
Abstract
First identified in 1974, m6A RNA methylation, which serves as a predominant internal modification of RNA in higher eukaryotes, has gained prodigious interest in recent years. Modifications of m6A are dynamic and reversible in mammalian cells, which have been proposed as another layer of epigenetic regulation similar to DNA and histone modifications. m6A RNA methylation is involved in all stages in the life cycle of RNA, ranging from RNA processing, through nuclear export, translation modulation to RNA degradation, which suggests its potential of influencing a plurality of aspects of RNA metabolism. All of the recent studies have pointed to a complicated regulation network of m6A modification in different tissues, cell lines, and space–time models. m6A methylation has been found to have an impact on tumor initiation and progression through various mechanisms. Furthermore, m6A RNA methylation has provided new opportunities for early stage diagnosis and treatment of cancers.
Herein, we review the chemical basis of m6A RNA methylation, its multiple functions and potential significance in cancer.
Similar content being viewed by others

Background
Termed as “epitranscriptome,” RNA harbors the potential of being dynamically and reversibly regulated by the addition and removal of distinct chemical moieties, which extends the RNA repertoire and alters its chemistry in various ways [1].
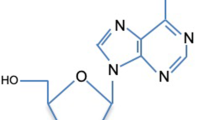
Among more than 100 types of post-transcriptional modifications identified in RNAs so far [2,3,4], N6-methyladenosine (m6A) RNA methylation is one of the most prevalent modifications, accounting for about 50% of total methylated ribonucleotides and 0.1–0.4% of all adenosines in total cellular RNAs [5, 6]. Most mammalian m6A sites are found within the consensus sequence Rm6ACH (R = G or A, H = A, C, or U), which is consistent with the enriched binding motifs observed in studies of methyltransferase-like 14 (METTL14), methyltransferase-like 3 (METTL3), and Wilms tumor 1-associated protein (WTAP) (GGAC, GGAC, and GACU respectively) [7]. There have been plenty of researches concerning DNA and histone methylation. DNA methylation is an important biochemical process which comprises the addition of a methyl group to the carbon 5 of the pyrimidine ring of cytosine or the nitrogen 6 of the purine ring of adenine. DNA methylation is brought about by a group of enzymes known as the DNA methyltransferases (DNMT) and can be de novo (when CpG dinucleotides on both DNA strands are unmethylated) or maintenance (when CpG dinucleotides on one strand are methylated). The enzymes that actively demethylate DNA include 5-methylcytosine glycosylase and MBD2b. Accumulating evidence has shown that DNA methylation plays an important role in all aspects of cellular processes including tumorigenesis [8].
Histone methylation in localized promoter regions is one kind of histone codes for chromatin packing and transcription. Generally, methylation of H3K4, H3K36, and H3K79 is linked to gene expression activation, whereas H3K9me2, H3K9me3, H3K27me3, and H4K20 are associated with gene repression [9,10,11,12]. Modifications of m6A RNA methylation are dynamic and reversible in mammalian cells, which have been proposed as another layer of epigenetic regulation similar to DNA and histone methylation. The biological function of m6A RNA methylation is highly variable depending on context and little is known about the underlying mechanisms; however, emerging evidence has suggested that m6A modification plays a pivotal role in pre-mRNA splicing, 3′-end processing, nuclear export, translation regulation, mRNA decay, and miRNA processing [13]. Growing appreciation of the biological significance of m6A RNA methylation has implied its important and diverse biological functions in mammals including tissue development, circadian rhythm, DNA damage response, sex determination, and tumorigenesis [14].
In this review, we will discuss how m6A RNA methylation participates in tumorigenesis, invasion, metastasis, and drug resistance and how to use m6A modification as new diagnostic biomarkers and therapeutic targets.
Chemical basis of m6A RNA methylation
The m6A methylase complex is consisted of at least five “writer” proteins where METTL3 acts as the catalytic core. METTL14 serves as structural support for METTL3, while WTAP stabilizes the core complex. RNA-binding motif protein 15 (RBM15) helps to recruit the complex to its target sites. The molecular function of KIAA1429 is still elusive [15]. On the other hand, “erasers” consisting of fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5) act as demethylases to reverse m6A modification [16, 17]. The functional interplay among the m6A methyltransferases and demethylases probably determines the dynamic regulation of m6A modification.
YT521-B homology (YTH) domain-containing proteins including YTHDF1-3, YTHDC1, and YTHDC2 have recently been identified as “readers” of m6A marks on mRNA. They display a 10- to 50-fold higher affinity for m6A-methylated mRNA than for unmethylated mRNA [18,19,20,21,22]. When perturbed in different cellular contexts, individual YTH domain family proteins interact with distinct subsets of m6A sites and produce different effects on gene expression. For example, YTHDF2, the first identified m6A reader, increases turnover of m6A-modified mRNA by promoting co-localization with decay factors [23, 24]. Conversely, YTHDF1 can be bound to m6A sites near the stop codon, which then interacts with the translation initiation factor eIF3, causing a stimulatory effect on translation in mammals [25]. Another mechanism for the regulation of translation by m6A RNA methylation comes into play during stress responses where preferential m6A methylation of the 5′-UTR of stress response genes leads to cap-independent translation, which involves nuclear relocalization of YTHDF2 to prevent m6A removal by FTO [26, 27]. YTHDF3 has been shown to interact cooperatively with YTHDF2 to accelerate mRNA decay while it also promotes translation of methylated RNAs in cooperation with YTHDF1 [28, 29]. YTHDC1 recruits the splicing factor Serine and arginine-rich splicing factor (SRSF) 3, promotes exon inclusion, and restricts binding of the exon-skipping factor SRSF10 [30].
As to the processing of pri-miRNAs, hnRNPA2B1 has the capability of recruiting DGCR8 to RNA by targeting m6A sites, indicating its important role in promoting pri-miRNA processing [31, 32].
A special combination of direct and indirect mediation has also been observed and studied. m6A RNA methylation has been shown to modify RNA structure, as a direct effect, in a way that improves accessibility for RNA-binding proteins such as hnRNPC, which in turn mediates an indirect effect. This phenomenon is termed as “m6A-switch” [33].
To sum up, the emerging new data and discoveries have revealed a complex picture concerning epitranscriptome (Fig. 1).

Chemical basis and diverse molecular functions of m6A RNA methylation. m6A RNA methylation is modulated by its “writers,” “erasers,” and “readers.” Writers refer to the m6A methylase complex which is consisted of METTL3, METTL14, WTAP, RBM15, and KIAA1429. Erasers are m6A demethylases including FTO and ALKBH5. Readers are proteins that bind to m6A modifications and exert various functions, some of whom identified so far are YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, and hnRNPA2B1. m6A RNA methylation is known to be involved in all stages in the life cycle of RNA including pre-mRNA splicing, pri-miRNA processing, through nuclear export, RNA translation modulation and RNA degradation
Methods of detecting m6A RNA methylation
The research related with m6A modification has been reinvigorated due to the advent of potent analytical methods, which harness the effects of m6A RNA methylation on RNA structure and metabolism, in combination with novel, high-throughput methods.
Several methods, including dot-blot and high-performance liquid chromatography coupled to triple–quadrupole mass spectrometry (LC–MS/MS) can be utilized to yield important quantitative information about existence and abundance of m6A modification, while they are not suitable for widespread identification and localization of modified sites [34].
Thus, an emerging method termed m6A-seq or MeRIP-seq has received considerable attention recently due to its accuracy and reproducibility. This novel method is based on the high specificity of one antibody raised against m6A in combination of high-throughput sequencing, making the mapping of m6A in the mammalian transcriptome possible [23, 35].
However, the immunoprecipitation-based approach m6A-seq localizes m6A residues to 100- to 200-nt-long regions of transcripts and is incapable of single-nucleotide-resolution detection of methylation sites. Contrary to other base modifications, such as 5-methylcytosine (5mC), which can make use of chemical conversion of cytosine as a mapping approach at single-nucleotide resolution in RNA [36], m6A RNA methylation has no such chemical conversion. Additionally, m6A does not introduce errors during reverse transcription that would allow direct mapping of its position [37]. A novel method called m6A individual-nucleotide-resolution crosslinking and immunoprecipitation (miCLIP) has marked a major step forward in the field, which augments m6A-seq with UV-induced crosslinking of the antibody to the immunoprecipitated RNA fragments [38]. Reverse transcription of crosslinked RNA then results in a highly specific pattern of mutations or truncations in the cDNA, and these specific mutational signatures are crucial for mapping m6A residues throughout the transcriptome at single-nucleotide resolution.
Moreover, it is currently possible to directly test the influence of altering any modification site in many organisms with the use of CRISPR-based genome engineering. CRISPR multiplexing strategies could potentially permit interrogation of many sites in parallel and hasten functional discoveries and would be valuable for the research of m6A RNA methylation as a complementary approach [39].
Multiple functions of m6A RNA methylation in cancer
Currently, m6A RNA methylation has been found to have diverse biological regulatory functions in cancer initiation and progression and dysregulated expression of m6A RNA methylation is closely associated with various kinds of cancers. Herein, we mainly discuss four aspects of these functions exerted by m6A RNA methylation in cancer (Table 1, Fig. 2).

Multiple functions of m6A RNA methylation in cancer. m6A RNA methylation exerts multiple functions in cancer initiation and progression. The circle in the middle represents the reversible process of m6A RNA methylation, which plays a pivotal role in modulating CSC pluripotency, cancer proliferation, cancer metastasis, and tumor immunity. Distinct mechanisms through which m6A RNA methylation exerts these four aspects of functions in various types of cancers are shown in a graphical form in this picture
m6A RNA methylation influences cancer stem cell pluripotency and cell differentiation
In recent years, a new dimension of intratumor heterogeneity and a hitherto-unappreciated subclass of neoplastic cells within tumors, termed cancer stem cells (CSCs), have aroused interests of researchers [40]. CSCs, typically rare within tumors, may prove to regenerate all facets of a tumor as a result of their stem cell-like capacity to self-renew, survive, and become dormant in protective microenvironments, representing a reservoir of self-sustaining cells that give rise to many types of cancers [41, 42].
Hematopoietic stem cells (HSCs) have well-defined developmental trajectories, and it is plausible to monitor and quantify their differentiation, providing an ideal model system in which to explore differentiation states. Abnormal or blocked differentiation is a common feature of myeloid hematological malignancies.
According to the recent research conducted by Ly P Vu and colleagues, leukemia cells show an elevated abundance of METTL3 as compared to normal hematopoietic cells. Utilizing miCLIP coupled with ribosome profiling, it is elucidated that METTL3 augments m6A levels of its target genes including myelocytomatosis (MYC), B cell lymphoma 2 (BCL2), and phosphatase and tensin homolog (PTEN) genes in the human acute myeloid leukemia (AML) MOLM-13 cell line, thereby promoting the translation of these mRNAs. These data suggest that AML cells regulate their translational state through m6A RNA methylation of specific transcripts to retain pluripotency properties and inhibit cell differentiation. Depletion of METTL3 in human myeloid leukemia cell lines induces cell differentiation and apoptosis and delays leukemia progression in recipient mice in vivo [43].
Consistent with METTL3, METTL14 is also highly expressed in AML cells carrying t(11q23), t(15;17), or t(8;21) and is downregulated during myeloid differentiation. Mechanistically, METTL14 exerts its oncogenic role by regulating its mRNA targets (e.g., myelocytomatosis (MYB) and MYC) through m6A modification, while the protein itself is negatively regulated by spleen focus-forming virus proviral integration oncogene (SPI1), collectively forming a SPI1/METTL14/MYB, MYC, et al. signaling axis in myelopoiesis and leukemogenesis. This highlights the critical roles of METTL14 and m6A modification in normal and malignant hematopoiesis [44].
RBM15, another component of the m6A writer complex, is linked to myeloid leukemia as well where AML initiation is mediated by a chromosomal translocation t (1;22) of RBM15 (also called OTT1) with the myelin and lymphocyte (MAL) gene [45]. Bansal and colleagues also suggested a role for m6A RNA methylation in myeloid leukemia that WTAP expression was elevated in cells derived from 32% of patients with AML and knockdown of WTAP resulted in reduced proliferation, increased differentiation, and increased apoptosis in a leukemia cell line [46].
However, upregulated m6A RNA methylation expression can result in anti-proliferation effects in some circumstances. Su and colleagues have elaborated that R-2-hydroxyglutarate (R-2HG) exhibits broad and variable anti-proliferation effects in leukemia and glioma since it increases global m6A RNA modification in the sensitive cells via suppressing FTO. The R-2HG/FTO/m6A axis regulates MYC and CCAAT/enhancer-binding protein alpha (CEBPA) gene expression and downstream pathways [47].
On the other hand, decreased m6A RNA methylation levels may exert oncogenic functions in some certain types of AML. For example, FTO, an m6A demethylase, is highly expressed in AML with t(11q23)/MLL rearrangements, t(15;17)/PML-RARA, FLT3-ITD, and/or NPM1 mutations and functions as an oncogene that promotes leukemic oncogene-mediated cell transformation and inhibits all-trans-retinoic acid (ATRA)-mediated leukemia cell differentiation. The oncogenic role of FTO is exerted through regulating expression of its target genes such as a suppressor of cytokine signaling box-2 (ASB2) and the retinoic acid receptor alpha (RARA) by reducing m6A RNA methylation levels in these mRNA transcripts. RNA stability assays have proposed that FTO-induced repression of ASB2 and RARA expression is at least in part due to the decreased stability of ASB2 and RARA mRNA transcripts upon FTO-mediated decrease of m6A RNA methylation levels. However, the reader(s) that targets m6A modification and impairs mRNA stability remains to be identified, which indicates an additional reading process controlling the stability of FTO target transcripts. Studies have [48,49,50,51] demonstrated the anti-leukemic effects of ASB2 and RARA, suggesting that the FTO/ASB2 or RARA axis likely plays a critical role in the pathogenesis of AML [52].
In breast cancer, Zhang and colleagues proposed that the exposure of breast cancer cells to hypoxia could stimulate hypoxia-inducible factor (HIF)-1α- and HIF-2α-dependent expression of ALKBH5, which induced m6A demethylation and stabilization of NANOG mRNA. Intratumoral hypoxia is a critical feature of the tumor microenvironment caused by dysregulated cell proliferation in combination with abnormal blood vessel formation and function, which drives cancer progression [53,54,55]. NANOG, as a pluripotency factor, is required for primary tumor formation and metastasis since they play a pivotal role in the maintenance and specification of cancer stem cells. This explains the correlation between decreased m6A RNA methylation level and promotion of breast cancer stem cell (BCSC) phenotype [56]. The researchers have also reported that the exposure of breast cancer cells to hypoxia induces zinc finger protein 217 (ZNF217)-dependent inhibition of m6A methylation of mRNAs encoding NANOG and Kruppel-like factor 4 (KLF4), which is another pluripotency factor that mediates BCSC speciation [57].
When it comes to glioblastoma (GBM), a study carried out by Cui et al. demonstrates that reduced mRNA m6A level is critical for maintaining glioblastoma stem-like cell (GSC) growth, self-renewal, and tumor development as downregulation of METTL3 or METTL14 expression reduces mRNA m6A levels of theirs target gene A disintegrin and metallopeptidase domain 19 (ADAM19) and promotes ADAM19 expression. ADAM19 is a metalloproteinase disintegrin gene that exhibits elevated expression in glioblastoma cells and promotes glioblastoma cell growth and invasiveness [58,59,60].
Similarly, Zhang and colleagues have identified that m6A demethylase ALKBH5 is highly expressed in GSCs and demethylates forkhead box protein M1 (FOXM1) nascent transcripts [61]. The nuclear RNA-binding protein HuR, which reportedly regulates both pre-mRNA splicing and expression [62, 63], has been shown to bind with RNAs with no m6A modification and exert stabilizing effects on its bound RNAs [24]. Recruiting HuR to the unmethylated 3′UTR makes FOXM1 nascent transcripts more stable and upregulates its expression. Accumulating evidence has shown that the transcription factor FOXM1 functions as a key cell-cycle molecule required for G1/S and G2/M transition and M-phase progression [64] and is overexpressed in GBM, playing a pivotal role in regulating GSC proliferation, self-renewal, and tumorigenicity [65,66,67]. Taken together, lower m6A level mediated by ALKBH5 in GBM helps to promote tumor progression.
m6A RNA methylation involves in cancer cell proliferation
m6A RNA methylation has been found involved in cancer cell proliferation in many kinds of cancers. There remains a coherent natural link between the oncoprotein hepatitis B virus X-interacting protein (HBXIP) and METTL3 in the development of breast cancer (BC). Mechanistically, HBXIP upregulates METTL3 in breast cancer cells by inhibiting miRNA let-7g, which downregulates the expression of METTL3 by targeting its 3′UTR. METTL3 then promotes the expression of HBXIP gene through m6A modification, forming a positive feedback loop of HBXIP/let-7g/METTL3/HBXIP, which leads to accelerated cell proliferation in breast cancer [68].
In human hepatocellular carcinoma (HCC), METTL3 has been elucidated to be frequently upregulated and contributes to HCC progression through a distinct mechanism. SOCS2, one of the members of suppressor of cytokine signaling (SOCS) family, functions as a tumor suppressor gene by negatively regulating the JAK/STAT pathway [69]. METTL3 substantially augments SOCS2 mRNA m6A modification and downregulates SOCS2 mRNA expression by degrading SOCS2 mRNA transcripts through m6A “reader” protein YTHDF2-dependent degradation pathway, suggesting a new dimension of epigenetic alteration in liver carcinogenesis [70].
m6A RNA methylation promotes cancer cell migration and tumor metastasis
The topic that m6A RNA methylation can act on cancer cell migration and tumor metastasis has now blossomed into a full-fledged field of research.
In HCC, especially in metastatic HCC, a decreased tendency of m6A modifications is observed and METTL14 is addressed to be the main factor involved in aberrant m6A modification [71]. It has been demonstrated by Alarcon and colleagues that m6A modification can mark pri-miRNAs for processing by recognizing DiGeorge critical region 8 (DGCR8) in a manner dependent on METTL3/m6A, highlighting the important role of m6A modification in RNA processing, including mRNAs and pri-miRNAs [32]. Similarly, METTL14 manipulates pri-miRNA processing by regulating the recognition and binding of DGCR8 to pri-miRNAs in an m6A-dependent manner and thus METTL14 depletion in HCC results in pri-miR126 processing arrest and reduces the expression of mature miR126 who has been identified as a metastasis suppressor, leading to advanced metastasis capability.
Besides the genetic background of cancer cells, alteration in microenvironment has emerged as a vital layer of regulating cancer metastasis. In colorectal carcinoma (CRC), serine proteases are important component in microenvironment and can selectively activate protease-activated receptor 2 (PAR2) through proteolysis of the receptor [72]. The research conducted by Yang and colleagues has unraveled that PAR2 activation decreases the level of miR-125b through NOP2/Sun RNA methyltransferase family, member 2 (NSun2)-mediated pre-miR-125b2 methylation in CRC. NSun2 is discovered to methylate precursor of miR-125b, interferes with its processing, and reduces the level of mature miR-125b [73]. The downregulation of miR-125b augments the expression of its target gene GRB2-associated-binding protein 2 (Gab2), thereby dramatically promoting cancer cell migration, which provides a novel epigenetic mechanism by which m6A modification on miRNAs promotes cancer metastasis [74].
In human pancreatic cancer (PC), the research conducted by Chen et al. showed that YTHDF2 was upregulated at both mRNA and protein levels and orchestrated proliferation and epithelial–mesenchymal transition (EMT) dichotomy [75]. Two of the main characteristics of tumor growth are uncontrolled proliferation and abnormal cell migration [76, 77]. However, cells are usually not supposed to respond to gene alterations by proliferating or migrating both at the same time, which is called migration–proliferation dichotomy [78]. YTHDF2 functions as the main regulator in this phenomenon who can promote the ability of proliferation via Akt/GSK3b/CyclinD1 pathway, while it can suppress the migration, invasion, and adhesion ability by inhibiting EMT probably via downregulation of yes-associated protein (YAP) gene. There exist two m6A RNA methylation sites in YAP mRNA transcript with one site in coding DNA sequences (CDS) and the other site in exon [35]. Therefore, it seems reasonable to come to the hypothesis that YTHDF2 might bind to m6A sites of YAP mRNA to decrease the stability of mRNA, while the direct link between YTHDF2 and YAP remains to be clarified.
m6A RNA methylation may contribute to tumor immunity
As elucidated by Li et al., m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathway. This prompts the very first attempt at studying m6A RNA methylation as key regulators of T cell differentiation, suggesting their involvement in tumor–immune system communication and importance in tumor growth and spread [79].
In other circumstances, toll-like receptors (TLRs) establish the first line of defense against besieging pathogens as the most conserved molecules of the innate immune system, which recognize pathogen-associated molecular patterns to facilitate an immune response. RNAs with m6A modification are found incapable of activating TLR3, and those with 5mC and/or m6A do not activate TLR7 or TLR8, leading to non-recognition of pathogens carrying these RNA modifications by TLR receptors (e.g., a viral nucleic acid). The methylation in m6A interferes with Watson–Crick base pairing; thus, its presence destabilizes RNA duplexes, which may explain why RNAs containing m6A modification are incapable of stimulating TLR3 [80]. Dendritic cells (DCs) exposed to such modified RNA express significantly less cytokines and activation markers than those treated with unmodified RNA. These undetected viral components may then stimulate a pathway involved in cancer development [81,82,83].
The future of m6A RNA methylation
m6A RNA methylation as diagnostic and therapeutic targets
Huang et al. have demonstrated a significant increase of m6A RNA methylation in circulating tumor cells (CTCs) compared with whole blood cells for the first time, constituting the first step for the investigation of RNA methylation in CTCs from lung cancer patients. This research outcome may facilitate uncovering the metastasis mechanism of cancers in the future. It is worth noting that early detection and characterization of upregulated m6A expression in CTCs can make contributions to monitoring and preventing the development of overt metastatic diseases [84].
Moreover, m6A RNA methylation represents brand new therapeutic targets. All the subtypes of AMLs with high levels of endogenous FTO expression, such as those carrying t(11q23), t(15;17), NPM1 mutations, and/or FLT3-ITD, are more sensitive to ATRA treatment than the other AML subtypes [52]. It is plausible to hypothesize that the proliferation of these subtypes of AML cells relies more on the FTO signaling, and thus they are more responsive to ATRA treatment, as ATRA can release the expression/function of ASB2 and RARA, two negative targets of FTO, and thereby trigger cell differentiation.
Meclofenamic acid (MA) is a US Food and Drug Administration (FDA)-approved non-steroidal anti-inflammatory drug primarily known for its inhibition of prostaglandins synthesis as well as for its more specific inhibition of cyclooxygenase enzymes and lipoxygenases [85]. Intriguingly, recently identified FTO inhibitors are also derived from dual cyclooxygenase/lipoxygenase inhibitors [86]. Mechanistic studies conducted by Huang et al. indicated that MA specifically competed with FTO binding for the m6A-containing nucleic acid and treatment of HeLa cells with the ethyl ester form of MA (MA2) led to elevated levels of m6A modification in mRNA [87]. The identification of MA as a highly selective inhibitor of FTO will certainly shed light on more research into specific inhibitors existing within our resources of known drugs. Studies of MA2 have presented possibilities for therapeutic targets in cancers. For example, as elucidated by Cui and colleagues, MA2 suppresses glioblastoma progression and prolongs lifespan of GSC-grafted animals, which suggests that m6A methylation could be a promising target for anti-glioblastoma therapy [58].
The cellular components of m6A regulatory complex themselves can also function as novel options for cancer treatment. For example, researchers have endeavored to design a peptide mimicking the critical METTL14 domain and investigate whether it has a potential therapeutic effect in HCC [71].
However, the correlation between m6A RNA methylation and clinical features including predict treatment-free survival (TFS) and overall survival (OS) in cancer still warrants further research.
Challenges of future research on m6A RNA methylation
As described above, the rapid development of m6A RNA methylation research has contributed to revealing underlying mechanisms of cancer initiation and progression. However, there remain a large number of challenges.
The first issue we should pay attention to is that whether the multiple functions proposed in cancer is actually dependent on m6A modification. Cellular components of m6A methylation complex are well established to be involved in cancer carcinogenesis and may function as tumor promoters or suppressors [56, 88,89,90,91,92,93]. It is identified by Lin and colleagues that METTL3 boosts translation of certain mRNAs including epidermal growth factor receptor (EGFR) and the Hippo pathway effector TAZ, thus promoting growth, survival, and invasion of human lung cancer cells. METTL3 activates mRNA translation through an interaction with translation initiation machinery rather than invoking m6A reader proteins downstream of nuclear METTL3, since both wild-type and catalytically inactive METTL3 promote translation when tethered to a reporter mRNA [56].
Additionally, the elevated level of the S-adenosyl methionine (SAM) donor of the methyl group in the m6A methylation process has been shown to suppress cell growth in cancer [94,95,96,97,98]. However, a direct causative link between m6A RNA methylation caused by SAM and its cancer growth-inhibitory effect remains to be established.
Secondly, we need to identify through which mechanism m6A RNA methylation is introduced. Despite the strong consensus, only a small fraction of RACH sites is detectably methylated in vivo, arguing that the sequence motif is not sufficient to determine the distribution of m6A [39]. It has been elucidated by Zhou and colleagues that miRNAs regulate m6A modification via a sequence pairing mechanism, through which miRNA sequences alter m6A modification levels via modulating the binding of METTL3 to mRNAs containing miRNA targeting sites. These results reveal how miRNAs involves in regulating the formation of m6A and partially explain the site selection mechanism of m6A [99]. Similarly, as reported by Zhang et al., the interaction between ALKBH5 and FOXM1 nascent transcripts is facilitated by a nuclear antisense lncRNA FOXM1-AS [61]. More than 70% of mammalian transcriptomes have antisense transcription [100]. It is worthwhile to determine whether there are other antisense lncRNAs that give rise to the specificity of m6A methylation so that the current view of methylation regulation can be completed.
Another mechanism by which the emplacement of m6A is regulated refers to that METTL3 can associate with chromatin and localize to the transcriptional start sites of active genes. Thereby, METTL3 induces m6A modification within the coding region of the associated mRNA transcript and enhances its translation by relieving ribosome stalling. One example is that METTL3 modulates nn n zSP1, an oncogene in several cancers which regulates c-MYC expression in this way [101], identifying a new paradigm for selecting RNAs to be modified, namely the stable recruitment of the RNA-modifying enzyme to specific genomic loci [102].
Iron metabolism also participates in m6A recruitment modulation. ALKBH5 belongs to the AlkB family of non-heme Fe(II)/a-ketoglutarate-dependent dioxygenases, whose activity is iron dependent [17]. Gene expression profiling carried out by Schonberg et al. revealed ferritin-dependent regulation of FOXM1 signaling. Given that Zhang et al. have proposed the interaction between ALKBH5 and FOXM1 nascent transcripts, it is plausible to indicate that ALKBH5 activity can be modulated by iron metabolism, consequently affecting FOXM1 expression [61, 66].
The third issue is that although there exist emerging links between m6A RNA methylation and transcript-specific fates in different cell types, it remains to be determined whether m6A is differentially recognized by distinct YTH domain proteins or whether m6A RNA methylation is used in a cell type-specific manner to either direct translational enhancement or mRNA decay.
Interestingly, METTL14 promotes stability of MYC mRNA through affecting m6A abundance mainly on the 3′-terminal exon, while FTO promotes stability of MYC mRNA through inhibition of YTHDF2-mediated RNA decay which can be attributed to decreased m6A abundance on the 5′-terminal and internal exons of MYC mRNA [52]. This phenomenon indicates that m6A modifications on different regions of the same mRNA transcript (e.g., MYC) may result in distinct fates likely due to recognition by different readers. It also leads to very open and constructive discussion that YTH domain family proteins may depend on sequence context to control their selectivity towards m6A sites and may be dynamically regulated.
However, much remains to be learned before we can understand the dynamic regulation of the co-transcriptional installation of m6A RNA methylation and its impacts on downstream mRNA processing events and cancer progression.
Conclusion
As the most prevalent modifications of mRNA, m6A RNA methylation has a deep root in modulating gene expression and is implicated in affecting many cellular processes and diseases, including cancers. Over the past few years, empowered by the availability of highly specific antibodies and the accessibility of high-throughput sequencing technologies, it has become feasible to identify the chemical basis and multiple functions of m6A RNA methylation. There remain many other kinds of RNA post-transcriptional modifications which may share similar properties with m6A RNA methylation, shedding light on the advancement of epitranscriptome.
Abbreviations
- 5mC:
-
5-Methylcytosine
- ADAM19:
-
A disintegrin and metallopeptidase domain 19
- ALKBH5:
-
alkB homolog 5
- AML:
-
Acute myeloid leukemia
- ASB2:
-
A suppressor of cytokine signaling box-2
- ATRA:
-
All-trans-retinoic acid
- BC:
-
Breast cancer
- BCL2:
-
B cell lymphoma 2
- BCSC:
-
Breast cancer stem cell
- CDS:
-
Coding DNA sequences
- CEBPA:
-
CCAAT/enhancer-binding protein alpha
- CRC:
-
Colorectal carcinoma
- CSCs:
-
Cancer stem cells
- CTCs:
-
Circulating tumor cells
- DC:
-
Dendritic cells
- DGCR8:
-
DiGeorge Critical Region 8
- DNMT:
-
DNA methyltransferases
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial–mesenchymal transition
- FDA:
-
Food and Drug Administration
- FOXM1:
-
Forkhead box protein M1
- FTO:
-
Fat mass and obesity-associated protein
- Gab2:
-
GRB2-associated-binding protein 2
- GBM:
-
Glioblastoma
- GSCs:
-
Glioblastoma stem-like cells
- HBXIP:
-
Hepatitis B virus X-interacting protein
- HCC:
-
Hepatocellular carcinoma
- HIF:
-
Hypoxia-inducible factor
- HSCs:
-
Hematopoietic stem cells
- KLF4:
-
Kruppel-like factor 4
- LC–MS/MS:
-
Liquid chromatography coupled to triple–quadrupole mass spectrometry
- m6A:
-
N6-Methyladenosine
- MA:
-
Meclofenamic acid
- MA2:
-
The ethyl ester form of MA
- MAL:
-
Myelin and lymphocyte
- METTL:
-
Methyltransferase-like
- miCLIP:
-
m6A individual-nucleotide-resolution crosslinking and immunoprecipitation
- MYB:
-
Myeloblastosis
- MYC:
-
Myelocytomatosis
- NSun2:
-
NOP2/Sun RNA methyltransferase family, member 2
- OS:
-
Overall survival
- PAR2:
-
Protease-activated receptor 2
- PC:
-
Pancreatic cancer
- PTEN:
-
Phosphatase and tensin homolog
- R-2HG:
-
R-2-hydroxyglutarate
- RARA:
-
Retinoic acid receptor alpha
- RBM15:
-
RNA-binding motif protein 15
- SAM:
-
S-Adenosyl methionine
- SOCS:
-
Suppressor of cytokine signaling
- SPI1:
-
Spleen focus-forming virus proviral integration oncogene
- SRSF:
-
Serine and arginine-rich splicing factor
- TFS:
-
Treatment-free survival
- TLRs:
-
toll-like receptors
- WTAP:
-
Wilms tumor 1-associated protein
- YAP:
-
yes-associated protein
- YTH:
-
YT521-B homology
- ZNF217:
-
Zinc finger protein 217
References
Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012;13(10):175.
Globisch D, Pearson D, Hienzsch A, Brückl T, Wagner M, Thoma I, et al. Systems-based analysis of modified tRNA bases. Angew Chem Int Ed Engl. 2011;50(41):9739–42.
Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, et al. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39(Database issue):D195–201.
He C. Grand challenge commentary: RNA epigenetics. Nat Chem Biol. 2010;6(12):863–5.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71(10):3971–5.
Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975;4(4):379–86.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Akhavan-Niaki H, Samadani AA. DNA methylation and cancer development: molecular mechanism. Cell Biochem Biophys. 2013;67(2):501–13.
Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12(2):142–8.
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–37.
Litt MD, Simpson M, Gaszner M, Allis CD, Felsenfeld G. Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science. 2001;293(5539):2453–5.
Noma K, Allis CD, Grewal SI. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science. 2001;293(5532):1150–5.
Roignant JY, Soller M. m6A in mRNA: an ancient mechanism for fine-tuning gene expression. Trends Genet. 2017;33(6):380–90.
Deng X, Su R, Feng X, Wei M, Chen J. Role of N6-methyladenosine modification in cancer. Curr Opin Genet Dev. 2017;48:1–7.
Ear J, Lin S. RNA methylation regulates hematopoietic stem and progenitor cell development. J Genet Genomics. 2017;44(10):473–4.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, et al. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540(7632):301–4.
Theler D, Dominguez C, Blatter M, Boudet J, Allain FH. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 2014;42(22):13911–9.
Wang X, He C. Reading RNA methylation codes through methyl-specific binding proteins. RNA Biol. 2014;11(6):669–72.
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10(11):927–9.
Zhu T, Roundtree IA, Wang P, Wang X, Wang L, Sun C, et al. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res. 2014;24(12):1493–6.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A -seq. Nature. 2012;485(7397):201–6.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5' UTR m(6)A promotes Cap-independent translation. Cell. 2015;163(4):999–1010.
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526(7574):591–4.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27(3):444–7.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–19.
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–308.
Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482–5.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Peer E, Rechavi G, Dominissini D. Epitranscriptomics: regulation of mRNA metabolism through modifications. Curr Opin Chem Biol. 2017;41:93–8.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40(11):5023–33.
Sugimoto Y, König J, Hussain S, Zupan B, Curk T, Frye M, et al. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biol. 2012;13(8):R67.
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–72.
Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: form, distribution, and function. Science. 2016;352(6292):1408–12.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8.
Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–91.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. 2018;22(2):191–205.e9.
Mercher T, Coniat MB, Monni R, Mauchauffe M, Nguyen KF, Gressin L, et al. Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A. 2001;98(10):5776–9.
Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Proia D, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28(5):1171–4.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. 2018;172(1–2):90–105.e23.
Glasow A, Prodromou N, Xu K, von LM ZA. Retinoids and myelomonocytic growth factors cooperatively activate RARA and induce human myeloid leukemia cell differentiation via MAP kinase pathways. Blood. 2005;105(1):341–9.
Guibal FC, Moog-Lutz C, Smolewski P, Di GY, Darzynkiewicz Z, Lutz PG, et al. ASB-2 inhibits growth and promotes commitment in myeloid leukemia cells. J Biol Chem. 2002;277(1):218–24.
Kohroki J, Fujita S, Itoh N, Yamada Y, Imai H, Yumoto N, et al. ATRA-regulated Asb-2 gene induced in differentiation of HL-60 leukemia cells. FEBS Lett. 2001;505(2):223–8.
Zhu J, Heyworth CM, Glasow A, Huang QH, Petrie K, Lanotte M, et al. Lineage restriction of the RARalpha gene expression in myeloid differentiation. Blood. 2001;98(8):2563–7.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell. 2017;31(1):127–41.
Vaupel P, Mayer A, Höckel M. Tumor hypoxia and malignant progression. Methods Enzymol. 2004;381:335–54.
Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47.
Pries AR, Höpfner M, le NF DMW, Secomb TW. The shunt problem: control of functional shunting in normal and tumour vasculature. Nat Rev Cancer. 2010;10(8):587–93.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113(14):E2047–56.
Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7(40):64527–42.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–34.
Wildeboer D, Naus S, Amy SQX, Bartsch JW, Pagenstecher A. Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J Neuropathol Exp Neurol. 2006;65(5):516–27.
Mochizuki S, Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007;98(5):621–8.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591–606.e6.
Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, et al. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43(3):340–52.
Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43(3):327–39.
Li Y, Zhang S, Huang S. FoxM1: a potential drug target for glioma. Future Oncol. 2012;8(3):223–6.
Kim SH, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. 2015;4(2):226–38.
Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28(4):441–55.
Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20(4):427–42.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9.
Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet. 2001;28(1):29–35.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase METTL3 promotes liver cancer progression through YTHDF2 dependent post-transcriptional silencing of SOCS2. Hepatol. 2017.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6-methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–43.
Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (PARs)—focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun Signal. 2013;11:86.
Yuan S, Tang H, Xing J, Fan X, Cai X, Li Q, et al. Methylation by NSun2 represses the levels and function of microRNA 125b. Mol Cell Biol. 2014;34(19):3630–41.
Yang L, Ma Y, Han W, Li W, Cui L, Zhao X, et al. Proteinase-activated receptor 2 promotes cancer cell migration through RNA methylation-mediated repression of miR-125b. J Biol Chem. 2015;290(44):26627–37.
Chen J, Sun Y, Xu X, Wang D, He J, Zhou H, et al. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16(23):2259–71.
O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417–30.
Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. 2015;5(9):932–43.
Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C, et al. A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol Biol Cell. 2010;21(13):2338–54.
Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548(7667):338–42.
Kierzek E, Kierzek R. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res. 2003;31(15):4472–80.
Chandola U, Das R, Panda B. Role of the N6-methyladenosine RNA mark in gene regulation and its implications on development and disease. Brief Funct Genomics. 2015;14(3):169–79.
Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23(2):165–75.
So EY, Ouchi T. The application of Toll like receptors for cancer therapy. Int J Biol Sci. 2010;6(7):675–81.
Huang W, Qi CB, Lv SW, Xie M, Feng YQ, Huang WH, et al. Determination of DNA and RNA methylation in circulating tumor cells by mass spectrometry. Anal Chem. 2016;88(2):1378–84.
Bartzatt R. Anti-inflammatory drugs and prediction of new structures by comparative analysis. Antiinflamm Antiallergy Agents Med Chem. 2012;11(2):151–60.
Zheng G, Cox T, Tribbey L, Wang GZ, Iacoban P, Booher ME, et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Neurosci. 2014;5(8):658–65.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43(1):373–84.
Linnebacher M, Wienck A, Boeck I, Klar E. Identification of an MSI-H tumor-specific cytotoxic T cell epitope generated by the (-1) frame of U79260(FTO). J Biomed Biotechnol. 2010;2010:841451.
Kaklamani V, Yi N, Sadim M, Siziopikou K, Zhang K, Xu Y, et al. The role of the fat mass and obesity associated gene (FTO) in breast cancer risk. BMC Med Genet. 2011;12:52.
Pierce BL, Austin MA, Ahsan H. Association study of type 2 diabetes genetic susceptibility variants and risk of pancreatic cancer: an analysis of PanScan-I data. Cancer Causes Control. 2011;22(6):877–83.
Machiela MJ, Lindström S, Allen NE, Haiman CA, Albanes D, Barricarte A, et al. Association of type 2 diabetes susceptibility variants with advanced prostate cancer risk in the Breast and Prostate Cancer Cohort Consortium. Am J Epidemiol. 2012;176(12):1121–9.
Long J, Zhang B, Signorello LB, Cai Q, Deming-Halverson S, Shrubsole MJ, et al. Evaluating genome-wide association study-identified breast cancer risk variants in African-American women. PLoS One. 2013;8(4):e58350.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–45.
Pascale RM, Simile MM, De Miglio MR, Feo F. Chemoprevention of hepatocarcinogenesis: S-adenosyl-L-methionine. Alcohol. 2002;27(3):193–8.
Pakneshan P, Szyf M, Farias-Eisner R, Rabbani SA. Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J Biol Chem. 2004;279(30):31735–44.
Guruswamy S, Swamy MV, Choi CI, Steele VE, Rao CV. S-adenosyl L-methionine inhibits azoxymethane-induced colonic aberrant crypt foci in F344 rats and suppresses human colon cancer Caco-2 cell growth in 3D culture. Int J Cancer. 2008;122(1):25–30.
Lu SC, Ramani K, Ou X, Lin M, Yu V, Ko K, et al. S-adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology. 2009;50(2):462–71.
Zhao Y, Li JS, Guo MZ, Feng BS, Zhang JP. Inhibitory effect of S-adenosylmethionine on the growth of human gastric cancer cells in vivo and in vitro. Chin J Cancer. 2010;29(8):752–60.
Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16(3):289–301.
Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309(5740):1564–6.
Geltinger C, Hörtnagel K, Polack A. TATA box and Sp1 sites mediate the activation of c-myc promoter P1 by immunoglobulin kappa enhancers. Gene Expr. 1996;6(2):113–27.
Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. 2017;552(7683):126–31.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (no. 81672896; no. 81772475), the Jiangsu Province Clinical Science and Technology Projects (Clinical Research Center, BL2012008), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (JX10231801).
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Author information
Authors and Affiliations
Contributions
YP, PM, and YL drafted the manuscript. YL and WL discussed and revised the manuscript. YS, WL, and PM designed the research and drafted the manuscript. All authors read and approved final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pan, Y., Ma, P., Liu, Y. et al. Multiple functions of m6A RNA methylation in cancer. J Hematol Oncol 11, 48 (2018). https://doi.org/10.1186/s13045-018-0590-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-018-0590-8