
Abstract
Myeloid-derived suppressor cells (MDSCs) are newly identified immature myeloid cells that are characterized by the ability to suppress immune responses and expand during cancer, infection, and inflammatory diseases. Although MDSCs have attracted a lot of attention in the field of tumor immunology in recent years, little is known about their multiple roles in hematological malignancies as opposed to their roles in solid tumors. This review will help researchers better understand the various characteristics and functions of MDSCs, as well as the potential therapeutic applications of MDSCs in hematological malignancies, including lymphoma, multiple myeloma, leukemia, and hematopoietic stem cell transplantation.
Similar content being viewed by others
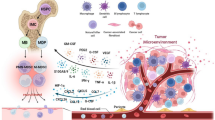


Background
Myeloid-derived suppressor cells (MDSCs) are a newly identified, heterogeneous population of immature myeloid cells that are characterized by the ability to suppress both innate and adaptive immune responses. The role of MDSCs in solid tumors has been extensively characterized as pro-tumorigenic [1,2,3]. In intensive clinical studies, circulating and/or infiltrating MDSCs at the tumor site were associated with poor prognosis in patients with solid tumors [4]. Removing MDSCs might contribute to restoring immune surveillance. Meanwhile, conflicting roles have been reported in hematological malignancies [5,6,7,8,9,10], especially in allogeneic hematopoietic stem cell transplantation (allo-HSCT) for hematological malignancies, which requires the balance between graft-versus-leukemia (GVL) effects and immune tolerance [11]. In this review, we aimed to provide a comprehensive summary of the multiple roles of MDSCs in hematological malignancies and to highlight the double-sided roles of MDSCs.
What are MDSCs?
In the past 10 years, MDSCs have been defined as a new group of myeloid cells with potent immune regulatory activity. Human MDSCs have been defined as “premature” because of their early-stage cell nature and because of their heterogeneous definitions and their unclear mechanisms of action in human beings. In contrast, the definition of MDSCs in mice is far clearer than in humans; in mice, MDSCs simultaneously express the two markers: CD11b and Gr-1. The expression of Ly-6C and Ly-6G further subdivide murine MDSCs into two different subsets: monocytic-MDSCs (M-MDSCs, CD11b+Ly6G−Ly6Chigh) and polymorphonuclear or granulocytic-MDSCs (PMN/G-MDSCs, CD11b+Ly6G+Ly6Clow) [1, 12]. To mimic these findings in mice, human MDSCs have also been identified by flow cytometry according to cellular markers, but these markers are far from uniform. Human G-MDSCs are defined as CD11b+CD15+CD14− or CD11b+CD14-CD66+ cells, as CD15 or CD66b is an activation marker for human granulocytes; however, minimal CD66b is upregulated during nonpathologic conditions. Human M-MDSCs are defined as CD11b+CD14+HLA-DRlow/−CD15− cells. CD14 is a typical surface marker for monocyte, while lower or negative HLA-DR help to distinguish M-MDSCs from the mature monocyte and negative CD15 distinguish M-MDSCs from G-MDSCs. The third group of MDSCs was identified as a group of more immature progenitors called Lin- (including CD3, CD14, CD15, CD16, CD19, CD56, HLA-DR-) CD33+ cells that are in an early development stage, and it has been proposed that these cells be defined properly as “early-stage MDSCs”(eMDSCs) [12]. In addition to the three main populations, various new definitions of MDSC have been identified in different environments, such as CXCR1+CD15−CD14+HLA-DR−/low [13] PD-L1+ CD11b+CD33+HLA-DR− [14] MDSC in tumor microenvironments secreted protein acidic and rich in cysteine (SPARC)-positive MDSC in inflammatory state [15], while it remains unknown whether these MDSCs are truly distinct from classical G-MDSCs, M-MDSCs, or eMDSCs.
How do MDSCs distinguish themselves?
As MDSCs are morphologically and phenotypically similar to neutrophils and monocytes, it is immune suppression that allows MDSCs to be distinguished from other myeloid cell populations. What is so special about these cells that would justify a separate name and what mechanism makes these cells different?
In response to a group of signals produced by tumors or stroma in chronic infection and inflammation, including granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor(G-CSF), and macrophage colony-stimulating factor (M-CSF), MDSCs accumulate in more pathological conditions compared with mature neutrophils and monocytes, which are then activated by the second group of signals, including interferon (IFN)-γ, interleukin (IL)-13, IL-4, and IL-6, which finally distinguishes MDSCs based on special gene expression profiles from mature myeloid cells in healthy donors [16]. The endoplasmic reticulum stress response has emerged in recent years as an important mechanism regulating the pathologic activation of MDSCs [17].
With these gene and protein expression profiles, now we know that MDSCs utilize a number of mechanisms to suppress both the innate and adaptive responses of anti-tumor immunity, mostly through the direct inhibition of T cell activation and expansion, including a high level of arginase 1 (ARG1), inducible nitric oxidase (iNOS) [18], or reactive oxygen species (ROS) [19] production, as well as indoleamine 2,3-dioxygenase (2,3-IDO) activity [20]. In addition, MDSCs also mediate immune suppression, including upregulation of regulatory T cells (Tregs) and immune-suppressive cytokines, such as TGF-β and IL-10 [21,22,23,24]. Altogether, these unique features of MDSCs allow for their identification and provide insight into their biological activity in clinical disease.
Are MDSCs always associated with poor outcomes in hematological malignancies?
The role of MDSCs was first established in mouse tumor models. In recent years, the clinical role of MDSCs has emerged, and numerous studies have suggested a positive correlation of MDSCs in peripheral blood or tumor infiltrating sites with high tumor burden, advanced stage tumors, and poor outcomes, which has been intensively reviewed elsewhere [1, 4, 16]. In hematological malignancies, are MDSCs only an enemy?
Lymphoma
Lymphomas comprise a large group of hematological tumors arising in the lymphatic system, and they share more characteristics with solid tumors than with other hematological malignancies.
In mice model, Serafini et al. demonstrated that MDSCs in a lymphoma animal model shared the same functional properties as MDSCs in solid tumors. By using a murine A20 B cell lymphoma model, a CD45.2+A20-HA-GFP tumor was injected intravenously into BALB/c mice. After 28 days, a cell population was found (CD45.2−/GFP+) with a phenotype consistent with other murine MDSC phenotypes described in solid tumors. CD45.2−/GFP+ cells showed high expression of CD11b, low expression of MHC class I and MHC class II molecules, and expression of Gr1, F4/80, and IL-4Rα, which could inhibit CD8+ T cell proliferation and induce pre-existing Tregs. By using arginase and NOS2 inhibitors (NOHA and L-NMMA, respectively), it was shown that arginase-1 and NO were responsible for the inhibition of CD8+ T cell proliferation, while only proliferation of CD4+ Treg cells was exclusively dependent on arginase-1 [25]. In light of these hopeful results, a series of clinical studies of MDSCs in lymphomas was carried out in the past 10 years.
In peripheral blood of both B cell non-Hodgkin lymphoma(B-NHL) and T-NHL patients, M-MDSCs (CD14+HLA-DRlow/− ± CD120blow) were observed accumulating compared with healthy donors, which were correlated with advanced lymphoma stage, refractory state, higher International Prognostic Index score (IPI), and disease-free survival (DFS). M-MDSCs might return to normal after patients achieve remission. MDSC-dependent T cell suppression was correlated with the upregulated expression of Arg-1, IL-10, programmed death-ligand 1(PD-L1), or S100A12 (a member of the S100 family of calcium-binding proteins involved in T cell suppression through increasing the PD-L1 expression on MDSC). Removing M-MDSCs from patients could restore T cell proliferation [26,27,28,29,30,31,32].
G-MDSCs (CD66b+CD33dimHLA-DR−) were also demonstrated to accumulate in HL and B-NHL patients compared with healthy donors, while depletion of CD66b+ cells could restore T cell proliferation similar to depletion of M-MDSCs [33]. In addition to circulating MDSCs in peripheral blood, Bontkes et al. found that high G-MDSCs (CD11b+CD15+CD33int) in the duodenum are associated with enteropathy-associated T cell lymphoma and its precursor lesions, which may contribute to the development of enteropathy-associated T cell lymphoma (EATL) through the suppression of anti-tumor T cell immunity [34].
In one study of only Hodgkin lymphoma (HL) patients, CD34+MDSCs rather than M or G-MDSCs were identified at diagnosis and were found to be the only independent variable for reducing disease-free survival [35]. In extranodal NK/T cell lymphoma (ENKL) patients, total MDSCs (CD33+CD11b+HLA-DR−) and M-MDSCs were independent prognostic factors for DFS and overall survival (OS). In addition to the elevated signal of arginase-1 and iNOS in MDSCs, IL-17, an inflammatory cytokine produced by CD4+ Th17 cells, may promote the induction of MDSCs and enhance the suppressive function of MDSCs on the inhibition of T cell proliferation [36] (Table 1). Regarding the other subtypes of NHL, such as mantle cell lymphoma or follicular lymphoma, no data were found regarding the role of MDSCs.
In sum, MDSCs, especially M-MDSCs, might contribute to tumorigenesis by inhibiting T cell surveillance in lymphoma, which shares similar mechanisms with solid tumors.
Multiple myeloma
Multiple myeloma (MM) is a malignant plasma cell disorder characterized by the accumulation of neoplastic plasma cells in the bone marrow (BM). Immune dysfunction is an important feature of MM patients and leads to infections and increased tumor growth. A variety of immune defects are observed in MM, including cellular abnormalities (e.g., B cells, T cells, and dendritic cells), secretion of immunosuppressive cytokines (e.g., TGF-b, VEGF, and HGF), and increased frequencies of immunosuppressive cell types (including Tregs and MDSCs) [37, 38].
In mice model, it was demonstrated that the percentage of Ly6Glow cells is significantly increased in MM-diseased BM compared with naive mice, indicating a skewing of myelopoiesis away from granulopoiesis in the course of MM tumor growth. The higher immunosuppressive activity of MM-derived MDSCs compared with normal MDSCs is accompanied by a higher gene expression of iNOS, Arg-1, and IL-10 [39]. In a similar 5TGM1 model, MDSC expansion in the blood, BM, and spleen could also be observed up to 28 days after MM cell inoculation [40]. In another immunocompetent mouse model, tumor cell lines derived from transgenic Bcl-xl/Myc mice were intravenously injected into syngeneic mice. A clear increased MDSCs (CD11b+Gr1+) in BM was shown the first week after MM cell inoculation with a similar increase in both M-MDSCs and G-MDSCs subsets. The BM is the primary tumor site for MM cells and is also the site where MDSCs are generated. The direct contact of the cancer cells with myeloid progenitor cells might explain the early MDSC conversion and accumulation [41]. In addition, CD11b+Gr1+ MDSCs in a mouse model were found contribute to MM chemotherapy resistance [42].
In the peripheral blood of MM patients at diagnosis, M-MDSCs (CD14+HLA-DRlow/−) was first reported to increase compared to healthy donors [43]. Later, it was demonstrated that the level of M-MDSCs was positively correlated with relapsed MM and was negatively related to the treatment response [44]. However, in contrast to the functions of M-MDSCs in lymphoma, G-MDSCs (CD11b+CD14-CD33+CD15+HLA-DRlow) have been suggested to play a key role in MM pathogenesis. It was reported that G-MDSCs were highly accumulated both in BM and PB in MM patients compared to healthy donors, and this accumulation was also positively associated with the activity of disease in MM [41, 45,46,47,48] (Table 2).
Similar to other tumor models, in MM patients, Arg-1, iNOS, ROS, and TNF-α were found to be overexpressed by MDSCs [45, 47]. For example, one recent study reported that PMN-MDSCs and their function through increased Arg-1 are associated with MM progression. Arg-1 is mainly expressed by G-MDSCs. PMN-MDSCs and arginase are increased in myeloma and may contribute to resistance to therapy [49]. Tregs could also be induced by MM MDSCs in a cell contact-dependent manner [46]. In addition, MDSCs and MM cells appear to interact in a bidirectional manner, in which MM cells are able to induce MDSCs, probably by mesenchymal stromal cells, and the latter provide a safe haven within the microenvironment for tumor growth and progression [44, 45, 47, 48] (Table 2).
The effect of new therapies, including bortezomib and lenalidomide, on MDSCs are conflicting. In the study by Wang et al., bortezomib combined with dexamethasone resulted in a gradual decrease of the number of MDSCs. When MM cells and PBMCs were cocultured in the presence of bortezomib, a significant decrease in M-MDSCs was observed compared to MM cells and PBMCs alone [44]. However, in the study by Görgün et al., the total number and the immune suppressive capacity of MDSCs did not change after exposure to bortezomib and lenalidomide in vitro [45].
MDSCs, especially G-MDSCs, might interact with MM cells in a bidirectional “win-win” manner, which suggests that treatments targeting M-MDSCs may improve therapeutic outcomes for MM patients.
Leukemia and myelodysplastic syndromes
In contrast with lymphoma and MM, studies on MDSCs in leukemia, including acute myeloid leukemia (AML), chronic myeloid leukemia (CML), acute lymphoblastic leukemia (ALL), and chronic lymphocytic leukemia (CLL), and studies on myelodysplastic syndromes (MDS) have been relatively limited.
In mice model of acute leukemia, engraftment of C57BL/6 mice with TIB-49 AML cell lines led to an expansion of CD11b+Gr1+MDSCs in bone marrow and spleen. Coculture of the AML cell lines or primary AML cells with donor peripheral blood mononuclear cells expanded MDSCs, probably by MUC1-mediated tumor-derived extracellular vehicles [50]. In clinical studies, it has been reported that more eMDSCs (CD11b+HLA-DR-CD33+Lin-) were accumulated in PB and BM of AML patients when compared with healthy donors [50]. In addition, V-domain Ig suppressor of T cell activation (VISTA), a recently defined negative regulator mediating immune evasion in tumors, was highly expressed on these MDSCs in AML patients; knockdown of VISTA by specific siRNA potently reduced the MDSCs-mediated inhibition of CD8 T cell activity in AML, suggesting a suppressive effect of VISTA on anti-leukemia T cell response [51]. G-MDSC (HLA-DR−/low CD11b+CD33int/high) was demonstrated to be significantly elevated in both the peripheral blood and BM of patients with B-ALL and was positively correlated with clinical therapeutic responses, such as minimal residual disease [52]. In acute promyelocytic leukemia (APL) patients, tumor-activated ILC2s secreted IL-13 to induce M-MDSCs (CD33+CD14+HLA-DR-) and to support tumor growth, while ATRA treatment reversed the increase of ILC2-MDSCs in APL [53] (Table 3).
In the BM of MDS patients, it was demonstrated that more MDSCs (Lin−HLA−DR–CD33+) accumulated compared with healthy donor, the interaction of CD33 with receptor S100A9 (bind to surface glycoprotein receptors on MDSC) promoted MDSCs and induced IL-10 and TGF-β secretion, while early forced maturation of MDSCs rescued the hematologic phenotype of MDS [54]. In addition, CD33-S100A9 initiated suppressive inflammatory signaling cascades that lead to the secretion of ROS, which was strongly correlated to DNA damage and accumulation of phosphorylated γH2AX, a main marker of genomic instability. Fc-engineered monoclonal antibody against CD33 was demonstrated to improves the bone marrow microenvironment by reducing MDSC, blocking CD33 downstream signaling, preventing immune-suppressive cytokine secretion, and reducing both ROS and the levels of double stranded breaks and adducts in MDS [55] (Table 3).
In Sokal high-risk CML patients, Christiansson, L. et al. found that MDSCs (CD11b+CD14−CD33+) and Arg-1 were increased, which upregulated PD-L1 and PD-1 on T cells [56]. Giallongo C. et al. identified that MDSCs (CD11b+CD33+CD14−HLA-DR−) were elevated at diagnosis and decreased to normal levels after imatinib therapy in CML patients [57]. In CLL patients, M-MDSCs (CD14+HLA-DRlow cells) were significantly increased at diagnosis, suppressing in vitro T cell activation and inducing suppressive regulatory T cells. The MDSC-mediated modulation of T cells could be attributed to their increased 2,3-IDO activity. CLL cells induced IDOhi MDSCs from healthy donor monocytes, suggesting bidirectional crosstalk between CLL-cells, MDSCs, and Tregs [58] (Table 3).
As characteristics and pathogenesis of leukemia and MDS are distinct from those of solid tumors, fewer shared mechanisms were found in these hematological malignancies compared with lymphoma and MM, which is in line with the fact that the enhancement of anti-tumor responses by blocking negative immune regulators is a more common mechanism in lymphoma and MM.
Hematopoietic stem cell transplantation
In the above models of hematological malignancies, the relationship between MDSCs and the tumor microenvironment is relatively simple, as only bidirectional regulations were involved. In contrast, in the unique model of HSCT for hematological malignancies, a far more complex triangular relationship that correlated the immune balance between GVL effects, graft-versus-host disease (GVHD), and hematological malignancies and MDSCs was noticed [59].
Several mice models have indicated that MDSCs modulate the function of alloreactive T cells and prevent GVHD without impairing the GVL effects. MacDonald first demonstrated MDSCs (CD11b+Gr-1+), previously described as the granulocyte-monocyte precursor population, could differentiate into class II+, CD80/CD86+, and CD40− APC during GVHD, which promoted transplant tolerance by MHC class II-restricted generation of IL-10-secreting, Ag-specific regulatory T cells, and more importantly, preserved GVL effects [24]. Messmann reported that MDSCs induced by GM-CSF/G-CSF in vitro inhibited GVHD-induced death and attenuated histologic GVHD by Th2 induction, whereas antitumor cytotoxicity of alloantigen-specific T cells was maintained [60]. Wang reported that adding functional MDSCs to the donor graft alleviated GVHD, whereas removal of MDSCs in vivo exacerbated GVHD. MDSCs from the recipients with GVHD showed much higher suppressive potency compared with those from recipients without GVHD. In addition, MDSC (CD11b+Gr-1+) accumulation was positively correlated with the severity of GVHD and further increased upon leukemia relapse, suggesting that there are different characteristics of MDSCs in graft and immune reconstitution [61]. Zhang suggested CD115+ MDSCs efficiently suppressed GVHD but did not significantly impair GVL effects, as MDSC exhibited cytolytic activities against allogeneic leukemia cells via induced NKG2D+ CD8 T cells, while suppressed GVHD by upregulating Tregs [62].
Clinical results have suggested that MDSCs could be expanded by G-CSF mobilization and that MDSCs in grafts are closely associated with lower risk of GVHD in allo-HSCT. Antonio et al. reported that systemic treatment with G-CSF induces the expansion of myeloid cells displaying the M-MDSC phenotype of (Linlow/negHLA-DR−CD11b+CD33+CD14+), which is the only graft parameter to predict acute GVHD (aGVHD) [63]. Lv showed that G-CSF induced the expansion of M-MDSCs (Lin−HLA−DR−/lowCD33+CD11b+CD14+CD15dimCD16−) and eMDSCs (Lin−HLA-DR−/lowCD33+CD11b−/lowCD14−CD15−CD16−) in the graft are negatively correlated with the incidence of acute and chronic GVHD without significant influence on relapse and survival [64]. Fan demonstrated that a higher frequency of MDSCs (Linlow/negHLA-DR−CD33+CD11b+) in the G-CSF primed BM than in the G-CSF peripheral blood stem cells harvest (G-PBSC) grafts lead to a better GVHD and relapse-free survival (GRFS) and less GVHD [65]. Wang identified a new subset of eMDSCs (HLA-DR−/lowCD33+CD16- cells) in a humanized mice model that may control acute GVHD in mice HSCT, and these cells were also identified as an independent factor that reduced the occurrence of grade II-IV aGvHD in allo-HSCT patients [66]. Schneidawind reported that administration of G-CSF- donor lymphocyte infusion (DLI) results in graft-versus-leukemia effects without exacerbating GVHD because of the M-MDSCs in the DLI component [67].
Hematological malignancy-derived MDSCs exerted negative effects on survival in most circumstances both in mice model and human being, similar to their roles in solid tumors microenvironment. However, in HSCT for hematological malignancies, MDSCs in grafts were regarded as helpful for reducing the risk of GVHD without interfering with the GVL effect for malignancies; meanwhile, the roles of reconstituted MDSCs might still be controversial and remain to be clarified.
What is behind these disparities?
Considering the disparities of MDSC activity between lymphoma/MM and leukemia/MDS, and in graft or reconstitution in HSCT, one might wonder what mechanisms are behind these disparities?
Disparities in tumor microenvironment
Take lymphoblastic malignancies as an example. The tumor microenvironment (TME) of lymphoma provides striking examples of a pivotal interaction of hematopoietic tumor cells with stromal cells compared with the TME of acute leukemia. Lymphoma originates and progresses in primary or secondary lymphoid organs where immune cells develop and reside and in which anti-tumor immune responses are typically initiated, suggesting that most are poorly immunogenic and fail to alert innate or adaptive immune sensing mechanisms. In addition, the degree of dependence on MDSCs varies widely among hematologic malignancies. For instance, the normal lymph node architecture is completely replaced by an inflammatory milieu rich in immune suppressive cells in classic HL, which is in contrast to Burkitt lymphoma, where there is a relative paucity of immune suppressive cells, and the normal nodal tissue is almost completely replaced by malignant cells. Thus, the heterogeneous roles of MSCSs could be identified between hematologic malignancies, as previously introduced (Tables 1, 2, and 3) [7].
On the other hand, acute leukemia disseminates rapidly after inception compared with lymphoma and MM, which may negatively impact the initiation and execution of anti-leukemia immunity. Leukemia-specific T cells are never properly activated but, rather, are deleted or anergized upon initial antigen encounter, which is contrary to what is observed in solid tumors and lymphoma, where tumor-specific T cells are primed but become functionally impaired by MDSCs and other components of TMEs [5].
Disparities in MDSC origins
The disparity of MDSCs in graft or reconstitution after HSCT might be contributed to the question “how these MDSCs are induced?”. Increasing evidence suggests that G-CSF affects different immune cells [24, 68] that modulate T cell responses either directly by inducing Th2 differentiation [69, 70] or by mobilizing functional regulatory T cells [71] or indirectly through monocytes [72,73,74], DCs [75], and neutrophils [76]. Thus, nearly all the MDSCs that accumulated in grafts of HSCT or DLIs were induced by G-CSF mobilization rather than by generation by the TME or imbalanced immune environment post-HSCT.
For example, dominated with IL-10/TGF-β signal and Treg induction, MDSCs in G-CSF mobilized grafts were different from the MDSCs cells accumulated during GVHD post-HSCT, which were expressing low pSTAT1 (phosphorylated signal transducer and activator of transcription) and high 2,3-IDO [63]. In addition, MDSC induced by G-CSF mobilization might also be distinct from MDSCs induced in TME of lymphoma, MM, and leukemia, which were characterized by high expression of arginase I, PD-L1, ROS, and STAT-3, etc. (Tables 1, 2, 3, and 4). Therefore, it is worth comparing the differences of MDSCs originated from different models on the single cell level in the future, which could help us to target MDSCs precisely in treatment of hematological malignancies and avoid potential risk of increasing relapse of malignancies post-MDSC-based cellular therapy.
Potential application of MDSCs
Considering the disparate roles of MDSCs in non-HSCT and HSCT models of hematological malignancies, it is reasonable to consider MDSCs as a valid therapeutic target in chemotherapy, and even in cellular therapies, since MDSCs contribute to distinct processes in tumor development, progression, and metastasis in the microenvironment [77]. In contrast, MDSCs would be regarded as useful cellular therapy products for prophylaxis or treatment of GVHD post-HSCT.
MDSC deactivation
Activated MDSCs express high amounts of arginase 1 and NOS2, and inhibitors of both enzymes (L-NMMA for NOS2 and nor NOHA for arginase-1) reversed MDSC suppressive mechanisms in MM and lymphoma models [39, 45, 78]. Consequently, sildenafil (phosphodiesterase-5 inhibitors) treatment of peripheral blood mononuclear cells isolated from MM patients could downregulate arginase 1 and nitric oxide synthase–2 expression, resulted in increased T cell proliferation in vitro [79]. Recently, Borello et al. demonstrated a reduction in Mspike by Tadalafil (phosphodiesterase-5 inhibitors) treatment in an end-stage relapsed/refractory MM patient. BM CD14+ cells decreased overtime with Tadalafil treatment. This decrease was associated with a decrease in IL-4Ra, iNOS, and arginase-1 expression in MDSC. BM nitrosylation was also decreased, and T cell activity was enhanced upon Tadalafil administration [80]. In addition, synthetic triterpenoid bardoxolone methyl (CDDO-Me), the activator of the NRF2 transcription factor that results in upregulation of several antioxidant genes, also reduced intracellular ROS and nitrotyrosine levels in EL4 mice, which was accompanied by an increased T cell response and reduced tumor load [81]. Cyclooxygenase-2 (COX2) inhibitors have also been described to reduce MDSC numbers and to have immunosuppressive function in solid tumors, but they have not been tested in hematological malignancies [82, 83]. Targeting IDO1 represents a therapeutic opportunity not only in MDSCs, but also in Treg [84].
MDSC depletion
Anti-GR1 antibodies bind with a high affinity to Ly6G molecules and have been extensively used to deplete G-MDSCs in tumor-bearing mice. The effect of anti-GR1 antibodies has been investigated in EL4 tumor-bearing mice, in which a complete elimination of MDSCs in the spleen and peripheral blood was observed [85]. In contrast to anti-GR1 specific antibodies, which predominantly target the granulocytic population, peptibodies were able to deplete both monocytic and granulocytic MDSCs. Intravenous peptibody injection was able to deplete blood, spleen, and intra tumoral MDSCs in distinct lymphoma models (A20, EG7, EL4) and delayed tumor growth in vivo, as determined by tumor size and tumor mass, without inducing effects on other immune cells, including DC and lymphocytes (T, B, and NK cells) [86]. 17-DMAG (HSP90 inhibitors), 5-fluorouracil, and gemcitabine have been explored in a preclinical model of MDSC depletion, with efficacy about 50–75% [87, 88]. Anti-CD33 antibody (gemtuzumab ozogamicin, etc.) have been proved useful in targeting human MDSC which could restore T cell immunity against cancers and enhance CAR-T therapy [55, 89].
MDSC differentiation and development
Another mechanism to target the MDSC population is the induction of MDSC differentiation into mature myeloid cells with no suppressive activities. MDSC differentiation can be triggered by distinct vitamins, including vitamin A, vitamin D3, or vitamin E [90,91,92,93]. ATRA (all-trans retinoic acid), a vitamin A metabolite, induces the differentiation of monocytic MDSC in DC and macrophages and causes apoptosis of the granulocytic MDSC population. As a consequence, ATRA improved immunotherapy in distinct murine models [93].
S100A8/S100A9 proteins are also involved in MDSC recruitment by the binding of S100 proteins to carboxylated N-glycan receptors. The anti-carboxylated glycan antibody mAbGB3.1 was able to reduce MDSC numbers in the blood and secondary lymphoid organs [94]. Furthermore, mAbGB3.1 was able to block tumor cell proliferation in colorectal cancer [95, 96]. In addition, tasquinimod, a quinoline-3-carboxamide derivative, binds to S100A9 and blocks the interaction with its ligands, receptor of advanced glycation end products (RAGE) and toll-like receptor 4 (TLR4). It has been demonstrated that tasquinimod reduced MDSC accumulation, modulated local tumor immunity, and reduced tumor growth and metastasis [97, 98]. In addition, the N-bisphosphonate zoledronic acid reduced MDSC number and osteoclast formation in MM disease [40]. JAK2/STAT3 inhibitors (JSI-124 and cucurbitacin B) [19, 99] and multikinase inhibitors (sunitinib and sorafenib) were also found to reduce MDSC levels [100, 101].
MDSC infusion
In contrast to the role of MDSCs in the chemotherapy of hematological malignancies, MDSCs were found to be beneficial in grafts of HSCT or DLI, which reduced GVHD and preserved the GVL effect [102]. Lim reported in a preclinical model that ex vivo-generated human cord blood MDSCs could attenuate clinical and pathologic cGVHD severity by preserving thymus function and regulating Th17 signaling, suggesting a possible therapeutic strategy for the clinical application of MDSC infusion [103]. Further, caution must be taken in utilizing MDSC infusion due to the conflicting role of reconstituted MDSCs, which might contribute to GVHD onset.
Conclusions
MDSCs are new but important regulators that hamper the host anti-tumor immune response by inhibition of T cell proliferation, cytokine secretion, and recruitment of regulatory T cells in hematological malignancies, similar to their actions in solid tumors, which would be a key target in chemotherapy and immune therapy. On the other hand, transfusing MDSCs would be a potentially beneficial therapy for reducing GVHD but for preserving GVL effect post-allo-HSCT for hematological malignancies. The double-sided roles of MDSCs need to be further clarified in future (Fig. 1).

Conflict roles of MDSCs in hematological malignancies
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- 2,3-IDO:
-
Indoleamine 2,3-dioxygenase
- ALL:
-
Acute lymphoblastic leukemia
- Allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- AML:
-
Acute myeloid leukemia
- APL:
-
Acute promyelocytic leukemia
- ARG1:
-
Arginase 1
- ATRA:
-
All-trans retinoic acid
- BM:
-
Bone marrow
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- COX2:
-
Cyclooxygenase-2
- DCs:
-
Dendritic cells
- DFS:
-
Disease-free survival
- DLI:
-
Donor lymphocyte infusion
- EATL:
-
Enteropathy-associated T cell lymphoma
- eMDSCs:
-
Early-stage MDSCs
- ENKL:
-
Extranodal NK/T cell lymphoma
- G-CSF:
-
Granulocyte colony-stimulating factor
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- G-PBSC:
-
G-CSF peripheral blood stem cells harvest
- GRFS:
-
GVHD and relapse-free survival
- GVHD:
-
Graft-versus-host disease
- GVL:
-
Graft-versus-leukemia
- IFN:
-
Interferon
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxidase
- IPI:
-
International Prognostic Index score
- M-CSF:
-
Macrophage colony-stimulating factor
- MDS:
-
Myelodysplastic syndromes
- MDSCs:
-
Myeloid-derived suppressor cells
- MM:
-
Multiple myeloma
- M-MDSCs:
-
Monocytic-MDSCs
- NHL:
-
Non-Hodgkin lymphoma
- OS:
-
Overall survival
- PD-L1:
-
Programmed death-ligand 1
- PMN/G-MDSCs:
-
Polymorphonuclear or granulocytic-MDSCs
- RAGE:
-
Receptor of advanced glycation end products
- ROS:
-
Reactive oxygen species
- TLR4:
-
Toll-like receptor 4
- TME:
-
Tumor microenvironment
- Tregs:
-
Regulatory T cells
- VISTA:
-
V-domain Ig suppressor of T cell activation
References
Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108–19.
Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. 2019;133:2159–67.
Tesi RJ. MDSC; the most important cell you have never heard of. Trends Pharmacol Sci. 2019;40:4–7.
Wang PF, Song SY, Wang TJ, Ji WJ, Li SW, Liu N, Yan CX. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: a meta-analysis of 40 studies. Oncoimmunology. 2018;7:e1494113.
Hopken UE, Rehm A. Targeting the tumor microenvironment of leukemia and lymphoma. Trends Cancer. 2019;5:351–64.
Betsch A, Rutgeerts O, Fevery S, Sprangers B, Verhoef G, Dierickx D, Beckers M. Myeloid-derived suppressor cells in lymphoma: the good, the bad and the ugly. Blood Rev. 2018;32:490–8.
Curran EK, Godfrey J, Kline J. Mechanisms of immune tolerance in leukemia and lymphoma. Trends Immunol. 2017;38:513–25.
Malek E, de Lima M, Letterio JJ, Kim BG, Finke JH, Driscoll JJ, Giralt SA. Myeloid-derived suppressor cells: the green light for myeloma immune escape. Blood Rev. 2016;30:341–8.
Vetro C, Romano A, Ancora F, Coppolino F, Brundo MV, Raccuia SA, Puglisi F, Tibullo D, La Cava P, Giallongo C, Parrinello NL. Clinical impact of the Immunome in lymphoid malignancies: the role of myeloid-derived suppressor cells. Front Oncol. 2015;5:104.
Giallongo C, Parrinello N, Brundo MV, Raccuia SA, Di Rosa M, La Cava P, Tibullo D. Myeloid derived suppressor cells in chronic myeloid leukemia. Front Oncol. 2015;5:107.
Koehn BH, Blazar BR. Role of myeloid-derived suppressor cells in allogeneic hematopoietic cell transplantation. J Leukoc Biol. 2017;102:335–41.
Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150.
Mastio J, Condamine T, Dominguez G, Kossenkov AV, Donthireddy L, Veglia F, Lin C, Wang F, Fu S, Zhou J, et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J Exp Med. 2019;216:2150–69.
Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. 2016;5:e1247135.
Sangaletti S, Talarico G, Chiodoni C, Cappetti B, Botti L, Portararo P, Gulino A, Consonni FM, Sica A, Randon G, et al. SPARC is a new myeloid-derived suppressor cell marker licensing suppressive activities. Front Immunol. 2019;10:1369.
Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–20.
Cubillos-Ruiz JR, Mohamed E, Rodriguez PC. Unfolding anti-tumor immunity: ER stress responses sculpt tolerogenic myeloid cells in cancer. J Immunother Cancer. 2017;5:5.
Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+HLA-DR−/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010;70:4335–45.
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–701.
Mougiakakos D, Jitschin R, von Bahr L, Poschke I, Gary R, Sundberg B, Gerbitz A, Ljungman P, Le Blanc K. Immunosuppressive CD14+HLA-DRlow/neg IDO+ myeloid cells in patients following allogeneic hematopoietic stem cell transplantation. Leukemia. 2013;27:377–88.
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, Korangy F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–43.
Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31.
Yang R, Cai Z, Zhang Y, Yutzy WH, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated gr-1+CD11b+ myeloid cells. Cancer Res. 2006;66:6807–15.
MacDonald KP, Rowe V, Clouston AD, Welply JK, Kuns RD, Ferrara JL, Thomas R, Hill GR. Cytokine expanded myeloid precursors function as regulatory antigen-presenting cells and promote tolerance through IL-10-producing regulatory T cells. J Immunol. 2005;174:1841–50.
Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68:5439–49.
Lin Y, Gustafson MP, Bulur PA, Gastineau DA, Witzig TE, Dietz AB. Immunosuppressive CD14+HLA-DR (low)/− monocytes in B-cell non-Hodgkin lymphoma. Blood. 2011;117:872–81.
Khalifa KA, Badawy HM, Radwan WM, Shehata MA, Bassuoni MA. CD14(+) HLA-DR low/(−) monocytes as indicator of disease aggressiveness in B-cell non-Hodgkin lymphoma. Int J Lab Hematol. 2014;36:650–5.
Xiu B, Lin Y, Grote DM, Ziesmer SC, Gustafson MP, Maas ML, Zhang Z, Dietz AB, Porrata LF, Novak AJ, et al. IL-10 induces the development of immunosuppressive CD14(+)HLA-DR (low/−) monocytes in B-cell non-Hodgkin lymphoma. Blood Cancer J. 2015;5:e328.
Azzaoui I, Uhel F, Rossille D, Pangault C, Dulong J, Le Priol J, Lamy T, Houot R, Le Gouill S, Cartron G, et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood. 2016;128:1081–92.
Tadmor T, Fell R, Polliack A, Attias D. Absolute monocytosis at diagnosis correlates with survival in diffuse large B-cell lymphoma-possible link with monocytic myeloid-derived suppressor cells. Hematol Oncol. 2013;31:65–71.
Wu C, Wu X, Zhang X, Chai Y, Guo Q, Li L, Yue L, Bai J, Wang Z, Zhang L. Prognostic significance of peripheral monocytic myeloid-derived suppressor cells and monocytes in patients newly diagnosed with diffuse large b-cell lymphoma. Int J Clin Exp Med. 2015;8:15173–81.
Wilcox RA, Feldman AL, Wada DA, Yang ZZ, Comfere NI, Dong H, Kwon ED, Novak AJ, Markovic SN, Pittelkow MR, et al. B7-H1 (PD-L1, CD274) suppresses host immunity in T-cell lymphoproliferative disorders. Blood. 2009;114:2149–58.
Marini O, Spina C, Mimiola E, Cassaro A, Malerba G, Todeschini G, Perbellini O, Scupoli M, Carli G, Facchinelli D, et al. Identification of granulocytic myeloid-derived suppressor cells (G-MDSCs) in the peripheral blood of Hodgkin and non-Hodgkin lymphoma patients. Oncotarget. 2016;7:27676–88.
Bontkes HJ, Jordanova ES, Nijeboer P, Neefjes-Borst EA, Cillessen S, Hayat A, Mulder CJ, Bouma G, von Blomberg BM, de Gruijl TD. High myeloid-derived suppressor cell frequencies in the duodenum are associated with enteropathy associated T-cell lymphoma and its precursor lesions. Br J Haematol. 2017;178:988–91.
Romano A, Parrinello NL, Vetro C, Forte S, Chiarenza A, Figuera A, Motta G, Palumbo GA, Ippolito M, Consoli U, Di Raimondo F. Circulating myeloid-derived suppressor cells correlate with clinical outcome in Hodgkin lymphoma patients treated up-front with a risk-adapted strategy. Br J Haematol. 2015;168:689–700.
Zhang H, Li ZL, Ye SB, Ouyang LY, Chen YS, He J, Huang HQ, Zeng YX, Zhang XS, Li J. Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator. Cancer Immunol Immunother. 2015;64:1587–99.
Romano A, Conticello C, Cavalli M, Vetro C, La Fauci A, Parrinello NL, Di Raimondo F. Immunological dysregulation in multiple myeloma microenvironment. Biomed Res Int. 2014;2014:198539.
Pratt G, Goodyear O, Moss P. Immunodeficiency and immunotherapy in multiple myeloma. Br J Haematol. 2007;138:563–79.
Van Valckenborgh E, Schouppe E, Movahedi K, De Bruyne E, Menu E, De Baetselier P, Vanderkerken K, Van Ginderachter JA. Multiple myeloma induces the immunosuppressive capacity of distinct myeloid-derived suppressor cell subpopulations in the bone marrow. Leukemia. 2012;26:2424–8.
Zhuang J, Zhang J, Lwin ST, Edwards JR, Edwards CM, Mundy GR, Yang X. Osteoclasts in multiple myeloma are derived from gr-1+CD11b+myeloid-derived suppressor cells. PLoS One. 2012;7:e48871.
Ramachandran IR, Martner A, Pisklakova A, Condamine T, Chase T, Vogl T, Roth J, Gabrilovich D, Nefedova Y. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J Immunol. 2013;190:3815–23.
Ramachandran IR, Condamine T, Lin C, Herlihy SE, Garfall A, Vogl DT, Gabrilovich DI, Nefedova Y. Bone marrow PMN-MDSCs and neutrophils are functionally similar in protection of multiple myeloma from chemotherapy. Cancer Lett. 2016;371:117–24.
Brimnes MK, Vangsted AJ, Knudsen LM, Gimsing P, Gang AO, Johnsen HE, Svane IM. Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR(−)/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand J Immunol. 2010;72:540–7.
Wang Z, Zhang L, Wang H, Xiong S, Li Y, Tao Q, Xiao W, Qin H, Wang Y, Zhai Z. Tumor-induced CD14+HLA-DR (−/low) myeloid-derived suppressor cells correlate with tumor progression and outcome of therapy in multiple myeloma patients. Cancer Immunol Immunother. 2015;64:389–99.
Gorgun GT, Whitehill G, Anderson JL, Hideshima T, Maguire C, Laubach J, Raje N, Munshi NC, Richardson PG, Anderson KC. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood. 2013;121:2975–87.
Favaloro J, Liyadipitiya T, Brown R, Yang S, Suen H, Woodland N, Nassif N, Hart D, Fromm P, Weatherburn C, et al. Myeloid derived suppressor cells are numerically, functionally and phenotypically different in patients with multiple myeloma. Leuk Lymphoma. 2014;55:2893–900.
Giallongo C, Tibullo D, Parrinello NL, La Cava P, Di Rosa M, Bramanti V, Di Raimondo C, Conticello C, Chiarenza A, Palumbo GA, et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget. 2016;7:85764–75.
Ai L, Mu S, Sun C, Fan F, Yan H, Qin Y, Cui G, Wang Y, Guo T, Mei H, et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol Cancer. 2019;18:88.
Romano A, Parrinello NL, La Cava P, Tibullo D, Giallongo C, Camiolo G, Puglisi F, Parisi M, Pirosa MC, Martino E, et al. PMN-MDSC and arginase are increased in myeloma and may contribute to resistance to therapy. Expert Rev Mol Diagn. 2018;18:675–83.
Pyzer AR, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, Fung J, Bryant MP, Cole L, Palmer K, et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. 2017;129:1791–801.
Wang L, Jia B, Claxton DF, Ehmann WC, Rybka WB, Mineishi S, Naik S, Khawaja MR, Sivik J, Han J, et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology. 2018;7:e1469594.
Liu YF, Chen YY, He YY, Wang JY, Yang JP, Zhong SL, Jiang N, Zhou P, Jiang H, Zhou J. Expansion and activation of granulocytic, myeloid-derived suppressor cells in childhood precursor B cell acute lymphoblastic leukemia. J Leukoc Biol. 2017;102:449–58.
Trabanelli S, Chevalier MF, Martinez-Usatorre A, Gomez-Cadena A, Salome B, Lecciso M, Salvestrini V, Verdeil G, Racle J, Papayannidis C, et al. Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat Commun. 2017;8:593.
Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N, Epling-Burnette P, Van Bijnen S, Dolstra H, Cannon J, et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest. 2013;123:4595–611.
Eksioglu EA, Chen X, Heider KH, Rueter B, McGraw KL, Basiorka AA, Wei M, Burnette A, Cheng P, Lancet J, et al. Novel therapeutic approach to improve hematopoiesis in low risk MDS by targeting MDSCs with the fc-engineered CD33 antibody BI 836858. Leukemia. 2017;31:2172–80.
Christiansson L, Soderlund S, Svensson E, Mustjoki S, Bengtsson M, Simonsson B, Olsson-Stromberg U, Loskog AS. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in Sokal high risk chronic myeloid leukemia. PLoS One. 2013;8:e55818.
Giallongo C, Parrinello N, Tibullo D, La Cava P, Romano A, Chiarenza A, Barbagallo I, Palumbo GA, Stagno F, Vigneri P, Di Raimondo F. Myeloid derived suppressor cells (MDSCs) are increased and exert immunosuppressive activity together with polymorphonuclear leukocytes (PMNs) in chronic myeloid leukemia patients. PLoS One. 2014;9:e101848.
Jitschin R, Braun M, Buttner M, Dettmer-Wilde K, Bricks J, Berger J, Eckart MJ, Krause SW, Oefner PJ, Le Blanc K, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood. 2014;124:750–60.
Luyckx A, Schouppe E, Rutgeerts O, Lenaerts C, Fevery S, Devos T, Dierickx D, Waer M, Van Ginderachter JA, Billiau AD. G-CSF stem cell mobilization in human donors induces polymorphonuclear and mononuclear myeloid-derived suppressor cells. Clin Immunol. 2012;143:83–7.
Messmann JJ, Reisser T, Leithauser F, Lutz MB, Debatin KM, Strauss G. In vitro-generated MDSCs prevent murine GVHD by inducing type 2 T cells without disabling antitumor cytotoxicity. Blood. 2015;126:1138–48.
Wang D, Yu Y, Haarberg K, Fu J, Kaosaard K, Nagaraj S, Anasetti C, Gabrilovich D, Yu XZ. Dynamic change and impact of myeloid-derived suppressor cells in allogeneic bone marrow transplantation in mice. Biol Blood Marrow Transplant. 2013;19:692–702.
Zhang J, Chen HM, Ma G, Zhou Z, Raulet D, Rivera AL, Chen SH, Pan PY. The mechanistic study behind suppression of GVHD while retaining GVL activities by myeloid-derived suppressor cells. Leukemia. 2019;33(8):2078–89.
Vendramin A, Gimondi S, Bermema A, Longoni P, Rizzitano S, Corradini P, Carniti C. Graft monocytic myeloid-derived suppressor cell content predicts the risk of acute graft-versus-host disease after allogeneic transplantation of granulocyte colony-stimulating factor-mobilized peripheral blood stem cells. Biol Blood Marrow Transplant. 2014;20:2049–55.
Lv M, Zhao XS, Hu Y, Chang YJ, Zhao XY, Kong Y, Zhang XH, Xu LP, Liu KY, Huang XJ. Monocytic and promyelocytic myeloid-derived suppressor cells may contribute to G-CSF-induced immune tolerance in haplo-identical allogeneic hematopoietic stem cell transplantation. Am J Hematol. 2015;90:E9–E16.
Fan Q, Liu H, Liang X, Yang T, Fan Z, Huang F, Ling Y, Liao X, Xuan L, Xu N, et al. Superior GVHD-free, relapse-free survival for G-BM to G-PBSC grafts is associated with higher MDSCs content in allografting for patients with acute leukemia. J Hematol Oncol. 2017;10:135.
Wang K, Lv M, Chang YJ, Zhao XY, Zhao XS, Zhang YY, Sun YQ, Wang ZD, Suo P, Zhou Y, et al. Early myeloid-derived suppressor cells (HLA-DR(−)/(low)CD33(+)CD16(−)) expanded by granulocyte colony-stimulating factor prevent acute graft-versus-host disease (GVHD) in humanized mouse and might contribute to lower GVHD in patients post Allo-HSCT. J Hematol Oncol. 2019;12:31.
Schneidawind C, Jahnke S, Schober-Melms I, Schumm M, Handgretinger R, Faul C, Kanz L, Bethge W, Schneidawind D. G-CSF administration prior to donor lymphocyte apheresis promotes anti-leukaemic effects in allogeneic HCT patients. Br J Haematol. 2019;186:60–71.
Rutella S, Zavala F, Danese S, Kared H, Leone G. Granulocyte colony-stimulating factor: a novel mediator of T cell tolerance. J Immunol. 2005;175:7085–91.
Pan L, Delmonte J Jr, Jalonen CK, Ferrara JL. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces severity of experimental graft-versus-host disease. Blood. 1995;86:4422–9.
Franzke A, Piao W, Lauber J, Gatzlaff P, Konecke C, Hansen W, Schmitt-Thomsen A, Hertenstein B, Buer J, Ganser A. G-CSF as immune regulator in T cells expressing the G-CSF receptor: implications for transplantation and autoimmune diseases. Blood. 2003;102:734–9.
Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, Wei S, David O, Curiel TJ, Zou W. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004;64:8451–5.
Mielcarek M, Graf L, Johnson G, Torok-Storb B. Production of interleukin-10 by granulocyte colony-stimulating factor-mobilized blood products: a mechanism for monocyte-mediated suppression of T-cell proliferation. Blood. 1998;92:215–22.
Mielcarek M, Martin PJ, Torok-Storb B. Suppression of alloantigen-induced T-cell proliferation by CD14+ cells derived from granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cells. Blood. 1997;89:1629–34.
Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, Ochoa JB, Ochoa AC. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171:1232–9.
Arpinati M, Green CL, Heimfeld S, Heuser JE, Anasetti C. Granulocyte-colony stimulating factor mobilizes T helper 2-inducing dendritic cells. Blood. 2000;95:2484–90.
Perobelli SM, Mercadante AC, Galvani RG, Goncalves-Silva T, Alves AP, Pereira-Neves A, Benchimol M, Nobrega A, Bonomo A. G-CSF-induced suppressor IL-10+ neutrophils promote regulatory T cells that inhibit graft-versus-host disease in a long-lasting and specific way. J Immunol. 2016;197:3725–34.
Tadmor T, Attias D, Polliack A. Myeloid-derived suppressor cells--their role in haemato-oncological malignancies and other cancers and possible implications for therapy. Br J Haematol. 2011;153:557–67.
Schouppe E, Mommer C, Movahedi K, Laoui D, Morias Y, Gysemans C, Luyckx A, De Baetselier P, Van Ginderachter JA. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur J Immunol. 2013;43:2930–42.
Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–702.
Noonan KA, Ghosh N, Rudraraju L, Bui M, Borrello I. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol Res. 2014;2:725–31.
Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, Meyer C, Becerra CR, Fishman M, Antonia S, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 2010;16:1812–23.
Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, Hegmans JP. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. 2010;10:464.
Grauers Wiktorin H, Nilsson MS, Kiffin R, Sander FE, Lenox B, Rydstrom A, Hellstrand K, Martner A. Histamine targets myeloid-derived suppressor cells and improves the anti-tumor efficacy of PD-1/PD-L1 checkpoint blockade. Cancer Immunol Immunother. 2019;68:163–74.
Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, Li Y. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. 2018;11:100.
Ma C, Kapanadze T, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. Anti-gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol. 2012;92:1199–206.
Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS, Qian J, Hailemichael Y, Nurieva R, Dwyer KC, et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med. 2014;20:676–81.
Rao A, Taylor JL, Chi-Sabins N, Kawabe M, Gooding WE, Storkus WJ. Combination therapy with HSP90 inhibitor 17-DMAG reconditions the tumor microenvironment to improve recruitment of therapeutic T cells. Cancer Res. 2012;72:3196–206.
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rebe C, Ghiringhelli F. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–61.
Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, Booth S, Lenton R, Noyvert B, Shannon-Lowe C, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine. 2019.
Kang TH, Knoff J, Yeh WH, Yang B, Wang C, Kim YS, Kim TW, Wu TC, Hung CF. Treatment of tumors with vitamin E suppresses myeloid derived suppressor cells and enhances CD8+ T cell-mediated antitumor effects. PLoS One. 2014;9:e103562.
Young MR, Day TA. Immune regulatory activity of vitamin d3 in head and neck cancer. Cancers (Basel). 2013;5:1072–85.
Kuwata T, Wang IM, Tamura T, Ponnamperuma RM, Levine R, Holmes KL, Morse HC, De Luca LM, Ozato K. Vitamin a deficiency in mice causes a systemic expansion of myeloid cells. Blood. 2000;95:3349–56.
Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–9.
Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–75.
Manitz MP, Horst B, Seeliger S, Strey A, Skryabin BV, Gunzer M, Frings W, Schonlau F, Roth J, Sorg C, Nacken W. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. 2003;23:1034–43.
Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008;29:2035–43.
Kallberg E, Vogl T, Liberg D, Olsson A, Bjork P, Wikstrom P, Bergh A, Roth J, Ivars F, Leanderson T. S100A9 interaction with TLR4 promotes tumor growth. PLoS One. 2012;7:e34207.
Raymond E, Dalgleish A, Damber JE, Smith M, Pili R. Mechanisms of action of tasquinimod on the tumour microenvironment. Cancer Chemother Pharmacol. 2014;73:1–8.
Lu P, Yu B, Xu J. Cucurbitacin B regulates immature myeloid cell differentiation and enhances antitumor immunity in patients with lung cancer. Cancer Biother Radiopharm. 2012;27:495–503.
Molina AM, Lin X, Korytowsky B, Matczak E, Lechuga MJ, Wiltshire R, Motzer RJ. Sunitinib objective response in metastatic renal cell carcinoma: analysis of 1059 patients treated on clinical trials. Eur J Cancer. 2014;50:351–8.
Cao M, Xu Y, Youn JI, Cabrera R, Zhang X, Gabrilovich D, Nelson DR, Liu C. Kinase inhibitor Sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Lab Investig. 2011;91:598–608.
Blazar BR, MacDonald KPA, Hill GR. Immune regulatory cell infusion for graft-versus-host disease prevention and therapy. Blood. 2018;131:2651–60.
Lim JY, Ryu DB, Park MY, Lee SE, Park G, Kim TG, Min CK. Ex vivo generated human cord blood myeloid-derived suppressor cells attenuate murine chronic graft-versus-host diseases. Biol Blood Marrow Transplant. 2018;24:2381–96.
Acknowledgements
We would also thank American Journal Experts (https://www.aje.com) for the assistance in editing this manuscript.
Funding
This work was supported by National Key Research and Development Plan of China (2017YFA0104500), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (No.81621001), National Natural Science Foundation of China (No.81530046, 81873445, 81930004), the Science and Technology Project of Guangdong Province of China (No.2016B030230003), and Peking University Clinical Scientist Program (BMU2019LCKXJ003).
Author information
Authors and Affiliations
Contributions
XJH designed the review. ML and KW drafted the manuscript and prepared the figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lv, M., Wang, K. & Huang, Xj. Myeloid-derived suppressor cells in hematological malignancies: friends or foes. J Hematol Oncol 12, 105 (2019). https://doi.org/10.1186/s13045-019-0797-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-019-0797-3