linked polyubiquitin chains on Akt and then inhibit Akt activation [105, 106]. Thus, the ying-yang balance of Akt activity can be maintained by Akt ubiquitination and deubiquitination.
Akt SUMOylation is another mechanism of Akt activation. The SUMO-conjugating enzyme Ubc9, SUMO-activating enzyme SAE1 and SUMO E3 ligase PIAS1 jointly promote Akt SUMOylation [107,108,109]. While the dependency on phosphorylation for Akt SUMOylation is controversial [110, 111], Akt SUMOylation enhances Akt activity [103]. Of note, SUMO and activation of SUMOylated AKT may be independent of PI3K or AKT distribution to the cell membrane [108]. On the other hand, activated Akt phosphorylates SUMO1 at T76 and Ubc9 at T35, leading to an increase in global SUMOylation [111].
Overall, it seems that Akt can be activated in PI3K-dependent or -independent manner. The binding of PIP3 to the PH domain, or co-operative modifications of Akt by protein kinases, ubiquitin ligases and SUMOylation enzymes can promote the membrane distribution of Akt and abrogate the intramolecular autoinhibition of Akt by the PH domain and further enhance the kinase activity of Akt.
Mechanisms of Akt inactivation and degradation
While PDK1, mTOR, TBK1 and MERTK are Akt-phosphorylating kinases, the lipid phosphatases such as phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and Src homology 2 domain-containing inositol-5-phosphatase (SHIP) can inhibit the phosphorylation of Akt by PI3K pathway [112,113,114]. Mechanistically, PTEN and SHIP convert PIP3 into PI(4,5)P2 and PI(3,4)P2, respectively. In addition, Akt can be directly dephosphorylated by PH domain leucine-rich repeat protein phosphatases (PHLPP) and protein phosphatase 2A (PP2A). Both PHLPP1 and PHLPP2 can dephosphorylate and inactivate Akt [115]. Meanwhile, the interaction between PHLPP and Akt is regulated by other proteins. ERBB receptor feedback inhibitor 1 interferes with the interaction between PHLPP and Akt thereby promoting Akt activation [116]. In contrast, sirtuin 7 promotes the interaction among FKBP51, Akt and PHLPP by deacetylating FKBP51 at lysines 28 and 155, leading to Akt dephosphorylation [117]. On the other hand, PP2A forms a complex with receptor for protein kinase 1 (RACK1) to dephosphorylate Akt [118]. Aldolase B facilitates the recruiting of PP2A to phosphorylated Akt and then promotes Akt dephosphorylation independent of the enzyme activity of adolase B [119]. Furthermore, the stability of PP2A subunits is enhanced by WNK lysine-deficient protein kinase 1, which interacts with protein phosphatase 2 scaffold subunit alpha [120]. However, Akt may reciprocally inactivate PP2A via microtubule-associated serine/threonine kinase-like (MASTL) [121]. Moreover, inhibitor 1 of PP2A (I1PP2A/ANP32A), inhibitor 2 of PP2A (I2PP2A/SET) and cellular inhibitor of PP2A (CIP2A) are oncoproteins that directly bind to PP2A and inhibit its activity [122, 123]. The micropeptide encoded by a so-called long non-coding RNA LIN00665 binds to CIP2A and inhibits its activity [124]. Hence, the activity of Akt may be determined by the balance among these regulatory elements. Except for inactivation of Akt by dephosphorylation, small ubiquitin-like modifier-specific protease 1 (SENP1), SENP2 and SENP3 can de-SUMOylate and inactivate Akt [125, 126].
While K63-linked ubiquitination of Akt positively regulates Akt activity, ubiquitinated Akt may be subject to proteasomal or lysosomal degradation. K48-linked polyubiquitination of Akt1 by zinc and ring finger 1, tetratricopeptide repeat domain 3, tripartite motif containing 13 and mitochondrial E3 ubiquitin protein ligase 1 leads to its degradation by the proteasome [127,128,129,130]. After Akt1 is sequentially ubiquitinated at lysines 284 and 214, the arginylated HSPA5 (GRP78/BIP) can promote lysosomal degradation of K48-linked ubiquitinated form of Akt [131]. In contrast, the deubiquitinase USP7 inhibits Akt ubiquitination at lysine 284/214 and lysosomal degradation [130]. Meanwhile, phosphorylation of Akt1 at T92/450 residues is essential for binding to the peptidyl-prolyl isomerase Pin1, which prevents the proteasomal degradation of Akt1 [131]. In addition, the deubiquitinase BRCA1-associated protein 1 (BAP1) reportedly stabilizes phosphorylated Akt by antagonizing its ubiquitination [132]. C-terminally truncated form of mutant ASCL1 (additional sex combs-like protein 1) interacts with BAP1 to deubiquitinate and stabilize phosphorylated Akt [132]. However, another study suggests that BAP1 may deubiquitinate and stabilize PTEN, leading to Akt inhibition [133]. Hence, the effect of BAP1 on Akt activity is inconclusive. Except for polyubiquitination, acetylation of Akt1 at lysine 14/20 by histone acetyltransferase P300 and lysine acetyltransferase 2B inhibits Akt activation [134]. Deacetylation of Akt1 by sirtunin 1 restores the activity of Akt1 [134]. Finally, caspase-mediated Akt cleavage is another mechanism of Akt inhibition [135]. Inhibition of Akt by caspase may promote cell apoptosis.
The targets and functions of Akt
Akt can directly phosphorylate many proteins that are involved in diverse cellular processes, including cell proliferation, survival, migration and metabolism (Fig. 3). The list of Akt substrates has been expanding. For most of the substrates, no Akt isoform specificity has been demonstrated. Although Akt isoforms have overlapping roles in cell signaling, previous studies have suggested that the three Akt isoforms may have some different functions, which may be due in part to isoform specific tissue distribution, subcellular localization and interaction with scaffold proteins that determine substrate selectivity [136]. While Akt1 is critical for cell survival, Akt2 is essential for glucose homeostasis [137,138,139]. Moreover, Akt isoforms have specific roles in cancer cells signaling. Opposing roles of Akt1 and Akt2 in cell cycle progression, migration and invasion have been detected in various types of human cancer [140, 141].

The targets and functions of Akt in tumorigenesis. Akt can directly phosphorylate its substrates in many signaling pathways and then regulate cell proliferation, survival, migration, glucose and lipid metabolism. The phosphorylation sites in the substrates of Akt are shown
Many substrates are activated upon phosphorylation by Akt, whereas some substrates are inhibited by Akt-catalyzed phosphorylation. A number of Akt substrates contain a minimal consensus motif P/R-X-R-X-X-S/T-F/L, in which X denotes any amino acid [142]. Recently, a chemical phosphoproteomics study identifies 276 phosphorylation sites in 185 proteins that can be inhibited by 5 Akt inhibitors [143], suggesting that there are a number of direct or indirect targets of Akt in cells. The full landscape of Akt-regulated networks remains to be explored.
Akt regulation of cell proliferation and survival
Akt can phosphorylate GSK3α and GSK3β at residues S21 and S9, respectively, thereby inactivating GSK3 and suppressing β-catenin degradation [144,145,146]. Similarly, Akt is able to phosphorylate and inactivate forkhead box protein O (FOXO) proteins. Akt phosphorylates FOXO1 at residues T24 and S256/319, FOXO3 at T32 and S253/315, and FOXO4 at T287 and S193/258 [147]. Phosphorylation of FOXO1/3/4 by Akt may inhibit their tanscriptional activity and/or promote their degradation [147, 148]. While both GSK3 and FOXO are inhibited by Akt, GSK3 is able to promote FOXO activation [149]. Other proteins that are phosphorylated and inhibited by Akt include p21waf1, p27kip1, BAD and caspase 9 [149,150,151,152,153,154]. In addition, Akt can phosphorylate YAP1 and sequester it in the cytoplasm, thereby disabling its induction of apoptosis [155]. Moreover, phosphorylation of the pro-apoptotic kinase MST2 by Akt may inactivate MST2 and inhibit apoptosis [156]. Hence, the inactivation of GSK3, FOXO, p21waf1, p27kip1, BAD, YAP1, MST2 and caspase 9 by Akt may promote cell proliferation and survival. While Akt reportedly phosphorylates and inactivates Raf1 [157], other studies suggest that Akt activates Raf1 to inhibit apoptosis [158]. Therefore, the regulation of Raf1 by Akt and its effect on apoptosis remains controversial.
The mechanistic target of rapamycin (mTOR) pathway is another Akt target that is involved in cell proliferation and survival [83]. Mechanistically, Akt directly phosphorylates the tuberous sclerosis 2 (TSC2) at S939/T1462 and impairs its stability, leading to the release of its inhibitory effect on mTOR [159]. In turn, activated mTOR transduces the growth factors signaling to downstream targets such as p70 ribosomal S6 kinase (S6K1) and eukaryotic translation initiation factor 4E binding protein 1, leading to increased protein translation. In addition, Akt is able to phosphorylate and activate cAMP responsive element binding protein 1, which also plays important roles in tumorigenesis [97, 160].
Akt regulation of cell migration and cancer metastasis
Akt not only promotes tumor growth but also accelerates metastasis. Phosphorylation of actin-binding protein Girdin at S1416 by Akt leads to the accumulation of Girdin at the leading edge of migrating cells, facilitation of lamellipodia formation and promotion of cell motility [161]. Thus, phosphorylation of Girdin by Akt may not only promote cancer metastasis but also enhance VEGF-induced angiogenesis [162]. In addition, phosphorylation of Twist1 by Akt promotes cancer metastasis via upregulating transforming growth factor-β2 expression [163]. Of note, the effect of Akt on cell migration may vary among the different isoforms of Akt and among different types of human cancers. Although Akt is generally considered as oncogene, both Akt1 and Akt2 paradoxically inhibit prostate cancer cell migration by downregulating β1-integrin activity [164]. Except for cancer cell autonormous Akt1, Akt1 deficiency in endothelial cells also facilitates prostate cancer metastasis due to activation of β-catenin [165]. Moreover, Akt1 inhibits head and neck carcinoma cells and NSCLC invasion and metastasis [166, 167]. While Akt1 still inhibits breast cancer cell migration, Akt2 promotes breast cancer cell migration [50]. However, Akt1 deficiency suppressed thyroid cancer invasion and metastasis in a murine model [168]. Also, Akt1 reportedly phosphorylates vimentin at S39 and then promotes soft tissue sarcoma cell migration and invasion [169]. Other studies demonstrate that Akt1 activation promotes melanoma metastasis [170, 171]. Whereas Akt2 promotes growth factors-induced epithelial–mesenchymal transition (EMT), a process that promotes cancer metastasis [172], Akt1 negatively regulates EMT [173]. On the other hand, activated Akt1 can sequestrate β-catenin to the cell membrane, thereby reducing ZEB1 transcription and reversing EMT [174].
The cytoskeleton is critical for cell migration and invasion. Akt1 inhibits NADPH oxidase 4 (Nox4)-derived reactive oxygen species (ROS) and then downregulates diaphanous-related formin expression and F-actin remodeling [175]. Akt1, but not Akt2, phoshorylates the S507 residue of palladin, another actin-binding protein [176]. Phosphorylation of palladin by Akt1 promotes F-actin bundling and inhibits breast cancer cell migration [176]. In contrast, Akt2 promotes palladin expression [177]. However, it is unclear how Akt2-induced palladin expression may affect cancer cell migration. As an actin-bundling protein and a scaffold, palladin has versatile effects on cell migration. It is possible that the phosphorylated and the unphosphorylated palladin have opposing effects on cell migration. In addition, Akt2 inhibits the expression of metastasis suppressor 1, a negative regulator of the actin nucleation-promoting factor cortacin [178]. Together, these studies indicate that the cytoskeleton is a key target of Akt in regulating cell migration, angiogenesis and cancer metastasis.
Similar to Akt1 inhibition, Akt3 deficiency also promotes breast cancer cells migration, invasion and metastasis due to upregulation of S100A4 and increased activation of HER2 and discoidin domain receptor tyrosine kinase 1/2 [179, 180]. In the murine breast cancer cell line PyMT, the cell–cell adhesion molecule N-cadherin downregulates Akt3 and then reduces cell motility [181]. While Akt1 promotes vascular tumor growth, Akt3 inhibits both tumor endothelial cell growth and migration [182]. However, Akt3 promotes prostate cancer metastasis by inhibiting chromosome maintenance region 1 that in turn promotes the nuclear localization of peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC1α) and mitochondrial biogenesis [183]. These data further support the versatile roles of Akt isoforms in different types of human cancers.
Akt regulation of cancer metabolism
Regulation of cellular metabolism is another critical function of Akt. While mTOR is an important mediator of Akt’s regulation of metabolism, there are many substrates of Akt that are involved in metabolism. Glucose metabolism is tightly regulated by the PI3K-Akt pathway. Glucose transporters (GLUT) are responsible for the uptake of glucose from extracellular milieu [184]. Insulin-regulated glucose uptake and whole-body glucose homeostasis are largely mediated by GLUT4. Akt phosphorylates TBC1D4 (AS160) and inhibits its GTPase-activating activity, thereby enabling GLUT4 trafficking to the plasma membrane [185]. In addition, phosphorylation of FYVE finger-containing phosphoinositide kinase (PIKFYVE), 250 kDa substrate of Akt (AS250) and myosin 5 by Akt promotes membrane localization of GLUT4 [186, 187]. Moreover, Akt may inhibit the expression of thioredoxin interacting protein, a negative regulator of the GLUT1 plasma membrane localization, which in turn promotes the plasma membrane distribution of GLUT1 and glucose uptake in cancer cells [188]. Of note, Akt not only promotes glucose uptake, but also enhances glucose metabolism. Phosphorylation of 6-phosphofructo-2-kinase at S483 by Akt accelerates glycolysis, a hallmark of metabolic reprogramming during tumor progression [189, 190]. Phosphorylation of the glyolytic enzyme hexokinase II at T473 enhances its protection of mitochondria and prevention of cell death [191]. In addition to direct regulation of glyolytic enzymes, Akt can promote glycolysis through other effectors such as HIF1A and mTOR. Furthermore, Akt acts downstream of the electrolytic enzymes phosphorescence isomerase and glyceraldehyde-3-phosphate dehydrogenase to mediate an antiapoptotic effect [192].
Except for regulating glucose metabolism, Akt may promote lipid synthesis by direct phosphorylation of acyl-CoA thioesterase 4 at S392 and ATP citrate lyase (ACLY) at S454 [193, 194]. Activation of ACLY promotes the production of acetyl-CoA, which can be hydrolyzed by acyl-CoA thioesterase to release free fatty acids. On the other hand, Akt may inhibit lipolysis by phosphorylating phosphodiesterase 3B [195]. Furthermore, Akt indirectly promotes lipogenic gene expression though mTOR-sterol regulatory element binding transcription factor pathway [196]. The complex roles of Akt in cancer metabolism are addressed in a recent review [197].
Akt inhibitors for precision cancer therapy
Since Akt has critical roles in many types of human tumors, the development of Akt inhibitors has been attractive for cancer therapy. Akt has been validated as a molecular target since 1990s. Overall, many allosteric Akt inhibitors, such as MK-2206 and miransertib, and ATP-competitive inhibitors (capivasertib, ipatasertib, etc.) have been developed [176]. The action of allosteric Akt inhibitors is dependent on the PH domain of Akt to maintain Akt in the inactive conformation, while ATP-competitive inhibitors directly target the kinase domain to inhibit its activity [198, 199]. Due to the differences in target binding, different classes of Akt inhibitors may have varied potency against clinically relevant Akt mutant variants [200]. Since the pan-Akt inhibitors may have some limitations such as adverse effects, isoform-specific inhibitors of Akt have been developed as potential therapeutics [201]. Structure-based design of small molecule agents that interact with various residues in PH and CAT domains of Akt isoforms may allow isoform-specific inhibition of Akt [202]. In addition, Akt1 or Akt2 nanobodies, the antigen-binding fragment of heavy-chain-only antibodies, have been developed to target Akt1 and Akt2, respectively [203, 204]. Alternatively, knockdown of individual Akt isoform in cancer cells may be achieved by small interfering RNA. Besides genetic interference of gene expression, proteolysis targeting chimera (PROTAC) is emerging as an approach to degrading proteins of interest [205]. PROTACs against specific Akt isoform may be another choice to achieve isoform specific inhibition of Akt.
Numerous Akt inhibitors have anticancer effects in preclinical investigation [206, 207] and thus move forward with clinical trials (Table 1). However, the advances in clinical evaluation of Akt inhibitors are somewhat slow. Since 2004, the results of various stages of clinical trials of Akt inhibitors have been reported [208]. Currently, several Akt inhibitors are being tested in the phase III stage (Table 2). Many Akt inhibitors have limited anticancer activity as a monotherapy in the clinic. Hence, the ongoing clinical trials on Akt inhibitors mainly explore their potential to improve the standard cancer therapy. A recent review explicitly introduced current patents on Akt inhibitors [209]. Here, we just update some recent advances in the clinical trials.
The phase I clinical trials demonstrate that the pan-Akt inhibitors are generally tolerable in cancer patients either as a monotherapy or in combination with chemotherapy, while these drugs have some side effects including diarrhea, hypertension, rash, hyperglycemia and fatigue [210, 211]. Combination of MK-2206 with the aromatase inhibitor anasterozole appears to be unable to enhance the efficacy of anasterozole in patients with PIK3CA-mutant ER+/Her2− breast cancer [212]. However, results from the I-SPY 2 trial demonstrate that the pathologic complete response rate in patients with HER2+ breast cancer treated with MK-2206 and standard neoadjuvant is improved when compared to neoadjuvant therapy alone, predicting a high probability of success in phase III trial [213]. In patients with locally advanced or metastatic gastric cancer and gastric-esophageal junction cancer, combination of the ATP-competitive pan-Akt inhibitor ipatasertib with leucovorin, 5-FU and oxalipatin does not improve the progression-free survival even in PTEN-low and PI3K/Akt pathway-activated subgroup [214]. For patients with metastatic triple negative breast cancer (TNBC), combination of ipatasertib with paclitaxel moderately increases the progression-free survival and overall survival when compared to treatment with placebo and paclitaxel in the LOTUS trial [215]. However, the FAIRLANE trial suggests that no significant improvement in pathologic complete response is achieved by combined treatment of stage I-IIIa TNBC with ipatasertib and paclitaxel compared to paclitaxel monotherapy, even though there is significant improvement in the MRI complete response rate [216]. In addition, combined treatment with ipatasertib and abiraterone may improve the radiographic progression-free survival in patients with metastatic castration-resistant prostate cancer when compared to abiraterone monotherapy, especially in PTEN-deficient tumors [217]. However, there is no association between tumor PTEN loss and RECIST overall response rate, circulating tumor cell reduction and prostate-specific antigen response [217]. It warrants further trials to evaluate this regimen in castration-resistant prostate cancer patients.
While the BEECH trial shows that combination of the ATP-competitive pan-Akt inhibitor capivasertib with paclitaxel, compared to paclitaxel monotherapy, has no significant improvement in the median progression-free survival of patients with advanced or metastatic ER+/HER2−/PIK3CA-mutant breast cancer [218], the FAKTION trial demonstrates that combined treatment of aromatase inhibitor-resistant, advanced or metastatic ER+/HER2− breast cancer with capivasertib and fulvestrant significantly increased the progression-free survival [219]. Further evaluation of the data from this phase II trial by restricted mean survival time analysis indicates that this regimen may also improve the overall survival compared to fulvestrant monotherapy [220, 221]. In addition, this regimen seems to be effective in both PI3K pathway-altered and PI3K pathway-unaltered groups, while it is unclear whether the changes in this pathway can be accurately detected [220, 221]. Another cell-free circulating tumor DNA (ctDNA) testing-guided clinical trial, which also includes the combined treatment of breast cancer with capivasertib and fulvestrant, indicates that ctDNA testing rapidly and accurately detects gene mutations [222]. Moreover, combined treatment of TNBC with capivasertib and paclitaxel significantly improves the progression-free survival and overall survival compared to treatment with placebo and paclitaxel [223]. Biomarker analysis shows that treatment of ER+ breast cancer with capivasertib (360 mg or 480 mg twice a day) for 4.5 days effectively inhibits the phosphorylation of Akt targets including GSK3 and PRAS40 [224].
Genomic profiling-guided precision cancer therapy is an attractive strategy. AKT1 mutation occurs in many cancers at a low prevalence (1% across diverse solid tumors) [225]. The frequency of AKT1 mutations may vary among different populations. The oncogenic AKT1E17K mutation is detected in only 1.4% of breast cancer patients who are primarily Caucasian and Hispanic [226]. While AKT1E17K is absent in the breast cancer subset of the Cancer Genome Atlas (TCGA) datasets, it is detected in appropriately 6% of a population of Chinese breast cancer patients and a cohort of endocrine-resistant and ER+ metastatic breast cancer [227, 228]. In addition, 12% of carcinosarcomas have AKT2 alterations [229]. In the TCGA datasets, AKT3 amplification is detected in approximately 10% of breast cancer patients [228, 230]. AKT1E17K- mutant tumors are resistant to allosteric Akt inhibitors, but sensitive to ATP-competitive inhibitors. A basket study of capivasertib in heavily pretreated patients with AKT1E17K-mutant tumors demonstrates that the median progression-free survival ranges from 4.2 to 6.6 months in patients with ER+ breast, gynecologic and other solid tumors [231]. The most common grade ≥ 3 adverse events were hyperglycemia (24%), diarrhea (17%) and rash (15.5%) [230]. In the subprotocol EAY131-Y of US National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH) trial, the effects of capivasertib in heavily pretreated patients with metastatic tumors harboring AKT1E17K mutation were evaluated [232]. Among 35 evaluable and analyzable patients in this trial, the objective response rate was 28.6% including one complete response, and approximately half of the patients had stable disease, while 2 (6%) had progressive disease [232]. Another phase I clinical trial of capivasertib monotherapy or combined treatment with fulvestrant in 63 heavily pre-treated patients with AKT1E17K/ER+ metastatic breast cancer demonstrated that the objective response rate was 20% in 20 patients receiving monotherapy, 36% in fulvestrant-pretreated patients with combined treatment with capivasertib and fulvestrant, and 20% in fulvestrant-naïve patients with combined treatment [233]. In addition, a more than 50% decrease in AKT1E17K at cycle 2 day 1 appears to be associated with improved progression-free survival [233]. Surprisingly, the adverse effects are less severe in patients treated with both capivasertib and fulvestrant when compared to caspivasertib monotherapy (Table 1).
In the VIKTORY Umbrella Trial, patients with metastatic gastric cancer were recruited to receive tumor genomic profiling-guided therapy [234]. While the response rate is less than 15% in PIK3CA-unmutated patients receiving capivaserib monotherapy, the objective response rate is 33.3% (8/24) in patients with PIK3CA-mutated/amplified tumors receiving capivasertib/paclitaxel therapy [234]. Of note, this study indicates that PIK3CAE542K patients may have a more profound decrease in tumor burden and increased response rate compared to patients with other point mutations in PIK3CA (Table 1). However, these data should be interpreted with caution, due to the small sample size in individual groups. Randomized clinical trials with larger sample size are warranted to validate these data.
Based on the results from some phase II clinical trials, several phase III clinical trials of capivasertib and ipatasertib have been initiated (Table 2). The CAPItello series of trials aim to evaluate the efficacy of combined treatment with capivasertib and paclitaxel, palbociclib, fulvestrant or abiraterone in breast and prostate cancer patients (clinicaltrials.gov; NCT03997123/NCT04862683/NCT04305496/NCT04493853). In parallel, the IPATunity 130/150 and FINER trials aim to evaluate the efficacy of combined treatment with ipatasertib and paclitaxel, palbociclib, fulvestrant or abiraterone/prednisone (clinicaltrials.gov; NCT03072238/NCT03337724/NCT04660862/NCT04650581/NCT04177108). Recently, the results of IPATential150, a randomized and double-blind trial, demonstrate that combination of ipatasertib with abiraterone and prednisolone may significantly improve radiographic progression-free survival compared with placebo plus abiraterone among patients with PTEN-loss, metastatic and castration-resistant prostate cancers (18.5 m for the ipatasertib arm versus 16.5 m for the placebo arm (hazard ratio, 0.77; 95%CI 0.61–0.98) [235]. The final overall survival analysis of IPATential150 trial and the results from other randomized phase III trials may provide more powerful evidence to determine whether Akt inhibitors can improve cancer therapy.
Biomarkers and molecular basis of response to Akt inhibitors in cancer
The sensitivity to molecular targeted therapy usually varies among individual cancer patients and at different stages of tumor progression. Elucidation of the mechanisms underpinning the sensitivity to molecular targeted agents may help identify the appropriate biomarkers to stratify patients for precision cancer therapy [236, 237]. Akt-dependent tumors may be vulnerable to therapeutic Akt inhibition. Currently, the PI3K/Akt/mTOR pathway activation in tumors has been taken into account in some clinical trials of Akt inhibitors. Either high or low levels of Akt pathway phosphoproteins may be associated with the responsiveness to Akt inhibition, depending on the cancer types. In general, it appears that the biomarker-assigned therapy has better effects than conventional therapy.
Alterations in PI3K/Akt /mTOR pathway as biomarkers for Akt inhibition
Among the potential biomarkers for precision treatment with Akt inhibitors, alterations in PI3K/Akt pathway are generally taken into account in multiple clinical trials (Table 3). In the ISPY-2 breast cancer trial, higher levels of Akt substrate phosphorylation in HER2+ tumors are associated with better response to MK2206, whereas lower levels of Akt pathway phosphoproteins correlate with better response in TNBC [238]. The oncogenic AKT1E17K, the most common AKT1 mutation, occurs infrequently in many types of cancers [226, 227, 232]. AKT1E17K-mutant is constitutively active and capable of promoting GLUT4 translocation [63]. Inhibition of Akt appears to be effective in treating tumors with AKT1E17K and AKT1Q79K mutants, even if these patients were heavily pre-treated [232, 233]. Notably, the imbalance of the AKT1E17K-mutant allele, most frequently present in breast and gynecological cancers, is associated with better response to capivasertib [232]. Except for Akt missense mutations, activating AKT1/2 indels are detected in breast, prostate and clear-cell renal cancers [239]. One of the Akt indels, AKT1 P68_C77dup, leads to high level of Akt activation and increased sensitivity to capivasertib in breast cells, indicating that this genomic alteration may be a potential biomarker of sensitivity to ATP-competitive Akt inhibitors [239].
Another study demonstrates that the presence of PIK3CA/AKT1 mutations and absence of alterations in MTOR or TSC1 were associated with sensitivity to capivasertib monotherapy in HER2− breast cancer [240]. The presence of AKT1E17K and coincident PI3K pathway hotspot mutations may render tumors more responsive to Akt inhibition [232]. Among the many point mutations of PIK3CA, the data from VIKTORY Umbrella Trial suggest that PIK3CAE542K-mutant is more significantly correlated with the response to capivaserib compared to other PIK3CA mutations [234]. The reason for this differential association among the PIK3CA mutations remains unclear, and this scenario may need validation in larger clinical trials. In addition, copy number loss, truncation or point mutations of phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1/p85α) are frequent events in some types of cancer. PIK3R1 deficiency leads to Akt activation due to an increase in the activity of P110 and a decrease in the activity of PTEN and growth factor receptor bound protein 2-associated protein 2 (GAB2) [241]. Preclinical study indicates that PIK3R1-depleted ovarian cancer cells are vulnerable to Akt inhibition [234]. While some clinical trials indicate that PTEN status is also associated with the benefit of Akt inhibition [215, 223], another study demonstrates that there is no difference in the sensitivity to capivasertib between PI3K/PTEN-altered and PI3K/PTEN non-altered cancer patients [209]. Taken into account of data from these clinical trials, the role of PTEN loss in dictating the response to Akt inhibitors remains to be clarified.
Diverse regulators of Akt as potential biomarkers for Akt inhibition
Except for the genetic changes in the PI3K/Akt pathway, other genetic aberration that may affect Akt activation should be considered. AT-rich interactive domain 1 (ARID1) is a component of the mammalian SWI/SNF chromatin-remodeling complex that regulates gene expression. Loss-of-function mutations of ARID1A are frequently detected in various types of cancer such as gastric cancer, endometriosis-associated ovarian cancer and endometrioid adenocarcinoma [242,243,244]. ARID1A deficiency leads to activation of Akt and hypersensitivity to Akt inhibitor [245, 246]. Moreover, loss-of-function mutation in the ARID1B gene or downregulation of ARID1B expression in medulloblastoma leads to an increase in PI3K/Akt signaling due to decreased expression of negative regulators of Akt such as dual specificity phosphatase 2, thyroid transcription factor 1, leucine zipper tumor suppressor 1, DAB2 interacting protein and protein tyrosine phosphatase receptor type U [247]. It warrants further studies to determine whether ARID1A/B mutation or expression may be a potential biomarker to predict the sensitivity to Akt inhibition.
In fact, there is a long list of genetic events that affect Akt activation in cancer cells. Whether some of these aberrant changes can serve as biomarkers to predict the sensitivity to Akt inhibitors remains to be determined in clinical setting. The metastasis suppressor NM23-H1 (NME1) interacts with the class I PI3K catalytic subunit p110α and then inhibits Akt activation [248]. It remains to know whether NM23-H1 mutation or deficiency renders cancer cells sensitive to Akt inhibitors. Moreover, there are frequent mutations of casitas B-lineage lymphoma (CBL), which encodes an E3 ubiquitin ligase that regulates many tyrosine kinases, in myeloid neoplasms [249]. Phosphorylation of mutant CBL by Lyn kinase promotes its interaction with PIK3R1 and activation of PI3K/Akt signaling [249]. It warrants further study to determine if CBL mutation can predict the sensitivity to Akt inhibitors. In addition, G protein pathway suppressor 2 (GPS2) is a significantly mutated gene in breast cancer, medulloblastoma and other tumors [250,251,252]. GPS2 depletion in breast cancer cells results in sustained PI3K/Akt signaling and increased cell proliferation, migration and invasion, which can be inhibited by MK2206 [250]. Thus, GPS2 mutation may be a potential biomarker for the responsiveness to Akt inhibitors [250].
In follicular lymphoma, inactivating mutations in Bruton tyrosine kinase (BTK) lead to augmented Akt activation [253]. Moreover, mutations in cancer-associated splicing factor 3b subunit 1 (SF3B1) have been detected in hematological malignancies, breast cancer, prostate cancer, pancreatic ductal carcinoma, melanoma and other types of cancer [254]. SF3B1 mutants cause aberrant mRNA splicing and suppression of PPP2R5A, leading to Akt activation and the hypersensitivity to Akt inhibitors [255]. Since mutations in another splicesomal gene, SURP and G-patch domain containing 1 (SUGP1), induce aberrant splicing identical or similar to that observed in mutant SF3B1 cancers, it remains to know whether SUGP1 mutations also confer hypersensitivity to Akt inhibitors [256]. Moreover, the frameshift mutations in nucleophosmin 1 (NPM1), a genetic event in about one-third of patients with acute myeloid leukemia (AML), lead to increased Akt activation that renders hypersensitivity to Akt inhibitors [257]. Whereas the wild-type NPM1 inhibits Akt phosphorylation, the AML-associated NPM1 mutants can prevent the nuclear localization of Akt and promote Akt phosphorylation [257]. Of note, overexpression of NPM1 has been detected in solid tumors including colorectal and hepatocellular carcinoma [258]. It warrants further studies to determine whether NPM1 overexpression may affect the sensitivity to Akt inhibitors. In addition, cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) deficiency in malignant pleural mesothelioma leads to PI3K/Akt activation [259]. It remains to know whether loss of CDKN2A/B renders a priority to treatment with Akt inhibitors.
Germline mutations as potential biomarkers for Akt-targeted cancer therapy
Individuals with Cowden syndrome, an autosomal dominant genetic disease, have a high risk of breast cancer in their lifetime. Approximately 80% of patients with Cowden syndrome have a germline inactivating mutation in PTEN. Kinston et al. reported an exceptional response to capivasertib in two cases with Cowden syndrome, breast cancer and germ-line PTEN mutations [260], indicating that Akt is a promising target for treating patients with germline PTEN mutations. In addition, germline mutations in cadherin 1 (CDH1) may lead to hereditary diffuse gastric cancer syndrome that is associated with upregulated Akt activity [261]. Preclinical study indicates that CDH1 deficiency renders gastric cancer cells vulnerable to allosteric Akt inhibitors, possibly due to the increased expression of Akt3 [262]. Of note, CDH1 mutations have been detected in many other types of cancer including breast, prostate, colorectal, ovarian and thyroid cancers [262]. It warrants clinical study to determine whether Akt inhibitors can effectively treat cancer patients with germline or somatic CDH1 mutations.
Germline mutations may lead to the development of familial and sporadic hematological malignancies [263]. The GAB2 P621fs frameshift mutation leads to increased interaction between GAB2 and the p85 regulatory subunit of PI3K, which promotes P3K/Akt signaling [264]. Except for GAB2 mutation, GAB2 amplification or overexpression has been detected in ovarian, lung, breast cancers and melanoma [265,266,267,268]. Hence, it warrants further studies to determine if Akt inhibitors may effectively treat GAB2 mutant-associated hematological malignancies and GAB2-amplified cancers. In addition, mothers against decapentaplegic homolog 4 (SMAD4) interacts with rictor and prevents the phosphorylation of Akt by mTORC2 [269]. Hence, loss-of-function SMAD4 mutations, which are present in some types of tumors such as mucinous adenocarcinoma of the colon and pancreatic carcinoma [270], may lead to Akt activation. SMAD4-deficient colon cancer may be vulnerable to combined treatment with Akt inhibitors and irinotecan [269]. Finally, germline mutations of SAMD4 are associated with juvenile polyposis syndrome [271, 272]. It remains to know whether Akt inhibition is a potential strategy to prevent or treat cancers in individuals with germline or somatic mutations of SMAD4.
Exploiting redox and metabolic vulnerability in Akt-driven cancer
While a significant proportion of patients with cancers harboring PI3K/Akt pathway alteration may be treated by Akt inhibitors, some of these patients may be insensitive to Akt inhibition due to primary or acquired drug resistance. In addition, Akt inhibitors may have severe adverse effects in about 30% of patients, which may lead to drug discontinuation. Although Akt activation may promote tumorigenesis, it may also make cancer cells vulnerable to some insults. Therefore, an alternative strategy to treat cancers with Akt hyperactivation may be exploiting its vulnerability.
Since Akt can promote both glycolysis and oxidative phosphorylation, the generation of ROS, a by-product of oxygen consumption and oxidative phosphorylation, may be increased in Akt-hyperactive cells [273, 274]. In addition, Akt may phosphorylate mitochondrial calcium uptake 1 (MICU1) at S124 and then inhibit mitochondrial calcium uniporter (MCU), leading to an increase in mitochondrial calcium levels and ROS [275]. NADPH is an important metabolite to maintain redox homeostasis. Activation of the NOX compromises the anti-oxidant effect of NOX and then promotes the rapid accumulation of ROS. Akt3 can activate NOX by directly phosphorylate p47phox, one of the NOX regulators, at S304 and S328, which results in increased levels of intracellular ROS [276]. On the other hand, previous studies demonstrate that all Akt isoforms can inhibit reactive oxygen species (ROS) scavenging, while Akt3 is the most potent isoform to induce ROS [276, 277]. In addition, the inactivation of FoxO transcriptional factors by Akt may result in decreased expression of FoxO-regulated ROS scavengers, such as SOD and sestrin 3 [274]. Hence, the intracellular levels of ROS may be increased in Akt-hyperactive cancer cells, depending on the plasticity of redox homeostasis.
ROS is a double-edge sword with both tumor-promoting effects and detrimental consequence on cell survival [278]. In fact, ROS can promote PI3K/Akt signaling through complex mechanisms including downregulation of PTEN, inhibition of protein tyrosine phosphatases and upregulation of receptor tyrosine kinases [277, 279, 280]. Akt is one of the mediators of ROS signaling [281]. On the one hand, ROS may promote cell senescence, growth arrest and cell death if the redox homeostasis is disrupted, and the cells fail to adapt to high levels of intracellular ROS [282]. Since Akt activation compromises some of the anti-oxidant elements, the growth of tumor cells expressing activated Akt may rely on alternative antioxidation system. In fact, the survival of Akt-activated cancer cells is dependent on FoxM1, which can not only downregulate ROS levels by upregulating the expression of anti-oxidant genes, but also prevent ROS-induced cell death [283]. During cancer evolution, increased ROS as a consequence of Akt activation may exert selective pressure to enable a reprogrammed antioxidant and detoxification system that allows for higher level of redox balance to favor the pro-tumorigenic effects of ROS. After acquiring a fitness advantage, Akt hyperactive cancer cells may expand over time.
Harnessing Akt-induced impairment of ROS scavenging for therapeutic development is an alternative strategy to treat tumors expressing hyperactive Akt. Further increasing intracellular ROS by β-phenylethyl isothiocyanate can kill cancer cells in which Akt activation is induced by rapamycin [274]. While Akt activates some pro-oxidant proteins and impairs selective antioxidant factors, the efficacy of ROS-increasing therapy for Akt-hyperactive tumors may depend on the plasticity of antioxidant system and the extent to which ROS levels are increased. Intracellular ROS levels may be a predictive biomarker for this therapy. The reliance of Akt-hyperactive cancer cells on antioxidant activity may be another vulnerability that can be exploited to treat cancer [284]. Further disabling key antioxidant proteins may render cancer cells more vulnerable to agents that induce the production of ROS.
Conclusions and perspectives
As a well-studied protein kinase, Akt has been suggested to play key roles in the regulation of development, glucose homeostasis, tumor growth and metastasis. Mounting studies have provided deep insight into the mechanisms of Akt activation and inactivation, and the diverse functions of Akt in tumor progression and drug resistance. As described above, Akt can be activated in PI3K-dependent or PI3K-independent manners. The identification of molecular events associated with Akt signaling has established its role in tumorigenesis and development of new therapeutics. The successful development of allosteric or ATP-competitive Akt inhibitors and clarification of their efficacy in preclinical studies pave the way to evaluate the potential of Akt-targeted cancer therapy in the clinic. Alternatively, inhibition of Akt by some natural agents may be valuable for cancer chemoprevention and therapy [285, 286]. Since Akt activation confers resistance to anticancer agents such as the multikinase inhibitor sorafenib, the ER antagonists tamoxifen and fulvestrant [219, 287,288,289], combination of Akt inhibitors and other anticancer agents may improve the efficacy of cancer therapy.
In view of the latest results from multiple clinical trials, Akt inhibitors such as ipatasertib and capivasertib hold promise in treating breast and prostate cancers, especially when combined with paclitaxel, fulvestrant, abiraterone, palbociclib and atezolizumab. Most of the finished trials were conducted in heavily pretreated and advanced cancer patients with relatively small sample size. Randomized clinical trials with large sample size are warranted to confirm the efficacy of biomarker-guided treatment with Akt inhibitors in cancer patients. In addition, it remains to know the efficacy of Akt inhibitors as first-line treatment for cancer. Even though Akt inhibitors may effectively treat selective cancer patients, it needs compare the efficacy of Akt inhibitors with other agents that target different members in the related pathways, such as PI3K, mTOR and CDK.
In the era of precision cancer therapy, predictive biomarkers are critical to stratify patients who most likely benefit from a given targeted therapy. Genetic, epigenetic and proteomic changes in tumors are associated with the sensitivity to many drugs. Overall, cancer patients receiving genomically matched precision therapy have increased objective tumor response rate and better overall survival when compared to unmatched therapy [290, 291]. The same may be true for Akt inhibitors. While PTEN loss and activating Akt mutations are promising biomarkers, and most of the clinical trials for Akt inhibitors are guided by changes in PTEN, PI3K, Akt and mTOR, it warrants further studies to validate these biomarkers and identify other potential biomarkers that can help select cancer patients who may be responsive to Akt–targeted therapy.
The genetic, epigenetic and proteomic changes in tumors can be identified by local testing of tumor tissues or central testing of plasma cell-free DNA (cfDNA) by high-throughput approaches [292]. Local testing of DNA from tumor tissues can be achieved by matrix-assisted laser desorption ionization time-of-flight mass spectrometry, whole-genome array-comparative genomic hybridization or next-generation sequencing (NGS), and exon-capture NGS [291, 293]. The approach for central testing of plasma cfDNA includes BEAMing, droplet digital polymerase chain reaction and NGS [231, 294, 295]. While local testing of archival tumor tissues may be more sensitive than central testing of plasma cfDNA [231], central testing of plasma cfDNA may not only help stratify cancer patients for matched precision therapy, but also capture additional tumor heterogeneity, monitor the dynamic changes in genetic markers and evaluate the therapeutic response or predict the prognosis [296].
Of note, pan-Akt inhibitors may have severe adverse effects in some patients, which even lead to drug discontinuation. Under these circumstances, an alternative strategy may be development of isoform-specific Akt inhibitors. However, this kind of Akt inhibitors still meets many challenges. The efficacy of isoform-specific Akt inhibitors may depend on whether tumors are addictive to a given isoform of Akt. On the other hand, tumors may evolve to adapt to the selective pressure of isoform-specific Akt inhibitors by rewiring to other isoforms. Exploiting the vulnerability of Akt-hyperactive cancer cells is another strategy to treat such cancers. Except for the redox disregulation, other potentially fatal weakness of Akt-hyperactive cancer remains to be identified.
Drug resistance is a severe problem in cancer therapy. Cancer cells may resist Akt inhibition via complex mechanisms. Since Akt induces inhibitory phosphorylation of RAF1 S259 and BRAF S365 [151], inhibition of Akt may result in derepressing RAF1 and BRAF. In addition, RAF/RAS mutations may lead to ERK hyperactivation, which confers resistance to Akt inhibition. Inhibition of Akt may also lead to FOX3A-mediated upregulation of ER and IGF-IR expression [143, 297]. Furthermore, serum/glucocorticoid regulated kinase (SGK) and Akt share many substrates. SGK can sustain Akt-independent activation of signaling molecules such as mTOR [298]. Meanwhile, cyclin D1 overexpression and loss of Akt1 E17K mutation are associated with acquired resistance to capivasertib [240]. Simultaneous inhibition of Akt and other compensatory pathways may improve the efficacy.
Akt plays important roles in many physiological and pathological processes. Hence, the systemic effects of Akt inhibitors must be taken into account when they are used to treat cancer. Targeted inhibition of PI3K/Akt/mTOR pathway often disrupts glucose uptake and metabolism in some tissues, which may lead to hyperglycemia, one of the common side effects of PI3K, Akt and mTOR inhibitors [299, 300]. Hyperglycemia usually feeds back to stimulate insulin release from the pancreas. The systemic glucose-insulin feedback contributes to PI3K inhibitors resistance during cancer therapy, which can be circumvented by ketogenic diet, metformin and SGLT2 inhibitors [301]. The same may be true for Akt-targeted cancer therapy. Meanwhile, the insulin-independent glucose consumption by tumor cells may also be involved in Akt inhibitors resistance as well. On the other hand, hepatic Akt inhibition may induce liver injury, inflammation and carcinogenesis and promote lung metastasis in mice [301]. It remains to know whether long-term treatment with Akt inhibitors may induce such adverse effects in human. Tumor-targeted delivery of Akt inhibitors may relieve the systemic adverse effects and enhance the anticancer activity.
Availability of data and materials
Not applicable.
Abbreviations
- ACLY:
-
ATP citrate lyase
- AML:
-
Acute myeloid leukemia
- ARID1:
-
AT-rich interactive domain 1
- BAP1:
-
BRCA1-associated protein 1
- CBL:
-
Casitas B-lineage lymphoma
- CDH1:
-
Cadherin 1
- CDK:
-
Cyclin-dependent kinases
- CDKN2A/B:
-
Cyclin-dependent kinase inhibitor 2A/B
- cfDNA:
-
Cell-free DNA
- CIP2A:
-
Cellular inhibitor of PP2A
- CML:
-
Chronic myeloid leukemia
- ctDNA:
-
Circulating tumor DNA
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial–mesenchymal transition
- ER:
-
Estrogen receptor
- FOXO:
-
Forkhead box protein O
- GAB2:
-
Growth factor receptor bound protein 2-associated protein 2
- GLUT:
-
Glucose transporters
- GPS2:
-
G protein pathway suppressor 2
- GSK3:
-
Glycogen synthase kinase 3
- HER2:
-
Epidermal growth factor receptor-2
- I1PP2A:
-
Inhibitor 1 of PP2A
- I2PP2A:
-
Inhibitor 2 of PP2A
- IGF-IR:
-
Insulin-like growth factor receptor
- IKKE:
-
IkappaB kinase epsilon
- InsR:
-
Insulin receptor
- MERTK:
-
Mer tyrosine kinase
- mTOR:
-
Mechanistic target of rapamycin
- mTORC2:
-
mTOR complex 2
- NGS:
-
Next-generation sequencing
- NSCLC:
-
Non-small-cell lung cancer
- PDK1:
-
3-Phosphoinositide-dependent protein kinase 1
- PGC1α:
-
Peroxisome proliferator-activated receptor gamma co-activator 1 alpha
- PI3K:
-
Phosphoinositide 3-kinase
- PIK3CA:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- PIK3R1:
-
Phosphoinositide-3-kinase regulatory subunit 1
- PIP2:
-
Phosphatidylinositol 3,4-bisphosphate
- PIP3:
-
Phosphatidylinositol 3,4,5-trisphosphate
- PHLPP:
-
PH domain leucine-rich repeat protein phosphatases
- PROTAC:
-
Proteolysis targeting chimera
- PP2A:
-
Protein phosphatase 2A
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome 10
- RACK1:
-
Receptor for protein kinase 1
- ROS:
-
Reactive oxygen species
- SENP:
-
Small ubiquitin-like modifier-specific protease
- SGK:
-
Serum/glucocorticoid regulated kinase
- SHIP:
-
Src homology 2 domain-containing inositol-5-phosphatase
- SKP2:
-
S-phase kinase-associated protein 2
- SMAD4:
-
Mothers against decapentaplegic homolog 4
- SUMO:
-
Small ubiquitin-like modifier
- TBK1:
-
TANK-binding kinase 1
- TCGA:
-
The cancer genome atlas
- TNBC:
-
Triple negative breast cancer
- TRAF:
-
Tumor necrosis factor receptor associated factor
- TSC1:
-
Tuberous sclerosis 1
- TSC2:
-
Tuberous sclerosis 2
- VEGFR:
-
Vascular endothelium growth factor receptor
References
Bellantoni AJ, Wagner LM. Pursuing precision: Receptor tyrosine kinase inhibitors for treatment of pediatric solid tumors. Cancers (Basel). 2021;13:3531.
Qian Y, Gong Y, Fan Z, Luo G, Huang Q, Deng S, et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J Hematol Oncol. 2020;13:130.
Wang Q, Yang S, Wang K, Sun SY. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J Hematol Oncol. 2019;12:63.
Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: Treating cancer with specificity. Eur J Pharmacol. 2018;834:188–96.
Yu B, Jiang T, Liu D. BCMA-targeted immunotherapy for multiple myeloma. J Hematol Oncol. 2020;13:125.
Bloom HJ, Boesen E. Antioestrogens in treatment of breast cancer: value of nafoxidine in 52 advanced cases. Br Med J. 1974;2:7–10.
Cole MP, Jones CTA, Todd DH. The treatment of advanced carcinoma of the breast with the antioestrogenic agent tamoxifen (ICI46,474)-a series of 96 patients. Adu Antineoplastic Chemother. 1972;2:529–31.
Nasrazadani A, Thomas RA, Oesterreich S, Lee AV. Precision medicine in hormone receptor-positive breast cancer. Front Oncol. 2018;8:144.
Soverini S, Bassan R, Lion T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: recent advances and remaining challenges. J Hematol Oncol. 2019;12:39.
Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–3.
de Klein A, van Kessel AG, Grosveld G, Bartram CR, Hagemeijer A, Bootsma D, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765–7.
Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6.
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7.
Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–53.
Samra B, Jabbour E, Ravandi F, Kantarjian H, Short NJ. Evolving therapy of adult acute lymphoblastic leukemia: state-of-the-art treatment and future directions. J Hematol Oncol. 2020;13:70.
Drebin JA, Link VC, Stern DF, Weinberg RA, Greene MI. Down-modulation of an oncogene protein product and reversion of the transformed phenotype by monoclonal antibodies. Cell. 1985;41:697–706.
Semba K, Kamata N, Toyoshima K, Yamamoto T. A v-erbB-related protooncogene, c-erbB-2, is distinct from the c-erbB-1/epidermal growth factor-receptor gene and is amplified in a human salivary gland adenocarcinoma. Proc Natl Acad Sci U S A. 1985;82:6497–501.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82.
Drebin JA, Link VC, Greene MI. Monoclonal antibodies specific for the neu oncogene product directly mediate anti-tumor effects in vivo. Oncogene. 1988;2:387–94.
Shalaby MR, Shepard HM, Presta L, Rodrigues ML, Beverley PC, Feldmann M, et al. Development of humanized bispecific antibodies reactive with cytotoxic lymphocytes and tumor cells overexpressing the HER2 protooncogene. J Exp Med. 1992;175:217–25.
Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA. 1992;89:4285–9.
Valone FH, Kaufman PA, Guyre PM, Lewis LD, Memoli V, Deo Y, et al. Phase Ia/Ib trial of bispecific antibody MDX-210 in patients with advanced breast or ovarian cancer that overexpresses the proto-oncogene HER-2/neu. J Clin Oncol. 1995;13:2281–92.
Pegram MD, Lipton A, Hayes DF, Weber BL, Baselga JM, Tripathy D, et al. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. J Clin Oncol. 1998;16:2659–71.
Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–26.
Downward J, Yarden Y, Mayes E, Scrace G, Totty N, Stockwell P, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307:521–7.
Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature. 1984;309:418–25.
Merlino GT, Xu YH, Ishii S, Clark AJ, Semba K, Toyoshima K, et al. Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. Science. 1984;224:417–9.
Riedel H, Massoglia S, Schlessinger J, Ullrich A. Ligand activation of overexpressed epidermal growth factor receptors transforms NIH 3T3 mouse fibroblasts. Proc Natl Acad Sci USA. 1988;85:1477–81.
Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol. 2020;13:143.
Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000;6:2053–63.
Liu D. CAR-T “the living drugs”, immune checkpoint inhibitors, and precision medicine: a new era of cancer therapy. J Hematol Oncol. 2019;12:113.
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer. J Clin Oncol. 2003;21:2237–46.
Kris MG, Natale RB, Herbst RS, Lynch TG Jr, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290:2149–58.
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32.
Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung cancer). Lancet. 2005;366:1527–37.
Blackhall F, Ranson M, Thatcher N. Where next for gefitinib in patients with lung cancer? Lancet Oncol. 2006;7:499–507.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39.
Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500.
Gridelli C, De Marinis F, Di Maio M, Cortinovis D, Cappuzzo F, Mok T. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating epidermal growth factor receptor mutation: review of the evidence. Lung Cancer. 2011;71:249–57.
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92.
Chen R, Manochakian R, James L, Azzouqa AG, Shi H, Zhang Y, et al. Emerging therapeutic agents for advanced non-small cell lung cancer. J Hematol Oncol. 2020;13:58.
Qin S, Li A, Yi M, Yu S, Zhang M, Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol. 2019;12:27.
Subbiah V, Dumbrava EI, Jiang Y, Thein KZ, Naing A, Hong DS, et al. Dual EGFR blockade with cetuximab and erlotinib combined with anti-VEGF antibody bevacizumab in advanced solid tumors: a phase 1 dose escalation triplet combination trial. Exp Hematol Oncol. 2020;9:7.
Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA. 1987;84:5034–7.
Belacossa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–7.
Jones PF, Jakubowicz T, Hemmings BA. Molecular cloning of a second form of rac protein kinase. Cell Regul. 1991;2:1001–9.
Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–36.
Konishi H, Kuroda S, Tanaka M, Matsuzaki H, Ono Y, Kameyama K, et al. Molecular cloning and characterization of a new member of the RAC protein kinase family: association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteins. Biochem Biophys Res Commun. 1995;216:526–34.
Masure S, Haefner B, Wesselink JJ, Hoefnagel E, Mortier E, Verhasselt P, et al. Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3. Eur J Biochem. 1999;265:353–60.
Hinz N, Jücker M. Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Commun Signal. 2019;17:154.
Degan SE, Gelman IH. Emerging roles for AKT isoform preference in cancer progression pathways. Mol Cancer Res. 2021. https://doi.org/10.1158/1541-7786.MCR-20-1066.
Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, et al. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA. 1992;89:9267–71.
Altomare DA, Tanno S, De Rienzo A, Klein-Szanto AJ, Tanno S, Skele KL, et al. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2002;87:470–6.
Dobashi Y, Kimura M, Matsubara H, Endo S, Inazawa J, Ooi A. Molecular alterations in AKT and its protein activation in human lung carcinomas. Hum Pathol. 2012;43:2229–40.
Ruggeri BA, Huang L, Wood M, Cheng JQ, Testa JR. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog. 1998;21:81–6.
Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–5.
Turner KM, Sun Y, Ji P, Granberg KJ, Bernard B, Hu L, et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc Natl Acad Sci USA. 2015;112:3421–6.
Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–10.
Roy HK, Olusola BF, Clemens DL, Karolski WJ, Ratashak A, Lynch HT, et al. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis. 2002;23:201–5.
Xu X, Sakon M, Nagano H, Hiraoka N, Yamamoto H, Hayashi N, et al. Akt2 expression correlates with prognosis of human hepatocellular carcinoma. Oncol Rep. 2004;11:25–32.
Stal O, Perez-Tenorio G, Akerberg L, Olsson B, Nordenskjold B, Skoog L, et al. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. 2003;5:37–44.
Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W, Weigel RJ, et al. Upregulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J Biol Chem. 1999;274:21528–32.
Bleeker FE, Felicioni L, Buttitta F, Lamba S, Cardone L, Rodolfo M. AKT1(E17K) in human solid tumours. Oncogene. 2008;27:5648–50.
Shoji K, Oda K, Nakagawa S, Hosokawa S, Nagae G, Uehara Y, et al. The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br J Cancer. 2009;101:145–8.
Dutt A, Salvesen HB, Greulich H, Sellers WR, Beroukhim R, Meyerson M. Somatic mutations are present in all members of the AKT family in endometrial carcinoma. Br J Cancer. 2009;101:1218–9.
Herberts C, Murtha AJ, Fu S, Wang G, Schönlau E, Xue H, et al. Activating AKT1 and PIK3CA mutations in metastatic castration-resistant prostate cancer. Eur Urol. 2020;78:834–44.
Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99:1265–8.
Sobočan M, Bračič S, Knez J, Takač I, Haybaeck J. The communication between the PI3K/AKT/mTOR pathway and Y-box binding protein-1 in gynecological cancer. Cancers (Basel). 2020;12:205.
Ma XL, Shen MN, Hu B, Wang BL, Yang WJ, Lv LH, et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/AKT signaling by inducing Rap1-mediated membrane localization of P110beta and predicts poor prognosis. J Hematol Oncol. 2019;12:37.
Gallyas F Jr, Sumegi B, Szabo C. Role of Akt activation in PARP inhibitor resistance in cancer. Cancers (Basel). 2020;12:532.
Smith U, Carvalho E, Mosialou E, Beguinot F, Formisano P, Rondinone C. PKB inhibition prevents the stimulatory effect of insulin on glucose transport and protein translocation but not the antilipolytic effect in rat adipocytes. Biochem Biophys Res Commun. 2000;268:315–20.
Saitoh M, Ishikawa T, Matsushima S, Naka M, Hidaka H. Selective inhibition of catalytic activity of smooth muscle myosin light chain kinase. J Biol Chem. 1987;262:7796–801.
Smyth JT, Dehaven WI, Bird GS, Putney JW Jr. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci. 2008;121:762–72.
Reuveni H, Livnah N, Geiger T, Klein S, Ohne O, Cohen I, et al. Toward a PKB inhibitor: modification of a selective PKA inhibitor by rational design. Biochemistry. 2002;41:10304–14.
Kozikowski AP, Sun H, Brognard J, Dennis PA. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J Am Chem Soc. 2003;125:1144–5.
Van Ummersen L, Binger K, Volkman J, Marnocha R, Tutsch K, Kolesar J, et al. A phase I trial of perifosine (NSC 639966) on a loading dose/maintenance dose schedule in patients with advanced cancer. Clin Cancer Res. 2004;10:7450–6.
Gambardella V, Tarazona N, Cejalvo JM, Lombardi P, Huerta M, Roselló S, et al. Personalized medicine: recent progress in cancer therapy. Cancers (Basel). 2020;12:1009.
Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP, Brandhuber BJ. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE. 2010;5:e12913.
Ebner M, Lučić I, Leonard TA, Yudushkin I. PI(3,4,5)P3 engagement restricts Akt activity to cellular membranes. Mol Cell. 2017;65:416–31.
James SR, Downes CP, Gigg R, Grove SJ, Holmes AB, Alessi DR. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J. 1996;315:709–13.
Chu N, Salguero AL, Liu AZ, Chen Z, Dempsey DR, Ficarro SB, et al. Akt kinase activation mechanisms revealed using protein semisynthesis. Cell. 2018;174:897–907.
Sugiyama MG, Fairn GD, Antonescu CN. Akt-ing up just about everywhere: compartment-specific Akt activation and function in receptor tyrosine kinase signaling. Front Cell Dev Biol. 2019;7:70.
Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12:71.
Liu SL, Wang ZG, Hu Y, Xin Y, Singaram I, Gorai S, et al. Quantitative lipid imaging reveals a new signaling function of phosphatidylinositol-3,4-bisphophate: isoform- and site-specific activation of Akt. Mol Cell. 2018;71:1092–104.
Yin Y, Hua H, Li M, Liu S, Kong Q, Shao T, et al. mTORC2 promotes type I insulin-like growth factor receptor and insulinreceptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016;26:46–65.
Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, et al. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci USA. 2011;108:6474–9.
Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–70.
Cooper JM, Ou YH, McMillan EA, Vaden RM, Zaman A, Bodemann BO, et al. TBK1 provides context-selective support of the activated AKT/mTOR pathway in lung cancer. Cancer Res. 2017;77:5077–94.
Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature. 2014;508:541–5.
Jiang Y, Zhang Y, Leung JY, Fan C, Popov KI, Su S, et al. MERTK mediated novel site Akt phosphorylation alleviates SAV1 suppression. Nat Commun. 2019;10:1515.
Matsuzaki H, Yamamoto T, Kikkawa U. Distinct activation mechanisms of protein kinase B by growth-factor stimulation and heat-shock treatment. Biochemistry. 2004;43:4284–93.
Konishi H, Matsuzaki H, Tanaka M, Ono Y, Tokunaga C, Kuroda S, et al. Activation of RAC-protein kinase by heat shock and hyperosmolarity stress through a pathway independent of phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1996;93:7639–43.
Konishi H, Fujiyoshi T, Fukui Y, Matsuzaki H, Yamamoto T, Ono Y, et al. Activation of protein kinase B induced by H(2)O(2) and heat shock through distinct mechanisms dependent and independent of phosphatidylinositol 3-kinase. J Biochem. 1999;126:1136–43.
Bensalma S, Turpault S, Balandre AC, De Boisvilliers M, Gaillard A, Chadéneau C, et al. PKA at a cross-road of signaling pathways involved in the regulation of glioblastoma migration and invasion by the neuropeptides VIP and PACAP. Cancers (Basel). 2019;11:123.
García-Morales V, Luaces-Regueira M, Campos-Toimil M. The cAMP effectors PKA and Epac activate endothelial NO synthase through PI3K/Akt pathway in human endothelial cells. Biochem Pharmacol. 2017;145:94–101.
Guerra DD, Bok R, Lorca RA, Hurt KJ. Protein kinase A facilitates relaxation of mouse ileum via phosphorylation of neuronal nitric oxide synthase. Br J Pharmacol. 2020;177:2765–78.
Zhang H, Kong Q, Wang J, Jiang Y, Hua H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp Hematol Oncol. 2020;9:32.
Maira SM, Galetic I, Brazil DP, Kaech S, Ingley E, Thelen M, et al. Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science. 2001;294:374–80.
Liu YP, Liao WC, Ger LP, Chen JC, Hsu TI, Lee YC, et al. Carboxyl-terminal modulator protein positively regulates Akt phosphorylation and acts as an oncogenic driver in breast cancer. Cancer Res. 2013;73:6194–205.
Chang JW, Jung SN, Kim JH, Shim GA, Park HS, Liu L, et al. Carboxyl-terminal modulator protein positively acts as an oncogenic driver in head and neck squamous cell carcinoma via regulating Akt phosphorylation. Sci Rep. 2016;6:28503.
Li W, Peng C, Lee MH, Lim D, Zhu F, Fu Y, et al. TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 2013;73:6938–50.
Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–8.
Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149:1098–111.
Wang G, Long J, Gao Y, Zhang W, Han F, Xu C, et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat Cell Biol. 2019;21:214–25.
Yang WL, Jin G, Li CF, Jeong YS, Moten A, Xu D, et al. Cycles of ubiquitination and deubiquitination critically regulate growth factor-mediated activation of Akt signaling. Sci Signal. 2013;6:ra3.
Goldbraikh D, Neufeld D, Eid-Mutlak Y, Lasry I, Gilda JE, Parnis A, et al. USP1 deubiquitinates Akt to inhibit PI3K-Akt-FoxO signaling in muscle during prolonged starvation. EMBO Rep. 2020;21:e48791.
Li R, Wei J, Jiang C, Liu D, Deng L, Zhang K, et al. Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res. 2013;73:5742–53.
de la Cruz-Herrera CF, Campagna M, Lang V, del Carmen G-S, Marcos-Villar L, Rodríguez MS, et al. SUMOylation regulates AKT1 activity. Oncogene. 2015;34:1442–50.
Yang Y, Liang Z, Xia Z, Wang X, Ma Y, Sheng Z, et al. SAE1 promotes human glioma progression through activating AKT SUMOylation-mediated signaling pathways. Cell Commun Signal. 2019;17:82.
Risso G, Pelisch F, Pozzi B, Mammi P, Blaustein M, Colman-Lerner A, et al. Modification of Akt by SUMO conjugation regulates alternative splicing and cell cycle. Cell Cycle. 2013;12:3165–74.
Lin CH, Liu SY, Lee EH. SUMO modification of Akt regulates global SUMOylation and substrate SUMOylation specificity through Akt phosphorylation of Ubc9 and SUMO1. Oncogene. 2016;35:595–607.
Villalobos-Ayala K, Ortiz Rivera I, Alvarez C, Husain K, DeLoach D, Krystal G, et al. Apigenin increases SHIP-1 expression, promotes tumoricidal macrophages and anti-tumor immune responses in murine pancreatic cancer. Cancers (Basel). 2020;12:3631.
Gkountakos A, Sartori G, Falcone I, Piro G, Ciuffreda L, Carbone C, et al. PTEN in lung cancer: dealing with the problem, building on new knowledge and turning the game around. Cancers (Basel). 2019;11:1141.
Luongo F, Colonna F, Calapà F, Vitale S, Fiori ME, De Maria R. PTEN tumor-suppressor: the dam of stemness in cancer. Cancers (Basel). 2019;11:1076.
Grzechnik AT, Newton AC. PHLPPing through history: a decade in the life of PHLPP phosphatases. Biochem Soc Trans. 2016;44:1675–82.
Cairns J, Fridley BL, Jenkins GD, Zhuang Y, Yu J, Wang L. Differential roles of ERRFI1 in EGFR and AKT pathway regulation affect cancer proliferation. EMBO Rep. 2018;19:e44767.
Yu J, Qin B, Wu F, Qin S, Nowsheen S, Shan S, et al. Regulation of serine-threonine kinase Akt activation by NAD(+)-dependent deacetylase SIRT7. Cell Rep. 2017;18:1229–40.
Li G, Ji XD, Gao H, Zhao JS, Xu JF, Sun ZJ, et al. EphB3 suppresses non-small-cell lung cancer metastasis via a PP2A/RACK1/Akt signalling complex. Nat Commun. 2012;3:667.
He X, Li M, Yu H, Liu G, Wang N, Yin C, et al. Loss of hepatic aldolase B activates Akt and promotes hepatocellular carcinogenesis by destabilizing the Aldob/Akt/PP2A protein complex. PLoS Biol. 2020;18:e3000803.
Chi RA, Wang T, Huang CL, Wu SP, Young SL, Lydon JP, et al. WNK1 regulates uterine homeostasis and its ability to support pregnancy. JCI Insight. 2020;5:e141832.
Reshi I, Nisa MU, Farooq U, Gillani SQ, Bhat SA, Sarwar Z, et al. AKT regulates mitotic progression of mammalian cells by phosphorylating MASTL, leading to protein phosphatase 2A inactivation. Mol Cell Biol. 2020;40:e00366-18.
Li M, Damuni Z. I1PP2A and I2PP2A. Two potent protein phosphatase 2A-specific inhibitor proteins. Methods Mol Biol. 1998;93:59–66.
Tseng LM, Liu CY, Chang KC, Chu PY, Shiau CW, Chen KF. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012;14:R68.
Guo B, Wu S, Zhu X, Zhang L, Deng J, Li F, et al. Micropeptide CIP2A-BP encoded by LINC00665 inhibits triple-negative breast cancer progression. EMBO J. 2020;39:e102190.
Chen Y, Xu T, Li M, Li C, Ma Y, Chen G, et al. Inhibition of SENP2-mediated Akt deSUMOylation promotes cardiac regeneration via activating Akt pathway. Clin Sci (Lond). 2021;135:811–28.
Xiao M, Bian Q, Lao Y, Yi J, Sun X, Sun X, et al. SENP3 loss promotes M2 macrophage polarization and breast cancer progression. Mol Oncol. 2021. https://doi.org/10.1002/1878-0261.12967.
Wakatsuki S, Saitoh F, Araki T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat Cell Biol. 2011;13:1415–23.
Suizu F, Hiramuki Y, Okumura F, Matsuda M, Okumura AJ, Hirata N, et al. The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev Cell. 2009;17:800–10.
Joo HM, Kim JY, Jeong JB, Seong KM, Nam SY, Yang KH, et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur J Cell Biol. 2011;90:420–31.
Kim HJ, Kim SY, Kim DH, Park JS, Jeong SH, Choi YW, et al. Crosstalk between HSPA5 arginylation and sequential ubiquitination leads to AKT degradation through autophagy flux. Autophagy. 2021;17:961–79.
Liao Y, Wei Y, Zhou X, Yang JY, Dai C, Chen YJ, et al. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene. 2009;28:2436–45.
Fujino T, Goyama S, Sugiura Y, Inoue D, Asada S, Yamasaki S, et al. Mutant ASXL1 induces age-related expansion of phenotypic hematopoietic stem cells through activation of Akt/mTOR pathway. Nat Commun. 2021;12:1826.
Deng R, Guo Y, Li L, He J, Qiang Z, Zhang H, et al. BAP1 suppresses prostate cancer progression by deubiquitinating and stabilizing PTEN. Mol Oncol. 2021;15:279–98.
Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, et al. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal. 2011;4:ra46.
Rokudai S, Fujita N, Hashimoto Y, Tsuruo T. Cleavage and inactivation of antiapoptotic Akt/PKB by caspases during apoptosis. J Cell Physiol. 2000;182:290–6.
Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502–8.
Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52.
Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKBbeta. J Clin Invest. 2003;112:197–208.
Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBbeta). Science. 2001;292:1728–31.
Heron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, et al. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol Cell Biol. 2006;26:8267–80.
Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ. Akt1 and Akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 2009;69:5057–64.
Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996;399:333–8.
Wiechmann S, Ruprecht B, Siekmann T, Zheng R, Frejno M, Kunold E, et al. Chemical phosphoproteomics sheds new light on the targets and modes of action of AKT inhibitors. ACS Chem Biol. 2021;16:631–41.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by proteinkinase B. Nature. 1995;378:785–9.
Zhu Q, Zhong AL, Hu H, Zhao JJ, Weng DS, Tang Y, et al. Acylglycerol kinase promotes tumour growth and metastasis via activating the PI3K/AKT/GSK3β signalling pathway in renal cell carcinoma. J Hematol Oncol. 2020;13:2.
Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–804.
Burgering BM, Medema RH. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J Leukoc Biol. 2003;73:689–701.
Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278:12361–6.
Huo X, Liu S, Shao T, Hua H, Kong Q, Wang J, et al. GSK3 protein positively regulates type I insulin-like growth factor receptor through forkhead transcription factors FOXO1/3/4. J Biol Chem. 2014;289:24759–70.
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–52.
Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–52.
del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–9.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41.
Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21.
Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11:11–23.
Romano D, Matallanas D, Weitsman G, Preisinger C, Ng T, Kolch W. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res. 2010;70:1195–203.
Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286:1741–4.
Majewski M, Nieborowska-Skorska M, Salomoni P, Slupianek A, Reiss K, Trotta R, et al. Activation of mitochondrial Raf-1 is involved in the antiapoptotic effects of Akt. Cancer Res. 1999;59:2815–9.
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–62.
Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–6.
Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, et al. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9:389–402.
Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, et al. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008;10:329–37.
Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, et al. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov. 2012;2:248–59.
Virtakoivu R, Pellinen T, Rantala JK, Perälä M, Ivaska J. Distinct roles of AKT isoforms in regulating β1-integrin activity, migration, and invasion in prostate cancer. Mol Biol Cell. 2012;23:3357–69.
Gao F, Alwhaibi A, Artham S, Verma A, Somanath PR. Endothelial Akt1 loss promotes prostate cancer metastasis via beta-catenin-regulated tight-junction protein turnover. Br J Cancer. 2018;118:1464–75.
Brolih S, Parks SK, Vial V, Durivault J, Mostosi L, Pouysségur J, et al. AKT1 restricts the invasive capacity of head and neck carcinoma cells harboring a constitutively active PI3 kinase activity. BMC Cancer. 2018;18:249.
Rao G, Pierobon M, Kim IK, Hsu WH, Deng J, Moon YW, et al. Inhibition of AKT1 signaling promotes invasion and metastasis of non-small cell lung cancer cells with K-RAS or EGFR mutations. Sci Rep. 2017;7:7066.
Saji M, Narahara K, McCarty SK, Vasko VV, La Perle KM, Porter K, et al. Akt1 deficiency delays tumor progression, vascular invasion, and distant metastasis in a murine model of thyroid cancer. Oncogene. 2011;30:4307–15.
Zhu QS, Rosenblatt K, Huang KL, Lahat G, Brobey R, Bolshakov S, et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene. 2011;30:457–70.
Kircher DA, Trombetti KA, Silvis MR, Parkman GL, Fischer GM, Angel SN, et al. AKT1(E17K) activates focal adhesion kinase and promotes melanoma brain metastasis. Mol Cancer Res. 2019;17:1787–800.
Cho JH, Robinson JP, Arave RA, Burnett WJ, Kircher DA, Chen G, et al. AKT1 activation promotes development of melanoma metastases. Cell Rep. 2015;13:898–905.
Dong Y, Zheng Q, Wang Z, Lin X, You Y, Wu S, et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J Hematol Oncol. 2019;12:112.
Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–34.
Gao SP, Kiliti AJ, Zhang K, Vasani N, Mao N, Jordan E, et al. AKT1 E17K inhibits cancer cell migration by abrogating β-catenin signaling. Mol Cancer Res. 2021;19:573–84.
Choi JA, Jung YS, Kim JY, Kim HM, Lim IK. Inhibition of breast cancer invasion by TIS21/BTG2/Pc3-Akt1-Sp1-Nox4 pathway targeting actin nucleators, mDia genes. Oncogene. 2016;35:83–93.
Chin YR, Toker A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Mol Cell. 2010;38:333–44.
Chin YR, Toker A. Akt2 regulates expression of the actin-bundling protein palladin. FEBS Lett. 2010;584:4769–74.
Agarwal E, Robb CM, Smith LM, Brattain MG, Wang J, Black JD, et al. Role of Akt2 in regulation of metastasis suppressor 1 expression and colorectal cancer metastasis. Oncogene. 2017;36:3104–18.
Grottke A, Ewald F, Lange T, Nörz D, Herzberger C, Bach J, et al. Downregulation of AKT3 increases migration and metastasis in triple negative breast cancer cells by upregulating S100A4. PLoS ONE. 2016;11:e0146370.
Hinz N, Baranowsky A, Horn M, Kriegs M, Sibbertsen F, Smit DJ, et al. Knockdown of AKT3 activates HER2 and DDR kinases in bone-seeking breast cancer cells, promotes metastasis in vivo and attenuates the TGFβ/CTGF axis. Cells. 2021;10:430.
Chung S, Yao J, Suyama K, Bajaj S, Qian X, Loudig OD, et al. N-cadherin regulates mammary tumor cell migration through Akt3 suppression. Oncogene. 2013;32:422–30.
Phung TL, Du W, Xue Q, Ayyaswamy S, Gerald D, Antonello Z, et al. Akt1 and Akt3 exert opposing roles in the regulation of vascular tumor growth. Cancer Res. 2015;75:40–50.
Galbraith LCA, Mui E, Nixon C, Hedley A, Strachan D, MacKay G, et al. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene. 2021;40:2355–66.
Moldogazieva NT, Mokhosoev IM, Terentiev AA. Metabolic heterogeneity of cancer cells: An interplay between HIF-1, GLUTs, and AMPK. Cancers (Basel). 2020;12:862.
Sano H. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–602.
Stöckli J, Fazakerley DJ, James DE. GLUT4 exocytosis. J Cell Sci. 2011;124:4147–59.
Berwick DC, Dell GC, Welsh GI, Heesom KJ, Hers I, Fletcher LM, et al. Protein kinase B phosphorylation of PIKfyve regulates the trafficking of GLUT4 vesicles. J Cell Sci. 2004;117:5985–93.
Hong SY, Yu FX, Luo Y, Hagen T. Oncogenic activation of the PI3K/Akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell Signal. 2016;28:377–83.
Novellasdemunt L, Tato I, Navarro-Sabate A, Ruiz-Meana M, Méndez-Lucas A, Perales JC, et al. Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) isoenzyme by amino acids. J Biol Chem. 2013;288:10640–51.
Song H, Li D, Wang X, Fang E, Yang F, Hu A, et al. HNF4A-AS1/hnRNPU/CTCF axis as a therapeutic target for aerobic glycolysis and neuroblastoma progression. J Hematol Oncol. 2020;13:24.
Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015;22:248–57.
Lincet H, Icard P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene. 2015;34:3751–9.
Ni C, Zheng K, Gao Y, Chen Y, Shi K, Ni C, et al. ACOT4 accumulation via AKT-mediated phosphorylation promotes pancreatic tumourigenesis. Cancer Lett. 2021;498:19–30.
Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277:33895–900.
DiPilato LM, Ahmad F, Harms M, Seale P, Manganiello V, Birnbaum MJ. The role of PDE3B phosphorylation in the inhibition of lipolysis by insulin. Mol Cell Biol. 2015;35:2752–60.
Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–81.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88.
Fang Z, Simard JR, Plenker D, Nguyen HD, Phan T, Wolle P, et al. Discovery of inter-domain stabilizers-a novel assay system for allosteric akt inhibitors. ACS Chem Biol. 2015;10:279–88.
Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e17.
Kostaras E, Kaserer T, Lazaro G, Heuss SF, Hussain A, Casado P, et al. A systematic molecular and pharmacologic evaluation of AKT inhibitors reveals new insight into their biological activity. Br J Cancer. 2020;123:542–55.
Quambusch L, Landel I, Depta L, Weisner J, Uhlenbrock N, Müller MP, et al. Covalent-allosteric inhibitors to achieve Akt isoform-selectivity. Angew Chem Int Ed Engl. 2019;58:18823–9.
Akhtar N, Jabeen I. Pharmacoinformatic approaches to design novel inhibitors of protein kinase B pathways in cancer. Curr Cancer Drug Targets. 2018;18:830–46.
Merckaert T, Zwaenepoel O, Gevaert K, Gettemans J. Development and characterization of protein kinase B/AKT isoform-specific nanobodies. PLoS ONE. 2020;15:e0240554.
Merckaert T, Zwaenepoel O, Gevaert K, Gettemans J. An AKT2-specific nanobody that targets the hydrophobic motif induces cell cycle arrest, autophagy and loss of focal adhesions in MDA-MB-231 cells. Biomed Pharmacother. 2021;133:111055.
He Y, Khan S, Huo Z, Lv D, Zhang X, Liu X, et al. Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J Hematol Oncol. 2020;13:103.
Song M, Bode AM, Dong Z, Lee MH. AKT as a therapeutic target for cancer. Cancer Res. 2019;79:1019–31.
Lang L, Shay C, Zhao X, Xiong Y, Wang X, Teng Y. Simultaneously inactivating Src and AKT by saracatinib/capivasertib co-delivery nanoparticles to improve the efficacy of anti-Src therapy in head and neck squamous cell carcinoma. J Hematol Oncol. 2019;12:132.
Lazaro G, Kostaras E, Vivanco I. Inhibitors in AKTion: ATP-competitive vs allosteric. Biochem Soc Trans. 2020;48:933–43.
Guo Y, Jin Y, Qu B, Zhang Y, Che J, Dong X. An updated patent review of Akt inhibitors (2016-present). Expert Opin Ther Pat. 2021. https://doi.org/10.1080/13543776.2021.1915291.
Schneeweiss A, Hess D, Joerger M, Varga A, Moulder S, Tsimberidou AM, et al. Phase 1 dose escalation study of the allosteric AKT inhibitor BAY 1125976 in advanced solid cancer-lack of association between activating AKT mutation and AKT inhibition-derived efficacy. Cancers (Basel). 2019;11:1987.
Smyth LM, Batist G, Meric-Bernstam F, Kabos P, Spanggaard I, Lluch A, et al. Selective AKT kinase inhibitor capivasertib in combination with fulvestrant in PTEN-mutant ER-positive metastatic breast cancer. NPJ Breast Cancer. 2021;7:44.
Ma CX, Suman V, Goetz MP, Northfelt D, Burkard ME, Ademuyiwa F, et al. A phase II trial of neoadjuvant MK-2206, an AKT Inhibitor, with anastrozole in clinical stage II or III PIK3CA-mutant ER-positive and HER2-negative breast cancer. Clin Cancer Res. 2017;23:6823–32.
Chien AJ, Tripathy D, Albain KS, Symmans WF, Rugo HS, Melisko ME, et al. MK-2206 and standard neoadjuvant chemotherapy improves response in patients with human epidermal growth factor receptor 2-positive and/or hormone receptor-negative breast cancers in the I-SPY 2 Trial. J Clin Oncol. 2020;38:1059–69.
Bang YJ, Kang YK, Ng M, Chung HC, Wainberg ZA, Gendreau S, et al. A phase II, randomised study of mFOLFOX6 with or without the Akt inhibitor ipatasertib in patients with locally advanced or metastatic gastric or gastroesophageal junction cancer. Eur J Cancer. 2019;108:17–24.
Kim SB, Dent R, Im SA, Espié M, Blau S, Tan AR, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18:1360–72.
Oliveira M, Saura C, Nuciforo P, Calvo I, Andersen J, Passos-Coelho JL, et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann Oncol. 2019;30:1289–97.
de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res. 2019;25:928–36.
Turner NC, Alarcón E, Armstrong AC, Philco M, López Chuken YA, Sablin MP, et al. BEECH: a dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann Oncol. 2019;30:774–80.
Jones RH, Casbard A, Carucci M, Cox C, Butler R, Alchami F, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020;21:345–57.
Ludmir EB, McCaw ZR, Kim DH, Tian L, Wei LJ. Fulvestrant plus capivasertib for metastatic breast cancer. Lancet Oncol. 2020;21:e233.
Jones RH, Casbard A, Carucci M, Foxley A, Howell SJ. Fulvestrant plus capivasertib for metastatic breast cancer—Authors’ reply. Lancet Oncol. 2020;21:e234.
Turner NC, Kingston B, Kilburn LS, Kernaghan S, Wardley AM, Macpherson IR, et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020;21:1296–308.
Schmid P, Abraham J, Chan S, Wheatley D, Brunt AM, Nemsadze G, et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: The PAKT Trial. J Clin Oncol. 2020;38:423–33.
Robertson JFR, Coleman RE, Cheung KL, Evans A, Holcombe C, Skene A, et al. Proliferation and AKT activity biomarker analyses after capivasertib (AZD5363) treatment of patients with ER+ invasive breast cancer (STAKT). Clin Cancer Res. 2020;26:1574–85.
Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19784 diverse solid tumors. JAMA Oncol. 2016;2:1565–73.
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91.
Wen L, Zhang G, Ren C, Li X, Mok H, Jia M, et al. Characterization of AKT somatic mutations in Chinese breast cancer patients. Cancer Manag Res. 2021;13:3055–65.
Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34:427–38.
Millis SZ, Jardim DL, Albacker L, Ross JS, Miller VA, Ali SM, et al. Phosphatidylinositol 3-kinase pathway genomic alterations in 60,991 diverse solid tumors informs targeted therapy opportunities. Cancer. 2019;125:1185–99.
Kumar S, Bal A, Das A, Loriya I, Khare S, Bhattacharya S, et al. Spectrum of PIK3CA/AKT mutations across molecular subtypes of triple-negative breast cancer. Breast Cancer Res Treat. 2021;187:625–33.
Hyman DM, Smyth LM, Donoghue MTA, Westin SN, Bedard PL, Dean EJ, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35:2251–9.
Kalinsky K, Hong F, McCourt CK, Sachdev JC, Mitchell EP, Zwiebel JA, et al. Effect of capivasertib in patients with an AKT1 E17K-mutated tumor: NCI-MATCH Subprotocol EAY131-Y Nonrandomized Trial. JAMA Oncol. 2021;7:271–8.
Smyth LM, Tamura K, Oliveira M, Ciruelos EM, Mayer IA, Sablin MP, et al. Capivasertib, an AKT kinase inhibitor, as monotherapy or in combination with fulvestrant in patients with AKT1 E17K-mutant, ER-positive metastatic breast cancer. Clin Cancer Res. 2020;26:3947–57.
Lee J, Kim ST, Kim K, Lee H, Kozarewa I, Mortimer PGS, et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY Umbrella Trial. Cancer Discov. 2019;9:1388–405.
Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2021;398:131–42.
Mulvey E, Ruan J. Biomarker-driven management strategies for peripheral T cell lymphoma. J Hematol Oncol. 2020;13:59.
Liu D. Cancer biomarkers for targeted therapy. Biomark Res. 2019;7:25.
Wolf DM, Yau C, Wulfkuhle J, Brown-Swigart L, Gallagher RI, Magbanua MJM, et al. Mechanism of action biomarkers predicting response to AKT inhibition in the I-SPY 2 breast cancer trial. NPJ Breast Cancer. 2020;6:48.
Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018;8:174–83.
Gris-Oliver A, Palafox M, Monserrat L, Brasó-Maristany F, Òdena A, Sánchez-Guixé M, et al. Genetic alterations in the PI3K/AKT pathway and baseline AKT activity define AKT inhibitor sensitivity in breast cancer patient-derived xenografts. Clin Cancer Res. 2020;26:3720–31.
Li X, Mak VCY, Zhou Y, Wang C, Wong ESY, Sharma R, et al. Deregulated Gab2 phosphorylation mediates aberrant AKT and STAT3 signaling upon PIK3R1 loss in ovarian cancer. Nat Commun. 2019;10:716.
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–43.
Bosse T, ter Haar NT, Seeber LM, v Diest PJ, Hes FJ, Vasen HF, et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol. 2013;26:1525–35.
Takeda T, Banno K, Okawa R, Yanokura M, Iijima M, Irie-Kunitomi H, et al. ARID1A gene mutation in ovarian and endometrial cancers. Oncol Rep. 2016;35:607–13.
Wiegand KC, Hennessy BT, Leung S, Wang Y, Ju Z, McGahren M, et al. A functional proteogenomic analysis of endometrioid and clear cell carcinomas using reverse phase protein array and mutation analysis: protein expression is histotype-specific and loss of ARID1A/BAF250a is associated with AKT phosphorylation. BMC Cancer. 2014;14:120.
Lee D, Yu EJ, Ham IH, Hur H, Kim YS. AKT inhibition is an effective treatment strategy in ARID1A-deficient gastric cancer cells. Onco Targets Ther. 2017;10:4153–9.
Deogharkar A, Singh SV, Bharambe HS, Paul R, Moiyadi A, Goel A, et al. Downregulation of ARID1B, a tumor-suppressor in the WNT subgroup medulloblastoma, activates multiple oncogenic signaling pathways. Hum Mol Genet. 2021. https://doi.org/10.1093/hmg/ddab134.
Pennino FP, Murakami M, Zollo M, Robertson ES. The metastasis suppressor protein NM23-H1 modulates the PI3K-AKT axis through interaction with the p110α catalytic subunit. Oncogenesis. 2021;10:34.
Belizaire R, Koochaki SHJ, Udeshi ND, Vedder A, Sun L, Svinkina T, et al. CBL mutations drive PI3K/AKT signaling via increased interaction with LYN and PIK3R1. Blood. 2021;137:2209–20.
Chan S, Smith E, Gao Y, Kwan J, Blum BC, Tilston-Lunel AM, et al. Loss of G-protein pathway suppressor 2 promotes tumor growth through activation of AKT signaling. Front Cell Dev Biol. 2021;8:608044.
Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–10.
Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575:210–6.
Hu N, Wang F, Sun T, Xu Z, Zhang J, Bernard D, et al. Follicular lymphoma-associated BTK mutations are inactivating resulting in augmented AKT activation. Clin Cancer Res. 2021;27:2301–13.
Zhou Z, Gong Q, Wang Y, Li M, Wang L, Ding H, et al. The biological function and clinical significance of SF3B1 mutations in cancer. Biomark Res. 2020;8:38.
Liu B, Liu Z, Chen S, Ki M, Erickson C, Reis-Filho JS, et al. Mutant SF3B1 promotes AKT- and NF-κB-driven mammary tumorigenesis. J Clin Invest. 2021;131:e138315.
Liu Z, Zhang J, Sun Y, Perea-Chamblee TE, Manley JL, Rabadan R. Pan-cancer analysis identifies mutations in SUGP1 that recapitulate mutant SF3B1 splicing dysregulation. Proc Natl Acad Sci USA. 2020;117:10305–12.
Ren Z, Shrestha M, Sakamoto T, Melkman T, Meng L, Cairns RA, et al. Opposing effects of NPM1wt and NPM1c mutants on AKT signaling in AML. Leukemia. 2020;34:1172–6.
Karimi Dermani F, Gholamzadeh Khoei S, Afshar S, Amini R. The potential role of nucleophosmin (NPM1) in the development of cancer. J Cell Physiol. 2021. https://doi.org/10.1002/jcp.30406.
Yang H, Xu D, Yang Z, Yao F, Zhao H, Schmid RA, et al. Systematic analysis of aberrant biochemical networks and potential drug vulnerabilities induced by tumor suppressor loss in malignant pleural mesothelioma. Cancers (Basel). 2020;12:2310.
Kingston B, Bailleux C, Delaloge S, Schiavon G, Scott V, Lacroix-Triki M, et al. Exceptional response to AKT inhibition in patients with breast cancer and germline PTEN mutations. JCO Precis Oncol. 2019;3:PO.19.00130.
Bougen-Zhukov N, Nouri Y, Godwin T, Taylor M, Hakkaart C, Single A, et al. Allosteric AKT inhibitors target synthetic lethal vulnerabilities in E-cadherin-deficient cells. Cancers (Basel). 2019;11:1359.
Massari G, Magnoni F, Favia G, Peradze N, Veronesi P, La Vecchia C, et al. Frequency of CDH1 germline mutations in non-gastric cancers. Cancers (Basel). 2021;13:2321.
Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21:122–37.
de Smith AJ, Lavoie G, Walsh KM, Aujla S, Evans E, Hansen HM, et al. Predisposing germline mutations in high hyperdiploid acute lymphoblastic leukemia in children. Genes Chromosomes Cancer. 2019;58:723–30.
Davis SJ, Sheppard KE, Anglesio MS, George J, Traficante N, Fereday S, et al. Enhanced GAB2 expression is associated with improved survival in high-grade serous ovarian cancer and sensitivity to PI3K inhibition. Mol Cancer Ther. 2015;14:1495–503.
Park YR, Bae SH, Ji W, Seo EJ, Lee JC, Kim HR, et al. GAB2 amplification in squamous cell lung cancer of non-smokers. J Korean Med Sci. 2017;32:1784–91.
Chernoff KA, Bordone L, Horst B, Simon K, Twadell W, Lee K, et al. GAB2 amplifications refine molecular classification of melanoma. Clin Cancer Res. 2009;15:4288–91.
Bocanegra M, Bergamaschi A, Kim YH, Miller MA, Rajput AB, Kao J, et al. Focal amplification and oncogene dependency of GAB2 in breast cancer. Oncogene. 2010;29:774–9.
Wong CK, Lambert AW, Ozturk S, Papageorgis P, Lopez D, Shen N, et al. Targeting RICTOR sensitizes SMAD4-negative colon cancer to irinotecan. Mol Cancer Res. 2020;18:414–23.
Singhi AD, Wood LD. Early detection of pancreatic cancer using DNA-based molecular approaches. Nat Rev Gastroenterol Hepatol. 2021;18:457–68.
Jee MJ, Yoon SM, Kim EJ, Choi HJ, Kim JW, Sung RH, et al. A novel germline mutation in exon 10 of the SMAD4 gene in a familial juvenile polyposis. Gut Liver. 2013;7:747–51.
Hashimoto Y, Yokoyama K, Kumagai H, Okada Y, Yamagata T. Juvenile polyposis syndrome-hereditary hemorrhagic telangiectasia associated with a SMAD4 mutation in a girl. Clin J Gastroenterol. 2020;13:1096–101.
Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–7.
Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–70.
Marchi S, Corricelli M, Branchini A, Vitto VAM, Missiroli S, Morciano G, et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J. 2019;38:e99435.
Polytarchou C, Hatziapostolou M, Yau TO, Christodoulou N, Hinds PW, Kottakis F, et al. Akt3 induces oxidative stress and DNA damage by activating the NADPH oxidase via phosphorylation of p47phox. Proc Natl Acad Sci USA. 2020;117:28806–15.
Koundouros N, Poulogiannis G. Phosphoinositide 3-kinase/Akt signaling and redox metabolism in cancer. Front Oncol. 2018;8:160.
Jan YH, Lai TC, Yang CJ, Lin YF, Huang MS, Hsiao M. Adenylate kinase 4 modulates oxidative stress and stabilizes HIF-1α to drive lung adenocarcinoma metastasis. J Hematol Oncol. 2019;12:12.
Covey TM, Edes K, Fitzpatrick FA. Akt activation by arachidonic acid metabolism occurs via oxidation and inactivation of PTEN tumor suppressor. Oncogene. 2007;26:5784–92.
Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–99.
Vidimar V, Gius D, Chakravarti D, Bulun SE, Wei JJ, Kim JJ. Dysfunctional MnSOD leads to redox dysregulation and activation of prosurvival AKT signaling in uterine leiomyomas. Sci Adv. 2016;2:e1601132.
Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21:363–83.
Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, et al. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009;28:2908–18.
Cockfield JA, Schafer ZT. Antioxidant defenses: a context-specific vulnerability of cancer cells. Cancers (Basel). 2019;11:1208.
Safdari Y, Khalili M, Ebrahimzadeh MA, Yazdani Y, Farajnia S. Natural inhibitors of PI3K/AKT signaling in breast cancer: emphasis on newly-discovered molecular mechanisms of action. Pharmacol Res. 2015;93:1–10.
Hua H, Zhang H, Kong Q, Wang J, Jiang Y. Complex roles of the old drug aspirin in cancer chemoprevention and therapy. Med Res Rev. 2019;39:114–45.
Fornari F, Giovannini C, Piscaglia F, Gramantieri L. Elucidating the molecular basis of sorafenib resistance in HCC: Current findings and future directions. J Hepatocell Carcinoma. 2021;8:741–57.
Yang S, Yang S, Zhang H, Hua H, Kong Q, Wang J, et al. Targeting Na+/K+-ATPase by berbamine and ouabain synergizes with sorafenib to inhibit hepatocellular carcinoma. Br J Pharmacol. 2021. https://doi.org/10.1111/bph.15616.
Maqbool SN, Lim SC, Park KC, Hanif R, Richardson DR, Jansson PJ, et al. Overcoming tamoxifen resistance in oestrogen receptor-positive breast cancer using the novel thiosemicarbazone anti-cancer agent. DpC Br J Pharmacol. 2020;177:2365–80.
Tsimberidou AM, Hong DS, Wheler JJ, Falchook GS, Janku F, Naing A, et al. Long-term overall survival and prognostic score predicting survival: the IMPACT study in precision medicine. J Hematol Oncol. 2019;12:145.
Stockley TL, Oza AM, Berman HK, Leighl NB, Knox JJ, Shepherd FA, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016;8:109.
Govindarajan M, Wohlmuth C, Waas M, Bernardini MQ, Kislinger T. High-throughput approaches for precision medicine in high-grade serous ovarian cancer. J Hematol Oncol. 2020;13:134.
Bertucci F, Gonçalves A, Guille A, Adelaïde J, Garnier S, Carbuccia N, et al. Prospective high-throughput genome profiling of advanced cancers: results of the PERMED-01 clinical trial. Genome Med. 2021;13:87.
Toledano-Fonseca M, Cano MT, Inga E, Rodríguez-Alonso R, Gómez-España MA, Guil-Luna S, et al. Circulating cell-free DNA-based liquid biopsy markers for the non-invasive prognosis and monitoring of metastatic pancreatic cancer. Cancers (Basel). 2020;12:1754.
Patel H, Okamura R, Fanta P, Patel C, Lanman RB, Raymond VM, et al. Clinical correlates of blood-derived circulating tumor DNA in pancreatic cancer. J Hematol Oncol. 2019;12:130.
Smyth LM, Reichel JB, Tang J, Patel JAA, Meng F, Selcuklu DS, et al. Utility of serial cfDNA NGS for prospective genomic analysis of patients on a phase I basket study. JCO Precis Oncol. 2021;5:PO.20.00184.
Ribas R, Pancholi S, Guest SK, Marangoni E, Gao Q, Thuleau A, et al. AKT antagonist AZD5363 influences estrogen receptor function in endocrine-resistant breast cancer and synergizes with fulvestrant (ICI182780) in vivo. Mol Cancer Ther. 2015;14:2035–48.
Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, et al. PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell. 2016;30:229–42.
Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170:605–35.
Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. 2018;560:499–503.
Wang Q, Yu WN, Chen X, Peng XD, Jeon SM, Birnbaum MJ, et al. Spontaneous hepatocellular carcinoma after the combined deletion of Akt isoforms. Cancer Cell. 2016;29:523–35.
Acknowledgements
We would like to thank Qiulin Tang for her assistance in preparing the manuscript.
Funding
This work was supported by Grant 81872388 from the National Natural Science Foundation of China.
Author information
Authors and Affiliations
Contributions
HH and YJ conceived the review and wrote the manuscript. HZ prepared the figures. JC and JW prepared the tables. JL edited the references. All authors participated in drafting and revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hua, H., Zhang, H., Chen, J. et al. Targeting Akt in cancer for precision therapy. J Hematol Oncol 14, 128 (2021). https://doi.org/10.1186/s13045-021-01137-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-021-01137-8