
Abstract
Due to their efficient recognition and lysis of malignant cells, natural killer (NK) cells are considered as specialized immune cells that can be genetically modified to obtain capable effector cells for adoptive cellular treatment of cancer patients. However, biological and technical hurdles related to gene delivery into NK cells have dramatically restrained progress. Recent technological advancements, including improved cell expansion techniques, chimeric antigen receptors (CAR), CRISPR/Cas9 gene editing and enhanced viral transduction and electroporation, have endowed comprehensive generation and characterization of genetically modified NK cells. These promising developments assist scientists and physicians to design better applications of NK cells in clinical therapy. Notably, redirecting NK cells using CARs holds important promise for cancer immunotherapy. Various preclinical and a limited number of clinical studies using CAR-NK cells show promising results: efficient elimination of target cells without side effects, such as cytokine release syndrome and neurotoxicity which are seen in CAR-T therapies. In this review, we focus on the details of CAR-NK technology, including the design of efficient and safe CAR constructs and associated NK cell engineering techniques: the vehicles to deliver the CAR-containing transgene, detection methods for CARs, as well as NK cell sources and NK cell expansion. We summarize the current CAR-NK cell literature and include valuable lessons learned from the CAR-T cell field. This review also provides an outlook on how these approaches may transform current clinical products and protocols for cancer treatment.
Similar content being viewed by others

Introduction
The human immune system has long been recognized as an important pillar of anticancer responses [1, 2]. As tumor cells originate from normal tissue, one of the main struggles in the design of anticancer therapies is the design of treatments that specifically target cancerous cells without affecting normal tissue. Ideally, these therapies have a strong potency against a particular malignancy that is characterized by rapid proliferation, resistance to apoptosis, and continuously changes through mutations and adaptations to the environment [3]. Recently, adoptive immune cell therapy (ACT) has shown promising clinical outcomes in patients with hematologic malignancies but remains difficult in treatment of solid tumors [4]. Unlike dispersed hematologic malignancies, solid tumors—especially late stage—exhibit an inhibitory tumor microenvironment due to hypoxia, a low pH, presence of suppressive cytokines, lactate, prostaglandins and others. These factors circumvent the ability of effector cells like NK cells to infiltrate and function in an optimal fashion [5]. Development of ACT should always be developed to overcome this induced tolerant state.
In ACT, cells are collected from autologous or allogenic donors and can be genetically engineered with a chimeric antigen receptor (CAR) that recognizing tumor-specific proteins on the cell surface. Often, this procedure also includes an ex vivo expansion step to obtain sufficient cell numbers [6, 7]. At this moment, the most successful form of ACT comprises of a CD19 CAR-T cell that cures up to 90% of patients with advanced refractory acute lymphoblastic leukemia (ALL). Here, complete remissions are attained within 4 weeks of treatment, as evident from several trials across multiple institutions [8, 9].
A CAR is composed of three domains: an ectodomain, the transmembrane region and a cytoplasmic activation tail [6, 10]. The ectodomain contains a single-chain variable fragment (scFv), usually derived from antibodies that provides the ability to specifically recognize tumor antigens expressed on cancerous cells [11]. The transmembrane domain anchors the CAR structure on the effector cell membrane [12]. Once the CAR recognizes and gets triggered by its specific antigen, the intracellular activation domain(s) of the CAR will signal, resulting in downstream processes that facilitate the killing of target cells [13, 14]. Numerous immune effector cells, like T cells, γδsT cells, natural killer (NK) cells, NKT cells, and macrophages, have been equipped with a CAR and were shown to be able to mediate anticancer responses in preclinical studies and clinical trials [4, 15, 16]. In 2017, the US Food and Drug Administration (FDA) approved the first two CAR-T treatments for CD19+ hematologic malignancies (axicabtagene ciloleucel and tisagenlecleucel) [1, 17].
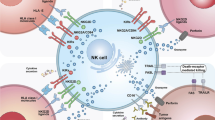
NK cells are innate immune effector cell that can rapidly identify and kill abnormal cells, virally infected cells and tumor cells [18]. In contrast to T cells, NK cells are lymphocytes that lack antigen-specific receptors, while abundantly expressing neural cell adhesion molecule (NCAM; also known as CD56). The unique mechanism of NK to distinguish pathologic cells from normal tissue cells is determined by the combination of surface stimulatory and inhibitory receptors that recognize a wide range of ligands on target cells [19]. T cells recognize peptide in the MHC (in human called human leukocyte antigen, HLA) molecules on an antigen-presenting cell (APC) and after receiving the proper danger signals, APC activate T cells that can kill MHC class I expressing tumor cells. However, tumor cells or stressed (e.g., virus-infected) cells often exhibit reduced or no expression of MHC class I molecules and thereby become susceptible to NK cell elimination [20]. MHC class I molecules bind a suite of inhibitory killer cell immunoglobulin-like receptors (KIRs). The inhibitory KIR cluster restrains NK cell activity and thereby prevents the damage to normal “self”-cells [21]. During NK cell development, the interaction between KIRs and autologous MHC molecules provides essential signals for NK cell maturation and contributes to the acquisition of functional competency, a process termed NK cell licensing [15]. This process is part of a sophisticated mechanism known as NK cell education, in which NK cells obtain functional competence and adapt to the host where they develop [21, 22]. NK cells, unlike T cells, can also become activated after antibody binding to the CD16 surface receptor. When an antibody binds to a specific tumor antigen, this complex is bound via the Fc tail to CD16 molecules on NK cells that become activated and will eliminate the tumor cells.
An interesting property of mature NK cells for adoptive cell therapy is that they can be transplanted into a new surrounding with different MHC expression patterns without losing their function [23, 24]. In great contrast to T lymphocytes, NK cells do not induce graft-versus-host disease but rather play a regulatory role in most cases (reviewed in [25]). With the development of genetic modification technologies, NK cells have been demonstrated they can be further tailored, including the introduction of CARs and knockout of inhibitory genes [26]. Using such techniques, NK cells from patients with hematologic malignancies can swiftly kill autologous tumor cells that were previously resistant to killing by the same NK cells without a CAR [11, 27]. Preclinical studies with CAR-NK cells unraveled in vivo activity similar to that of CAR-T cells in xenograft murine models. Interestingly, the CAR-NK groups show less cytokine release and better overall survival rates [28, 29]. A first CAR-NK study in men showed a promising anti-tumor response without the development of major toxic effects, such as cytokine release storm (CRS) and graft-versus-host disease (GVHD) [30]. These encouraging results pave the way for further development of CAR-NK as an attractive modality for cancer therapy [11].
In this review, we summarize information from preclinical and clinical studies, reporting on 72 CAR-NK cell line and 35 primary CAR-NK cell investigations. We try to interpret the paradigm for CAR-NK by focusing the design of a CAR and engineering of NK cells. We describe in detail the structure of the CAR, several detection methods of the CAR on the NK cells, the ideal NK cell source for CAR-NK therapy and the techniques for ex vivo expansion of NK cells. We also discuss the approaches to deliver the CAR-containing transgene to NK cells and the methods that have been used to enhance the transduction efficiency. Finally, we will provide a comprehensive outlook on how future CAR-NK-based therapies can be used to eradicate cancer.
A global overview of current CAR-NK cell studies
We found 72 CAR-NK cell line studies and 35 primary CAR-NK preclinical studies based on “chimerical antigen receptor and natural killer cells” from PubMed and Global data® (until March 2021). All these investigations introduce an external antigen binding motif derived from a tumor-targeting monoclonal antibody (moAb) into the cells. In Tables 1 and 2, we provide an overview of these studies using cell lines and primary NK cells, respectively. Due to space constraints, further details on the CAR design and study are made available online as interactive tables on www.carnkreview.com and are provided as Additional file 1: Table S1 and Additional file 2: Table S2.
The number of CAR-NK preclinical studies is increasing year by year, describing both CAR-NK cell lines and primary CAR-NK cells (Fig. 1a). In this review, we will zoom in on the various CAR elements and the techniques required to generate CAR-NK cells for clinical applications. At first glance, the overview of the current CAR-NK studies already shows interesting trends. In CAR-NK cell line studies, Her2 (expressed on a subset of breast cancer cells) is the most used target for solid tumors, while the CD19 antigen (B cell malignancies) is the most popular in hematological cancers (Fig. 1b). Of the primary NK cell studies, 65% use primary CAR-NK cells investigating B cell malignancies with CD19 as the most favorite target (Fig. 1c). Interestingly, the number of solid tumor CAR-NK cell lines studies are over 2 times higher than of hematological malignancies. In the following chapters, we will discuss the various components of that make up a successful CAR-NK cell.

CAR-NK cells: growing interest and diversity of tumor targets. a Bar graph showing the number of manuscripts reporting experimental data on human CAR-NK cells until December 2020. Stacked bars show the number of publications per year with CAR-NK cells derived from cell lines (black) and primary NK cells (pink). b Tumor targets for cell line-derived (black) and primary NK cell-derived (pink) CAR-NK cells. Each symbol represents one study with experimental data, with blood drops for hematological malignancies and rectangles for solid malignancies. Numbers per tumor type are shown, with annotated boxes highlighting the most important tumor targets
CAR structure design
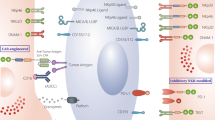
Functional CAR molecules expressed on NK cells consist of three parts: the ectodomain, transmembrane region and the endodomain (Fig. 2a). The ectodomain is made up of a signal peptide, the single-chain fragment variant (scFv) with a linker between the heavy chain and light chain and the hinge region that connects this structure to the transmembrane region. This latter region docks the CAR molecule to the cell membrane. It also connects intracellularly to the endodomain that encompasses the activating signals. The combination of these building blocks, together with regulatory elements that are situated outside the open reading frame (e.g., promotor), will determine the efficacy of the CAR. Successful CAR design is achieved by a combination of careful in silico design and functional testing. In this section, we will discuss the various building blocks of a CAR in detail and will summarize the current knowledge of successful combinations of these components. This information, often combined with software that assists in the analysis of expression levels, protein folding and spatial confirmation resulting from a certain DNA and protein sequence [31], will aid in successful CAR design. Before discussing CAR design, we will focus on the vector that carries the transgene and facilitates the insertion into the effector cell.

CAR-NK cell design. a Schematic overview of the building blocks for CAR-NK cells. From left to right: the extracellular single-chain variable fragment (scFv) consists linker-bound heavy and light chain and it determines the specificity of the CAR. The hinge region connects the tumor antigen-binding domain to the transmembrane region, ensures CAR stability and provides the flexibility for easy access to the target protein. The transmembrane region joins the extracellular and intracellular CAR domains, docks the CAR to the cell membrane and transduces activation signals to the intracellular activation signals. The number of intracellular activation domains determines the CAR generation. Various combinations of activating domains are used to mount a strong anti-tumor response. One example of a “fourth-generation” CAR is shown, co-expressing stimulating cytokines. Other examples of next-generation CAR constructs are discussed in Table 3. b and c Analysis of the most frequently used hinge (b) and transmembrane (c) regions for CAR-NK construction. Only studies reporting on the hinge region were included for this analysis. d Quantification of experimental studies with the indicated CAR-NK generations for cell line-derived (black) and primary NK cell-derived (pink) CAR-NK cells. e Pie charts showing the activation signals used for cell line-derived and primary cell CAR-NK cells, separated by CAR generation. Only the most frequently used combinations are shown
Vector backbone and promoter
The vector backbone contains all elements needed for expression of the CAR such as the promoter, the polyadenine signal and transcriptional regulation fragments.
As promoters initiate CAR transcription, the choice for a certain promoter has a direct influence on expression levels of the transgene. Furthermore, Allan et al. have recently reported that the size of the promoter influences the viral transduction efficiency and subsequent GFP expression in a negative correlation [32]. In their study, the shorter EFS, PGK, SV40 and CMV promotors are good promoters that can maintain the GFP expression for 17 days [32]. CAR genes are usually introduced using viral plasmids with an exogenous promoter. Currently, there is only one comparison of different promotors on CAR expression and function in NK cell lines published, while no comparative data are available for primary NK cells [33]. From the single report on CAR-NK cells, no definitive statements can be made about the best promotor for CAR-NK cells [33]. In primary T cells, various comparisons of the effect of promoters on CAR expression levels were performed. One study shows that the EF1α promotor induced higher CAR expression levels compared to other commonly used promotors, such as CMV, UbiC and PGK [34]. Another study showed that the MSCV outperformed both EF1α and PGK in terms of expression levels and CAR expression stability [35]; however, the promotor efficacy may be influenced by the scFv sequence itself [36] (see section Single-chain fragment variant (scFv)). Given these and other studies [37] that report different optimal promoters, it is important to determine the optimal promoter for a given construct (that may also contain additional elements that influence expression levels, such as reporters) and viral vector. The latter is important, since the choice of a promoter also influences the virus titer. For instance, one study reports that the CMV promoter sequence yielded higher virus titers compared to RSV promoter sequences when located within the SIN 5′ LTR lentiviral vector [38]. Besides baseline CAR expression in the immune cell, the dynamic ubiquitination, down-regulation and re-expression kinetics following antigen exposure will have large impact on the efficacy of CAR cells [39].
Compared with the retroviral LTR promoter-based CAR that integrates “randomly,” including in potential oncogenic sites, it may be desirable to make use of site directed insertion into a target cell specific promotor area. An interesting example is the use of the endogenous TRAC promotor where the CAR gene was deliberately integrated into the TCR alpha chain gene locus [40]. This concept was already introduced as a way to avoid GVHD in T cell-based adoptive cell therapy [41]. Eyquem and colleagues took this a step further by demonstrating that CAR-transduced lymphocytes integrated in the TRAC locus persist longer in murine tumor models [40]. In addition, more effector cells infiltrated into the tumor and exhibited a less exhausted cell phenotype [40]. TRAC-CAR-T outperformed both conventional retroviral transduced CAR T cells (RV-CAR) as well as T cells with the CAR inserted in the β2M locus (β2M-CAR) in an in vivo mouse model, based on tumor eradication and expression of exhaustion markers, including PD1, TIM3 and LAG3 [40]. In addition, TRAC-CAR increased CAR expression levels after antigen stimulation, while CARs driven by an exogenous promotor showed important downregulation. Although this CAR expression under an endogenous NK cell-specific promoter has not been reported, GFP or HA epitopes have successfully been expressed in primary NK cells by CRISPR/Cas9-mediated knock-in into the CLTA [42], AAVS1 [43], CD96, ACTB and RAB11A gene loci [26]. Thus, the use of endogenous promotors should also be further explored in the CAR-NK cell field.
Current reports on CAR-NK cells show a wide variety of promoters being used for driving CAR expression, both for cell line-derived and primary NK cells. In both primary CAR-NK and CAR-NK cell lines, viral promoters (CMV, MPSV, MMLV, SFFV, etc.) are more frequently used to generate CAR-NK than constitutively active promoters, such as EF1α, CMV and PGK (80% and 78% for primary and cell line CAR-NK, respectively; Additional file 1: Table S1 and Additional file 2: Table S2).
Signal peptide for CAR-NK
The CAR sequence starts with a short signal peptide (SP). These are short peptides located at the N terminus of proteins, carrying information for protein post-translational assembly in the endoplasmic reticulum (ER) and Golgi organelle and expression on the membrane [44]. The final CAR protein on NK cells will be a type I transmembrane protein that anchors on the cell membrane. CAR, like many other proteins, is also secreted by immune cells via the co-translational translocation pathway [45]. In eukaryotic cells, SP located at the N terminus of nascent proteins are identified by the signal recognition particle (SRP) while the protein is still translating in the ribosome. After the nascent protein crosses the ER membrane, the signal peptide is cleaved off by a signal peptide peptidase (SPP), and then protein assembly and folding will commence [46].
There is enormous heterogeneity in signal peptides, which directly translates into different levels of protein secretion efficiency. For both CAR-NK and CAR-T cells, we did not find comparative studies that determined the optimal signal peptide for CAR expression and function. At present, CD8a SP is the most used peptide sequence for primary NK (16%, with data missing in 71% of the studies; Additional file 2: Table S2) and immunoglobulin heavy or light chain SP for NK cell lines (29%) (Additional file 1: Table S1). While the SP sequence is likely to influence CAR expression levels, there are currently no data to support claims about the optimal SP for CAR-NK generation.
Single-chain fragment variant (scFv)
The single-chain fragment variant (scFv), a fusion protein of the variable regions of the heavy and light chain of an antibody, is the tumor antigen-binding domain of the CAR. Different scFv could bind to different epitopes of the same protein, and therefore, this domain will determine both the specificity and the function of the CAR-NK cell [47]. For instance, targeting Mucin 1 (MUC1) which is a glycosylated, abundant protein, many antibodies have been described and various scFvs have been created in CAR constructs [48, 49]. Diverse scFvs lead to various anti-tumor capacities but also distinct side effects, such as binding to normal tissue. Therefore, the target of a CAR needs to be selected carefully to be specific for the tumor-associated antigen, while at the same time not recognizing self-antigens that could induce severe side effects [1].
However, the numerous efforts undertaken to find a suitable target on solid tumors illustrate how difficult the process of scFv selection can be. This is largely due to the expression of most so-called tumor-associated antigens on both tumors and—often at lower levels—in healthy tissue. There already have been reports of the potential damaging effects of CAR-engineered T cells, such as the 4D5-scFv anti-ERBB2 [50] and G250-scFv anti-CAIX [51].
As the scFv is not a naturally occurring form of an antibody, it is always synthesized, and therefore the order of the heavy and light chain is artificially determined. Thus far, most groups prefer a VH-VL orientation over VL-VH for CAR-NK design (Additional file 1: Table S1 and Additional file 2: Table S2). Fujiwara et al. demonstrated that the order of H and L chain did not influence the expression level of anti-KDR CAR expression on T cells [36]. In addition, scFv performance in terms of antigen specificity and association constant in a CAR is sometimes found to be lower compared to its originating antibody, mostly due to the changed connectivity of the VL and VH domains. This may also have implications for the dynamics and relative orientation of the two chains. Computational-assisted design of a scFv may help in the development of functional scFv by analyzing the amino acid sequence of the CDR region of the VH and VL, and predicting the interaction of the scFv with its target [52]. Interestingly, VH and VL chains from different antibodies against the same epitope can also be mixed, sometimes leading to increased CAR affinity [53].
The importance of scFv design was underlined by a study that shows distinct scFvs targeting the same antigen may yield very different results in terms of CAR expression [36]. In this study, CAR-T cells that expressed functional CARs were detected, but also CAR-T cells with no or very low levels of antigen binding. Here, changes in the CDR region accounted for the observed differences, while there were no effects due to a specific VH/VL order [36]. Another example was published very recently and showed that from the 8 differently derived scFvs specific for CD19 when lentiviral transduced as CAR into the NK cell line KHYG-1, only two of them were efficiently expressed on the cell surface and showed improved cytotoxicity towards CD19 + tumor cell lines [54]. However, whether this was due to the specific sequence of the scFv as the CDR3 regions do vary, or a different spacer and transmembrane domain remains to be elucidated [54].
In addition, immune cells can be equipped with multiple scFv, thus broadening the antigen recognition capacity of a CAR effector cell. Here, there are various options: CARs can be transduced with vectors with bi-cistronic elements, inducing expression of two CAR constructs that may or may not have the same intracellular structure. In addition, two scFvs can be fused together in one construct, generating a tandem scFv or “single stalk” CAR. While these techniques have been employed for the generation of CAR-T cells [55], we are not aware of such attempts for generating CAR-NK cells.
Most current clinical CAR T cell trials have utilized scFv derived from murine antibodies, thereby increasing the risk for an anti-mouse IgG cell host-versus-graft disease, resulting in toxicity or shortened persistence of CAR-bearing cells [6]. This issue may be attenuated by humanizing murine scFv or by deriving scFv from fully human antibodies. In previous studies that employed murine scFv-based anti-CD19-CAR or anti-CD5-CAR, the NK-92 cells exhibited strong anti-tumor activity to ALL malignant cells both ex vivo and in a mouse model [56, 57]. An anti-GD2-CAR with humanized scFv showed similar expression levels as its murine counterpart, while increasing cytokine release and T cell proliferation [58]. Unfortunately, owing to the chimeric character of these CAR receptors, even humanized scFv constructs still may induce a host anti-idiotype immune responses [59]. Fortunately, in the limited number of CAR-NK clinical trials so far, no major side effects related to the anti-CAR immune responses were found [30]. Above all, from these studies, both murine and humanized svFv derived CAR-NK exhibit robust cytotoxicity against antigen-expressing tumor cells both in vitro and in vivo.
Linkers
The linker between the heavy and light chain contributes to the conformation of the scFv and therefore partly determines how well a CAR recognizes the target epitope. Linkers that are too short induce the formation of multimers, because they prevent association of the VH and VL domains. Too long domains, on the other hand, may induce proteolysis or weak association between the VH and VL domains [6]. For CAR-NK cells, multimers of the pentapeptide GGGGS (glycine-serine) are most widely used, often as a 15-mer (G4S)3. Another linker designed to enhance proteolytic stability is the Whitlow “218” linker: GSTGSGSKPGSGEGSTKG. In one study, this linker enhanced scFv affinity, possibly altering CAR-NK cell function as well [60]. Fujiwara et al. compared G4S and 218 linkers, but, in their anti-KDR-CAR-T, there were no expressional and functional differences between (G4S)5, (G4S)3 and 218 linkers [36].
Currently, there are insufficient data available to draw conclusions about the best linker for CAR. We found that 18 studies used G4S linkers, whereas 2 publications applied 218 linkers among the CAR-NK cell lines (Additional file 1: Table S1). While most primary CAR-NK reports did not provide specifics on the linker, all 4 studies that provided these details used G4S linkers to generate CAR-NK (Additional file 1: Table S2).
Hinge region (CD8α, Ig CH2CH3)
The hinge region, also referred to as a spacer, is the extracellular structural region of the CAR that connects the scFv units to the transmembrane domain. These spacers generally maintain the stability required for robust CAR expression and activity in effector cells. The hinge region also supplies flexibility to allow easy access to the targeted antigen. The majority of CAR-NK constructs use derivatives of CD8α or CD28 extracellular domains or immunoglobulin G (IgG)-based hinges (Fig. 2b). Accumulating evidence shows that the type and length of the spacer importantly influence the functional activity of the CAR [6]. As no specific data are available for CAR-NK cells, this part of information is all derived from the CAR-T field, and it remains to be shown if this can be directly translated to CAR-NK design.
In a direct comparison between the CD28 and CD8α hinge domains, it was found that CD28 hinge domains are more likely to promote dimerization of CAR molecules [61]. As a result, the activation stimulus derived from CD28 hinge-bearing CARs is stronger [61]. While this may be beneficial, this could also induce cytokine release syndrome, one of the most severe side effects of CAR-based therapy. Indeed, in a clinical study aiming to improve the safety of anti-CD19-CAR-T therapy, it was found that an optimized CD8α hinge region, together with other modifications, importantly improved the safety profile of the therapy [31].
Immunoglobulin G-based hinges are also widely used in CAR constructs. A major advantage of an IgG-based hinge region is the flexibility of the construct that is typically composed of IgG1 or IgG4 and the CH2/CH3 domains of IgG Fc. The use of the latter domains is optional and thus allows for modulation of the length of the spacer. While a wide range of spacer lengths will accommodate antigen recognition, studies have found that shorter spacers were associated with increased cytokine production, CAR-T cell proliferation and better persistence and antitumor effects in vivo [62].
For CAR-NK cells, we found that most studies employ a CD8α hinge region, both in primary NK cell (16/35) and CAR-NK cell lines (41/72) (Fig. 2b). Other spacers, such as CD28, IgG Fc domains and DAP12 were also used in CAR-NK.
Transmembrane domain (CD3, CD8, CD28, NKG2D, 2B4)
The transmembrane (TM) domain connects the ectodomain of the CAR to the intracellular activation signaling domains and docks the receptor to the NK cell membrane. The most commonly used TM parts of CAR-NK have been adapted from CD3ζ, CD8 and CD28, but others (e.g., NKG2D, 2B4, DNAM1) have been explored as well (Fig. 2c). The choice of the TM domain was shown to influence the functionality of the CAR construct in the extent of cell activation. The TM domain of CD28, CD16, NKp44, NKp46, NKG2D, DNAM-1 and 2B4 have been used to screen for CAR function using the NK-92 cell line. Interestingly, the TM from molecules typically expressed on NK cells, like DNAM-1, 2B4 and NKG2D leads to more CD107a degranulation and higher cytotoxicity. Thus, the specific source of the TM will determine the activity of CAR-NK [28].
One important aspect of the TM domain is that the optimal TM region should follow the natural orientation (N- to C-terminal order) of the protein from the transmembrane protein on the T cells or NK cells [63]. In this paper, Guo et al. addressed the use of the NK-native molecule NKG2D as a TM region and activator signal for NK-92 cells [63]. NKG2D is a powerful activator of NK cells and thus constitutes a suitable backbone for NK-focused CAR design. However, natural NKG2D has the transmembrane region in the C- to N-terminal order with a short cytoplasmic tail. Interestingly, the use of a signal peptide to forcibly reverse the NKG2D TM region to the N- to C-terminal order, showed in combination with 2B4 and DAP10 the strongest cytotoxic effects in iPSC-derived CAR-NK cells [28]. But whether this is really due to the reversed order alone cannot be concluded as the Kaufmann group focused on identifying the best combination of intracellular signaling domains.
These CAR-NK studies underline that it is important to consider the TM region in its function as a linker to the intracellular activation signal, as the type of TM may influence signal transduction and CAR dimerization. In CAR-T cells, CD28-derived TM domain was shown to promote activation-induced cell death (AICD) and cytokine production, whereas CARs with a CD8α-derived TM assisted in CAR dimerization with endogenous TCRs, leading to first activation signal of T cells [61]. At this moment, CD8α and CD28-dervied TM are most popular in primary CAR-NK cells, while CD28 was the preferred TM region for CAR-NK cell lines (Fig. 2c).
Activation signal for CAR-NK
CARs are often identified by their respective generation. The number of intracellular activating signals determines this “generation”: First-generation CARs have one activating signal, second-generation CARs have two, and so forth (Fig. 2a, d).
The activating domains are responsible for the activation of the NK cell upon recognition of the target antigen. In contrast to T lymphocytes, NK cells use a variety of different, non-rearranging receptors for activation. This also includes a large variety of cytokine receptors that are important regulators of development, maturation and activation of NK cells [64]. Most of these receptors share common adapter molecules and signaling pathways. For instance, NKp30 and CD16 both signal via CD3ζ [65]. The cytokines IL-2, IL-7, IL-12, IL-15, IL-18, IL-21, IL-27, and IFN-α/β signal via the highly conserved JAK/STAT pathway [64]. Thus, even though the first activation signal in T cells is provided through a clonally rearranged antigen receptor, many of the downstream signaling pathways are shared between T and NK cells. Therefore, some insights from the CAR-T field also apply to CAR-NK cells. At the same time, it is important to realize that CAR-based signaling differs significantly from canonical activation pathways. For instance, in normal T lymphocytes, activation depends on the stabilization of multimolecular complexes (TCR-peptide-MHC with co-receptors) and recruitment of co-stimulatory molecules, whereas these signals are provided "in line" in CAR-equipped cells. This has direct consequences for the temporal availability of the various activating molecules involved. We have only just begun to grasp the importance of these differences between canonical signaling and CAR signaling. The findings from the CAR-T field (summarized by Lindner and colleagues [66]) will again serve as an important starting point for studying these differences in CAR-NK cells as well.
First-generation CAR-NK cells, like CAR-T cells, only contain the CD3ζ signal. The second- and third-generation CAR-NK bear one and two additional co-stimulatory signals, respectively. The costimulatory molecules are usually derived from the CD28 family (including CD28 and ICOS), the tumor necrosis factor receptor (TNFR) family of genes (including 4-1BB, OX40 and CD27) or signaling lymphocytic activation molecule (SLAM)-related receptor family (comprising 2B4) [67]. To these activating domains, safety switches can be added to quickly eliminate the infused CAR-containing cells in the case of adverse reactions occur, such as the CRS [30, 68] (this aspect will be further discussed in the Prospective and outlook section). In addition to this example, other modifications to the effector cells have been developed that improve persistence, enhance tumor activity, prevent antigen escape, allow for control of CAR expression or combinations of these (Table 3). An in-depth discussion of these next-generation CAR constructs is beyond the scope of this review, also because almost all development took place in the CAR-T field. The only published CAR-NK clinical trial so far employed a second-generation CAR-NK construct that was enhanced with IL-15 expression and inducible Caspase 9 [30]. CAR-T cells that include cytokines enhancing persistence and anti-tumor activity are popularly referred to as fourth-generation CARs (Fig. 2a) or TRUCKs: T cells redirected for antigen‐unrestricted cytokine‐initiated killing. Two recent examples of CAR-NK enhancements show that the CAR-NK field follows these developments, adapting T cell-specific signals to NK cells where needed. A first example of the translation of a CAR-T cell enhancements to CAR-NK cells was recently provided by Wang and colleagues [69]. They developed CAR-NK cells with a protein switch that enhances NK cell proliferation and survival, while at the same time coupling CAR expression with ectopic IL-15 expression and an inducible suicide gene [69]. In recently published other study, the expression of a CAR/IL-15 construct (“fourth-generation CAR”) is coupled with CRISPR/Cas9-mediated knockout of CIS, a negative regulator of IL-15 signaling [70]. This interesting strategy boosted CAR-NK cell function in vitro and in xenograft models, at least in part through increased aerobic glycolysis. This double enhancement of IL-15 signaling is likely to be beneficial in the tumor microenvironment that is often limited in IL-15 levels [70]. Modification of cytokine signaling in CAR-NK cells may thus provide further possibilities for CAR-NK cell improvement. For instance, cytokines like IL-12, IL-15, IL-18 are responsible for the induction of memory-like NK activity following CMV infections [71, 72] or even anticancer responses [73]. Future studies will provide more insight in the other enhancement strategies work best for CAR-NK cells, helping them to overcome challenges posed by the tumor microenvironment (also see section Challenges).
Most current CAR configurations depend on the CD3ζ chain signaling domain, yielding 3 immunoreceptor tyrosine-based activation motifs (ITAMs) per CAR. In turn, these ITAMs will recruit and activate the Syk or ZAP70 tyrosine kinases, or induce PI3-kinase signaling [22]. As CAR-NK cells are usually also designed around the CD3ζ domain, and again specific studies regarding activation signals in CAR-NK cells are currently lacking, we must rely on findings from the CAR-T field regarding the molecular makeup of downstream activation.
Strong activation signals are important to induce a potent anti-tumor response, but can also lead to quick exhaustion of the effector cells. Therefore, the combination of co-stimulatory domains can be used to calibrate the desired immune cell response. Compared to 4-1BB-based CARs, CD28-based CARs exhibit an effector profile that is faster and provokes larger-magnitude changes in lymphocyte-specific protein tyrosine kinase (Lck) phosphorylation in the signaling pathway [74]. In this way, CD28-triggered signals induce higher levels of interferon-γ (IFN-γ), granzyme B, tumor necrosis factor α (TNF-α) [74]. However, it is known from CD28-based CAR-T cells that this strong co-stimulatory signal also causes activation-induced cell death (AICD) and that the weaker CD3ζ ITAM signals lead to better CAR-T cell function and longer persistence [75, 76]. On the other hand, 4-1BB-CD3ζ signals preferentially induce memory-associated genes and sustained antitumor activity [74]. The reason may be that antigen-independent tonic signaling through CD28 domains increases T cell exhaustion while the presence of the 4-1BB domain ameliorate this [77]. The implications of tonic signaling through CAR structures were covered in an excellent review by Ajina and Maher [78].
Moreover, activation signals may have an impact on the metabolism in immune cells. CD28-CAR cells are predominantly dependent on glycolytic metabolism, while 4-1BB-containing CAR cells exhibited superior persistence as a result from increased oxidative metabolism [79]. It was recently found that calibration of the CD3 ITAM region importantly changes the functional phenotype of CAR-T: Introduction of mutations in different parts of the CD3ζ ITAM motifs leads to a naïve-like T cell phenotype, with high proliferative capacity and longer persistence [76]. On the other hand, NK cells may be induced into cells with a memory-like phenotype after activation with IL-12, IL-15 and IL-18 [80]. When an anti-CD19 CAR was introduced in NK cells activated a memory phenotype, these cells showed enhanced in vitro and in vivo anti-tumor activity [80].
Another study shows that there is a lot of room for improvement of the activation signals in CAR cells. In CAR-equipped T cells, proximal signaling downstream of the antigen receptor was significantly reduced compared to normal T cells, due to inefficient recruitment of ZAP-70, resulting in important deficiencies in downstream signaling [81]. Thus, the choice of activation signal will have great impact on the functional and persistence of CAR-bearing cells. As these studies were conducted in T cells, there is an important need for structured analysis of the optimal combination of activation domains for NK cells.
We found 72 publications describing CAR-NK cell lines using 90 constructs and 35 studies with 43 different constructs using primary CAR-NK cells (Fig. 2e). Both in CAR-NK cell line and primary CAR-NK cell studies, CD3ζ is almost universally used as the main activation domain of which about half carry one additional domain, usually adding 4-1BB or CD28. As for third-generation constructs, combination of CD28/4-1BB/CD3ζ is most often used. These constructs thus provide a means for NK cells to directly receive co-stimulation signals once the CAR binds the tumor antigens. Given the disparities in both antigen sensitivity and number of ITAMs, it is plausible that the smaller number of ITAMs per CAR receptor could result in a comparatively longer persistence of CAR-T cells [76].
To our knowledge, the only direct comparison of different activation signals in CAR-NK was performed by Li et al.[28]. In that study, among 9 different constructs, a 2B4-CD3ζ-based CAR-NK construct showed the most robust CD107a degranulation and antigen specific cytotoxicity. Moreover, a point mutation of Arg-to-Ala in the transmembrane or Tyr-to-Phe in the ITAM/ITSM of NKG2D-2B4-CD3ζ construct sections reduces phosphate recruitment of downstream activation molecules like pSyk, pPLC-γ2 and pERK1/2. This will decrease degranulation, cytokine release and antigen-specific lysis capacities of the CAR-NK cells. This indicates that the transmembrane and intracellular domains are also crucial parts that determine CAR-NK cell functionality.
In addition, the combination CD28-CD3ζ was also shown to provide strong activation signals and is able to support CAR-NK cell survival for one year in vivo in the first published clinical trial with CAR-NK cells [30]. In general, the CD3ζ domain is most often used (Fig. 2e), but it still remains to be determined which is the best combination of domains, in which order and in which situation.
Detection tags (GFP, cMyc-tag, FLAG, LNGFR)
Introduction of molecular tags and fluorescent proteins together with CAR genes allows for easy approaches to enrich, quantify and trace the CAR-NK. For instance, various groups use c-Myc or Flag tags directly situated before or after the scFv, which are then expressed together with the CAR on NK cells [82]. Fluorescent proteins are also commonly used in CAR-NK plasmids using bi-cistronic elements, and allow researchers to conveniently trace the expression level of CAR in NK cells [83]. The disadvantage of this latter system is that the detection of the tag does not always reflect the CAR expression (our own unpublished data). A second issue in this case is that fluorescent proteins and other big epitopes are foreign proteins to the human body and could thus induce an immune response directed to this exogenous epitope. Therefore, these tags are generally used in the laboratory setting and are removed when clinical studies are started.
Synthetic biology: codon optimization and scFv humanization
Codon optimization is a technique used to alter the use of nucleotides without changing the amino acid sequence. For CAR-NK design, many researchers rely on the use of DNA synthesis for some of the building blocks for their plasmids. Because of technical limitations during synthesis, it may be necessary to change the DNA sequence to allow for successful production. Indeed, this codon optimization was mentioned in 8 out of 72 CAR-NK cell line and 5 out of 33 primary CAR-NK reports. It is important to realize that the use of a certain codon set may improve CAR-NK expression in mammalian cells, but may have negative effects on virus production [84]. In one direct comparison of CAR-T cells with and without codon optimization, no differences were found in CAR surface expression, tumor eradication and cytokine production [85].
The starting point for the development of new CARs often is murine antibodies recognizing a tumor-associated antigen. However, murine antibodies cannot directly be used for clinical use, as the non-human regions of the mouse antibody are often immunogenic. Therefore, these regions can be replaced with corresponding human sequences in a process called “humanization.” Indeed, this technique is also successfully applied for the development of CAR scFv without negative effects on CAR-NK killing capacity [58, 86].
Transfection or transduction vehicle for CAR expression
With the progress in gene modification technologies, numerous approaches have been applied to generate CAR-NK. The two main methods are viral transduction (using lenti- or retroviruses), or transfection with either naked plasmid DNA, transposase DNA-mediated integration or mRNA by electroporation [14] (Table 4). A total of 64 preclinical studies describing CAR-NK cell lines using viral transduction and 11 studies applying electroporation or nanoparticle-mediated transfection have been published to date. In primary CAR-NK cell studies, 29 described viral transduction and 9 publications conducted electroporation as method of choice (Additional file 1: Table S1, Additional file 2: Table S2 and Fig. 3e). Monoclonal or polyclonal NK cell lines can be generated using FACS or (Clini-) MACS, but this creates a more complex good manufacturing practice (GMP)-compliant production process. An important difference between the various technologies is the duration of stable CAR expression. For longer expression (multiple weeks) in primary cells, viral transduction is usually employed, while mRNA electroporation results in transient expression lasting for about one week. CAR expression levels in primary NK cells strongly vary (20–70%) and are thus sometimes low, but most studies still report highly specific and effective killing of target-positive tumor cells.

Optimization strategies for CAR-NK cell generation. a Different vectors can be used to transduce NK cell lines and primary NK cells with CAR constructs. Quantification of the vectors used for CAR-NK generation. b Details of the various vectors used for CAR-NK generation. In blue, lentivirus generation, with third-generation viruses considered the safest option with minimal risk of in vivo recombination with other lentiviruses. In purple, the genus of retrovirus (alpha or gamma, or unknown). In green, the electroporated vector. c Various compounds have been identified that can be used to enhance virus-mediated transduction. For studies that employ virus-based strategies for CAR-NK generation, the methods used for enhancing virus transduction were quantified. d Primary cells for generation of CAR-NK cells are derived from different sources. Quantification of the four sources for primary NK cells for CAR-NK generation
Lentiviruses
Lentiviruses have been widely used in the gene therapy space for decades, as they are able to transduce cycling and non-cycling cells with high transduction efficiency. They have been successfully applied in curing people from diseases including HIV and cancer [87]. Lentiviruses have the advantages that they integrate into the host genome, a feature that can lead to permanent expression of the transgene, and low intrinsic immunogenicity [88]. To date, there are 14 reports on primary CAR-NK cells and 44 papers on CAR-NK cell lines that have successfully used a lentivirus as vehicle (Fig. 3a). Among preclinical studies, 21 studies used a second-generation virus and 6 studies used a third-generation lentivirus to generate a CAR-expressing NK cell line (17 unknown) (Fig. 3b). In the primary CAR-NK cell studies, 5 studies used third-generation lentivirus while 7 reports describe second-generation lentiviral vectors (2 unknown) (Fig. 3b). The third-generation lentiviral vectors are being considered safer than the second-generation due to gag/pol and rev viral genes that are located on separate plasmids, thus making wild-type recombinant virus generation even more unlikely [89]. In order to improve the fraction of CAR-expressing cells after transduction, CAR-NK cells can be enriched using fluorescence-activated cell sorting (FACS) or through antibiotic selection. Most studies applied a third-generation lentivirus under GMP conditions for clinical application and often showed a transduction efficiency lower than 10% on NK cells [90], explained by the low binding capacity of the VSV-G envelope to the LDL-R, which is the receptor facilitating entry of the virus. Changing the virus pseudotype (e.g., to BaEV) was suggested to enhance the viral transduction efficiency as it targets other receptors expressed at higher levels in NK cells [91, 92]. Furthermore, stimulation with cytokines or certain compounds can facilitate higher transduction rates in NK cells [92,93,94] (discussed further in Chapter 5). All combined, these results have demonstrated that lentiviruses are a powerful vehicle to manufacture CAR-NK cells for clinical disease.
Retroviruses
Retroviruses have been used as gene therapy vectors for decades [95]. Retrovirus-based vectors require NK cells to be actively dividing to obtain integration of the vector into the genome [96]. There are 19 studies using CAR-NK cell lines and 16 reports using primary NK cells published that employ retroviruses (Fig. 3a). One recent Phase I clinical trial targeted CD19+ non-Hodgkin's lymphoma and chronic lymphocytic leukemia by infusion of retrovirus-transduced anti-CD19 CAR cord blood NK cells. In this study, 73% of patients responded with 7 out 8 patients attaining complete remission. Moreover, responses were fast and seen within 30 days after CAR-NK infusion at all dose levels. After one-year follow-up, expanded CAR-NK cells could still be detected by qPCR [30]. After infusion, CAR-NK DNA copy numbers remained stable for up to one year in peripheral blood and measured between 102 and 104 copies of the CAR-NK vectors per μg genomic DNA. These results show for the first time that retrovirally transduced CAR-NK cells can persist in vivo for a long time.
Different genera of retroviruses have been used to generate CAR-NK cells (Fig. 3b). Alpha retroviruses carrying the RD114 envelope are superior in transduction efficiency of primary NK cells compared to gamma retrovirus and lentivirus [97]. Recently, Muller et al. showed that RD114 alpha-retroviruses result in 3 times higher CAR-NK transduction efficiencies (around 45%) than VSV-G lentiviruses on day 3 in primary NK cells (about 15%) [91]. However, from day 7 onwards, retroviruses and lentiviruses showed equal performance [91]. Retrovirally transduced CAR-NK cells can be expanded with sustained CAR expression for at least 2 weeks. Although long and stable CAR expression in NK cell can be achieved using different retroviruses, safety of the retrovirus system is still a concern, especially when compared to safer lentiviruses.
Electroporation of mRNA
Electroporation of CAR encoding mRNA is a swift and efficient but transient approach to generate CAR-NK cells. Some companies already developed equipment to facilitate electroporation in a closed system compatible with CAR-NK cell generation, which is especially interesting for GMP-compliant production starting from primary NK cells. There are 11 studies in CAR-NK cell lines and 10 primary CAR-NK cells that rely on electroporation (Fig. 3a). The electroporated cargo mainly consists of CAR-encoding mRNA or plasmids (Fig. 3b). With the current technological advances and the use of high-purity CAR mRNA instead of cDNA in a plasmid, transfection efficiencies in NK cells have increased dramatically, achieving up to 95% with minimal negative effects on cell viability [98, 99]. Generally speaking, mRNA transfection efficiencies are much higher in expanded or activated NK cells (more than 60%) than in freshly isolated NK cells (about 40%) [100]. As mRNA synthesis is compatible with GMP regulations, and electroporation can be done in a clean room, it is thus feasible to generate GMP-compliant CAR-NK via mRNA electroporation. However, the main disadvantage of this method is the narrow, transient window of CAR expression: After electroporation, the CAR-NK cells should be transfused back into patients within 7 days.
Sleeping beauty transposon
Transposon-based systems can introduce CAR transgenes with higher efficiency and at predefined locations, which is an important advantage over conventional methods that do not possess an integrating element. Transposons are mainly introduced into NK cells by electroporation followed by integration into the host genome through transposase enzymes [101, 102]. Two studies applied the transposon system to generate CAR-NK cells: One used NK-92-MI cells [101], the other study described transposon transfection into iPSC cells followed by differentiation into NK cells [28] (Fig. 3a). Both studies used the 4D-Nucleofector electroporator to introduce the transposase encoding plasmids into cell nucleus (Fig. 3b). After enrichment, anti-mesothelin-CARs were stably expressed on iPSC-derived NK cells and were functional in a murine model of ovarian cancer [28]. Although there are many studies using the transposon system to generate primary CAR-T cells [53, 103], primary NK cells are much more difficult to transduce using transposons. We expect that with further advances in transposon and transfection methods, generation of CAR-NK cells using transposon will become a more viable approach.
CRISPR/Cas9-mediated strategies
CRISPR/Cas9 has recently emerged as a powerful technique for genetic modification. This technology relies on the introduction of Cas9 protein in conjunction with guide RNA into the NK cells. Initially, this technique was used in primary NK cells to disrupt the CD38 gene, aiming to prevent fratricide of NK cells when they were used in combination with daratumumab (anti-CD38), as CD38 is expressed both on NK cells and multiple myeloma and AML cells [104]. More recently, CRISPR/Cas9 has also been applied to introduce new genes [105]. Here, in parallel to CRISPR/Cas9, a homologous donor DNA template is introduced in the same cell via transfection [106]. This DNA template replaces the targeted gene, thus allowing for the introduction of genes that promote the anti-tumor effects. This technique was first successfully applied in primary T cells: CRISPR/Cas9 was used to target the TCR alpha gene, relying on homology-directed repair (HDR) to knock-in a CD19-CAR cassette. The expression of the CD19 CAR, now under the control of the endogenous TCR alpha promoter, manifested much stronger expression that also persisted longer compared to lentiviral vectors using a viral promoter [40, 107]. Following this paradigm, one can apply this concept to generate more potent and persistent CAR-NK cells. In several studies that employ HDR templates, knock-in efficiencies of more than 75% were obtained using K562-mIL-21-expanded NK cells (see also the section on Cytokines and K562-mIL-21/4-1BBL feeder cells) [43]. However, in fresh NK cells, the Cas9 knock-in efficiency is only 3–16% [42]. When the CAR gene is delivered via viral vectors or using transposon-based random insertion, expression levels are variable. CAR expression driven by an endogenous promotor following CRISPR/Cas9-specific locus knock-in strategies could result in long CAR-T persistence in vivo, with a memory-like phenotype and less expression of exhaustion markers [40]. Thus, the CRISPR/Cas9 strategy is a versatile technique that should be further explored for the generation of CAR-NK cells. It can be used to precisely delete, repair or introduce genes in a specific locus, hence holding promise to generate powerful anti-tumor NK cells [43].
CAR-expression detection methods
Following transduction of the chimeric antigen receptor, various techniques are available to assess CAR expression levels on the cell surface and to determine the efficacy of the developed CAR-NK cells. The most employed experimental modalities include flow cytometry, (fluorescence) microscopy and analysis of gene expression levels. In addition, flow cytometry and gene copy number analysis are used to monitor CAR-NK cells in patients. Here, we discuss the advantages and disadvantages of the various approaches.
Flow cytometry
Flow cytometry is widely used to detect CAR expression, since it is a fast and reliable detection method that provides quantitative data on protein expression at the single cell level. This technique relies on the use of fluorescently labeled antibodies that recognize parts of the CAR complex on the membrane. In addition, flow cytometry can be used to detect fluorescent tags or tags that were specifically added to the transduction cassette to facilitate expression recognition as is introduced in section Detection tags (GFP, cMyc-tag, FLAG, LNGFR).
Labeled antibodies or their derivates, such as antigen-binding fragments (Fab), can be used to detect components of the CAR complex on the cell membrane. Commonly, these antibodies target the antigen-recognizing scFv part of the CAR. In addition, the CAR ligand with a fragment crystallizable region (Fc) tail is used in conjunction with Fc-recognizing labeled antibodies. While these methods have the advantage that they are very specific for the detection of a specific CAR, this is also the main drawback of this approach: Some antibodies are generally not commercially available [108].
To overcome this challenge, universal detection methods have also been developed. For instance, protein L, a bacterial-derived product that recognizes the variable part of the light chain, binds to most immunoglobulin classes, including scFv that are used in CAR development [109]. Protein L has successfully been used to detect the expression of murine and human CAR on peripheral blood cells, including NK cells [110].
Another strategy comprises the incorporation of tags specifically included to facilitate the detection of the CAR. For this, molecular tags such as polyhistidine-tag (“His-Tag”), FLAG, or c-Myc tags have been incorporated into the CAR construct to facilitate easy detection using commercially available monoclonal antibodies [111]. Even though these tags are small and considered lowly immunogenic [112], it is undesirable to include non-essential and foreign sequences in products designed for clinical use.
Indeed, in the experimental setting other detection methods can also be used to help in CAR detection. For instance, fluorescent tags (e.g., GFP) or non-native receptor proteins, such as LNGFR [113], can be added to the CAR construct. While these methods simplify CAR detection, it is important to realize that fluorescent proteins or additional receptors are included in the CAR construct using bi-cistronic elements, or their expression is driven by additional promotors. Thus, the expression of the tags may not reflect CAR expression and may only be a marker of successful transduction.
Quantitative PCR
Quantitative PCR is a well-established technique for quantification of CAR transgene expression levels. As the technique requires disruption of individual cells to isolate genomic material, this technique does not provide direct information about CAR expression at the single cell level. However, surrogate markers that are expressed at relatively stable levels (housekeeping genes) can be used to estimate the number of cells and to follow copy numbers over time. Another limitation of PCR-based techniques is that they do not provide information on viable effector cells, nor do they take into account that not all transferred cells will be bone fide CAR-expressing effector cells.
Given these limitations, qRT-PCR is mostly used for follow-up studies in clinical trials in which flow cytometry cannot be used due to the low frequency of circulating CAR-positive effector cells. As an example, in a recently published phase I/II trial using CAR-NK to target CD19-expressing malignancies, gene copy number analysis was used to measure in vivo expansion of CAR-NK [30]. The viral vector transgene copy numbers were normalized to the amount of genomic DNA. Using this technique, the authors were able to show expansion of CAR-NK cells as early as 3 days after infusion and demonstrated that CAR-NK cells persisted in patients for at least 12 months.
Novel techniques, such as droplet digital PCR (ddPCR), allow for absolute quantification of single molecules using an advanced microfluidics system [114, 115]. The main advantage of this technique is that it eliminated the need for a standard curve, and therefore allows for reproducible analysis of very low copy numbers. This is likely to be important of the CAR-NK field, as the number of CAR-NK cells in a patient is relatively low, especially at later time points. However, at this moment, ddPCR has important disadvantages: Sample handling is much more complicated, time-consuming, expensive and relies on small-size chips or cartridges, disqualifying the technique for large numbers of samples.
Western blot
Western blots that employ antibodies recognizing an antibody, as for flow cytometry, can also be used to detect CAR expression in a cell population [111]. Immunoblots do not provide information at the single cell level, are time-consuming, and therefore are not routinely used to confirm CAR incorporation. However, they can provide valuable information about the size of the complex in which CAR is incorporated and are thus often used in CAR optimization studies. For instance, several studies have shown that CARs need to form dimers to be responsive and that they need to engage in a complex with signaling domains to be effective [116].
Confocal microscopy
Fluorescent and electron microscopy can be used to visualize CAR molecules on the surface of effector cells. Similar to flow cytometry and western blot, this technique relies on the use of antibodies that can be CAR-specific, or universal antibodies that recognize common CAR components. Indeed, fluorescent and electron microscopy have been used to detect CAR molecules on the surface of transduced NK cells [117]. Given that microscopic analysis is low-throughput, these techniques may not routinely be used to confirm CAR expression, but can be valuable tools to visualize the correct incorporation of the CAR in the plasma membrane and to determine the number of CAR molecules per cell.
Molecular imaging
In addition to follow-up by flow cytometry and determination of gene copy numbers, various molecular imaging strategies are currently under investigation for experimental and clinical use. Following transduction, cells are loaded with tags that can be detected using optical, nuclear or magnetic resonance techniques [118]. While fluorescent and bioluminescence techniques are only viable for use with laboratory animals, positron or photon-emission tomography (PET/SPECT) and magnetic resonance imaging can also be used in humans. These techniques can be used to gain insight in the NK cell biology at the cellular and molecular level, as they allow for quantitative tracking of (CAR-) NK cell biology in vivo [118]. However, most of these techniques are still in the very early phases, requiring complicated and expensive labeling procedures and long acquisition times.
Approaches for enhancing NK cell virus-mediated transduction
Lentiviruses and retroviruses are the most commonly used systems to induce stable expression of chimeric antigen receptors in NK cells. Encouraging results have been shown for the transduction of NK cell lines and primary NK cells, including their use in clinical trials [89, 119]. However, attaining high viral transduction efficiency while maintaining good cell viability remains a challenge, especially for primary NK cells. Various strategies have been developed to enhance viral transduction of NK cells (Fig. 3c).
These approaches often target the entry of a vector into NK cells, either via changing electric charges of cells or by targeting the viral envelope or the receptor on the target cell [120, 121]. Therefore, changes to the envelope on the viral surface and up-regulation of receptors on the target cell are the main approaches to boost transduction [89, 120].
Enhancing NK cell transduction via upregulation of LDLR using statins
Most lentiviruses used for gene transductions are pseudotyped with a VSV-G envelope glycoprotein. The main receptors for this envelope protein are the low-density lipoprotein receptor (LDLR) and phosphatidylserine [122]. NK cell lines and primary NK cells express low levels of LDLR. Interestingly, statins that are used in the clinic to lower blood cholesterol levels were shown to induce LDLR on B and T lymphocytes [123]. Similarly, we found that statins also induced LDLR levels in NK-92 and primary NK cells [93]. Consequently, transduction efficiency increased after treatment with statins. However, statins have also been reported to suppress the cytotoxic capacity of NK cells, which can be completely reversed by geranylgeranyl-pyrophosphate (GGPP). Not all statins are potent boosters of viral transduction: Rosuvastatin in combination with GGPP most potently improved viral transduction without affecting the cytotoxic properties of the NK cells [93].
Other small molecular compounds to enhance NK cell transduction
In addition to statins, other compounds have been tested to enhance the viral transduction of NK cells. Negative charges existing on both the viral envelope glycoproteins and the target cell receptors can be detrimental for transduction [124]. Therefore, cationic polymers, such as protamine sulfate, polybrene [125] and dextran [126], can be used during transduction to positively charge the cell surface [124] (Fig. 3c). Using high-throughput analyses, other compounds that enhance lentiviral transduction have also been identified. Phorbol 12-myristate 13-acetate (PMA) [127], rapamycin [128], Cyclosporin A [129], P338 poloxamer [130], prostaglandin E2 (PGE2) [131], vectofusin-1 [132] and RetroNectin [92] have all been reported to promote lentiviral transduction on hematopoietic stem cell (HSC), T cells or B cells. However, in our analysis on the effects of these compounds on NK cell transduction, we found that only statins consistently promote viral transduction without major negative effects on cell viability [93].
Next to compounds that promote viral transduction, substances that inhibit antiviral responses in NK cells can also be used to enhance transduction. Indeed, BX795, an inhibitor of the TBK1/IKKε complex that controls antiviral responses in NK cells, was demonstrated to promote lentiviral gene transduction efficiency by 3.8-fold [133].
Alternative lentiviral pseudotypes for NK cell transduction
VSV-G has long been used as the major glycoprotein for pseudotyping lentiviruses because of its broad tropism, facilitating the transduction of a wide range of cell types [134]. VSV-G lentiviruses have also been used to generate genetically modified NK cells for decades, and make stable NK cells line [135, 136]. However, as alluded to above, the low expression levels of the VSV-G receptor LDLR on fresh human NK cell hinder efficient and easy lentiviral transduction of NK cells [93, 94]. As an alternative to boosting LDLR expression levels, other glycoproteins can be used to pseudotype the viral particle [121].
Colamartino et al. showed that BaEV lentiviruses outperform VSVG-, MV- and RD114-pseudotyped viruses in human NK cells, even using activated NK cells [92]. Their study supports the idea that the abundance of the lentivirus receptor on the target NK cell may correlate with the efficiency of the integration of the gene of interest. The receptors for BaEV are ASCT1 and ASCT2, which are highly expressed on activated NK cells, both at the mRNA and protein level [92, 94]. Therefore, BaEV lentiviruses are potentially superior to VSV-G lentiviruses for gene therapy in NK cells [92].
RD114 pseudotyped retroviruses have also been used to generate human NK cells expressing an anti-CD19-CAR [137]. Suerth et al. demonstrated that efficient modification of human NK cells by using an alpha-retroviral vector with a RD114/TR envelope [97]. VSV-G-pseudotyped lentiviral particles normally outperform VSV-G pseudotyped alpha-retroviral and gamma-retroviral particles. However, when alpha-retroviral particles were pseudotyped with RD114/TR, this resulted in superior transduction performance. These results show that the compatibility of virus particles and the surface glycoprotein will ultimately determine the efficiency of viral transduction [97]. With these concepts, further studies towards modifying the component of the viral glycoprotein specific for NK cells may improve the LVs transduction efficiency [121].
Cytokines and K562-mIL-21/4-1BBL feeder cells
The cytokines and feeder cell lines that are used to expand NK (see section NK cell expansion) cells may also have positive effects on transduction with retro- and lentiviruses. Previous studies demonstrated that IL-2 induced upregulation of the viral receptor LDLR in NK cells, resulting in enhanced transduction, proliferation and cytotoxicity [138]. The addition of soluble IL-2, IL-12 and IL-21 to the NK cell medium has also been reported to enhance VSV-G lentiviral transduction, with a possible synergistic effect of additional PHA [133, 135]. Other reports have indicated that culturing primary human NK cells with K562 feeder cells with membrane-bound IL-21 and 4-1BBL also boosts lentiviral and retroviral transduction of NK cells [92, 139]. Thus, these methods are already used and could be easily implemented to further enhance viral transduction.
Microfluidic mechanic devices
The titer of the virus is a crucial parameter of the viral transduction. Although with the advancement of viral vectors and transfection technologies, it remains difficult to fully standardize the method to obtain a high enough and consistent titer to perform a successful transduction. Even though this system has not been tested for NK cell transduction, the microfluidic platform systems developed by Luni et al. [140] and Tran et al. [141] may develop into a new versatile tool to improve the transduction efficiency while working at low viral titers in a sequential manner.
Cell sources for CAR-NK generation
NK cell-based immunotherapy requires relatively large numbers of effector cells. The life span and proliferative capacity of NK cells is limited, and extensive ex vivo handling of NK cells might influence their cytotoxic capacity. Multiple sources have been used to isolate and generate sufficient NK cells for CAR-NK applications (Fig. 3d and Table 5). In this section, we will discuss the advantages and disadvantages of these NK cell sources for the development of CAR-NK cells.
Peripheral blood NK cells (PB-NK)
NK cells can be isolated relatively easily from the patient itself (autologous PB-NK) or from healthy donors (allogeneic PB-NK), and therefore most (31/33) preclinical CAR-NK studies use PB-NK. The majority of these studies use allogeneic NK cells from healthy donors (Fig. 3d).
For cancer immunotherapy, autologous NK cells are typically not very effective, as they are functionally silenced when they encounter self-MHC antigens. Furthermore, the function of patient-derived NK cells is often compromised by the underlying disease or previous treatment. Therefore, allogeneic PB-NK are often preferred for immunotherapy purposes, even though this requires careful depletion of the T lymphocytes from the product, as these latter cells could induce GVHD [142].
Since NK cell numbers in peripheral blood are relatively low, NK cells are routinely expanded after isolation. Even though the number of CAR-NK cells required for a meaningful response has not been established, previous NK cell-based therapies typically infused 105–108 cells/kg, thus requiring as many as 1010 NK cells for a patient weighing 100 kg. Therefore, various expansion protocols have been developed, which will be discussed further in this review (section NK cell expansion).
The main advantage of PB-NK cell sources for cancer immunotherapy is that the cells are already mature, and do not need to undergo a lengthy differentiation protocol, as for stem cell-derived sources. On the other hand, the transduction efficiency of PB-NK is relatively low (discussed in sections “Transfection or transduction vehicle for CAR expression” and “Approaches for enhancing NK cell virus-mediated transduction”), and prolonged culture often leads to telomerase shortening and reduced cytotoxicity due to exhaustion, even though these problems can be largely circumvented by using the correct combination of stimulatory molecules [143,144,145].
Cord blood-derived NK cells (CB-NK)
A possible limitation of PB-NK cells is the dependency on the availability of healthy donors at the time the NK cell expansion process needs to be started. In a clinical setting where the new diagnosis of cancer warranting CAR-NK treatment cannot be planned, this may pose a problem in logistics and planning. Therefore, alternatives starting from frozen material, becoming off-the-shelf products, are an attractive alternative. However, it has been reported that freezing of mature PB-NK cells importantly decreases their viability and cytotoxic capacity [146, 147]. In general, large numbers of NK cells can be obtained from umbilical cord blood samples, due to the high proliferative capacity of these cells. For instance, only 10% of one cord blood unit is required to generate an almost pure pool of more than 109 NK cells in two weeks [117], which is usually sufficient for one treatment cycle. Another advantage of CB-NK cells is that the haplotype of the sample can be determined at the time of collection, allowing the generation of a cell bank from which HLA-mismatched NK cells can be selected on demand.
There are also some concerns about the cytotoxic capabilities of CB-NK cells. The expression of natural cytotoxicity receptors is lower in CB-NK cells that were expanded in an artificial antigen-presenting cell system compared to IL-2-expanded cells. However, the expression levels of KIRs, NKG2A, CD94 and NKG2C were similar and no differences in cytotoxicity in killing multiple myeloma cells were observed [148].
Although only a minority of the preclinical CAR-NK studies uses cord blood as a cell source (Additional file 2: Table S2), the potency of CB-NK was recently confirmed in the first publication of a clinical trial using CAR-NK cells [30]. Here, CB-NK cells were expanded on K562-mbIL21 and 4-1BB ligand feeder cells and endowed with a CD19-directed CAR, ectopic IL-15 production and an inducible suicide gene. Remarkably, qPCR showed that these CB-CAR-NK cells persisted more than 270 days in vivo [30]. It is yet to be established if this finding correlates with long-lasting clinical responses as well.
Stem cell-derived NK cells: differentiation of NK cells from hESC and iPSC
Disadvantages of PB-NK and CB-NK as cell sources for CAR-NK development include the natural heterogeneity between donors, resulting in variance in performance of the final NK cell product. For that reason, stem cell-derived NK cell expansion is an attractive alternative to develop a standardized, off-the-shelf therapeutic product.
Indeed, clinical-scale NK cell production starting from human embryonic stem cells (hESC) or induced pluripotent stem cells (iPSC) has been demonstrated [149,150,151]. The use of commonly available hESC/iPSC cell lines is often preferred over the use of stem cells from bone marrow biopsies, G-CSF mobilization or human embryos [144, 145]. Next to ethical and practical issues attached to the use of these sources, the outcome of NK cell production starting from primary stem cells is less predictable and often less efficient (reviewed in [152]). The production of NK cells starting from hESC or iPSCs takes 3–5 weeks and is therefore significantly longer than NK cell expansion starting from peripheral blood- or cord blood-derived NK cells.
When considering the use of hESC-derived NK cells, it is important to note that NK cells derived from the H9 hESC cell line showed limited allogeneic immune response, while they showed a more mature cytotoxic phenotype compared to CB-NK [150]. While we are not aware of any preclinical or clinical studies using hESC to generate CAR-NK cells, it is probably technically feasible.
On the other hand, iPSC-derived NK cells that were already shown to be functional against tumors [151, 153] have recently been used to generate CAR-NK cells. After optimization of the activation domains for the use in iPSC-derived NK cells, mesothelin-targeting iPSC-CAR-NK cells were shown to be as potent as CAR-T cells [28]. Interestingly, tumor-bearing mice treated with CAR-iPSC-NK showed less pathogenic organ damage and lower IFN-γ and IL-6 levels than CAR-T-treated in a murine xenograft model.
There are still challenges ahead before iPSC-NK cells can be safely used for the generation of CAR-NK for clinical application. Firstly, CAR-iPSC-NK cells ceased proliferating in vivo after the exogenous administration of cytokines was stopped in the murine model. Systemic administration of cytokines in a clinical setting is highly undesirable, since it is not only costly, but also potentially dangerous. Secondly, iPSC-derived cells always bear the potential of malignant transformation. Even though there were no signs of transformation in the present studies, this point may not be neglected during long-term observations. Lastly, iPSC-derived cells are potentially immunogenic, which may lead to the destruction of the effector cells, or even to adverse immune reactions, such as cytokine release storms.
NK cell lines: NK-92
The aforementioned NK cell sources have one major disadvantage in common: Obtaining large numbers of NK cells is relatively cumbersome and time-consuming. In general, cell lines circumvent most of these problems, as they are easy to maintain and expand. Therefore, it is not surprising that 72 experimental studies have reported the use of NK cell lines for the development of CAR-NK cells. More than 80% of these studies use the lymphoma-derived NK-92 cell line [154]. Out of the six NK cell lines available, NK-92 has high anti-tumor activity and is capable of direct cytotoxicity. When NK-92 cells are transfected with CD16, this cell line can also trigger antibody-dependent cell-mediated cytotoxicity (ADCC) [155].
For CAR-NK cell development, NK-92 cells have another main advantage: the cell line also can be easily genetically modified using non-viral methods, including electroporation [156]. So far, NK-92 cells have been tested in preclinical CAR-NK studies targeting AML (directed to CD33), lymphoma (CD19), myeloma (CS1), prostate cancer (EpCAM), breast cancer (Her2), neuroblastoma (GD2), glioblastoma (EGFR) and ovarian cancer (mesothelin). Details on these trials can be found in Table 1 and Additional file 1: Table S1.
One of the major disadvantages of the NK-92 cell line is that the cells are tumor-derived and are aneuploid [154]. Therefore, they need to be irradiated before infusion, preventing in vivo proliferation and limiting their lifespan. In phase I trials, NK-92 administration was shown to be safe with only minor reactions [157, 158]. The number of head-to-head comparisons of CAR-equipped primary NK cells versus NK-92 cells is limited. In a study using a CD123-targeting CAR, primary NK cells were shown to be less effective than NK-92 cells in the eradication of AML cells in vitro [159]. This was possibly due to lower surface expression levels of the CAR in primary NK cells. However, it is difficult to draw a concrete conclusion whether the irradiated CAR-NK-92 cells is outperforming the primary CAR-NK cells on overall survival of patients, since the NK-92 cells are replication-deficient. Therefore, their persistence needs further investigation.
With only one CAR-NK clinical trial completed, it is impossible to draw conclusions on the usability of NK cell lines for CAR-NK therapy. Results from the CAR-T field indicate that long-term persistence is important for achieving and attaining complete remission [160]. While NK-92-CAR cells will not sustain in vivo, it is relatively easy to perform multiple infusions of this cell product. Further studies will be necessary to compare how NK-92-CAR can be used in the clinic.
NK cell expansion
Allogeneic NK cells have been proven safe and efficient to treat patients with AML and ALL [23, 24]. However, to achieve the number for clinical therapy a large-scale GMP expansion of NK cells ex vivo is necessary as the numbers present in peripheral blood of a donor is not sufficient. Several methodologies have been described to facilitate a successful NK cell expansion (Table 6). Expansion can be achieved culturing the cells in various cytokines mixtures (IL-2, IL-12, IL-15, IL-18 and/or IL-21], K562 cell-based cytokine transfected cells (e.g., with IL-15 or IL-21 in combination with 4-1BBL), or autologous PBMC stimulated cells. With the development of automatic and closed expansion equipment, like Miltenyi’s Prodigy, Lonza’s Wave and G-Rex static bioreactors, it has become feasible to expand NK cells according to GMP requirements.
However, the time it takes to reach clinically relevant numbers may be very long as exemplified by Lapteva et al. who showed that their cytokine mixture needed expansion of NK cells during 4–10 weeks [161]. Although this is biologically relevant, for large-scale production the time it takes is too long and thus not commercially and practically interesting.
It is also important to note that NK cells are generally easier to genetically modify after they have been activated by cytokines. While this is an important advantage, it also implies the need for an increased amount of the transduction agent. Obviously, a balance needs to be sought here, which is also clear from the fact that most studies report that viral transduction of NK cells was conducted after 7 days of activation and expansion.
The advancement of NK cell expansion technologies also facilitates CAR-NK cell generation. Co-culture of NK cells with the leukemia cell line K562, often transduced with membrane bound cytokines, recently gained attention as a relatively easy way to obtain large numbers of NK cells. After 7 days of expansion with K562-mIL-21/4-1BB-L feeder cells, the transduction efficiency of expanded NK cells can be above 80% compared to below 20% on freshly isolated NK cells [92]. In total, at this moment there are 13 studies published that used the K562 cells as feeder cells for CAR-NK cells to expand, including 12 studies using K562-mIL-15 feeder cells, 3 groups used K562-mIL-21 cells and 1 publication used the parental K562 cell. NK cells expanded with mbIL15 showed reduced telomere lengths, while those propagated with mbIL21 showed more elongated telomere lengths than freshly isolated peripheral blood NK cells [143]. This suggests that mbIL-21 is an attractive option to expand cells over many generations to reach clinically relevant cell numbers. Clinically relevant expansion levels have been reached in 2 weeks [162] (and our own unpublished data).
Prospective and outlook
Progression in CAR-NK clinical trials
The number of clinical trials in the CAR-NK cell space is limited (Table 7). Although in recent years, the first 2 papers showing evidence of clinical successes have been published [30, 163], many of the investigations are still in early planning or recruiting into clinical trials. Most of these trials are company-driven, with the majority coming from China (Table 7). In these trials, various diseases are targeted, with an anti-CD19 CAR (B cell malignancies) on NK cells initially being compared the same CAR on T cells. Furthermore, different tumor antigens, like HER2, Mucin-1, EpCAM or PMSA, are targeted on a variety of solid and hematological tumors (Table 7). In the clinical studies currently conducted, CAR-NK cells are derived from PBMC, the NK-92 cell line or a newly generated uniform cell line NK101. In contrast, Rezvani and colleagues have used expanded umbilical cord NK cells, selected on a KIR mismatch between donor and recipient and promising results were reported [30].
The treatment was safe as no CRS, neurotoxicity or GVHD occurred. From the 11 treated patients, 8 responded with 7 patients going into complete remission, although 3 patients had minimal residual disease at final assessment. CAR-NK cells were tracked in vivo and could be detected rather stable over time and for at least 12 months. It was suggested that in vivo persistence was driven by proliferation of the CAR-NK cells, which could be caused by the addition of IL-15 in the CAR construct (hence one could define this as a fourth-generation CAR (see section Activation signal for CAR-NK and Table 3). At least in mice, it has been shown that IL-15 aids in the persistence of (dual-switch CD123 or BCMA) CAR-NK cells for minimal 40–50 days [69], when the experiments were stopped.
In addition, the first patient treated with iPSC-derived CAR-NK cells has recently been reported, also suggesting that CAR-NK cells can outperform CAR-T cells [28, 164]. Still, the clinical trial is ongoing and the results have to be awaited. While many questions regarding CAR-NK cell therapy in humans remain, the large number of preclinical investigations imply that it is only a matter of time before more clinical trials are conducted.
Technological advances in CAR-NK generation: biomaterials and in vivo transduction
CAR-NK development was built on the knowledge gained from the CAR-T field. To further the development of CAR-NK for clinical use, technological advances will importantly speed up the process. Here, we consider two developments that will aid in the generation of sufficient CAR-NK cells for clinical use and for the in vivo generation of CAR-NK cells.
Biomaterials have shown promise as a concomitant agent together with effector cells for immunotherapy that can aid in mounting a stronger response, while simultaneously lowering the risk for side effects [165]. In NK cells, biomaterial micelles are able to facilitate the formation of an immune synapse with tumor cells, aiding in the eradication of solid malignant lumps in vivo [166]. A three-dimensional engineered hyaluronic acid-based niche (3D-ENHANCE), instead of the classic two-dimensional culture methods, was shown to promote CAR-NK cell expansion and to enhance cytokine release, while maintaining killing capacity of EGFR-CAR-NK92 towards solid tumors in vivo [167]. In another study, biomaterial-modified Fc fragments on antibodies showed a strong potential to broaden NK cell recognition of heterogeneous antigens on solid tumors [168]. Moreover, Smit et al. reported that DNA-carrying nanoparticles can efficiently introduce leukemia-targeting CAR genes into T-cells [169], which was also reported for NK-92MI cells in vitro [170]. Thus, various promising inventions from the bio-engineering field are likely to enter the CAR field as important improvements in the design and engineering of cancer immunotherapy.
Various groups are trying to harness autologous lymphocytes, including NK cells for immunotherapy without the need for ex vivo modifications [171]. This presents various challenges, such as achieving proper activation of NK cells without haplotype-dependent, allogeneic activation. Furthermore, in vivo proliferation and maintenance is required to obtain sufficient cell numbers to mount a successful anti-tumor response. Recently, it has been shown that the engineering of the glycoprotein on the VSV-G virus with anti-CD8 scFV makes it possible to specifically transduce CD8 T cells in vivo [171]. Using this concept, Pfeiffer et al. showed another approach of in vivo CAR-T generation by injecting lentivirus directly into laboratory mice. Indeed, in vivo injection of the CD8 specific-CD19 CAR-encoding lentiviruses resulted in transduction solely of the CD8+ T cells with a CD19-CAR [172]. However, this method also led to signs of a CRS, thus warranting further optimization before this in vivo CAR-T generation concept could be implemented in the clinic. These methods have not been tried on NK cells; we envisage that the concepts of these techniques can easily be applied to CAR-NK cell generation as well.
Challenges
Still many aspects on CAR constructs will have to be improved and many challenges lie ahead of the field. Besides the constructs, other aspects have to be taken into account. Here, we discuss three important challenges for the CAR-NK field that will need to be addressed in the near future regarding clinical safety, cryopreservation of CAR-NK cells and challenges posed by the tumor microenvironment.
The potential of CAR-based therapy has been clearly demonstrated, but it is not without risks. Indeed, patients treated with CAR-T cells have died because of cytokine release syndrome and off-target cytotoxic effects [1, 173]. While improvements in CAR design have made CAR-based therapy safer by reducing these severe side effects [31, 174], the incorporation of safety switches that quickly eliminate the CAR-NK infused cells, provides another level of safety [69, 175]. CAR-NK cells are in general considered safer than CAR-T cells, although there is very limited clinical experience with CAR-NK therapy in patients, justifying additional safety measures, including the use of “suicide genes” [117].
Indeed, inducible caspase 9 (iCasp9) was included as a safety switch in the first reported clinical trial of CAR-NK [30]. While no severe side effects warranting the need to use this safety switch were reported, it might be necessary to use it at a later time point, since CAR-NK cells were still detected after one year in vivo. The inducible safety switch caspase 9 (iCasp9) suicide gene is a modified caspase 9 gene fused to the human FK506 binding protein gene (FKBP). Addition of the inducer of dimerization (CID) (AP1903) leads to dimerization and recruits the downstream caspase molecules, thereby initiating apoptosis [176].
Recently, Ruella et al. demonstrated that anti-CAR19 idiotype chimeric antigen receptor (aCAR19) cells could eliminate CAR19+ T cells [177]. This CAR-T eliminating other CAR-T cells is not only useful for eliminating infused cells in the case of side effects, but also at the same time it depletes all transduced cells (including leukemic blasts) present during CAR-T manufacturing, minimizing the potential clinical effect, but preventing side effects. Additionally, these cells can be used to deplete CAR-T cells to reduce long-term side effects, such as B cell aplasia in the case of CD19-targeting CARs.
Truncated EGFR [178], CD19, CD20 or CD34 epitopes together with the CAR gene have been developed for CAR-T therapies [179]. Once the CAR-transduced cells cause undesirable effects, an additional administration of a specific antibody against these epitopes will induce ADCC to eliminate the CAR-NK cells in vivo [29, 69, 180, 181].
Not all issues related to the use of lentivirus for clinical use have already been resolved. For instance, it has been demonstrated that lentivirus-mediated p53 activation can lead to delayed cell proliferation, cell cycle arrest and slightly but significantly increased apoptosis during cell culture [182]. In a study using autologous CAR-T manufacturing, it was reported that the CAR gene was inadvertently transduced into a single leukemic B cell. The anti-CD19-scFv combined with the CD19 molecule on the surface of all leukemic B cells derived from this clone, camouflaging it from recognition by the CAR-T and becoming insensitive to the aimed effect [183]. Another case illustrated a patient suffering from chronic lymphocytic leukemia, in which after infusion with engineered autologous T cells, 94% of the CAR-T cells were found to originate from a single clone in which the lentiviral vector with the CAR transgene disrupted the methylcytosine dioxygenase TET2 gene [184]. Fortunately, the TET2 disruption by the lentivirus integration altered the epigenetic profile of the T cells that resulted in enhanced differentiation and expansion and exerted a central memory phenotype [184].
Retrovirus-mediated gene transduction has been reported to lead to random insertions creating the risk of leukemia [185, 186]. However, with advances in the development of viral vectors and detection technology, June’s group published 17 vector lots, 375 manufactured T cell products, and 308 treated patients with both oncologic and HIV-related indications for treatment (2001–2016), showing no evidence of generation of replication-competent retroviruses or lentiviruses [187].
Genome editing by CRISPR–Cas9 has been reported to induce p53-mediated DNA damage responses and cell cycle arrest in human cells [188]. Moreover, the potential of pre-existing humoral and cell-mediated adaptive immune responses to the Cas9 system in humans should be taken seriously when this system is applied in vivo during clinical trials [189]. All these issues will have to be addressed to guarantee the safe use of CAR-T and CAR-NK in patients.
An important technical issue is the cryopreservation of NK cell. For clinical application and infusion of cells into patients, large-scale expansion of CAR-NK cells under good manufacturing practice (GMP) is required by authorities. It is hardly possible that a freshly expanded CAR-NK cell product can be administered into patients at a suitable time point taking into account the status of the cells and the logistics and timing to prepare the patient [162]. Cryopreservation of CAR-NK cells is a necessity, but also allows to use NK cells as “off-the-shelf” products. This measure should guarantee the recipient always receives a similar quality of CAR-NK cells, thus disregarding the natural variety in NK cell donors [190]. Numerous groups are trying to optimize the ingredients of the freezing medium and procedures for expanded CAR-NK cells and have achieved encouraging outcomes [162, 190, 191]. A recent approach uses nanoparticle-mediated intracellular protection of NK cells which can avoid cryoinjury and maintain the killing potential of NK-92 cells, which could completely replace DMSO as a freezing protector [192]. This technique still has to be tried on and optimized for primary NK cells.
Even if the best CAR-NK cells can be made, they always will encounter the suppressive tumor microenvironment, possibly the biggest challenge of all to overcome [193]. So far, much effort is put in to designing CARs or combinations of treatments in overcoming the negative effects of the TME. CARs have been combined with checkpoint inhibitors like PD-1 [194, 195] and were successful in mice [194], but less in human [195]. Depletion of soluble suppressive factors like TGF-β, IDO and IL-4 can be obtained by expressing of dominant negative mutants or gene editing of the respective genes [196, 197]. Many solid tumors are characterized by hypoxic cores which may influence the function of NK cells, or on the other hand using and an HIF1α-driven CAR one could take advantage of the hypoxic environment [198]. A reduction of immune suppressor cells that are abundantly present in the TME (Treg [199], MDSC [200], TAM [201]) also is directed towards an improved clinical outcome and increased overall survival. The challenges posed by the tumor microenvironment and possible solutions using CAR-engineered cells have been covered extensively in various recent reviews [193, 202].
Conclusion
In this review, we performed a comprehensive analysis of preclinical and clinical studies from the CAR-NK field with an emphasis on the design of the CAR and engineering of NK cells. Recommendations for usage of gene elements have been described, as well as the state of the art, including important lessons from CAR-T development. This detailed information will pave the way to more robust CAR-NK cells studies with the aim of getting a stronger anti-tumor response with fewer side effects compared to CAR-T cells. Overcoming the suppressive tumor microenvironment remains an important challenge on the design of CARs before CAR-NK cells, in combination with other therapies, will fulfill their promise.
Availability of data and materials
The interactive tables for CAR-NK design are on www.carnkreview.com. All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Abbreviations
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- APC:
-
Antigen-presenting cell
- BCMA:
-
B cell maturation antigen
- CAR:
-
Chimeric antigen receptor
- Cas9:
-
CRISPR-associated system 9
- CCR:
-
CC chemokine receptor
- CD:
-
Cluster of differentiation
- CLL:
-
Chronic lymphocytic leukemia
- CR:
-
Complete remission
- CRS:
-
Cytokine release syndrome
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DLBCL:
-
Diffuse large B cell lymphoma
- EGFR:
-
Epidermal growth factor receptor
- FasL:
-
Fas ligand
- FDA:
-
US Food and Drug Administration
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- GVHD:
-
Graft-versus-host disease
- Her2:
-
Human epidermal growth factor receptor 2
- HLA:
-
Human leukocyte antigen
- HSPC:
-
Hematopoietic stem/progenitor cell
- IFN-γ:
-
Interferon-γ
- iPSC:
-
Induced pluripotent stem cell
- IS:
-
Immunologic synapse
- ITAM:
-
Immunoreceptor tyrosine-based activation motif
- KIR:
-
Killer immunoglobulin receptor
- MHC:
-
Major histocompatibility complex
- MoAb:
-
Monoclonal antibody
- MM:
-
Multiple myeloma
- MOI:
-
Multiplicity of infection
- NCR:
-
Natural cytotoxicity receptor
- NK:
-
Natural killer
- NSG:
-
NOD scid gamma
- PB:
-
Peripheral blood
- PR:
-
Partial remission
- PSMA:
-
Prostate-specific membrane antigen
- PSCA:
-
Prostate stem cell antigen
- RT-PCR:
-
Real-time PCR
- scFv:
-
Single-chain antibody variable fragment
- TAA:
-
Tumor-associated antigen
- TCR:
-
T cell receptor
- TGF-β:
-
Transforming growth factor-beta
- TME:
-
Tumor microenvironment
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- UCB:
-
Umbilical cord blood
References
June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73.
Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. 2018;359(6372):eaan4672.
Sordo-Bahamonde C, Vitale M. Mechanisms of resistance to NK cell immunotherapy. Cancers (Basel). 2020;12(4):893.
Weber EW, Maus MV, Mackall CL. The emerging landscape of immune cell therapies. Cell. 2020;181(1):46–62.
Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol. 2020;10:3038.
Stoiber S, Cadilha BL, Benmebarek M-R, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 2019;8(5):472.
Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–26.
Wei J, Han X, Bo J, Han W. Target selection for CAR-T therapy. J Hematol Oncol. 2019;12(1):62.
Fleischer LC, Spencer HT, Raikar SS. Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. J Hematol Oncol. 2019;12(1):141.
Walsh Z, Yang Y, Kohler ME. Immunobiology of chimeric antigen receptor T cells and novel designs. Immunol Rev. 2019;290(1):100–13.
Wang W, Jiang J, Wu C. CAR-NK for tumor immunotherapy: clinical transformation and future prospects. Cancer Lett. 2020;472:175–80.
Pfefferle A, Huntington ND. You have got a fast CAR: chimeric antigen receptor NK cells in cancer therapy. Cancers (Basel). 2020;12(3):706.
Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, Tonn T, Steinbach JP, Wels WS. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019;10:2683.
Hu Y, Tian Z, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. 2018;39(2):167–76.
Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19(3):200–18.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, Schmierer M, Gabrusiewicz K, Anderson NR, Petty NE, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–53.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5.
Fang F, Xiao W, Tian Z. Challenges of NK cell-based immunotherapy in the new era. J Front Med. 2018;12(4):440–50.
Hu Y, Tian Z, Zhang C. Natural killer cell-based immunotherapy for cancer: advances and prospects. Engineering. 2019;5(1):106–14.
Carotta S, Targeting NK. Cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
He Y, Tian Z. NK cell education via nonclassical MHC and non-MHC ligands. Cell Mol Immunol. 2017;14(4):321–30.
Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010;142(6):847–56.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–7.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100.
Simonetta F, Alvarez M, Negrin RS. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front Immunol. 2017;8:465.
Huang R-S, Lai M-C, Shih H-A, Lin S. A robust platform for expansion and genome editing of primary human natural killer cells. J Exp Med. 2021;218(3):e20201529.
Suen WC-W, Lee WY-W, Leung K-T, Pan X-H, Li G. Natural skiller cell-based cancer immunotherapy: a review on 10 years completed clinical trials. Cancer Investig. 2018;36(8):431–57.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181–92.
Quintarelli C, Sivori S, Caruso S, Carlomagno S, Falco M, Boffa I, Orlando D, Guercio M, Abbaszadeh Z, Sinibaldi M, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia. 2020;34(4):1102–15.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, Guo X, Liu H, Ding N, Zhang T, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25(6):947–53.
Allan DSJ, Chakraborty M, Waller GC, Hochman MJ, Poolcharoen A, Reger RN, Childs RW. Systematic improvements in lentiviral transduction of primary human natural killer cells undergoing ex vivo expansion. Mol Ther Methods Clin Dev. 2021;20:559–71.
Kulemzin SV, Matvienko DA, Sabirov AH, Sokratyan AM, Chernikova DS, Belovezhets TN, Chikaev AN, Taranin AV, Gorchakov AA. Design and analysis of stably integrated reporters for inducible transgene expression in human T cells and CAR NK-cell lines. BMC Med Genomics. 2019;12(2):44.
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–64.
Jones S, Peng PD, Yang S, Hsu C, Cohen CJ, Zhao Y, Abad J, Zheng Z, Rosenberg SA, Morgan RA. Lentiviral vector design for optimal T cell receptor gene expression in the transduction of peripheral blood lymphocytes and tumor-infiltrating lymphocytes. Hum Gene Ther. 2009;20(6):630–40.
Fujiwara K, Masutani M, Tachibana M, Okada N. Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem Biophys Res Commun. 2020;527(2):350–7.
Rad SMAH, Poudel A, Tan GMY, McLellan AD. Promoter choice: Who should drive the CAR in T cells? PLoS ONE. 2020;15(7):e0232915–e0232915.
Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, Sandalcioglu IE, Neyazi B, Moritz T, Meyer J, et al. Design and characterization of an “All-in-One” lentiviral vector system combining constitutive anti-GD2 CAR expression and inducible cytokines. Cancers (Basel). 2020;12(2):375.
Li W, Qiu S, Chen J, Jiang S, Chen W, Jiang J, Wang F, Si W, Shu Y, Wei P, et al. Chimeric antigen receptor designed to prevent ubiquitination and downregulation showed durable antitumor efficacy. Immunity. 2020;53(2):456–70.
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gonen M, Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–7.
Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, Huls H, Miller JC, Kebriaei P, Rabinovich B, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119(24):5697–705.
Nguyen DN, Roth TL, Li PJ, Chen PA, Apathy R, Mamedov MR, Vo LT, Tobin VR, Goodman D, Shifrut E, et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat Biotechnol. 2020;38(1):44–9.
Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, Lonetree C, Yamamoto K, Bendzick L, Miller JS, et al. A genetically engineered primary human natural killer cell platform for cancer immunotherapy. Mol Ther. 2020;28(1):52–63.
Owji H, Nezafat N, Negahdaripour M, Hajiebrahimi A, Ghasemi Y. A comprehensive review of signal peptides: structure, roles, and applications. Eur J Cell Biol. 2018;97(6):422–41.
Nyathi Y, Wilkinson BM, Pool MR. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim Biophys Acta. 2013;1833(11):2392–402.
Voss M, Schröder B, Fluhrer R. Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochimica et Biophysica Acta BBA Biomembranes. 2013;1828(12):2828–39.
Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165–74.
Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, Arif S, Mather SJ, Taylor-Papadimitriou J, Burchell JM, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180(7):4901–9.
Posey AD, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44(6):1444–54.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–51.
Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20-22.
Krokhotin A, Du H, Hirabayashi K, Popov K, Kurokawa T, Wan X, Ferrone S, Dotti G, Dokholyan NV. Computationally guided design of single-chain variable fragment improves specificity of chimeric antigen receptors. Mol Ther Oncolytics. 2019;15:30–7.
Thokala R, Olivares S, Mi T, Maiti S, Deniger D, Huls H, Torikai H, Singh H, Champlin R, Laskowski T, et al. Redirecting specificity of T cells using the sleeping beauty system to express chimeric antigen receptors by mix-and-matching of VL and VH domains targeting CD123+ tumors. PLoS ONE. 2016;11:e0159477.
Kang CH, Kim Y, Lee HK, Lee SM, Jeong HG, Choi SU, Park CH. Identification of potent CD19 scFv for CAR T cells through scFv screening with NK/T-cell line. Int J Mol Sci. 2020;21:23.
Fernández de Larrea C, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, Purdon TJ, Ponomarev V, Wendel H-G, Brentjens RJ, et al. Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven relapse in multiple myeloma. Blood Cancer Discov. 2020;1(2):146–54.
Liu Q, Xu Y, Mou J, Tang K, Fu X, Li Y, Xing Y, Rao Q, Xing H, Tian Z et al. Irradiated chimeric antigen receptor engineered NK-92MI cells show effective cytotoxicity against CD19(+) malignancy in a mouse model. Cytotherapy. 2020;22(10):552–62.
Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, Xing H, Tian Z, Tang K, Liao X, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12(1):49.
Thomas S, Straathof K, Himoudi N, Anderson J, Pule M. An optimized GD2-targeting retroviral cassette for more potent and safer cellular therapy of neuroblastoma and other cancers. PLoS ONE. 2016;11:e0152196.
Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, Lin AA, Schlom J, June CH, Sherwin SA. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5(1):22.
Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, Rollence ML, Wood JF, Schott ME, Milenic DE, et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. 1993;6(8):989–95.
Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, Yang S, Kochenderfer JN. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452–65.
Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125–35.
Guo C, Wang X, Zhang H, Zhi L, Lv T, Li M, Lu C, Zhu W. Structure-based rational design of a novel chimeric PD1-NKG2D receptor for natural killer cells. Mol Immunol. 2019;114:108–13.
Gotthardt D, Trifinopoulos J, Sexl V, Putz EM. JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Front Immunol. 2019;10:2590.
Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9(5):495–502.
Lindner SE, Johnson SM, Brown CE, Wang LD. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci Adv. 2020;6(21):eaaz3223.
MacKay M, Afshinnekoo E, Rub J, Hassan C, Khunte M, Baskaran N, Owens B, Liu L, Roboz GJ, Guzman ML, et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. 2020;38(2):233–44.
Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, Seifried E, Wels WS, Tonn T. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol Immunother. 2018;67(1):25–38.
Wang X, Jasinski DL, Medina JL, Spencer DM, Foster AE, Bayle JH. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020;4(9):1950–64.
Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, Mendt M, Kerbauy LN, Banerjee PP, Shanley M, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137(5):624–36.
Hammer Q, Rückert T, Borst EM, Dunst J, Haubner A, Durek P, Heinrich F, Gasparoni G, Babic M, Tomic A, et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat Immunol. 2018;19(5):453–63.
Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, Cooper MA, Fehniger TA. Cytokine activation induces human memory-like NK cells. Blood. 2012;120(24):4751–60.
Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123.
Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, Voytovich UJ, Lin C, Sommermeyer D, Liu L, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11(544):eaat6753.
Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, Morgan RA. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183(9):5563.
Feucht J, Sun J, Eyquem J, Ho Y-J, Zhao Z, Leibold J, Dobrin A, Cabriolu A, Hamieh M, Sadelain M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25(1):82–8.
Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–90.
Ajina A, Maher J. Strategies to address chimeric antigen receptor tonic signaling. Mol Cancer Ther. 2018;17(9):1795–815.
Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, Patel PR, Guedan S, Scholler J, Keith B, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–90.
Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, Schappe T, Meng W, Tran J, Schaettler M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136(20):2308–18.
Gudipati V, Rydzek J, Doel-Perez I, Gonçalves VDR, Scharf L, Königsberger S, Lobner E, Kunert R, Einsele H, Stockinger H et al. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat Immunol. 2020;21(8):848–56.
Oelsner S, Friede ME, Zhang C, Wagner J, Badura S, Bader P, Ullrich E, Ottmann OG, Klingemann H, Tonn T, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19(2):235–49.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, Peng Y, Mao H, Yi L, Ghoshal K, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27.
Chen F, Wu P, Deng S, Zhang H, Hou Y, Hu Z, Zhang J, Chen X, Yang J-R. Dissimilation of synonymous codon usage bias in virus–host coevolution due to translational selection. Nat Ecol Evol. 2020;4(4):589–600.
Schutsky K, Song D-G, Lynn R, Smith JB, Poussin M, Figini M, Zhao Y, Powell DJ Jr. Rigorous optimization and validation of potent RNA CAR T cell therapy for the treatment of common epithelial cancers expressing folate receptor. Oncotarget. 2015;6:30.
Qian L, Li D, Ma L, He T, Qi F, Shen J, Lu X. The novel anti-CD19 chimeric antigen receptors with humanized scFv (single-chain variable fragment) trigger leukemia cell killing. Cell Immunol. 2016;304–305:49–54.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin JR, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2018;380(1):45–56.
June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol. 2009;9(10):704–16.
Milone MC, O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529–41.
Gándara C, Affleck V, Stoll EA. Manufacture of third-generation lentivirus for preclinical use, with process development considerations for translation to good manufacturing practice. Hum Gene Ther Methods. 2018;29(1):1–15.
Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, Koehl U, Schambach A, Wels WS, Modlich U, et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol. 2020;10:3123.
Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, Roméro H, Selleri S, Béland K, Guiot M, et al. Efficient and robust NK-cell transduction with baboon envelope pseudotyped lentivector. Front Immunol. 2019;10:2873.
Gong Y, Klein Wolterink RGJ, Janssen I, Groot AJ, Bos GMJ, Germeraad WTV. Rosuvastatin enhances VSV-G lentiviral transduction of NK cells via upregulation of the low-density lipoprotein receptor. Mol Ther Methods Clin Dev. 2020;17:634–46.
Bari R, Granzin M, Tsang KS, Roy A, Krueger W, Orentas R, Schneider D, Pfeifer R, Moeker N, Verhoeyen E, et al. A distinct subset of highly proliferative and lentiviral vector (LV)-transducible NK cells define a readily engineered subset for adoptive cellular therapy. Front Immunol. 2019;10:2001.
Suerth JD, Labenski V, Schambach A. Alpharetroviral vectors: from a cancer-causing agent to a useful tool for human gene therapy. Viruses. 2014;6(12):4811–38.
Poletti V, Mavilio F. Interactions between retroviruses and the host cell genome. Mol Ther Methods Clin Dev. 2018;8:31–41.
Suerth JD, Morgan MA, Kloess S, Heckl D, Neudorfl C, Falk CS, Koehl U, Schambach A. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J Mol Med (Berl). 2016;94(1):83–93.
Carlsten M, Childs RW. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front Immunol. 2015;6:266.
Chu Y, Yahr A, Huang B, Ayello J, Barth MS, Cairo M. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. OncoImmunology. 2017;6(9):e1341031.
Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, Leung W, Campana D. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14(7):830–40.
Batchu RB, Gruzdyn OV, Tavva PS, Kolli BK, Dachepalli R, Weaver DW, Gruber SA. Engraftment of mesothelin chimeric antigen receptor using a hybrid Sleeping Beauty/minicircle vector into NK-92MI cells for treatment of pancreatic cancer. Surgery. 2019;166(4):503–8.
Wang J, Lupo KB, Chambers AM, Matosevic SJ. Purinergic targeting enhances immunotherapy of CD73+ solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. 2018;6(1):136.
Hudecek M, Izsvák Z, Johnen S, Renner M, Thumann G, Ivics Z. Going non-viral: the Sleeping Beauty transposon system breaks on through to the clinical side. Crit Rev Biochem Mol Biol. 2017;52(4):355–80.
Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, Zweegman S, van de Donk NWCJ, Mutis T, Szegezdi E et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2020. Online ahead of print.
Afolabi LO, Adeshakin AO, Sani MM, Bi J, Wan X. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology. 2019;158(2):63–9.
Bailey SR, Maus MV. Gene editing for immune cell therapies. Nat Biotechnol. 2019;37(12):1425–34.
Stenger D, Stief TA, Käuferle T, Willier S, Rataj F, Schober K, Vick B, Lotfi R, Wagner B, Grünewald TGP et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood. 2020;136(12):1407–18.
De Oliveira SN, Wang J, Ryan C, Morrison SL, Kohn DB, Hollis RP. A CD19/Fc fusion protein for detection of anti-CD19 chimeric antigen receptors. J Transl Med. 2013;11(1):23.
Nilson BH, Solomon A, Björck L, Akerström B. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J Biol Chem. 1992;267(4):2234–9.
Zheng Z, Chinnasamy N, Morgan RA. Protein L: a novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J Transl Med. 2012;10(1):29.
Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, Wels WS. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:533.
Chiarella P, Edelmann B, Fazio VM, Sawyer AM, de Marco A. Antigenic features of protein carriers commonly used in immunisation trials. Biotechnol Lett. 2010;32(9):1215–21.
Casucci M, Falcone L, Camisa B, Norelli M, Porcellini S, Stornaiuolo A, Ciceri F, Traversari C, Bordignon C, Bonini C, et al. Extracellular NGFR spacers allow efficient tracking and enrichment of fully functional CAR-T cells co-expressing a suicide gene. Front Immunol. 2018;9:507.
Härmälä SK, Butcher R, Roberts CH. Copy number variation analysis by droplet digital PCR. Methods Mol Biol. 2017;1654:135–49.
Pinheiro LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S, Emslie KR. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem. 2012;84(2):1003–11.
Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018;14(3):317–24.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31.
Gangadaran P, Ahn B-C. Molecular imaging: a useful tool for the development of natural killer cell-based immunotherapies. Front Immunol. 2017;8:1090.
Gandara C, Affleck V, Stoll EA. Manufacture of third-generation lentivirus for preclinical use, with process development considerations for translation to good manufacturing practice. Hum Gene Ther Methods. 2018;29(1):1–15.
Joglekar AV, Sandoval S. Pseudotyped lentiviral vectors: one vector, many guises. Hum Gene Ther Methods. 2017;28(6):291–301.
Verhoeyen E, Cosset F-L. Surface-engineering of lentiviral vectors. J Gene Med. 2004;6(S1):S83–94.
Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci USA. 2013;110(18):7306–11.
Amirache F, Lévy C, Costa C, Mangeot P-E, Torbett BE, Wang CX, Nègre D, Cosset F-L, Verhoeyen E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood. 2014;123(9):1422–4.
Davis HE, Rosinski M, Morgan JR, Yarmush ML. Charged polymers modulate retrovirus transduction via membrane charge neutralization and virus aggregation. Biophys J . 2004;86(2):1234–42.
Davis HE, Morgan JR, Yarmush ML. Polybrene increases retrovirus gene transfer efficiency by enhancing receptor-independent virus adsorption on target cell membranes. Biophys Chem. 2002;97(2):159–72.
Nanbakhsh A, Best B, Riese M, Rao S, Wang L, Medin J, Thakar MS, Malarkannan S. Dextran enhances the lentiviral transduction efficiency of murine and human primary NK cells. J Vis Exp. 2018;131(131):55063.
Johnston JM, Denning G, Moot R, Whitehead D, Shields J, Le Doux JM, Doering CB, Spencer HT. High-throughput screening identifies compounds that enhance lentiviral transduction. Gene Therapy. 2014;21:1008.
Wang CX, Sather BD, Wang X, Adair J, Khan I, Singh S, Lang S, Adams A, Curinga G, Kiem H-P, et al. Rapamycin relieves lentiviral vector transduction resistance in human and mouse hematopoietic stem cells. Blood. 2014;124(6):913–23.
Petrillo C, Cesana D, Piras F, Bartolaccini S, Naldini L, Montini E, Kajaste-Rudnitski A. Cyclosporin A and rapamycin relieve distinct lentiviral restriction blocks in hematopoietic stem and progenitor cells. Mol Ther. 2015;23(2):352–62.
Anastasov N, Höfig I, Mall S, Krackhardt AM, Thirion C. Optimized lentiviral transduction protocols by use of a poloxamer enhancer, spinoculation, and scFv-antibody fusions to VSV-G. Methods Mol Biol. 2016;1448:49–61.
Heffner GC, Bonner M, Christiansen L, Pierciey FJ, Campbell D, Smurnyy Y, Zhang W, Hamel A, Shaw S, Lewis G, et al. Prostaglandin E2 increases lentiviral vector transduction efficiency of adult human hematopoietic stem and progenitor cells. Mol Ther. 2018;26(1):320–8.
Radek C, Bernadin O, Drechsel K, Cordes N, Pfeifer R, Sträßer P, Mormin M, Gutierrez-Guerrero A, Cosset F-L, Kaiser A et al. Vectofusin-1 improves transduction of primary human cells with diverse retroviral and lentiviral pseudotypes, enabling robust, automated closed-system manufacturing. Hum Gene Ther. 2019;30(12):1477–93.
Sutlu T, Nyström S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther. 2012;23(10):1090–100.
Hastie E, Cataldi M, Marriott I, Grdzelishvili VZ. Understanding and altering cell tropism of vesicular stomatitis virus. Virus Res. 2013;176(1):16–32.
Micucci F, Zingoni A, Piccoli M, Frati L, Santoni A, Galandrini R. High-efficient lentiviral vector-mediated gene transfer into primary human NK cells. Exp Hematol. 2006;34(10):1344–52.
Tijani M, Munis AM, Perry C, Sanber K, Ferraresso M, Mukhopadhyay T, Themis M, Nisoli I, Mattiuzzo G, Collins MK, et al. Lentivector producer cell lines with stably expressed vesiculovirus envelopes. Mol Ther Methods Clin Dev. 2018;10:303–12.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83.
De Sanctis JB, Blanca I, Radzioch D, Bianco NE. Expression and function of low-density lipoprotein receptors in CD3-CD16+CD56+ cells: effect of interleukin 2. Cell Immunol. 1996;167(1):18–29.
Streltsova MA, Barsov E, Erokhina SA, Kovalenko EI. Retroviral gene transfer into primary human NK cells activated by IL-2 and K562 feeder cells expressing membrane-bound IL-21. J Immunol Methods. 2017;450:90–4.
Luni C, Michielin F, Barzon L, Calabrò V, Elvassore N. Stochastic model-assisted development of efficient low-dose viral transduction in microfluidics. Biophys J. 2013;104(4):934–42.
Tran R, Myers DR, Denning G, Shields JE, Lytle AM, Alrowais H, Qiu Y, Sakurai Y, Li WC, Brand O, et al. Microfluidic transduction harnesses mass transport principles to enhance gene transfer efficiency. Mol Ther. 2017;25(10):2372–82.
Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S, Lemberg K, Hurley CK, Kleiner DE, Merchant MS, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125(5):784–92.
Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, Singh H, Hurton L, Maiti SN, Huls MH, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE. 2012;7(1):e30264.
Fujisaki H, Kakuda H, Imai C, Mullighan CG, Campana D. Replicative potential of human natural killer cells. Br J Haematol. 2009;145(5):606–13.
Lapteva N, Durett AG, Sun J, Rollins LA, Huye LL, Fang J, Dandekar V, Mei Z, Jackson K, Vera J, et al. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy. 2012;14(9):1131–43.
Voshol H, Dullens HF, Den Otter W, Vliegenthart JF. Human natural killer cells: a convenient purification procedure and the influence of cryopreservation on cytotoxic activity. J Immunol Methods. 1993;165(1):21–30.
Fujiwara S, Akiyama M, Yamakido M, Seyama T, Kobuke K, Hakoda M, Kyoizumi S, Jones SL. Cryopreservation of human lymphocytes for assessment of lymphocyte subsets and natural killer cytotoxicity. J Immunol Methods. 1986;90(2):265–73.
Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJ, Decker WK, Li S, Robinson SN, Sekine T, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS ONE. 2013;8(10):e76781.
Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol. 2005;175(8):5095–103.
Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, Kaufman DS. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009;113(24):6094–101.
Ni Z, Knorr DA, Clouser CL, Hexum MK, Southern P, Mansky LM, Park IH, Kaufman DS. Human pluripotent stem cells produce natural killer cells that mediate anti-HIV-1 activity by utilizing diverse cellular mechanisms. J Virol. 2011;85(1):43–50.
Luevano M, Madrigal A, Saudemont A. Generation of natural killer cells from hematopoietic stem cells in vitro for immunotherapy. Cell Mol Immunol. 2012;9(4):310–20.
Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, Lee DA, Kaufman DS. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2(4):274–83.
Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8(4):652–8.
Snyder KM, Hullsiek R, Mishra HK, Mendez DC, Li Y, Rogich A, Kaufman DS, Wu J, Walcheck B. Expression of a recombinant high affinity IgG Fc receptor by engineered NK cells as a docking platform for therapeutic mAbs to target cancer cells. Front Immunol. 2018;9:2873.
Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, Tonn T. NK-92: an ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. 2016;65(4):485–92.
Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10(6):625–32.
Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, Suttorp M, Seifried E, Ottmann OG, Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15(12):1563–70.
Kloess S, Oberschmidt O, Dahlke J, Vu XK, Neudoerfl C, Kloos A, Gardlowski T, Matthies N, Heuser M, Meyer J, et al. Preclinical assessment of suitable natural killer cell sources for chimeric antigen receptor natural killer-based “Off-the-Shelf” acute myeloid leukemia immunotherapies. Hum Gene Ther. 2019;30(4):381–401.
Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. 2019;25(9):1341–55.
Lapteva N, Szmania SM, van Rhee F, Rooney CM. Clinical grade purification and expansion of natural killer cells. Crit Rev Oncog. 2014;19(1–2):121–32.
Lee DA. Cellular therapy: adoptive immunotherapy with expanded natural killer cells. Immunol Rev. 2019;290(1):85–99.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, Yin J, You F, Zhu M, Shen W, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, Abujarour R, Bonello GB, Wu J, Tsai P-F, et al. Pluripotent stem cell–derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. 2020;135(6):399–410.
Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol. 2020;20(5):321–34.
Im S, Jang D, Saravanakumar G, Lee J, Kang Y, Lee YM, Lee J, Doh J, Yang ZY, Jang MH, et al. Harnessing the formation of natural killer-tumor cell immunological synapses for enhanced therapeutic effect in solid tumors. Adv Mater. 2020;32(22):e2000020.
Ahn YH, Ren L, Kim SM, Seo S-H, Jung C-R, Kim DS, Noh J-Y, Lee SY, Lee H, Cho MY, et al. A three-dimensional hyaluronic acid-based niche enhances the therapeutic efficacy of human natural killer cell-based cancer immunotherapy. Biomaterials. 2020;247:119960.
Ji T, Lang J, Ning B, Qi F, Wang H, Zhang Y, Zhao R, Yang X, Zhang L, Li W, et al. Enhanced natural killer cell immunotherapy by rationally assembling Fc fragments of antibodies onto tumor membranes. Adv Mater. 2019;31(6):e1804395.
Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, Bonagofski E, Wohlfahrt ME, Pillai SPS, Stephan MT. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017;12(8):813–20.
Kim K-S, Han J-H, Park J-H, Kim H-K, Choi SH, Kim GR, Song H, An HJ, Han DK, Park W, et al. Multifunctional nanoparticles for genetic engineering and bioimaging of natural killer (NK) cell therapeutics. Biomaterials. 2019;221:119418.
Mhaidly R. Verhoeyen E 2019 The future: in vivo CAR T cell gene therapy. Mol Ther. 2019;27(4):707–9.
Pfeiffer A, Thalheimer FB, Hartmann S, Frank AM, Bender RR, Danisch S, Costa C, Wels WS, Modlich U, Stripecke R, et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol Med. 2018;10(11):e9158.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28.
Brudno JN, Lam N, Vanasse D, Shen Y-w, Rose JJ, Rossi J, Xue A, Bot A, Scholler N, Mikkilineni L, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. 2020;26(2):270–80.
Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13(1):168.
Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. 2019;18(1):125.
Ruella M, Barrett DM, Shestova O, Perazzelli J, Posey AD Jr, Hong SJ, Kozlowski M, Lacey SF, Melenhorst JJ, June CH, et al. A cellular antidote to specifically deplete anti-CD19 chimeric antigen receptor–positive cells. Blood. 2020;135(7):505–9.
Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–63.
Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975.
Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, Nakane K, Saito K, Takahashi M, Sasaki A, et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111(5):1478–90.
Ingegnere T, Mariotti FR, Pelosi A, Quintarelli C, De Angelis B, Tumino N, Besi F, Cantoni C, Locatelli F, Vacca P, et al. Human CAR NK cells: a new non-viral method allowing high efficient transfection and strong tumor cell killing. Front Immunol. 2019;10:957.
Piras F, Riba M, Petrillo C, Lazarevic D, Cuccovillo I, Bartolaccini S, Stupka E, Gentner B, Cittaro D, Naldini L, et al. Lentiviral vectors escape innate sensing but trigger p53 in human hematopoietic stem and progenitor cells. EMBO Mol Med. 2017;9(9):1198–211.
Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, Klichinsky M, Shestova O, Patel PR, Kulikovskaya I, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499–503.
Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, Cogdill AP, Morrissette JJD, DeNizio JE, Reddy S, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558(7709):307–12.
Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–50.
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–9.
Marcucci KT, Jadlowsky JK, Hwang W-T, Suhoski-Davis M, Gonzalez VE, Kulikovskaya I, Gupta M, Lacey SF, Plesa G, Chew A, et al. Retroviral and lentiviral safety analysis of gene-modified T cell products and infused HIV and oncology patients. Mol Ther. 2018;26(1):269–79.
Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24(7):927–30.
Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, Vakulskas CA, Collingwood MA, Zhang L, Bode NM, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019;25(2):249–54.
Li R, Johnson R, Yu G, McKenna DH, Hubel A. Preservation of cell-based immunotherapies for clinical trials. Cytotherapy. 2019;21(9):943–57.
Min B, Choi H, Her JH, Jung MY, Kim H-J, Jung M-Y, Lee E-K, Cho SY, Hwang YK, Shin E-C. Optimization of large-scale expansion and cryopreservation of human natural killer cells for anti-tumor therapy. Immune network. 2018;18(4):e31–e31.
Yao X, Jovevski JJ, Todd MF, Xu R, Li Y, Wang J, Matosevic S. Nanoparticle-mediated intracellular protection of natural killer cells avoids cryoinjury and retains potent antitumor functions. Adv Sci. 2020;7(9):1902938.
Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ Jr, Guedan S. CAR-T cells hit the tumor microenvironment: strategies to overcome tumor escape. Front Immunol. 2020;11:1109.
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–44.
Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017;25(9):2214–24.
Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, Wei XF, Han W, Wang H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5(4):e133977.
Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, June CH. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855–66.
Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, Duchateau P, Poirot L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017;7:39833.
Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, Semmrich M, Teige I, Mårtensson L, Penfold CA, et al. Antibodies to costimulatory receptor 4-1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity. 2018;49(5):958-970.e957.
Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, Gottschalk SM, Rooney CM. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res. 2019;7(3):363–75.
Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, Feldman M, Wasik MA, June CH, Gill S. Overcoming the immunosuppressive tumor microenvironment of hodgkin lymphoma using chimeric antigen receptor T cells. Cancer Discov. 2017;7(10):1154–67.
Jo Y, Ali LA, Shim JA, Lee BH, Hong C. Innovative CAR-T cell therapy for solid tumor; current duel between CAR-T spear and tumor shield. Cancers (Basel). 2020;12(8):66.
Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4(3):e994446.
Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133–41.
Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, Balyasnikova IV, Gottschalk S. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;5(7):571–81.
Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, Park H, Purdon TJ, Daniyan AF, Spitzer MH, et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018;23(7):2130–41.
Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, Zhao Y, June CH. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20(13):3025–33.
Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, Butler MO, Minden MD, Hirano N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352–9.
Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, Yi Z, Sauer T, Liu D, Parihar R, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7(11):1238–47.
Brown CE, Vishwanath RP, Aguilar B, Starr R, Najbauer J, Aboody KS, Jensen MC. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol. 2007;179(5):3332–41.
Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33(8):780–8.
Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, Powell DJ Jr, Riley JL, June CH, Albelda SM. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719–30.
Rapp M, Grassmann S, Chaloupka M, Layritz P, Kruger S, Ormanns S, Rataj F, Janssen KP, Endres S, Anz D, et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. Oncoimmunology. 2016;5(3):e1105428.
Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–402.
Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M, Peshwa MV, Childs RW. Efficient mRNA-based genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front Immunol. 2016;7:105.
Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, Wang E, Young HA, Murphy PM, Hwu P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 2002;13(16):1971–80.
Hillerdal V, Essand M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs. 2015;29(2):75–89.
Chen KH, Wada M, Pinz KG, Liu H, Shuai X, Chen X, Yan LE, Petrov JC, Salman H, Senzel L, et al. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia. 2018;32(2):402–12.
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–5.
Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KK, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–52.
Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, Wu MF, Orange JS, Sumazin P, Man TK, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20(4):506–18.
Caratelli S, Sconocchia T, Arriga R, Coppola A, Lanzilli G, Lauro D, Venditti A, Del Principe MI, Buccisano F, Maurillo L, et al. FCγ chimeric receptor-engineered T cells: methodology, advantages, limitations, and clinical relevance. Front Immunol. 2017;8:457.
Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, Davila E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18(23):6436–45.
Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR.” Oncotarget. 2017;8(19):31368–85.
Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172.
Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, Lim WA. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016;164(4):780–91.
Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. 2017;7(11):1306–19.
Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255–66.
Acknowledgements
The authors regret that it was not possible to include many interesting studies in the field due to limited space.
Funding
Y.G. thanks the China Scholarship Council (CSC) (No. 201707720056) for fellowship support. R.G.J.K.W. is supported by a Kootstra Talent Fellowship (Maastricht University), a Marie Skłodowska-Curie Individual fellowship (799810-TOPNIN; European Union), a Cancer Research Institute/Irvington Postdoctoral Fellowship and a fellowship from the “la Caixa” Foundation (ID 100010434; LCF/BQ/PR20/11770004). This research study was supported by Cancer Research Foundation Limburg (2012-03 KOFL) and GROW School of Maastricht University.
Author information
Authors and Affiliations
Contributions
All authors wrote and revised the initial draft, finalized the last version of the manuscript, and approved the final version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
G.M.J.B. and W.T.V.G. are the founders of CiMaas BV. Y.G., R.G.J.K.W. and J.X.W declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1:
Cell line-derived CAR-NK cells (extended version)
Additional file 2. Table S2:
Primary cell-derived CAR-NK cells (extended version)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gong, Y., Klein Wolterink, R.G.J., Wang, J. et al. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol 14, 73 (2021). https://doi.org/10.1186/s13045-021-01083-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-021-01083-5