
Abstract
Mechanistic target of rapamycin (mTOR) is a protein kinase regulating cell growth, survival, metabolism, and immunity. mTOR is usually assembled into several complexes such as mTOR complex 1/2 (mTORC1/2). In cooperation with raptor, rictor, LST8, and mSin1, key components in mTORC1 or mTORC2, mTOR catalyzes the phosphorylation of multiple targets such as ribosomal protein S6 kinase β-1 (S6K1), eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), Akt, protein kinase C (PKC), and type-I insulin-like growth factor receptor (IGF-IR), thereby regulating protein synthesis, nutrients metabolism, growth factor signaling, cell growth, and migration. Activation of mTOR promotes tumor growth and metastasis. Many mTOR inhibitors have been developed to treat cancer. While some of the mTOR inhibitors have been approved to treat human cancer, more mTOR inhibitors are being evaluated in clinical trials. Here, we update recent advances in exploring mTOR signaling and the development of mTOR inhibitors for cancer therapy. In addition, we discuss the mechanisms underlying the resistance to mTOR inhibitors in cancer cells.
Similar content being viewed by others


Introduction
The mechanistic target of rapamycin (mTOR) is a dual-specificity protein kinase phosphorylating serine/threonine as well as tyrosine residues [1]. Since the catalytic domain of mTOR resembles that of lipid kinases such as phosphoinositide 3-kinase (PI3K), mTOR is considered as an atypical protein kinase belonging to the PI3K-related kinase family [2]. As a core component of several distinct complexes including mTOR complex 1 (mTORC1), mTOR complex 2 (mTORC2), and a putative mTOR complex 3 (mTORC3), mTOR has critical roles in diverse biological processes, such as cell proliferation, survival, autophagy, metabolism, and immunity [2, 3]. While mTOR and mammalian lethal with SEC13 protein 8 (mLST8) are common members of both mTORC1 and mTORC2, regulatory-associated protein of mTOR (raptor), the 40 kDa proline-rich Akt substrate (PRAS40), and DEP domain-containing protein 6 (DEPTOR) are specific members of mTORC1 [1, 2]. Instead, rapamycin-insensitive companion of mTOR (rictor) and mammalian stress-activated protein kinase-interacting protein 1 (mSIN1 or MAPKAP1) are unique components in mTORC2 but not mTORC1 [1]. Another rapamycin-insensitive complex, mTORC3, consists of ETV7, mTOR, and other undefined components [3]. mTORC1 senses nutrients, growth factors, and cellular energy to orchestrate nucleotide, lipid, and protein synthesis; inhibit autophagy; and stimulate cell growth [2]. mTORC2 is not only regulated by growth factors, but also activates type I insulin-like growth factor receptor (IGF-IR) and insulin receptor (InsR) through the tyrosine kinase activity of mTOR [1]. Besides, mTORC2 regulates the actin polarization and endocytosis [4, 5].
The mTOR signaling pathway has critical roles in mammalian metabolism and physiology. The de-regulated activity of mTOR is involved in many pathophysiological conditions, such as aging, Alzheimer’s disease, diabetes, obesity, and cancer [2]. As a natural inhibitor of mTORC1, rapamycin is able to increase lifespan in mice [6, 7]. mTOR activity is frequently de-regulated in a variety of human cancers, such as breast, prostate, lung, liver, and renal carcinomas. Upregulation of mTOR signaling can promote tumor growth and progression through diverse mechanisms including the promotion of growth factor receptor signaling, angiogenesis, glyolytic metabolism, lipid metabolism, cancer cell migration, and suppression of autophagy [1, 2]. Hence, mTOR is a promising target for cancer therapy. In this review, we discuss the roles of mTOR in human cancer and the rationales and challenges for developing mTOR inhibitors to treat cancer.
The assembly of mTOR complexes
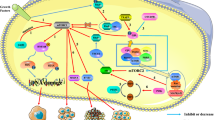
The studies of mTORC1 structure demonstrate that mTORC1 adopts a dimeric architecture with an overall size of (280~300) × (200~210) × (100~130) Å3 [8, 9]. mTOR and LST8 form the core of mTOR complex that contains raptor and other regulatory proteins [8]. The human mTOR contains 2549 amino acids that form several domains including the NH2-terminal HEAT (N-HEAT), middle HEAT (M-HEAT), FAT, and kinase domain with a FRB insertion (Fig. 1). Raptor also contains a HEAT domain, as well as WD40 and caspase-like domain [8, 9]. Besides, LST8 has WD40 domain. The HEAT motifs have conserved Asp and Arg residues at positions 19 and 25, respectively. A signature motif of WD40 repeats is ~ 40 amino acids often ending with a tryptophan-aspartic acid (W-D) dipeptide [10]. The HEAT repeats 12–13 in one mTOR interact with the HEAT repeats 20–23 in the M-HEAT domain of another mTOR, thereby forming a dimer [8]. Raptor may stabilize the dimer by binding the HEAT repeats 11–13 in one mTOR and repeats 20–22 in another mTOR [8, 11]. In addition, raptor is required for recruiting substrates to mTORC1 [12, 13]. Both mTOR and raptor are subjected to phosphorylation at multiple residues (Fig. 1a), which positively or negatively regulates mTORC1 activity.

The domains in key components of mTORC1 and mTORC2. a The molecular weight, domains, and phosphorylation sites in key components of mTORC1, including mTOR, LST8, and raptor. b The molecular weight, domains, and phosphorylation sites in key components of mTORC2, including mTOR, mSin1, and rictor
The assembly of mTORC2 and Saccharomyces cerevisiae TORC2 follows a similar principle to mTORC1. The human mTORC2 structure reveals a hollow rhombohedral fold with overall dimensions of ~ 220 × 200 × 130 (Å3) [14]. A dimer of mTOR is located in the core of this complex, while each mTOR or TOR heterodimerizes with rictor and mSIN1 [14, 15]. Rictor has an NH2-terminal armadillo (ARM) repeat cluster (~ 900 residues), and the rest of the rictor is largely unstructured (Fig. 1b) [16]. Interestingly, ARM and HEAT domains have similar conserved residues that form the hydrophobic domain core and may have a common phylogenetic origin [17]. In addition, mSin1 has a CRIM, a Ras-binding domain (RBD), and a pleckstrin homology (PH) domain [18]. During the assembly of mTORC2, the FRB domain of mTOR binds to mSin1 and the carboxy terminal region of rictor, while the NH2-terminal portion (residues 506–516) of rictor interacts with the COOH-terminal region (residues 1186-1218) of M-HEAT of mTOR [14]. In addition, mSin1 directly binds to rictor. Both rictor and mSin1 are responsible for recruiting substrates to mTORC2. Of note, both rictor and mSin1 have mTOR-independent partners. For example, rictor interacts with integrin-linked kinase and promotes its phosphorylation of Akt [19], while mSin1 interacts with Ras and inhibits ERK1/2 phosphorylation [20]. Thus, the outcome from the manipulation of rictor or mSin1 alone may not exactly reflect the function of mTORC2.
Regulation of mTORC1 activity
The activity of mTORC1 is regulated by growth factors, cellular energy, stresses and nucleotides, etc. The lysosomes are primary sites for mTORC1 activation. The activation of mTORC1 by growth factors is dependent on Ras homolog enriched in the brain (RHEB), a lysosomal GTPase that directly interacts with mTOR and activates it [21]. Upon binding to growth factors such as epidermal growth factor (EGF) and insulin-like growth factor (IGF), the growth factor receptors (EGFR, IGFR, etc.) are activated, which in turn activate PI3K-PDK1-Akt signaling pathway. Active Akt phosphorylates tuberous sclerosis complex 2 (TSC2) and inhibits the TSC complex, a GTPase-activating protein (GAP) complex consisting of TSC1/2 and TRE2-BUB2-CDC16 domain family member 7 (TBC1D7) [22, 23]. The TSC complex can inactivate RHEB thereby inhibiting mTOR [24]. Therefore, the activation of Akt leads to the depression of RHEB and then activates mTORC1. Moreover, the ubiquitination of RHEB regulates its ability to activate mTORC1 [21]. The E3 ubiquitin ligase RNF152 catalyzes RHEB ubiquitination, leading to an increase in the interaction between RHEB and TSC [21]. In contrast, Akt can phosphorylate the deubiquitinase USP4 that promotes RHEB deubiquitination thereby releasing RHEB from TSC [21].
Downstream of the growth factor receptors, the mitogen-activated protein kinase (MAPK) also upregulates mTORC1 activity. Mechanistically, MEK1/2 promotes raptor phosphorylation through ERK1/2 and p90 ribosomal S6 kinase (RSK1/2). ERK1/2 directly phosphorylates raptor at S8, S696, and S863, while RSK1/2 phosphorylates raptor at S719/722 [25, 26]. Meanwhile, the intestinal cell kinase (ICK), a MAPK-related kinase, phosphorylates raptor at T908 [27]. Phosphorylation of raptor by ERK/RSK/ICK promotes the activation of mTORC1.
mTORC1 not only senses growth factors, but also responds to cellular energy. Low cellular energy results in an increase in AMP/ATP ratio, which activates the energy sensor AMP-dependent kinase (AMPK). AMPK stimulates the GAP activity of TSC and then promotes the inhibition of RHEB by TSC, leading to the downregulation of mTORC1 [28]. In addition, the TCA cycle metabolite ketoglutarate inhibits mTORC1 through repressing ATP synthase, increasing AMP/ATP ratio and activating AMPK [29]. Cellular energy deficiency usually leads to endoplasmic reticulum stress, which in turn induces the unfolded protein response (UPR). Ire1, ATF6, and PERK are three major mediators of the UPR. Upon ER stress, ATF6 can induce RHEB expression, which in turn promotes mTORC1 activation and cell survival [30]. However, overactivated mTORC1 is also harmful to cell survival under ER stress. Mutations in TSC1/2 or activation of RHEB renders cells hypersensitive to ER stress-induced apoptosis, which may be due to the downregulation of ATF4/6 by mTOR [31]. Therefore, mTORC1 may have versatile effects on cell survival under ER stress.
While the regulation of mTORC1 by growth factors is dependent on RHEB and the TSC complex, amino acids can stimulate mTORC1 independent of TSC. The regulation of mTORC1 by amino acids is very complicated, involving multiple amino acid sensors and protein machinery [32]. The lysosomal Ragulator (RAG) guanosine triphosphatases (GTPases) play key roles in the activation of mTORC1 by amino acids. RAGA or RAGB heterodimerizes with RAGC or RAGD [33]. Further, RAG proteins form a large complex with LAMTOR1/2/3/4/5, which recruit RAG and mTORC1 to the lysosomal surface [34]. The activity of RAG is regulated by two complexes, GATOR1 and GATOR2. GATOR1, which is composed of DEPDC5, NPRL2, and NPRL3, inhibits the GTPase-activated protein (GAP) activity of RAGA/B thereby repressing the activation of mTORC1 by amino acids [35]. Instead, GATOR2, a protein complex consisting of MIOS, WDR24, WDR59 SEH1L, and SECB, negatively regulates GATOR1 by inducing DEPDC5 degradation [35]. Furthermore, KICSTOR, a large complex consisting of KPTN, ITFG2, C12ORF66, and seizure threshold 2 (SZT2), recruits GATOR1 to the lysosomal surface and mediates the interaction between GATOR1 and RAG [36, 37].
Sestrin (SESN) is another category of negative inhibitors of amino acid-induced mTORC1 activation. Mechanistically, SESNs interact with GATOR2, leading to the release of GATOR1 from GATOR2. The released GATOR1 in turn inhibits RAG and mTORC1 [38,39,40]. Of note, SESN2 is known as a leucine sensor in mTORC1 signaling. Leucine directly binds to SESN2, leading to the dissociation of SESN2 from GATOR2. The released GATOR2 binds to GATOR1 and then prevents the inhibition of RAG by GATOR1. These sequential processes result in RAG-mediated mTORC1 activation [41]. To prevent the overactivation of mTORC1 by amino acids, there are negative feedback pathways to RAG-mediated mTORC1 activation. Two E3 ubiquitin ligases, RNF152 and SKP2, reportedly induce RAGA ubiquitination and potentiate the binding of RAGA to GATOR1 [42, 43]. While leucine sufficiency is sensed by SESN2, the stimulation of mTORC1 by arginine is mediated by SLC38A9 [44]. Moreover, the ubiquitin ligase TRAF6 can catalyze K63 ubiquitination of both Akt and mTOR thereby promoting the activation of Akt and mTORC1 by amino acids [45, 46].
In addition, mTOR may be activated by lipid and cholesterol. Fatty acid metabolism leads to the de novo synthesis of phosphatidic acid (PA), which stabilizes both mTORC1 and mTORC2 [47]. Moreover, cholesterol can stimulate mTORC1 activation and growth signaling. Mechanistically, SLC38A9 acts as a lysosomal cholesterol sensor to stimulate the activation of mTORC1 by RAG complex [48]. Recently, it was reported that mTORC1 is also responsive to the levels of purine nucleotides [49]. While adenylate stimulates mTORC1 by inhibiting TSC, guanylate downregulates RHEB and then inhibits mTORC1 [49]. The mechanisms underlying the regulation of TSC and RHEB by adenylate and guanylate remain to be known.
Regulation of mTORC2 activity
Although mTORC1 and mTORC2 are distinct complexes, there is a crosstalk between these two complexes. On one hand, mTORC2 can activate IGF-IR-Akt axis thereby upregulating mTORC1 [1]. On the other hand, mTORC1 feeds back to inhibit mTORC2 via S6K1, one of the substrates of mTORC1. Once activated by mTORC1, S6K1 phosphorylates rictor and mSin1 on T1135 and T86/398, respectively, leading to the impairment of mTORC2 integrity [50,51,52].
While mTORC2 directly activates IGF-IR and InsR, receptor tyrosine kinases such as EGFR, PDGFR, and IGF-IR can activate mTORC2 via PI3K. Mechanistically, PI3K-induced PtdIns (3,4,5) P3 (PIP3) binds to the PH domain of mSin1 and then disables the inhibition of mTOR kinase domain by mSin1, thereby activating mTORC2 [18]. In addition, PI3K promotes the association of mTORC2 with ribosome, where mTORC2 is activated [53]. Therefore, mTORC2 also responds to growth factors. Notably, another study suggests that mTORC2 activity is localized in the plasma membrane, mitochondria, and endosomal vesicles, and the activity of mTORC2 via the mSin1-PH domain at the plasma membrane is PI3K- and growth factor-independent [54]. In addition, IKKα interacts with mTORC2 and enhances its kinase activity towards Akt [55]. These data suggest that the activation of mTORC2 involves multiple location and different mechanisms.
How does mTORC2 respond to cellular energy and nutrients? The energy sensor AMPK inhibits mTORC1 and then releases the suppression of mTORC2 by mTORC1, leading to the activation of mTORC2 [56]. Thus, upregulation of mTORC2 may help cells adapt to low levels of cellular energy. Moreover, mTORC2 is activated by glutamine starvation. Activated mTORC2 upregulates the expression and phosphorylation of glutamine:fructose-6-phosphate amidotransferase 1 (GFAT1), the rate-limiting enzyme of the hexosamine biosynthesis pathway (HBP) [57, 58]. A study of budding yeast demonstrates that the LKB1-ELM1-GIN4/HSL1 axis is required for coordinating TORC2 signaling to the changes in carbon source [59]. It remains to know if similar pathway works in human cancer cells.
Similar to mTORC1, mTORC2 is also stabilized by phosphatidic acid (PA), a central metabolite in the synthesis of membrane phospholipids [60]. The generation of PA is catalyzed by the phospholipase D, diacylglycerol kinases, and lysophosphatidic acid acyltransferases. Moreover, the activity of mTORC1 and mTORC2 is regulated by mLST8 ubiquitination. It has been reported that the E3 ubiquitin ligase TRAF2 positively regulates K63-linked polyubiquitination of mLST8, which impairs its interaction with mSin1 and compromises the mTORC2 integrity, but enhances the assembly of mTORC1 [61]. On the contrary, the deubiquitinase OTUDB7 removes polyubiquitin chains from GβL to promote GβL interaction with mSin1 and the integrity of mTORC2 [61]. Besides, the exchange factor found in platelets, leukemic, and neuronal tissues (XPLN) interacts with mTORC2 and negatively regulates mTORC2 activity [62]. Lastly, mTOR is a target of proteasomal degradation when it is ubiquitinated by FBXW7 [63].
Targets of mTORC1 and mTORC2
As a protein kinase, mTOR catalyzes the phosphorylation of its targets and regulates their activity. mTORC1 and mTORC2 have different substrates. While the repertoire of mTOR substrates keeps increasing, there are more targets remaining to be identified. S6K1 and 4E-BP1 are two well-known mTORC1 targets. mTORC1 phosphorylates S6K1 at T389 and 4E-BP1 at multiple residues [64]. Phosphorylation of S6K1 by mTORC1 leads to increased protein and nucleotide synthesis. While 4E-BP1 is a negative regulator of 5′cap-dependent mRNA translation, phosphorylation of 4E-BP1 by mTORC1 induces its dissociation from eIF4E, thereby relieving its inhibition of protein synthesis [65]. To cope with increased protein synthesis, mTORC1 also promote ribosome biogenesis by inducing ribosomal RNA transcription. Mechanistically, mTORC1 may translocate to the nucleus, where it binds to ribosomal DNA promoter [66,67,68]. Nuclear mTOR also phosphorylates TFIIIC and Maf1, thereby promoting tRNA gene transcription [69]. In fact, nuclear mTOR regulates RNA polymerase 1/2/3-driven transcription. In addition, mTORC1 phosphorylates the E3 ubiquitin ligase SKP2 at S64 and then inhibits SKP2 ubiquitination and degradation [70]. Given that SKP2 promotes the degradation of many proteins, mTORC1 may regulate the turnover of SKP2 substrates indirectly. Thus, mTORC1 not only promotes protein synthesis, but also regulates protein degradation.
Following the identification of mTORC2, it was found that protein kinase C (PKC) α/β were the substrates of mTORC2 that regulates the actin cytoskeleton [4, 71]. Moreover, mTORC2 phosphorylates and activates other AGC kinases, such as serum and glucocorticoid-induced kinase (SGK) and Akt. mTORC2 phosphorylates Akt at S473, leading to allosteric activation of Akt in cooperation with the catalytic activation by PDK1, which phosphorylates Akt at T308 [72]. During the synthesis of nascent proteins, mTORC2 can co-translationally phosphorylate some polypeptides while they are attached to the ribosome. IGF2 mRNA-binding protein (IMP) is responsible for the splicing and translation of IGF2 mRNA. mTORC2 co-translationally phosphorylates IMP1 at S181 and then promotes IMP1 binding to the untranslated region of IGF2 mRNA and enables translational initiation by internal ribosomal entry [73]. mTORC2 not only enhances the production of IGF2 protein, but also phosphorylates and activates IGF-IR and insulin receptor [1]. In contrast to mTORC1’s activity as a ser/thr kinase, mTORC2 has tyrosine kinase activity towards IGF-IR/InsR [1].
mTOR inhibitors for cancer therapy
The activity of mTOR is frequently upregulated in human cancer. The aberrant activation of mTOR in human cancer may be attributed to mTOR pathway-activating mutations, amplification, or overexpression of the components of mTOR complexes and mutations or loss of negative regulators of mTOR. PIK3CA mutations are frequently detected in human cancer. Activation of PI3K promotes both mTORC1 and mTORC2 activation. In addition, mutations in KRAS and BRAF may lead to mTORC1 activation. Especially, KRAS can directly bind to PIK3CA (p110α) and activates PI3K pathway, leading to mTOR activation [74]. mTOR-activating mutations are observed in kidney cancer. While mTOR activity is usually upregulated by growth factors and amino acids, activating mutations in mTOR may result in RAG- and RHEB-independent mTOR hyperactivation, thus loss of the dependency on growth factors and amino acids [75]. Point mutations in RHEB and GATOR1 were also detected in renal cancer and endometrial cancer [76]. RHEB1 is overexpressed in acute myeloid leukemia (AML) and promotes AML progression [77]. Whereas mTOR amplification is rare in human cancer, rictor amplification is detected in various kinds of cancer, such as breast cancer, gastric cancer, and liver cancer [78, 79]. Moreover, rictor is overexpressed in human cancers of the brain, breast, lung, gastric, colon, liver, and tongue [80, 81].
Given that mTOR has critical roles in tumor progression, mTOR inhibitors hold promise in cancer therapy. Indeed, rapamycin analogs (rapalog) have been approved for treating cancer in the clinic. In addition, many mTOR inhibitors with different mechanisms of action have been developed, some of which are undergoing clinical trials in variety types of human cancer.
Rapalog
Rapamycin was originally identified as an antifungal, immunosuppressive, and antiproliferative agent. Later studies revealed that rapamycin binds to the 12 kDa FK506-binding protein (FKBP12) and then inhibits mTORC1 [82]. Since rapamycin has poor solubility and pharmacokinetics, it is not suitable for treating human cancer. So far, several water-soluble rapamycin analogs have been developed. For example, temsirolimus and everolimus exhibit tumor-suppressive effects in vivo. Both temsirolimus and everolimus have been used to treat advanced renal cell carcinoma (RCC) in the clinic. Moreover, everolimus is prescribed for treating pancreatic neuroendocrine tumors and advanced breast cancer [83]. Besides, there are many clinical trials to evaluate the efficacy of rapalogs in treating other types of human cancer, such as advanced gastric cancer, hepatocellular carcinoma, non-small cell lung cancer, endometrial cancer, and mantle cell lymphoma (clinicaltrials.gov).
Of particular note, the effect of rapalog monotherapy on solid tumors is modest in the clinic. The incomplete inhibition of mTOR by rapalogs may result in limited clinical success. On the other hand, inhibition of mTORC1 may lead to feedback activation of IGF-IR and Akt, which compromises the anti-cancer effect of rapalogs [1]. Taking into account the complexity of mTOR signaling networks, it is not hard to understand that the response to rapalogs varies in patients with cancer, such as metastatic RCC. It is desirable that there are biomarkers to predict the responses to mTOR inhibition. KRAS, BRAF, and TSC mutations are known as resistant markers for mTOR inhibitors, whereas PIK3CA mutations are sensitive marker [84, 85]. However, the roles of TSC1/2 and mTOR mutations in responding to rapalogs remain controversial. Although it has been reported that mutations in TSC1/2 and mTOR are more frequent in RCC patients who respond well to rapalogs, the majority of rapalog responders have no mutations in mTOR pathway, suggesting that other factors are also involved in rapalog sensitivity [86]. Notably, rapalogs usually arrest cell proliferation but does not induce apoptosis. Despite the initial response, tumors frequently develop resistance to these agents.
ATP-competitive mTOR inhibitors
To more completely inhibit mTOR, a number of ATP-competitive mTOR inhibitors have been developed to target both mTORC1 and mTORC2. Tumors that are addicted to the mTOR signaling pathway may be sensitive to this kind of inhibitors. Unlike rapalogs, ATP-competitive mTOR inhibitors can not only arrest cell growth, but also induce apoptosis. MLN0128 (also called INK128, sapanisertib, TAK-228) is a pan-mTOR inhibitor that has potent in vitro and in vivo anti-tumor effects, and has underwent clinical trials for solid tumors such as bone and soft tissue sarcoma, breast cancer, and primary effusion lymphoma, a non-Hodgkin B cell lymphoma that usually results from infection of Kaposi sarcoma-associated herpesvirus [87,88,89,90]. MLN0128 also reduces tumor growth in CD44-high HCC xenografts and resensitizes HCC to sorafenib [91]. Of note, MLN0128 is an effective agent even in tumors that are resistant to rapamycin or chemotherapy. A recent study demonstrates that MLN0128 can overcome resistance to everolimus and reduce tumor size by 20% in PIK3CA-mutant colorectal cancers [92]. In addition, MLN0128 can induce tumor shrinkage in patient-derived xenograft model of pancreatic neuroendocrine tumors, even in everolimus-resistant tumors [93].
PP242 (Tokinib) is another selective ATP-competitive inhibitor of mTOR that has a promising anti-cancer activity over several cancer types, such as leukemia, gastric cancer, and colon cancer [94, 95]. Given that the Akt-mTOR signaling pathway is upregulated in platinum-resistant cancer cells, studies demonstrate that mTORC1/2 inhibitor, such as PP242 and MLN0128, can re-sensitize platinum-resistant ovarian cancer cells to carboplatin in vitro and in vivo [96, 97]. Mechanistically, mTOR inhibition leads to a sharp decrease in the translation of DNA damage and repair response and pro-survival mRNAs, including CHK1 [98]. Consistent with the inhibition of DNA repair, mTOR inhibitors are also effective in enhancing radiosensitivity or restoring radiosensitivity in radioresistant tumors [99, 100]. Moreover, inhibition of mTORC1/C2 signaling improves anti-leukemia efficacy of JAK/STAT blockade in CRLF2-rearranged and/or JAK-driven Philadelphia chromosome-like acute B cell lymphoblastic leukemia [101].
Both AZD2014 (vistusertib) and its analog AZD8055, two ATP-competitive mTORC1/2 inhibitors, are highly effective in treating estrogen receptor (ER)-positive breast cancer. Moreover, AZD2014 and AZD8055 can suppress breast cancer with acquired resistance to endocrine therapy, rapalogs, and paclitaxel [102, 103]. In addition, a combination of AZD2014 with paclitaxel reduces tumor volume in cisplatin-resistant ovarian cancer model [104]. Similar to PP242, AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells [105]. Based on the above-described studies, it appears that the pan-mTORC1/2 inhibitors generally reverse rapalog resistance, endocrine resistance, chemoresistance, and radioresistance.
Dual PI3K/mTOR inhibitors
Although inhibition of mTORC1 and mTORC2 can downregulate Akt S473 phosphorylation, mTOR inhibitors may paradoxically enhance the PI3K/PDK1 axis. Thus, an inhibitor targeting both PI3K and mTOR may have better anti-cancer activity compared to targeting mTOR alone [106, 107]. Due to the similarity between PI3K and mTOR, some chemicals can inhibit both PI3K and mTOR. NVP-BEZ235 (dactolisib) inhibits the activity of multiple class I PI3K isoforms, mTOR and ataxia telangiectasia, and Rad3-related protein (ATR) and has potent anti-cancer activity [108]. Notably, NVP-BEZ235 can penetrate the blood-brain barrier after systemic administration [109]. Therefore, it can be used to treat glioma and reverse temozolomide resistance [110]. In addition, NVP-BEZ235 can suppress paclitaxel-resistant gastric cancer, which exhibits increased PI3K/mTOR activity [111].
LY3023414, a complex fused imidazoquinolinone, is an oral PI3K/mTOR and DNA-PK inhibitor that has anti-tumor effects in animal models. Combination of LY3023414 with standard chemotherapeutic drugs has additive anti-tumor activity [112, 113]. Another dual PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765), a pyridopyrimidinone derivative, significantly inhibits tumor growth in multiple human xenograft models [114]. Combination of voxtalisib and the MEK inhibitor pimasertib synergistically inhibits certain endometrial cancer cells growth [115]. Other dual PI3K/mTOR inhibitors include PQR309, XH00230381967, SN20229799306, GSK2126458 (omipalisib), and PKI-587.
Of note, PQR309 is a 4,6-dimorpholino-1,3,5-triazine-based, brain-penetrant, and orally bioavailable PI3K/mTOR inhibitor [116]. PQR309 effectively inhibits lymphoma in monotherapy and in combination therapy with other drugs, such as the BCL2 inhibitor venetoclax, the HDAC inhibitor panobinostat, the Bruton’s tyrosine kinase inhibitor ibrutinib, lenalidomide, the BET proteolysis-targeting chimera ARV-825, the proteasome inhibitor marizomib, and the anti-CD20 monoclonal antibody rituximab [117]. Moreover, PQR309 can suppress cancer cells with primary or secondary resistance to the PI3Kδ. PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood-brain barrier [118].
The dual specificity PI3K/mTOR inhibitor gedatolisib (PKI-587, PF05212384) is a bis(morpholino-1,3,5-triazine) derivative [119]. Gedatolisib inhibits tumor growth in breast, colon, lung, and glioma xenograft models and displays efficacy against T cell acute lymphoblastic leukemia (T-ALL) and Philadelphia chromosome (Ph)-like B cell acute lymphoblastic leukemia (Ph-like ALL) [107, 120]. Combination of gedatolisib with ruxolitinib or dasatinib has superior efficacy than a single agent in CRLF2/JAK-mutant models and ABL/PDGFR-mutant models, respectively [120]. In addition, gedatolisib sensitizes head, neck, and nasophageal carcinoma to radiation therapy [121, 122] and sensitizes EGFR-resistant head and neck carcinoma to cetuximab [123]. Thus, gedatolisib may be a candidate sensitizer to radiotherapy and targeted therapy.
GSK2126458 (omipalisib) is an orally bioavailable inhibitor of PI3Kα and mTOR [124]. Omipalisib potently inhibits FGFR4-V550E tumor-derived cell and human rhabdomyosarcoma cell viability and reduces the growth of rhabdomyosarcoma in vivo [125]. In addition, a combination of the PI3K/mTOR inhibitor VS-5584 and the Wnt inhibitor ICG-001 synergistically inhibits AML with high PRL-3 expression [126]. Finally, the efficacy of mTOR inhibitor may be enhanced by linking the kinase inhibitor to rapamycin (RapaLink) [127]. EZH2 (Y641X)-mutant lymphomas show increased sensitivity to RapaLink-1 [128]. Given that RapaLink integrates the activity of both rapamycin and mTOR kinase inhibitor, it is worthwhile looking forward to the efficacy in clinical trials. Lastly, there are many drugs that may indirectly inhibit mTOR, such as aspirin and metformin [129,130,131].
Principle mechanisms of mTOR inhibitor resistance in cancer
Drug resistance is a serious problem in treating cancer. Although there may be an initial response, long-lasting treatment with chemotherapeutic or molecular-targeted drugs often faces the challenge of drug resistance. Due to the tumor heterogeneity, some tumors do not respond to a given drug at all. Clonal selection, adaptive evolution, and resistance to cell death are general mechanisms for drug resistance. Due to the complexity and crosstalk in signaling networks, cancer cells may adapt to an inhibitor that targets a given signaling pathway via the compensatory activation of other pathways. Although mTOR inhibitors exhibit potent anti-cancer effects in many preclinical models, resistance does occur. As described below, there are multiple mechanisms underlying the resistance to mTOR inhibitors (Fig. 2).

The mechanisms for resistance to mTOR inhibitors in cancer cells. ABC transporters, ATP binding cassette transporters; EMT, epithelial-mesenchymal transition
Drug efflux by ATP binding cassette transporters
ATP-binding cassette (ABC) transporters constitute drug efflux pumps that decrease the intracellular levels of drugs, leading to poor treatment outcome. Overexpression of ABC transporters is a general mechanism for multi-drug resistance in cancer. The same may be true for mTOR inhibitor resistance. In fact, the mTOR inhibitors rapamycin and NVP-BEZ235 are substrates of ABCB1 (P-glycoprotein) and ABCG2 (also called breast cancer resistance protein, BCRP), respectively [132]. In addition, AZD8055 is transported by both ABCB1 and ABCG2 [132].
Studies show that ABCB1 is overexpressed in luminal breast cancer cell lines that are resistant to everolimus [133]. Also, ABCB1 inhibits brain accumulation of everolimus [134]. Overexpression of ABCG2 in cancer cells confers significant resistance to PF-4989216, which can be reversed by an inhibitor or competitive substrate of ABCG2 [135]. Moreover, GDC-0980 is subject to active efflux by ABCB1 and BCRP, which limits its efficacy [136]. The affinity for ABC transporters may vary among different mTOR inhibitors. Lowering the affinity for ABC transporters or inhibiting ABC transporters may enhance the efficacy of mTOR inhibitors.
Cancer stem cells
Cancer stem cells (CSCs) are a subpopulation in tumor mass that is extremely resistant to standard cancer therapy. Slow-cycling CSC is one of the major obstacles to eradicate tumor [137]. It is generally thought that the mTOR pathway is hyperactivated in CSC. Transforming growth factor-β (TGF-β) can induce epithelial-mesenchymal transition (EMT), which enhances cancer stem cell generation. mTOR is one of the mediators in TGF-β signaling pathways that enhances cancer stemness and drug resistance [138]. The inhibitory effect on CSCs has already been shown for some mTOR inhibitors [139]. Rapamycin, everolimus, and PF-04691502 suppress tamoxifen-induced activation of breast cancer stem cells [140]. Inhibition of mTOR restores tamoxifen resistance in breast cancer cells [141]. Moreover, the ATP-competitive mTOR inhibitor Torin1 and PI3K/mTOR inhibitor VS-5584 preferentially reduce CSC levels in multiple mouse xenograft models of human cancer [142, 143].
However, the interplay between mTOR inhibitors and CSC is complex. Previous studies show that expansion of CSC promotes the resistance to mTOR inhibitor in leiomyosarcoma [144]. PDK1 signaling toward PLK1-MYC activation leads to tumor-initiating cell activation and resistance to mTOR inhibition [145]. Inhibition of EZH2, a catalytic component of polycomb repressive complex which plays a critical role in stem cell maintenance, restores sensitivity to PI3K/mTOR pathway inhibition. It appears that the sensitivity to mTOR inhibitors in CSC may be context- or cell type-dependent. Of note, one study demonstrates that TP53 mutation and BCL2 phosphorylation affect the sensitivity of glioblastoma stem-like cells to mTOR inhibitor [146]. BCL2 (T56/S70) phosphorylation in TP53 wild-type glioblastoma stem-like cells is responsible for the lower sensitivity to the mTORC1/2 inhibitor AZD8055, as compared to TP53-mutated glioblastoma stem-like cells [146]. In addition, while mTOR inhibitors reportedly suppress CSC, one study demonstrates that treatment of TNBC cell lines with PI3K/mTOR inhibitor or TORC1/2 inhibitor expands CSC population through upregulating FGF1-FGFR-Notch1 axis [147]. Blocking FGFR or Notch1 may prevent resistance to TORC1/2 inhibitors by abrogating the expansion of drug-resistant CSCs in TNBC [49]. Moreover, another dual PI3K/mTOR inhibitor PF-04691502 can induce a stem cell-like gene expression signature in KRAS-mutant colorectal cancer models [148]. Together, these data suggest that the effects of mTOR inhibitors on CSC may be dependent on the genetic background and rewiring of cancer stemness pathways.
Assembly of the translation machinery
Eukaryotic protein synthesis is regulated by several mechanisms including cap-dependent and cap-independent translation. The cap-dependent pathway involves many eukaryotic initiation factors (eIF), such as eIF1, eIF2, eIF3, eIF4A, eIF4B, eIF4E, eIF4H, eIF5, and eIF6. The protein synthesis is initiated by the association of the 40S ribosome subunit with eIF1A and eIF3, followed by binding of the eIF2-GTP-methionine tRNA complex to 40S subunit and then forming a 43S subunit [149]. The eIF4F complex, which consists of eIF4E, eIF4A, and eIF4G, binds to the m7G cap at the 5′ end of mRNA and then activates mRNA. The activated mRNA is recruited to the 43S complex and then subjected to ATP-dependent scanning of mRNA to locate the initiating AUG code [150]. Finally, the 60S ribosome subunit is associated with the 40S subunit to form the 80S initiation complex, possibly assisted by eIF5. For the initiation of cap-independent protein synthesis, the 40S ribosome subunit binds to an internal region of mRNA, which is referred to as internal ribosome entry sites (IRES), or the untranslated regions of mRNA.
Given that stimulation of cap-dependent translation is one of the major functions of mTORC1, the status of the translation machinery and modes of protein translation may impact on the efficacy of mTOR inhibitors. 4E-BPs are phosphorylated and inactivated by mTORC1. The sensitivity to PP242 is correlated with the extent to which 4E-BP1 phosphorylation is inhibited by this drug [151]. Loss of 4E-BPs in tumor cells results in the resistance to mTOR inhibition. The transcription factor Snail directly represses 4E-BP1 transcription and compromises the anti-cancer effects of mTOR inhibitors [152]. Of note, Snail is translationally regulated by eIF4E, which is exactly the target of 4E-BP. Phosphorylation of eIF4E (S209, etc.) promotes Snail synthesis [153]. Therefore, 4E-BP and eIF-4E can disable each other. Overexpression of eIF4E or phosphorylation of eIF4E (S209) by MAP kinase-interacting kinase 1 (Mnk1/2) leads to a shift from cap-dependent to cap-independent translation and then renders cancer cells insensitive to mTOR inhibition [154, 155]. Thus, inhibition of Mnk1/2 or its upstream kinase ERK1/2 may restore cap-dependent translation and the sensitivity of mTOR inhibitors [155]. On the other hand, inhibition of mTORC1 may lead to paradoxical phosphorylation of eIF4E in PI3K- and Mnk-dependent manner and promote cap-independent translation [156]. Hence, a combination of mTOR and Mnk inhibitors is an effective therapeutic strategy for cancer [157].
Notably, 4E-BP1 is not only phosphorylated by mTORC1, but also phosphorylated and inactivated by other kinases such as CDK1, CDK12, and GSK3 . CDK1 can substitute mTORC1 to phosphorylate 4E-BP1 and activate cap-dependent translation, which is resistant to mTOR inhibition [158]. In addition, CDK12 cooperates with mTORC1 to phosphorylate 4E-BP1 and releases it from mTORC1 target mRNAs thereby promoting their translation [159]. Therefore, combinatorial inhibition of mTOR and CDK1/12 may be synthetically lethal to cancer cells. Furthermore, GSK3β can directly phosphorylate4E-BP1 at the same residues (T37/46) that are phosphorylated by mTOR and CDK1 [160]. Given that mTORC2 positively regulates Akt, the negative regulator of GSK3β, mTOR kinase inhibitor may paradoxically activate GSK3. Hence, combinatorial inhibition of mTOR and GSK3β may synergistically suppress tumorigenesis.
mTOR mutations
Gene mutations may affect the sensitivity of a drug that targets the protein encoded by this gene. More than 30 activating mutations of mTOR have been reported in human cancer, such as L1460P, C1483F, E1799K, F1888L, T1977R, V2006I, V2046A, S2215Y, L2230V, E2388Q, I2500F, R2505P, and D2512H [127, 161]. Cancer cells that harbor a subset of those mutations, including C1483F, E1799K, and S2215Y, are hypersensitive to rapamycin, whereas three mutations (A2034V, F2018L, and S2035F) in the FRB domain of mTOR are associated with rapamycin resistance [162, 163]. While tumor cells with mutations in the kinase domain are still responsive to rapalogs [161], mutations in the kinase domain of mTOR, such as M2327I, S2215Y, L2230V, E2388Q, and V2046A, may be responsible for the resistance to the ATP-competitive inhibitor MLN0128 [127]. It remains to know whether activating mutations in the kinase domain of mTOR are responsible for the resistance to allosteric mTOR kinase inhibitors other than MLN0128. In addition, there are recurrent mutations in other mTOR pathway genes, such as raptor, rictor, and RHEB [163]. RHEB-Y35N mutant gains the function to activate mTORC1 [161]. It warrants further studies to clarify which cancer-associated mutations in raptor, rictor, and RHEB may be associated with mTOR inhibitors resistance.
Rewiring of oncogenic or metabolic pathways
The sensitivity to mTOR inhibitors is regulated by other oncogenic pathways, such as PI3K, MAPK, AURKA, and NF-kB signaling [164, 165]. Both the Ras/MAPK and PI3K/Akt/mTOR pathways are tightly involved in tumorigenesis. While tumors with PIK3CA/PTEN mutations or Akt hyperactivation usually are sensitive to mTOR inhibitors, KRAS/BRAF mutations are predictive biomarkers of mTOR inhibitor resistance [148, 166,167,168,169]. In addition, mTOR inhibition may lead to the activation of the MEK-Erk pathway. Combination of RAF/MEK inhibitors and mTOR inhibitors may be a strategy to treat KRAS-mutated cancer [170, 171]. Besides, the activation of Erk in response to mTOR inhibition can be abrogated by the CDK4/6 inhibitor palbociclib [172]. Combination of CDK4/6 and mTOR inhibitors synergistically inhibits tumor growth [172, 173]. Alternatively, combined inhibition of wee1, a protein kinase that regulates the G2 checkpoint in the cell cycle, with mTOR inhibition may selectively treat RAS-mutated cancer [174]. Lastly, treatment with everolimus or AZD8055 increases epidermal growth factor receptor (EGFR) activation in tumor cells, leading to drug resistance [175].
Although PIK3CA-mutated cancer is usually sensitive to mTOR inhibition, activation of GSK3β in response to PI3K/mTOR inhibition may lead to the resistance to PI3K/mTOR inhibitors in PIK3CA-mutated cancer [176]. A recent study demonstrates that lung squamous cell carcinoma adapt to chronic mTOR inhibition through the GSK3α/β signaling pathway, which involves the metabolic reprogramming via increased glutaminolysis [177]. One study also reveals that glutaminase (GLS) and glutamate levels are elevated in glioblastoma after treating with mTOR inhibitor [178]. Treatment with GSK3 inhibitors or the glutaminase inhibitor effectively overcomes the resistance to mTOR inhibition [176,177,178]. Moreover, the activation of the purine salvage pathway due to increased expression of hypoxanthine phosphoribosyl transferase 1 leads to the resistance to the dual PI3K/mTOR inhibitor gedatolisib [179]. In fact, mTOR is tightly involved in purine metabolism. mTORC1 is not only activated by purine nucleobases or nucleosides [49], but also promotes purine synthesis by ATF4-mediated upregulation of the mitochondrial tetrahydrofolate (mTHF) cycle enzyme methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) [180]. Moreover, mTORC1 promotes de novo pyrimidine biosynthesis by S6K1-mediated phosphorylation of carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD) [181, 182]. Therefore, the increased expression of hypoxanthine phosphoribosyl transferase 1 may rescue the defect in purine synthesis due to mTOR inhibition and help cancer cells adapt to mTOR inhibition.
Another compensatory response to mTORC1 inhibition is the upregulation of transglutaminase 2, a multifunctional enzyme that is involved in cross-linking polypeptide chains with e-(c-glutamyl)-lysine, apoptosis, signal transduction, cell migration, cell adhesion, and extracellular matrix remodeling [183,184,185]. Inhibition of transglutaminase 2 potently sensitizes mTORC1-hyperactive cancer cells to rapamycin in vitro and in vivo [183]. Moreover, mitochondria homeostasis is critical for cell growth and survival. Mitochondrial hyperfusion is an adaptive response to mTOR inhibition. Mechanistically, the translation of mitochondrial fission process 1 (MTFP1) is suppressed by mTOR inhibitors, which eventually results in mitochondrial hyperfusion, a process that antagonizes apoptosis [186].
Clinical testing of mTOR inhibitors
Given that preclinical studies demonstrate the anti-cancer efficacy of mTOR inhibitors alone or in combination with chemotherapy, radiotherapy, and targeted therapy, there are many completed or ongoing clinical trials to test the efficacy of mTOR inhibitors for treating various types of human cancer (Table 1). In general, most of mTOR inhibitors are well tolerated, while there are some common adverse effects including fatigue, rash, mucositis, and metabolic complications. mTOR inhibitors are associated with a significantly increased risk of hyperglycemia, hypertriglyceridemia, and hypercholesterolemia [187]. Other adverse events of everolimus are thrombocytopenia, anemia, nausea, and stomatitis [188]. Ridaforolimus is orally bioavailable and better tolerated in children than the adults [189]. Deforolimus was well tolerated and showed encouraging anti-tumor activity across a broad range of malignancies when administered intravenously, and a dose of 12.5 mg/day is being evaluated in phase II trials [190].
Moreover, MLN0028-treated patients may suffer from anorexia, dyspenea and macunopapular rash [191]. In clinical trials of solid tumors, the PI3K/mTOR inhibitor NVP-BEZ235 (twice daily) is poorly tolerated, which leads to treatment discontinuation in some patients and limits its efficacy in treating cancer [192, 193]. Apitolisib (GDC-0980), another dual pan-PI3K/mTOR inhibitor, also has grade 3–4 adverse effects and is less effective than everolimus [194]. GSK2126458 (GSK458) plus trametinib has poor tolerability, due to skin and gastrointestinal toxicities such as diarrhea [195]. Daily oral administration of PF-04691502 (8 mg/day) has adverse events including fatigue, nausea, vomiting, hyperglycemia, and rash [196]. The occurrence of the above-mentioned adverse effects following treatment with mTOR inhibitors may be due to the critical roles of mTOR in metabolism and immunity.
mTOR inhibitors monotherapy
Everolimus has been approved by the FDA for the treatment of advanced renal cell carcinoma, pancreatic neuroendocrine tumors, and advanced breast cancer [83]. Everolimus significantly improves progression-free survival (PFS) among patients with progressive advanced pancreatic neuroendocrine tumors [197]. As registered in clinicaltrials.gov, there are more than 80 clinical trials for mTOR inhibitor monotherapy in cancer patients. A phase 2 trial of everolimus in patients with recurrent adult low-grade gliomas demonstrates a high degree of disease stability [198]. Moreover, everolimus has a promising effect in patients with heavily pretreated, relapsed, or refractory classical Hodgkin’s lymphoma, with an overall response rate (ORR) of 45.6%, a median PFS of 8 months, and a long-term response (≥ 12 months) rate of 12% [188]. Of note, everolimus exhibits clinical activity as the first-line monotherapy in a phase 2 clinical trial in 27 patients with advanced biliary tract cancer [199]. Another phase 2 clinical trial in 35 patients with thyroid cancer demonstrates that everolimus has clinical benefit in patients with advanced differentiated thyroid cancer [200]. Also, single-agent ridaforolimus has anti-tumor activity and acceptable tolerability in advanced endometrial cancer patients [201]. These observations need to be validated in a large scale of randomized clinical trials.
Based on a phase 2 trial in 167 patients, oral administration of the mTOR kinase inhibitor voxtalisib (50 mg, twice daily) exhibits a promising efficacy in patients with follicular lymphoma but limited efficacy in patients with mantle cell lymphoma, diffuse large B cell lymphoma, or chronic lymphocytic leukemia/small lymphocytic lymphoma [202]. Of note, serious adverse events occurred in 58.1% of patients [202]. In contrast, the clinical efficacy of MLN0128 in patients with metastatic castration-resistant prostate cancer is limited, possibly due to the dose reductions secondary to toxicity [191]. Although it is expected that mTOR kinase inhibitor may have superior efficacy than rapalogs, a randomized phase 2 trial in patients with metastatic clear cell renal cancer demonstrated that the PFS and OS of AZD2014 were less than that of everolimus [203]. While the PI3K/mTOR inhibitor NVP-BEZ235 is poorly tolerated in cancer patients, a clinical trial in patients with recurrent endometrial cancer demonstrated that weekly intravenous administration of another P3K/mTOR inhibitor gedatolisib achieved moderate anti-cancer activity with tolerable toxicity [204].
mTOR inhibitors in combination therapy
While mTOR inhibitor monotherapy has efficacy in some type of cancer, preclinical studies demonstrate strong rationales for combinatorial treatment with mTOR inhibitors and other drugs. For example, inhibition of both Akt/mTOR and WNT/β-catenin pathways synergistically suppresses AML [205]. As registered in clinicaltrials.gov, there are many clinical trials to test the efficacy of mTOR inhibitors in combination with other molecular targeted or chemotherapeutic agents. For example, everolimus is combined with one or several chemotherapeutic agents, such as taxol, cisplatin, carboplatin, oxaliplatin, irinotecan, temozolomide, and gemcitabine.
The phase 3 BOLERO-2 trial in patients with ER-positive/HER2-negative advanced or metastatic breast cancer demonstrates that a combination of everolimus and the aromatase inhibitor exemestane significantly improves PFS, while the OS is not improved [206, 207]. Accordingly, a combination of everolimus and exemestane has been approved as a guideline for treating ER-positive/HER2-negative advanced or metastatic breast cancer [208]. In a phase 2 clinical trial, a combination of everolimus and the aromatase inhibitor letrozole achieved a 12-week PFS rate of 47% in patients with ER-positive relapsed high-grade ovarian cancer [209]. In addition, the combination of everolimus with trastuzumab and paclitaxel has a promising efficacy in patients with highly resistant HER2-positive advanced breast cancer (Table 1). This combination is currently under investigation in the BOLERO-1 phase 3 trial [210]. Moreover, a combination of everolimus with carboplatin is efficacious in treating metastatic triple-negative breast cancer, with a median PFS of 3 months (95% CI 1.6 to 4.6 months) and overall survival (OS) of 16.6 months [211]. In contrast, a combination of everolimus with gemcitabine/cisplatin has no synergistic effect in patients with metastatic triple-negative breast cancer. Hence, this combination still needs validation in more patients.
The CD20-targeted monoclonal antibody rituximab is a treatment for low-grade or follicular CD20-positive non-Hodgkin’s lymphoma. Diffuse large B cell lymphoma (DLBCL) is the most common type of non-Hodgkin’s lymphoma. A phase 2 study of everolimus (10 mg/day) in combination with rituximab demonstrated an overall response rate of 38%, a complete response rate of 12.5%, and a partial response rate of 25% among 24 patients with heavily pretreated DLBCL [212]. In addition, the combination of everolimus with rituximab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) was well tolerated in DLBCL patients [212, 213]. It warrants further study to determine if the combination of everolimus with R-CHOP has a better response in patients with DLBCL. In addition, the combination of mTORC1/2 inhibitor with other targeted cancer drugs has been tested in clinical trials. Among 54 cancer patients who were treated with MLN0128 and trastuzumab/paclitaxel, 14.8% (8/54) of them achieved a partial response, and near 11% (6/54) cases had stable disease for more than 6 months [87]. According to a phase 1 trial (NCT02193633), the combination of paclitaxel and vistusertib is highly active and well tolerated in patients with high-grade serous ovarian cancer and squamous non-small cell lung cancer [214].
Given that IGF-IR signaling may induce mTORC1 inhibitor resistance, the combination of cixutumumab, a humanized monoclonal antibody against IGF-1R, and temsirolimus was tested in a clinical setting. This combination shows clinical activity in patients with sarcoma and adrenocortical carcinoma [215, 216]. In addition, a combination of everolimus (5 mg daily) and the multi-kinase inhibitor sorafenib (400 mg twice daily) exhibits anti-tumor activity in previously untreated patients with metastatic renal cell carcinoma with tolerable toxicity [217]. However, a combination of sorafenib and everolimus fails to achieve the target of 6 month PFS of 50% or greater among patients with unresectable high-grade osteosarcoma progressing after standard treatment [218]. For patients with recurrent glioblastoma, a combination of sorafenib (200 mg twice daily) and temsirolimus (20 mg weekly) is associated with considerable toxicity and poor efficacy [219].
In patients with metastatic castration-resistant prostate cancer, a combination of everolimus and the EGFR inhibitor gefitinib has no significant anti-tumor activity [220]. According to a phase 2 trial, a combination of sunitinib and everolimus as the first-line therapy exhibits poor efficacy in treating advanced renal cell carcinoma [221]. However, another phase 2 trial in patients with metastatic renal carcinoma demonstrates that the first-line sunitinib treatment followed by everolimus achieves a longer OS than the first-line everolimus followed by sunitinib, suggesting that the sequence may affect the outcome [222]. Moreover, a combination of imatinib and everolimus has limited activity in the treatment of patients with advanced chordoma [223]. The combination of pimasertib and voxtalisib showed a poor long-term tolerability and limited anti-tumor activity in patients with advanced solid tumors [224].
Concluding remarks
The discovery of TOR in yeast and mTOR in mammals is a fundamental breakthrough in understanding cell and organism growth, metabolism, and diseases. In-depth studies to clarify the regulators and effectors of mTOR signaling have revealed multiple networks that work together to integrate growth factors, nutrients, sterols, and nucleotides signaling. The identification of the critical roles of mTOR and its regulators in tumorigenesis has driven the development of the ever-growing list of mTOR inhibitors. While some of the mTOR inhibitors have been approved to treat cancer patients, more mTOR inhibitors are under check to fulfill their promise for cancer therapy.
It appears that mTOR inhibitors have mixed efficacy in patients with distinct kinds of cancer and among patients with the same kind of cancer. Recent studies reveal that tumor organoids may help drug testing [225, 226]. Tumor organoids may be used to test the response of a given tumor to mTOR inhibitors. Alternatively, patient-derived tumor grafts may be transplanted to animals, followed by testing their response to mTOR inhibitors [227]. It would be of interest to determine if these emerging technologies are clinically relevant.
In the era of precise medicine, it needs to determine if there are predictive biomarkers that may guide the stratification of patients in clinical trials or help identify the patients who most likely benefit from treatment with mTOR inhibitors in a clinical setting. Gene testing is a promising approach to achieve this goal. The candidates for gene testing may include mTOR, PIK3CA, GATOR, KRAS, and BRAF. Mutations in PIK3CA and GATOR have been associated with higher sensitivity to mTOR inhibition in preclinical studies. Hence, PIK3CA mutations may be potential sensitive markers. In contrast, KRAS/BRAF mutations may be resistant biomarkers. Both DNA from tumor samples and ctDNA from the blood may be subject to testing of gene mutations. In addition, gene mutations in the tumors may be dynamic during cancer evolution or regression [228]. It remains to determine if dynamic testing of ctDNA during the course of therapy may monitor cancer evolution and better predict drug resistance, thereby adjusting the treatment regimen in time. Recent progress in liquid biopsy may help address this critical issue [229, 230]. In addition to gene testing, the solvable factors in the blood may be potential biomarkers as well. Of particular note, the mechanisms underlying the varied responsiveness to mTOR inhibitors in cancer patients may be complex. Rather than a single or few biomarkers, a set of biomarkers may be more powerful and accurate to meet the challenge.
Moreover, toxicity is a critical problem that precludes the clinical administration of drugs. Although mTOR inhibitors exhibit a promising efficacy in preclinical studies, some inhibitors have serious adverse effects in patients and have to be discontinued. Hence, elucidation of the mechanisms underlying these adverse effects may help manage them in the clinic.
Drug resistance is a serious challenge to successful cancer therapy. As discussed above, the mechanisms for mTOR inhibitor resistance are complex. Further studies to elucidate the diverse mechanisms may help design strategies to overcome the resistance to mTOR inhibition. Mechanism-based combination of mTOR inhibitors with chemotherapeutic agents or molecular-targeted drugs may be practical in the clinic. We expect the results from many ongoing clinical trials to validate the most powerful regimens that include mTOR inhibitors.
Availability of data and materials
Not applicable.
Abbreviations
- 4E-BP1:
-
Eukaryotic translation initiation factor 4E binding protein 1
- DEPTOR:
-
DEP domain-containing protein 6
- IGF-IR:
-
Type-I insulin-like growth factor receptor
- MAPK:
-
Mitogen-activated protein kinase
- mLST8:
-
Mammalian lethal with SEC13 protein 8
- mSIN1:
-
Mammalian stress-activated protein kinase-interacting protein 1
- mTOR:
-
Mechanistic target of rapamycin
- PI3K:
-
Phosphoinositide 3-kinase
- PKC:
-
Protein kinase C
- PRAS40:
-
40 kDa proline-rich Akt substrate
- Raptor:
-
Regulatory-associated protein of mTOR
- RCC:
-
Renal cell carcinoma
- RHEB:
-
Ras homolog enriched in the brain
- Rictor:
-
Rapamycin-insensitive companion of mTOR
- S6K1:
-
Ribosomal protein S6 kinase β-1
- TSC:
-
Tuberous sclerosis complex
References
Yin Y, Hua H, Li M, Liu S, Kong Q, Shao T, et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016;26:46–65.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–76.
Harwood FC, Klein Geltink RI, O’Hara BP, Cardone M, Janke L, Finkelstein D, et al. ETV7 is an essential component of a rapamycin-insensitive mTOR complex in cancer. Sci Adv. 2018;4:eaar3938.
Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8.
Rispal D, Eltschinger S, Stahl M, Vaga S, Bodenmiller B, Abraham Y, et al. Target of rapamycin complex 2 regulates actin polarization and endocytosis via multiple pathways. J Biol Chem. 2015;290:14963–78.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5.
Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13:468–77.
Yang H, Wang J, Liu M, Chen X, Huang M, Tan D, et al. 4.4 Å resolution cryo-EM structure of human mTOR complex 1. Protein Cell. 2016;7:878–87.
Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010;38:768–74.
Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300.
Aylett CH, Sauer E, Imseng S, Boehringer D, Hall MN, Ban N, et al. Architecture of human mTOR complex 1. Science. 2016;351:48–52.
Schalm SS, Fingar DC, Sabatini DM, Blenis J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol. 2003;13:797–806.
Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461–4.
Chen X, Liu M, Tian Y, Li J, Qi Y, Zhao D, et al. Cryo-EM structure of human mTOR complex 2. Cell Res. 2018;28:518–28.
Karuppasamy M, Kusmider B, Oliveira TM, Gaubitz C, Prouteau M, Loewith R, et al. Cryo-EM structure of Saccharomyces cerevisiae target of rapamycin complex 2. Nat Commun. 2017;8:1729.
Zhou P, Zhang N, Nussinov R, Ma B. Defining the domain arrangement of the mammalian target of rapamycin complex component Rictor protein. J Comput Biol. 2015;22:876–86.
Andrade MA, Perez-Iratxeta C, Ponting CP. Protein repeats: structures, functions, and evolution. J Struct Biol. 2001;134:117–31.
Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, et al. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov. 2015;5:1194–209.
McDonald PC, Oloumi A, Mills J, Dobreva I, Maidan M, Gray V, et al. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res. 2008;68:1618–24.
Schroder WA, Buck M, Cloonan N, Hancock JF, Suhrbier A, Sculley T, et al. Human Sin1 contains Ras-binding and pleckstrin homology domains and suppresses Ras signalling. Cell Signal. 2007;19:1279–89.
Deng L, Chen L, Zhao L, Xu Y, Peng X, Wang X, et al. Ubiquitination of Rheb governs growth factor-induced mTORC1 activation. Cell Res. 2019;29:136–50.
Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57.
Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–65.
Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–68.
Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, et al. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J Biol Chem. 2011;286:567–77.
Carrière A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, et al. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol. 2008;18:1269–77.
Wu D, Chapman JR, Wang L, Harris TE, Shabanowitz J, Hunt DF, et al. Intestinal cell kinase (ICK) promotes activation of mTOR complex 1 (mTORC1) through phosphorylation of Raptor Thr-908. J Biol Chem. 2012;287:12510–9.
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–68.
Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;510:397–401.
Schewe DM, Aguirre-Ghiso JA. ATF6 alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA. 2008;105:10519–24.
Kang YJ, Lu MK, Guan KL. The TSC1 and TSC2 tumor suppressors are required for proper ER stress response and protect cells from ER stress-induced apoptosis. Cell Death Differ. 2011;18:133–44.
Shimobayashi M, Hall MN. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016;26:7–20.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303.
de Araujo MEG, Naschberger A, Fürnrohr BG, Stasyk T, Dunzendorfer-Matt T, Lechner S, et al. Crystal structure of the human lysosomal mTORC1 scaffold complex and its impact on signaling. Science. 2017;358:377–81.
Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–6.
Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543:438–42.
Peng M, Yin N, Li MO. SZT2 dictates GATOR control of mTORC1 signalling. Nature. 2017;543:433–7.
Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9:1–8.
Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014;9:1281–91.
Peng M, Yin N, Li MO. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell. 2014;159:122–33.
Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351:43–8.
Deng L, Jiang C, Chen L, Jin J, Wei J, Zhao L, et al. The ubiquitination of rag A GTPase by RNF152 negatively regulates mTORC1 activation. Mol Cell. 2015;58:804–18.
Jin G, Lee SW, Zhang X, Cai Z, Gao Y, Chou PC, et al. Skp2-mediated RagA ubiquitination elicits a negative feedback to prevent amino-acid-dependent mTORC1 hyperactivation by recruiting GATOR1. Mol Cell. 2015;58:989–1000.
Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347:188–94.
Song P, Yang S, Hua H, Zhang H, Kong Q, Wang J, et al. The regulatory protein GADD34 inhibits TRAIL-induced apoptosis via TRAF6/ERK-dependent stabilization of myeloid cell leukemia 1 in liver cancer cells. J Biol Chem. 2019;294:5945–55.
Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell. 2013;51:283–96.
Menon D, Salloum D, Bernfeld E, Gorodetsky E, Akselrod A, Frias MA, et al. Lipid sensing by mTOR complexes via de novo synthesis of phosphatidic acid. J Biol Chem. 2017;292:6303–11.
Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–11.
Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, et al. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep. 2017;21:1331–46.
Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–70.
Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–21.
Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013;15:1340–50.
Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–68.
Ebner M, Sinkovics B, Szczygieł M, Ribeiro DW, Yudushkin I. Localization of mTORC2 activity inside cells. J Cell Biol. 2017;216:343–53.
Dan HC, Antonia RJ, Baldwin AS. PI3K/Akt promotes feedforward mTORC2 activation through IKKα. Oncotarget. 2016;7:21064–75.
Gao M, Kong Q, Hua H, Yin Y, Wang J, Luo T, et al. AMPK-mediated up-regulation of mTORC2 and MCL-1 compromises the anti-cancer effects of aspirin. Oncotarget. 2016;7:16349–61.
Moloughney JG, Kim PK, Vega-Cotto NM, Wu CC, Zhang S, Adlam M, et al. mTORC2 responds to glutamine catabolite levels to modulate the hexosamine biosynthesis enzyme GFAT1. Mol Cell. 2016;63:811–26.
Moloughney JG, Vega-Cotto NM, Liu S, Patel C, Kim PK, Wu CC, et al. mTORC2 modulates the amplitude and duration of GFAT1 Ser-243 phosphorylation to maintain flux through the hexosamine pathway during starvation. J Biol Chem. 2018;293:16464–78.
Alcaide-Gavilán M, Lucena R, Schubert KA, Artiles KL, Zapata J, Kellogg DR. Modulation of TORC2 signaling by a conserved Lkb1 signaling axis in budding yeast. Genetics. 2018;210:155–70.
Foster DA. Phosphatidic acid and lipid-sensing by mTOR. Trends Endocrinol Metab. 2013;24:272–8.
Wang B, Jie Z, Joo D, Ordureau A, Liu P, Gan W, et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature. 2017;545:365–9.
Khanna N, Fang Y, Yoon MS, Chen J. XPLN is an endogenous inhibitor of mTORC2. Proc Natl Acad Sci U S A. 2013;110:15979–84.
Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499–502.
Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–64.
Wang L, Rhodes CJ, Lawrence JC Jr. Activation of mammalian target of rapamycin (mTOR) by insulin is associated with stimulation of 4EBP1 binding to dimeric mTOR complex 1. J Biol Chem. 2006;281:24293–303.
Gentilella A, Kozma SC, Thomas G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim Biophys Acta. 1849;2015:812–20.
Giguère V. Canonical signaling and nuclear activity of mTOR-a teamwork effort to regulate metabolism and cell growth. FEBS J. 2018;285:1572–88.
Iadevaia V, Liu R, Proud CG. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin Cell Dev Biol. 2014;36:113–20.
Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, White RJ. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc Natl Acad Sci U S A. 2010;107:11823–8.
Geng Q, Liu J, Gong Z, Chen S, Chen S, Li X, et al. Phosphorylation by mTORC1 stabilizes Skp2 and regulates its oncogenic function in gastric cancer. Mol Cancer. 2017;16:83.
Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–302.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101.
Dai N, Christiansen J, Nielsen FC, Avruch J. mTOR complex 2 phosphorylates IMP1 cotranslationally to promote IGF2 production and the proliferation of mouse embryonic fibroblasts. Genes Dev. 2013;27:301–12.
Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–32.
Xu J, Pham CG, Albanese SK, Dong Y, Oyama T, Lee CH, et al. Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J Clin Invest. 2016;126:3526–40.
Ghosh AP, Marshall CB, Coric T, Shim EH, Kirkman R, Ballestas ME, et al. Point mutations of the mTOR-RHEB pathway in renal cell carcinoma. Oncotarget. 2015;6:17895–910.
Gao Y, Gao J, Li M, Zheng Y, Wang Y, Zhang H, et al. Rheb1 promotes tumor progression through mTORC1 in MLL-AF9-initiated murine acute myeloid leukemia. J Hematol Oncol. 2016;9:36.
El Shamieh S, Saleh F, Moussa S, Kattan J, Farhat F. RICTOR gene amplification is correlated with metastasis and therapeutic resistance in triple-negative breast cancer. Pharmacogenomics. 2018;19:757–60.
Kim ST, Kim SY, Klempner SJ, Yoon J, Kim N, Ahn S, et al. Rapamycin-insensitive companion of mTOR (RICTOR) amplification defines a subset of advanced gastric cancer and is sensitive to AZD2014-mediated mTORC1/2 inhibition. Ann Oncol. 2017;28:547–54.
Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, et al. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007;67:11712–20.
Gkountakos A, Pilotto S, Mafficini A, Vicentini C, Simbolo M, Milella M, et al. Unmasking the impact of Rictor in cancer: novel insights of mTORC2 complex. Carcinogenesis. 2018. https://doi.org/10.1093/carcin/bgy086.
Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–8.
Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res. 2019. https://doi.org/10.1016/j.phrs.2019.03.006.
Yeung Y, Lau DK, Chionh F, Tran H, Tse JWT, Weickhardt AJ, et al. K-Ras mutation and amplification status is predictive of resistance and high basal pAKT is predictive of sensitivity to everolimus in biliary tract cancer cell lines. Mol Oncol. 2017;11:1130–42.
Bodnar L, Stec R, Cierniak S, Synowiec A, Wcisło G, Jesiotr M, et al. Clinical usefulness of PI3K/Akt/mTOR genotyping in companion with other clinical variables in metastatic renal cell carcinoma patients treated with everolimus in the second and subsequent lines. Ann Oncol. 2015;26:1385–9.
Kwiatkowski DJ, Choueiri TK, Fay AP, Rini BI, Thorner AR, de Velasco G, et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2016;22:2445–52.
Burris HA 3rd, Kurkjian CD, Hart L, Pant S, Murphy PB, Jones SF, et al. TAK-228 (formerly MLN0128), an investigational dual TORC1/2 inhibitor plus paclitaxel, with/without trastuzumab, in patients with advanced solid malignancies. Cancer Chemother Pharmacol. 2017;80:261–73.
Slotkin EK, Patwardhan PP, Vasudeva SD, de Stanchina E, Tap WD, Schwartz GK. MLN0128, an ATP-competitive mTOR kinase inhibitor with potent in vitro and in vivo antitumor activity, as potential therapy for bone and soft-tissue sarcoma. Mol Cancer Ther. 2015;14:395–406.
Gökmen-Polar Y, Liu Y, Toroni RA, Sanders KL, Mehta R, Badve S, et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat. 2012;136:673–82.
Caro-Vegas C, Bailey A, Bigi R, Damania B, Dittmer DP. Targeting mTOR with MLN0128 overcomes rapamycin and chemoresistant primary effusion lymphoma. MBio. 2019;10:e02871–18.
Badawi M, Kim J, Dauki A, Sutaria D, Motiwala T, Reyes R, et al. CD44 positive and sorafenib insensitive hepatocellular carcinomas respond to the ATP-competitive mTOR inhibitor INK128. Oncotarget. 2018;9:26032–45.
Fricke SL, Payne SN, Favreau PF, Kratz JD, Pasch CA, Foley TM, et al. MTORC1/2 inhibition as a therapeutic strategy for PIK3CA mutant cancers. Mol Cancer Ther. 2019;18:346–55.
Chamberlain CE, German MS, Yang K, Wang J, VanBrocklin H, Regan M, et al. A patient-derived xenograft model of pancreatic neuroendocrine tumors identifies sapanisertib as a possible new treatment for everolimus-resistant tumors. Mol Cancer Ther. 2018;17:2702–9.
Rashid MM, Lee H, Jung BH. Metabolite identification and pharmacokinetic profiling of PP242, an ATP-competitive inhibitor of mTOR using ultra high-performance liquid chromatography and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1072:244–51.
Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38.
Musa F, Alard A, David-West G, Curtin JP, Blank SV, Schneider RJ. Dual mTORC1/2 inhibition as a novel strategy for the resensitization and treatment of platinum-resistant ovarian cancer. Mol Cancer Ther. 2016;15:1557–67.
David-West G, Ernlund A, Gadi A, Schneider RJ. mTORC1/2 inhibition re-sensitizes platinum-resistant ovarian cancer by disrupting selective translation of DNA damage and survival mRNAs. Oncotarget. 2018;9:33064–76.
Koppenhafer SL, Goss KL, Terry WW, Gordon DJ. mTORC1/2 and protein translation regulate levels of CHK1 and the sensitivity to CHK1 inhibitors in Ewing sarcoma cells. Mol Cancer Ther. 2018;17:2676–88.
Nam HY, Han MW, Chang HW, Lee YS, Lee M, Lee HJ, et al. Radioresistant cancer cells can be conditioned to enter senescence by mTOR inhibition. Cancer Res. 2013;73:4267–77.
Hayman TJ, Wahba A, Rath BH, Bae H, Kramp T, Shankavaram UT, et al. The ATP-competitive mTOR inhibitor INK128 enhances in vitro and in vivo radiosensitivity of pancreatic carcinoma cells. Clin Cancer Res. 2014;20:110–9.
Zhang Q, Shi C, Han L, Jain N, Roberts KG, Ma H, et al. Inhibition of mTORC1/C2 signaling improves anti-leukemia efficacy of JAK/STAT blockade in CRLF2 rearranged and/or JAK driven Philadelphia chromosome-like acute B-cell lymphoblastic leukemia. Oncotarget. 2018;9:8027–41.
Guichard SM, Curwen J, Bihani T, D’Cruz CM, Yates JW, Grondine M, et al. AZD2014, an inhibitor of mTORC1 and mTORC2, is highly effective in ER+ breast cancer when administered using intermittent or continuous schedules. Mol Cancer Ther. 2015;14:2508–18.
Jordan NJ, Dutkowski CM, Barrow D, Mottram HJ, Hutcheson IR, Nicholson RI, et al. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res. 2014;16:R12.
Wong Te Fong AC. Thavasu P, Gagrica S, Swales KE, Leach MO, Cosulich SC, et al. Evaluation of the combination of the dual m-TORC1/2 inhibitor vistusertib (AZD2014) and paclitaxel in ovarian cancer models. Oncotarget. 2017;8:113874–84.
Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K, et al. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol. 2014;16:29–37.
Hall CP, Reynolds CP, Kang MH. Modulation of glucocorticoid resistance in pediatric T-cell acute lymphoblastic leukemia by increasing BIM expression with the PI3K/mTOR inhibitor BEZ235. Clin Cancer Res. 2016;22:621–32.
Gazi M, Moharram SA, Marhäll A, Kazi JU. The dual specificity PI3K/mTOR inhibitor PKI-587 displays efficacy against T-cell acute lymphoblastic leukemia (T-ALL). Cancer Lett. 2017;392:9–16.
Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci. 2015;36:124–35.
Fròsina G, Profumo A, Marubbi D, Marcello D, Ravetti JL, Daga A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat Oncol. 2018;13:76.
Yu Z, Xie G, Zhou G, Cheng Y, Zhang G, Yao G, et al. NVP-BEZ235, a novel dual PI3K-mTOR inhibitor displays anti-glioma activity and reduces chemoresistance to temozolomide in human glioma cells. Cancer Lett. 2015;367:58–68.
Chen D, Lin X, Zhang C, Liu Z, Chen Z, Li Z, et al. Dual PI3K/mTOR inhibitor BEZ235 as a promising therapeutic strategy against paclitaxel-resistant gastric cancer via targeting PI3K/Akt/mTOR pathway. Cell Death Dis. 2018;9:123.
Zaidi AH, Kosovec JE, Matsui D, Omstead AN, Raj M, Rao RR, et al. PI3K/mTOR dual inhibitor, LY3023414, demonstrates potent antitumor efficacy against esophageal adenocarcinoma in a rat model. Ann Surg. 2017;266:91–8.
Smith MC, Mader MM, Cook JA, Iversen P, Ajamie R, Perkins E, et al. Characterization of LY3023414, a novel PI3K/mTOR dual inhibitor eliciting transient target modulation to impede tumor growth. Mol Cancer Ther. 2016;15:2344–56.
Yu P, Laird AD, Du X, Wu J, Won KA, Yamaguchi K, et al. Characterization of the activity of the PI3K/mTOR inhibitor XL765 (SAR245409) in tumor models with diverse genetic alterations affecting the PI3K pathway. Mol Cancer Ther. 2014;13:1078–91.
Inaba K, Oda K, Ikeda Y, Sone K, Miyasaka A, Kashiyama T, et al. Antitumor activity of a combination of dual PI3K/mTOR inhibitor SAR245409 and selective MEK1/2 inhibitor pimasertib in endometrial carcinomas. Gynecol Oncol. 2015;138:323–31.
Beaufils F, Cmiljanovic N, Cmiljanovic V, Bohnacker T, Melone A, Marone R, et al. 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a potent, brain-penetrant, orally bioavailable, pan-class I PI3K/mTOR inhibitor as clinical candidate in oncology. J Med Chem. 2017;60:7524–38.
Tarantelli C, Gaudio E, Arribas AJ, Kwee I, Hillmann P, Rinaldi A, et al. PQR309 is a novel dual PI3K/mTOR inhibitor with preclinical antitumor activity in lymphomas as a single agent and in combination therapy. Clin Cancer Res. 2018;24:120–9.
Brandt C, Hillmann P, Noack A, Römermann K, Öhler LA, Rageot D, et al. The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood-brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology. 2018;140:107–20.
Venkatesan AM, Dehnhardt CM, Delos Santos E, Chen Z, Dos Santos O, Ayral-Kaloustian S, et al. Bis(morpholino-1,3,5-triazine) derivatives: potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J Med Chem. 2010;53:2636–45.
Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017;129:177–87.
Leiker AJ, DeGraff W, Choudhuri R, Sowers AL, Thetford A, Cook JA, et al. Radiation enhancement of head and neck squamous cell carcinoma by the dual PI3K/mTOR inhibitor PF-05212384. Clin Cancer Res. 2015;21:2792–801.
Liu T, Sun Q, Li Q, Yang H, Zhang Y, Wang R, et al. Dual PI3K/mTOR inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radiosensitivity in nasopharyngeal carcinoma. Mol Cancer Ther. 2015;14:429–39.
D’Amato V, Rosa R, D’Amato C, Formisano L, Marciano R, Nappi L, et al. The dual PI3K/mTOR inhibitor PKI-587 enhances sensitivity to cetuximab in EGFR-resistant human head and neck cancer models. Br J Cancer. 2014;110:2887–95.
Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett. 2010;1:39–43.
McKinnon T, Venier R, Yohe M, Sindiri S, Gryder BE, Shern JF, et al. Functional screening of FGFR4-driven tumorigenesis identifies PI3K/mTOR inhibition as a therapeutic strategy in rhabdomyosarcoma. Oncogene. 2018;37:2630–44.
Zhou J, Toh SH, Chan ZL, Quah JY, Chooi JY, Tan TZ, et al. A loss-of-function genetic screening reveals synergistic targeting of AKT/mTORand WTN/β-catenin pathways for treatment of AML with high PRL-3 phosphatase. J Hematol Oncol. 2018;11:36.
Fan Q, Aksoy O, Wong RA, Ilkhanizadeh S, Novotny CJ, Gustafson WC, et al. A kinase inhibitor targeted to mTORC1 drives regression in glioblastoma. Cancer Cell. 2017;31:424–35.
Oricchio E, Katanayeva N, Donaldson MC, Sungalee S, Pasion JP, Béguelin W, et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma. Sci Transl Med. 2017;9:eaak9969.
Hua H, Zhang H, Kong Q, Wang J, Jiang Y. Complex roles of the old drug aspirin in cancer chemoprevention and therapy. Med Res Rev. 2019;39:114–45.
Boyle KA, Van Wickle J, Hill RB, Marchese A, Kalyanaraman B, Dwinell MB. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J Biol Chem. 2018;293:14891–904.
Ling S, Xie H, Yang F, Shan Q, Dai H, Zhuo J, et al. Metformin potentiates the effect of arsenic trioxide suppressing intrahepatic cholangiocarcinoma: roles of p38 MAPK, ERK3, and mTORC1. J Hematol Oncol. 2017;10:59.
Lin F, de Gooijer MC, Hanekamp D, Chandrasekaran G, Buil LC, Thota N, et al. PI3K-mTOR pathway inhibition exhibits efficacy against high-grade glioma in clinically relevant mouse models. Clin Cancer Res. 2017;23:1286–98.
Hurvitz SA, Kalous O, Conklin D, Desai AJ, Dering J, Anderson L, et al. In vitro activity of the mTOR inhibitor everolimus, in a large panel of breast cancer cell lines and analysis for predictors of response. Breast Cancer Res Treat. 2015;149:669–80.
Tang SC, Sparidans RW, Cheung KL, Fukami T, Durmus S, Wagenaar E, et al. P-glycoprotein, CYP3A, and plasma carboxylesterase determine brain and blood disposition of the mTOR Inhibitor everolimus (Afinitor) in mice. Clin Cancer Res. 2014;20:3133–45.
Wu CP, Murakami M, Hsiao SH, Chou AW, Li YQ, Huang YH, et al. Overexpression of ATP-binding cassette subfamily G member 2 confers resistance to phosphatidylinositol 3-kinase inhibitor PF-4989216 in cancer cells. Mol Pharm. 2017;14:2368–77.
Becker CM, Oberoi RK, McFarren SJ, Muldoon DM, Pafundi DH, Pokorny JL, et al. Decreased affinity for efflux transporters increases brain penetrance and molecular targeting of a PI3K/mTOR inhibitor in a mouse model of glioblastoma. Neuro Oncol. 2015;17:1210–9.
Begicevic RR, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci. 2017;18:e2362.
Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K, et al. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal. 2019;12:eaau8544.
Francipane MG, Lagasse E. Therapeutic potential of mTOR inhibitors for targeting cancer stem cells. Br J Clin Pharmacol. 2016;82:1180–8.
Karthik GM, Ma R, Lövrot J, Kis LL, Lindh C, Blomquist L, et al. mTOR inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett. 2015;367:76–87.
de Graffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. 2004;10:8059–67.
Kolev VN, Wright QG, Vidal CM, Ring JE, Shapiro IM, Ricono J, et al. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res. 2015;75:446–55.
Francipane MG, Lagasse E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget. 2013;4:1948–62.
Fourneaux B, Bourdon A, Dadone B, Lucchesi C, Daigle SR, Richard E, et al. Identifying and targeting cancer stem cells in leiomyosarcoma: prognostic impact and role to overcome secondary resistance to PI3K/mTOR inhibition. J Hematol Oncol. 2019;12:11.
Tan J, Li Z, Lee PL, Guan P, Aau MY, Lee ST, et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013;3:1156–71.
Venkatesan S, Hoogstraat M, Caljouw E, Pierson T, Spoor JK, Zeneyedpour L, et al. TP53 mutated glioblastoma stem-like cell cultures are sensitive to dual mTORC1/2 inhibition while resistance in TP53 wild type cultures can be overcome by combined inhibition of mTORC1/2 and Bcl-2. Oncotarget. 2016;7:58435–44.
Bhola NE, Jansen VM, Koch JP, Li H, Formisano L, Williams JA, et al. Treatment of triple-negative breast cancer with TORC1/2 inhibitors sustains a drug-resistant and Notch-dependent cancer stem cell population. Cancer Res. 2016;76:440–52.
Belmont PJ, Jiang P, McKee TD, Xie T, Isaacson J, Baryla NE, et al. Resistance to dual blockade of the kinases PI3K and mTOR in KRAS-mutant colorectal cancer models results in combined sensitivity to inhibition of the receptor tyrosine kinase EGFR. Sci Signal. 2014;7:ra107.
Merrick WC. Cap-dependent and cap-independent translation in eukaryotic systems. Gene. 2004;332:1–11.
Shatsky IN, Terenin IM, Smirnova VV, Andreev DE. Cap-independent translation: what’s in a name? Trends Biochem Sci. 2018;43:882–95.
Ducker GS, Atreya CE, Simko JP, Hom YK, Matli MR, Benes CH, et al. Incomplete inhibition of phosphorylation of 4E-BP1 as a mechanism of primary resistance to ATP-competitive mTOR inhibitors. Oncogene. 2014;33:1590–600.
Wang J, Ye Q, Cao Y, Guo Y, Huang X, Mi W, et al. Snail determines the therapeutic response to mTOR kinase inhibitors by transcriptional repression of 4E-BP1. Nat Commun. 2017;8:2207.
Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2015;34:2032–42.
Cope CL, Gilley R, Balmanno K, Sale MJ, Howarth KD, Hampson M, et al. Adaptation to mTOR kinase inhibitors by amplification of eIF4E to maintain cap-dependent translation. J Cell Sci. 2014;127:788–800.
D’Abronzo LS, Bose S, Crapuchettes ME, Beggs RE, Vinall RL, Tepper CG, et al. The androgen receptor is a negative regulator of eIF4E phosphorylation at S209: implications for the use of mTOR inhibitors in advanced prostate cancer. Oncogene. 2017;36:6359–73.
Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, et al. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol. 2007;27:7405–13.
Wen Q, Wang W, Luo J, Chu S, Chen L, Xu L, et al. CGP57380 enhances efficacy of RAD001 in non-small cell lung cancer through abrogating mTOR inhibition-induced phosphorylation of eIF4E and activating mitochondrial apoptotic pathway. Oncotarget. 2016;7:27787–801.
Shuda M, Velásquez C, Cheng E, Cordek DG, Kwun HJ, Chang Y, et al. CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proc Natl Acad Sci U S A. 2015;112:5875–82.
Choi SH, Martinez TF, Kim S, Donaldson C, Shokhirev MN, Saghatelian A, et al. CDK12 phosphorylates 4E-BP1 to enable mTORC1-dependent translation and mitotic genome stability. Genes Dev. 2019;33:418–35.
Shin S, Wolgamott L, Tcherkezian J, Vallabhapurapu S, Yu Y, Roux PP, et al. Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene. 2014;33:1690–9.
Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014;4:554–63.
Hassan B, Akcakanat A, Sangai T, Evans KW, Adkins F, Eterovic AK, et al. Catalytic mTOR inhibitors can overcome intrinsic and acquired resistance to allosteric mTOR inhibitors. Oncotarget. 2014;5:8544–57.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501.
Donnella HJ, Webber JT, Levin RS, Camarda R, Momcilovic O, Bayani N, et al. Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nat Chem Biol. 2018;14:768–77.
Balaji S, Ahmed M, Lorence E, Yan F, Nomie K, Wang M. NF-κB signaling and its relevance to the treatment of mantle cell lymphoma. J Hematol Oncol. 2018;11:83.
Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013;73:276–84.
Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F, et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res. 2012;18:1777–89.
Citi V, Del Re M, Martelli A, Calderone V, Breschi MC, Danesi R. Phosphorylation of AKT and ERK1/2 and mutations of PIK3CA and PTEN are predictive of breast cancer cell sensitivity to everolimus in vitro. Cancer Chemother Pharmacol. 2018;81:745-54.
Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858–66.
Mohan S, Vander Broek R, Shah S, Eytan DF, Pierce ML, Carlson SG, et al. MEK inhibitor PD-0325901 overcomes resistance to PI3K/mTOR inhibitor PF-5212384 and potentiates antitumor effects in human head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21:3946–56.
Xu DQ, Toyoda H, Qi L, Morimoto M, Hanaki R, Iwamoto S, et al. Induction of MEK/ERK activity by AZD8055 confers acquired resistance in neuroblastoma. Biochem Biophys Res Commun. 2018;499:425–32.
Olmez I, Brenneman B, Xiao A, Serbulea V, Benamar M, Zhang Y, et al. Combined CDK4/6 and mTOR inhibition is synergistic against glioblastoma via multiple mechanisms. Clin Cancer Res. 2017;23:6958–68.
Song X, Liu X, Wang H, Wang J, Qiao Y, Cigliano A, et al. Combined CDK4/6 and pan-mTOR inhibition is synergistic against intrahepatic cholangiocarcinoma. Clin Cancer Res. 2019;25:403–13.
Hai J, Liu S, Bufe L, Do K, Chen T, Wang X, et al. Synergy of WEE1 and mTOR inhibition in mutant KRAS-driven lung cancers. Clin Cancer Res. 2017;23:6993–7005.
Fujishita T, Kojima Y, Kajino-Sakamoto R, Taketo MM, Aoki M. Tumor microenvironment confers mTOR inhibitor resistance in invasive intestinal adenocarcinoma. Oncogene. 2017;36:6480–9.
Park YL, Kim HP, Cho YW, Min DW, Cheon SK, Lim YJ, et al. Activation of WNT/β-catenin signaling results in resistance to a dual PI3K/mTOR inhibitor in colorectal cancer cells harboring PIK3CA mutations. Int J Cancer. 2019;144:389–401.
Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell. 2018;33:905–21.
Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest. 2015;125:1591–602.
Makinoshima H, Umemura S, Suzuki A, Nakanishi H, Maruyama A, Udagawa H, et al. Metabolic determinants of sensitivity to phosphatidylinositol 3-kinase pathway inhibitor in small-cell lung carcinoma. Cancer Res. 2018;78:2179–90.
Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–33.
Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339:1320–3.
Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–8.
Cao J, Huang W. Compensatory increase of transglutaminase 2 is responsible for resistance to mTOR inhibitor treatment. PLOS One. 2016;11:e0149388.
Mehta K, Kumar A, Kim HI. Transglutaminase 2: a multi-tasking protein in the complex circuitry of inflammation and cancer. Biochem Pharmacol. 2010;80:1921–9.
Park D, Choi SS, Ha KS. Transglutaminase 2: a multi-functional protein in multiple subcellular compartments. Amino Acids. 2010;39:619–31.
Morita M, Prudent J, Basu K, Goyon V, Katsumura S, Hulea L, et al. mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol Cell. 2017;67:922–35.
Lew S, Chamberlain RS. Risk of metabolic complications in patients with solid tumors treated with mTOR inhibitors: meta-analysis. Anticancer Res. 2016;36:1711–8.
Johnston PB, Pinter-Brown LC, Warsi G, White K, Ramchandren R. Phase 2 study of everolimus for relapsed or refractory classical Hodgkin lymphoma. Exp Hematol Oncol. 2018;7:12.
Pearson AD, Federico SM, Aerts I, Hargrave DR, DuBois SG, Iannone R, et al. A phase 1 study of oral ridaforolimus in pediatric patients with advanced solid tumors. Oncotarget. 2016;7:84736–47.
Mita MM, Mita AC, Chu QS, Rowinsky EK, Fetterly GJ, Goldston M, et al. Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J Clin Oncol. 2008;26:361–7.
Graham L, Banda K, Torres A, Carver BS, Chen Y, Pisano K, et al. A phase II study of the dual mTOR inhibitor MLN0128 in patients with metastatic castration resistant prostate cancer. Invest New Drugs. 2018;36:458–67.
Antonuzzo L, Del Re M, Barucca V, Spada F, Meoni G, Restante G, et al. Critical focus on mechanisms of resistance and toxicity of m-TOR inhibitors in pancreatic neuroendocrine tumors. Cancer Treat Rev. 2017;57:28–35.
Carlo MI, Molina AM, Lakhman Y, Patil S, Woo K, DeLuca J, et al. A phase Ib study of BEZ235, a dual inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR), in patients with advanced renal cell carcinoma. Oncologist. 2016;21:787–8.
Powles T, Lackner MR, Oudard S, Escudier B, Ralph C, Brown JE, et al. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2016;34:1660–8.
Grilley-Olson JE, Bedard PL, Fasolo A, Cornfeld M, Cartee L, Razak AR, et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Invest New Drugs. 2016;34:740–9.
Britten CD, Adjei AA, Millham R, Houk BE, Borzillo G, Pierce K, et al. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Invest New Drugs. 2014;32:510–7.
Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–23.
Wahl M, Chang SM, Phillips JJ, Molinaro AM, Costello JF, Mazor T, et al. Probing the phosphatidylinositol 3-kinase/mammalian target of rapamycin pathway in gliomas: a phase 2 study of everolimus for recurrent adult low-grade gliomas. Cancer. 2017;123:4631–9.
Lau DK, Tay RY, Yeung YH, Chionh F, Mooi J, Murone C, et al. Phase II study of everolimus (RAD001) monotherapy as first-line treatment in advanced biliary tract cancer with biomarker exploration: the RADiChol Study. Br J Cancer. 2018;118:966–71.
Schneider TC, de Wit D, Links TP, van Erp NP, van der Hoeven JJ, Gelderblom H, et al. Everolimus in patients with advanced follicular-derived thyroid cancer: results of a phase II clinical trial. J Clin Endocrinol Metab. 2017;102:698–707.
Colombo N, McMeekin DS, Schwartz PE, Sessa C, Gehrig PA, Holloway R, et al. Ridaforolimus as a single agent in advanced endometrial cancer: results of a single-arm, phase 2 trial. Br J Cancer. 2013;108:1021–6.
Brown JR, Hamadani M, Hayslip J, Janssens A, Wagner-Johnston N, Ottmann O, et al. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: an open-label, phase 2 trial. Lancet Haematol. 2018;5:e170–80.
Powles T, Wheater M, Din O, Geldart T, Boleti E, Stockdale A, et al. A randomised phase 2 study of AZD2014 versus everolimus in patients with VEGF-refractory metastatic clear cell renal cancer. Eur Urol. 2016;69:450–6.
Del Campo JM, Birrer M, Davis C, Fujiwara K, Gollerkeri A, Gore M, et al. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol Oncol. 2016;142:62–9.
Zhou J, Toh SH, Chan ZL, Quah JY, Chooi JY, Tan TZ, et al. A loss-of-function genetic screening reveals synergistic targeting of AKT/mTOR and WTN/β-catenin pathways for treatment of AML with high PRL-3 phosphatase. J Hematol Oncol. 2018;11:36.
Yardley DA, Noguchi S, Pritchard KI, Burris HA 3rd, Baselga J, Gnant M, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870–84.
Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2†. Ann Oncol. 2014;25:2357–62.
O’Shaughnessy J, Thaddeus Beck J, Royce M. Everolimus-based combination therapies for HR+, HER2- metastatic breast cancer. Cancer Treat Rev. 2018;69:204–14.
Colon-Otero G, Weroha SJ, Foster NR, Haluska P, Hou X, Wahner-Hendrickson AE, et al. Phase 2 trial of everolimus and letrozole in relapsed estrogen receptor-positive high-grade ovarian cancers. Gynecol Oncol. 2017;146:64–8.
Hurvitz SA, Dalenc F, Campone M, O’Regan RM, Tjan-Heijnen VC, Gligorov J, et al. A phase 2 study of everolimus combined with trastuzumab and paclitaxel in patients with HER2-overexpressing advanced breast cancer that progressed during prior trastuzumab and taxane therapy. Breast Cancer Res Treat. 2013;141:437–46.
Singh J, Novik Y, Stein S, Volm M, Meyers M, Smith J, et al. Phase 2 trial of everolimus and carboplatin combination in patients with triple negative metastatic breast cancer. Breast Cancer Res. 2014;16:R32.
Barnes JA, Jacobsen E, Feng Y, Freedman A, Hochberg EP, LaCasce AS, et al. Everolimus in combination with rituximab induces complete responses in heavily pretreated diffuse large B-cell lymphoma. Haematologica. 2013;98:615–9.
Johnston PB, LaPlant B, McPhail E, Habermann TM, Inwards DJ, Micallef IN, et al. Everolimus combined with R-CHOP-21 for new, untreated, diffuse large B-cell lymphoma (NCCTG 1085 [Alliance]): safety and efficacy results of a phase 1 and feasibility trial. Lancet Haematol. 2016;3:e309–16.
Basu B, Krebs MG, Sundar R, Wilson RH, Spicer J, Jones R, et al. Vistusertib (dual m-TORC1/2 inhibitor) in combination with paclitaxel in patients with high-grade serous ovarian and squamous non-small-cell lung cancer. Ann Oncol. 2018;29:1918–25.
Schwartz GK, Tap WD, Qin LX, Livingston MB, Undevia SD, Chmielowski B, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013;14:371–82.
Naing A, Kurzrock R, Burger A, Gupta S, Lei X, Busaidy N, et al. Phase I trial of cixutumumab combined with temsirolimus in patients with advanced cancer. Clin Cancer Res. 2011;17:6052–60.
Harzstark AL, Small EJ, Weinberg VK, Sun J, Ryan CJ, Lin AM, et al. A phase 1 study of everolimus and sorafenib for metastatic clear cell renal cell carcinoma. Cancer. 2011;117:4194–200.
Grignani G, Palmerini E, Ferraresi V, D’Ambrosio L, Bertulli R, Asaftei SD, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol. 2015;16:98–107.
Schiff D, Jaeckle KA, Anderson SK, Galanis E, Giannini C, Buckner JC, et al. Phase 1/2 trial of temsirolimus and sorafenib in the treatment of patients with recurrent glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer. 2018;124:1455–63.
Rathkopf DE, Larson SM, Anand A, Morris MJ, Slovin SF, Shaffer DR, et al. Everolimus combined with gefitinib in patients with metastatic castration-resistant prostate cancer: phase 1/2 results and signaling pathway implications. Cancer. 2015;121:3853–61.
Davis ID, Long A, Yip S, Espinoza D, Thompson JF, Kichenadasse G, et al. EVERSUN: a phase 2 trial of alternating sunitinib and everolimus as first-line therapy for advanced renal cell carcinoma. Ann Oncol. 2015;26:1118–23.
Knox JJ, Barrios CH, Kim TM, Cosgriff T, Srimuninnimit V, Pittman K, et al. Final overall survival analysis for the phase II RECORD-3 study of first-line everolimus followed by sunitinib versus first-line sunitinib followed by everolimus in metastatic RCC. Ann Oncol. 2017;28:1339–45.
Stacchiotti S, Morosi C, Lo Vullo S, Casale A, Palassini E, Frezza AM, et al. Imatinib and everolimus in patients with progressing advanced chordoma: a phase 2 clinical study. Cancer. 2018;124:4056–63.
Schram AM, Gandhi L, Mita MM, Damstrup L, Campana F, Hidalgo M, et al. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer. 2018;119:1471–6.
Xu H, Jiao Y, Qin S, Zhao W, Chu Q, Wu K. Organoid technology in disease modelling, drug development, personalized treatment and regeneration medicine. Exp Hematol Oncol. 2018;7:30.
Xu H, Lyu X, Yi M, Zhao W, Song Y, Wu K. Organoid technology and applications in cancer research. J Hematol Oncol. 2018;11:116.
Chen Z, Huang W, Tian T, Zang W, Wang J, Liu Z, et al. Characterization and validation of potential therapeutic targets based on the molecular signature of patient-derived xenografts in gastric cancer. J Hematol Oncol. 2018;11:20.
Frenel JS, Carreira S, Goodall J, Roda D, Perez-Lopez R, Tunariu N, et al. Serial next-generation sequencing of circulating cell-free DNA evaluating tumor clone response to molecularly targeted drug administration. Clin Cancer Res. 2015;21:4586–96.
Yang M, Topaloglu U, Petty WJ, Pagni M, Foley KL, Grant SC, et al. Circulating mutational portrait of cancer: manifestation of aggressive clonal events in both early and late stages. J Hematol Oncol. 2017;10:100.
Rossi G, Ignatiadis M. Promises and pitfalls of using liquid biopsy for precision medicine. Cancer Res. 2019. https://doi.org/10.1158/0008-5472.
Acknowledgements
We would like to thank Qiulin Tang for her assistance in preparing the manuscript.
Funding
This work was supported by grants 81672814 and 81872388 from the National Natural Science Foundation of China and grant 2018SCUH0009 from the Fundamental Research Fund for the Central Universities.
Author information
Authors and Affiliations
Contributions
HH and YJ conceived the review and wrote the manuscript. QK and JW prepared the figures. HZ edited the references. TL was involved in editing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hua, H., Kong, Q., Zhang, H. et al. Targeting mTOR for cancer therapy. J Hematol Oncol 12, 71 (2019). https://doi.org/10.1186/s13045-019-0754-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-019-0754-1