
Abstract
Inhibitory interneurons are critical for maintaining the excitatory/inhibitory balance. During the development cortical interneurons originate from the ganglionic eminence and arrive at the dorsal cortex through two tangential migration routes. However, the mechanisms underlying the development of cortical interneurons remain unclear. 3-Phosphoinositide-dependent protein kinase-1 (PDK1) has been shown to be involved in a variety of biological processes, including cell proliferation and migration, and plays an important role in the neurogenesis of cortical excitatory neurons. However, the function of PDK1 in interneurons is still unclear. Here, we reported that the disruption of Pdk1 in the subpallium achieved by crossing the Dlx5/6-Cre-IRES-EGFP line with Pdk1fl/fl mice led to the severely increased apoptosis of immature interneurons, subsequently resulting in a remarkable reduction in cortical interneurons. However, the tangential migration, progenitor pools and cell proliferation were not affected by the disruption of Pdk1. We further found the activity of AKT-GSK3β signaling pathway was decreased after Pdk1 deletion, suggesting it might be involved in the regulation of the survival of cortical interneurons. These results provide new insights into the function of PDK1 in the development of the telencephalon.
Similar content being viewed by others

Introduction
The cognitive function of the cerebral cortex relies on highly organized neural circuits that consist of excitatory neurons and γ-aminobutyric acid (GABA) ergic inhibitory interneurons. Despite comprising only 20–30% of the cortical neurons, interneurons play crucial roles in maintaining the balance between excitation and inhibition (E/I) and modulating cortical function [1,2,3,4,5]. Previous studies have demonstrated that cortical interneuron abnormalities are closely associated with a variety of neuropsychiatric disorders, such as schizophrenia, epilepsy, and autism spectrum disorders [6,7,8,9]. During the development cortical interneurons originate from the ganglionic eminence in the ventral telencephalon [10,11,12]. Somatostatin (SST)- and parvalbumin (PV)-expressing interneurons are derived from the medial ganglionic eminence (MGE) [11], while the caudal ganglionic eminence (CGE) produces PROX1-expressing interneurons including vasoactive intestinal peptide (VIP+) and Reelin+ interneurons [13, 14]. Newborn interneurons migrate to the dorsal cortex following two well-defined tangential routes: a superficial route in the marginal zone (MZ) and another route in the intermediate zone/subventricular zones (SVZ) [15, 16]. After reaching the dorsal cortex, interneurons subsequently undergo radial migration to populate to the final destinations and establish connections with their targets, forming functional neural circuits [17, 18]. However, the mechanisms underlying the development of cortical interneurons are still poorly understood.
The serine/threonine kinase 3-Phosphoinositide-dependent protein kinase-1 (PDK1) belongs to the cAMP-dependent, cGMP-dependent and protein kinase C (AGC) kinase family [19,20,21,22]. PDK1, the key downstream effector of PI3K signaling that is activated by various extracellular stimuli, is involved in many biological processes, such as cell proliferation and growth, neural differentiation, cell migration, cell polarization and cell survival [23,24,25,26,27,28]. Conditional deletion of Pdk1 results in increased apoptosis and migration defects in excitatory neurons [29, 30]. Previously, we reported that PDK1 controls the transition of apical progenitors to basal progenitors through regulating asymmetric cell division, causing subsequent increased cell proliferation and neural output in the developing cortex [31]. We also found that the loss of PDK1 in the developing dentate gyrus causes unbalanced cell proliferation and neurogenesis [32]. A growing amount of evidence suggests that PDK1 is upregulated in cancer cells and required for cell survival [33,34,35]. All these studies demonstrate that PDK1 plays different roles during different biological processes. Previously, Oishi K et al. reported that PDK1 promotes the differentiation of neuronal progenitor cells (NPCs) into GABAergic interneurons but not glutamatergic neurons [36]. However, the precise role of PDK1 in cortical interneuron development is not well understood. In this study, we found that the conditional deletion of Pdk1 in the subpallium resulted in the severely increased apoptosis of immature interneurons and subsequently led to a significantly decreased number of cortical interneurons. No obvious defects in tangential migration and cell proliferation were detected. We further demonstrated that PDK1 regulates the survival of the developing cortical interneurons.
Materials and methods
Animals
The Pdk1fl/fl mice [37] were obtained from Dr. Zhongzhou Yang (Model Animal Research Center of Nanjing University) and Dr. Jun Gao (Nanjing Medical University at Nanjing). Conditional disruption of Pdk1 in the subpallium was achieved by crossing the Dlx5/6-Cre-IRES-EGFP line [38] with Pdk1fl/fl mice. The Dlx5/6-Cre-IRES-EGFP; Pdk1fl/fl mice are referred to as the Pdk1 cKO mice. The Pdk1fl/fl and Dlx5/6-Cre-IRES-EGFP; Pdk1fl/+ mice are referred to as control mice. The primer pairs 5′- ATCCCAAGTTAC TG AGTTGTGTTGGAA G-3′ and 5′- TGTGGACAAACAGCAATGAACAT ACACGC-3′, which amplified a 202-bp band for the control and a 236-bp band for Pdk1 cKO were used. All mice were maintained on a C57/B6 background and bred in the animal facility of Southeast University (Nanjing, Jiangsu, China). The day of vaginal plug detection was defined as embryonic day (E) 0.5, and the day of birth was defined as postnatal day 0 (P0). All experiments were performed according to the guidelines approved by Southeast University.
Quantitative real-time PCR
Total RNA from the GE at E16.5 was isolated using an RNeasy Plus mini kit for RNA isolation (Qiagen, Cat. No. 74134) according to the manufacturer’s instructions [39]. Each sample was reverse transcribed using a PrimeScript™ RT reagent kit (TakaRa, Cat. No. RR047A). Quantitative real-time PCR reactions was performed using SYBR Green fluorescent master mix (Roche, Cat. No. 04707516001) on a StepOne-Plus Real-Time PCR System (Applied Biosystems). Relative gene expression was compared between samples after the expression was normalized to the expression of the most reliable endogenous gene (glyceraldehyde 3-phosphate dehydrogenase, Gapdh). At least three brain pairs were obtained from 3 different litters and used for analysis. Statistical analysis was performed using Student’s t-test. The primers used are listed in Table 1.
Western blotting
GEs were collected at E16.5 and prepared as described previously [32]. Statistical analysis was performed using Student’s t-test. The PDK1 antibody is listed in Table 2.
Immunostaining
Brains were fixed by transcardiac perfusion with PBS followed by cold 4% paraformaldehyde (PFA). The brains were then postfixed overnight, cryoprotected in 30% sucrose at 4 °C for 12 to 16 h, and then embedded in optimum cutting temperature (OCT) compound. Brains at embryonic stages were coronally sectioned to 12 μm, while those at postnatal stages were coronally sectioned to 25 μm using a Leica CM 1950 cryostat. Immunohistochemical analysis was then performed as previously reported [40]. The antibodies used are listed in Table 2. DAPI was purchased from Sigma-Aldrich (D9564).
In situ hybridization
Brains were harvested, fixed and hybridized as previously described [41, 42]. Information regarding the primers used to generate probes for Sst, Pv, and CyclinD2 are listed in Table 3.
BrdU administration
5-Bromo-2-deoxyUridine (BrdU, Sigma-Aldrich) was dissolved in physiological saline at a concentration of 10 mg/ml. A single intraperitoneal injection of BrdU (50 mg/kg) was administered to pregnant mice at E12.5, E14.5, and E16.5. Then, embryonic brains were then harvested 0.5 h after the injection for acute labeling analysis.
Microscopy and image analysis
The tissue sections were viewed under a confocal microscope (Olympus FV1000), and images were collected and analyzed with FV10-ASW image analysis software.
Statistical analysis
Student’s t-tests were performed to compare changes in the developing telencephlon in absolute and relative percentages of cell numbers. To measure GFP+, SST+, PV+, and PROX1+ cells at P6 and P15, and PROX1+ cells at E18.5 brains, a minimum of 3 successive coronal sections were examined at the level at which the hippocampal structure is almost horizontal. Cells within a radial column of 200 μm width and spanning from the pial surface to the white matter in the somatosensory cortex were counted. For the measurement of GFP+ cells, cells in the whole E12.5 dorsal cortex, a 150 μm × 200 μm area in the dorsal cortex at E13.5 and a 150 μm × 600 μm area at E18.5 were counted. For SST+ cells, the whole ventral telencephalon at E14.5 and an area with 800 μm × 600 μm in the dorsal cortex at E18.5 were examined, respectively. For tangential migration ability analysis, a minimum of 3 successive coronal sections at the level at which the hippocampal structure is almost horizontal were selected at E18.5, GFP+ cells were counted in a 150 μm × 600 μm area near the pallia-subpallium boundery in the ventral telencephalon and an 150 μm × 600 μm area in the dorsal cortex were examined. Similar dorsal/ventral ratio analysis was performed at E13.5 with a 150 μm × 200 μm area examined. To measure cell proliferation, PH3+, BrdU+, and Ki67+ cells in the MGE were counted at E12.5 (in a 100 μm × 500 μm area), E14.5150 μm × 600 μm), and E16.5 (150 μm × 320 μm), respectively. As for cell apoptosis, at P6 a minimum of 3 successive coronal sections at the level at which the hippocampal structure is almost horizontal were selected, caspase3+ cells were then counted in the whole dorsal cortex. At embryonic stages of E12.5, E14.5 and E16.5, caspase3+ cells were counted in the whole subpallium. The caspase3+ cells in controls were normalized to 100%. All quantitative results are shown as the mean ± standard error of the mean (SEM). Student’s t-tests were used to analyze the level of Pdk1 transcription at E16.5 (Fig. 1a), the total number of GFP+ cortical interneurons at P6 (Fig. 1e), the number of SST+ interneurons at P6 (Fig. 1h), the number of PV+ interneurons at P15 (Fig. 1k), the number of PROX1+ interneurons at P6 and E18.5 (Fig. 1 and 2n and s), and the relative protein expression levels of AKT-GSK3β pathway (Fig. 6b、6D、6F、6H 、6 J and 6 L). Significance for comparisons was defined by Student’s t-tests as follows: *P < 0.05, **P < 0.01, and ***P < 0.001. Values of P < 0.05 indicated statistically significant differences. Multiple t-tests were used to analyze the number of GFP+ interneurons at E12.5, E13.5 and E18.5 (Fig. 2t), the dorsal/ventral ratio of the numbers of GFP+ interneurons at E13.5 and E18.5 (Fig. 2u), the number of SST+ interneurons at E14.5 and E18.5 (Fig. 2v), the number of PH3+, BrdU+ and Ki67+ cells at E12.5, E14.5 and E16.5 (Fig. 4s、4T and 4 U), and the number of Caspase-3+ cells at E12.5, E14.5, E16.5 and P6 (Fig. 5i). Bonferroni correction was applied to multiple t-tests in this study. Significance for comparisons was defined by Multiple t-tests as follows: *P < 0.0332, **P < 0.0021, ***P < 0.0002, and ****P < 0.0001. Values of P < 0.05/n (n = t-tests were done times) indicated statistically significant differences. At least 3 paired brains from 3 different litters were used for each analysis.

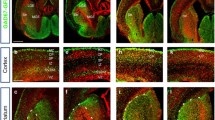
The reduction in cortical interneurons in the postnatal cortex after Pdk1 ablation. (a) Real-time PCR showed that the level of Pdk1 transcription was decreased in the Pdk1 cKO subpallium at E16.5 (Ctrl: N = 4, Pdk1 cKO: N = 4, P = 0.004, Student’s t-test, with alpha = 0.05). Gapdh was used as an internal control. (b) Western blot analysis revealed decreased levels of PDK1 in the Pdk1 cKO subpallium at E16.5 (N = 4). GAPDH was used as an internal control. (c-d’) Immunostaining for GFP showed that the total number of cortical interneurons in the Pdk1 cKO cortex was decreased at P6. (e) Statistical analysis of GFP+ interneurons (Ctrl: N = 3, Pdk1 cKO: N = 3, P = 0.0013, Student’s t-test, with alpha = 0.05). (f-g’) In situ hybridization of SST showed a reduction in SST+ interneurons in the Pdk1 cKO cortex at P6. (h) Statistical analysis of SST+ interneurons (Ctrl: N = 3, Pdk1 cKO: N = 3, P = 0.023, Student’s t-test, with alpha = 0.05). (i-j’) In situ hybridization of PV at P15. (k) Statistical analysis of PV+ interneurons (Ctrl: N = 4, Pdk1 cKO: N = 4, P = 0.0013, Student’s t-test, with alpha = 0.05). (l-m’) Immunostaining for PROX1 revealed that the number of CGE-derived cortical interneurons in the Pdk1 cKO cortex was decreased at P6. (n) Statistical analysis of PROX1+ interneurons (Ctrl: N = 3, Pdk1 cKO: N = 3, P = 0.0013, Student’s t-test, with alpha = 0.05). The data are presented as the mean ± SEM. Scale bar, 100 μm

Embryonic loss of cortical interneurons with unchanged tangential migration after Pdk1 deletion. (a-d”) Immunofluorescence for GFP in coronal sections showed comparable distribution patterns and numbers of cortical interneurons at E12.5 and E13.5 between Pdk1 cKO and control mice. (e-f) Immunofluorescence for GFP revealed that the number of GFP+ cortical interneurons in the SVZ was reduced in the Pdk1 cKO cortex at E14.5. (g-j”) The number of GFP+ cortical interneurons in the Pdk1 cKO cortex gradually decreased from E16.5 to E18.5. (t) Statistical analysis of GFP+ interneurons (E12.5 Ctrl: N = 3, Pdk1 cKO: N = 3, P = 0.9912; E13.5 Ctrl: N = 4, Pdk1 cKO: N = 4, P = 0.9089; E18.5 Ctrl: N = 3, Pdk1 cKO: N = 3, P = 0.00034, Multiple t-test, with alpha = 0.05/3 = 0.0167). (u) Statistical analysis the dorsal/ventral ratio of the numbers of GFP+ interneurons between Pdk1 cKO and control mice at E13.5 and E18.5 (E13.5 Ctrl: N = 4, Pdk1 cKO: N = 4, P = 0.1945; E18.5 Ctrl: N = 3, Pdk1 cKO: N = 3, P = 0.5371, Multiple t-test, with alpha = 0.05/2 = 0.025). (k-n) In situ hybridization showed that the number of SST+ interneurons in the Pdk1 cKO subpallium gradually decreased from E14.5 to E16.5. (o-p′) The number of SST+ interneurons in the Pdk1 cKO cortex was decreased at E18.5. (V) Statistical analysis of SST+ interneurons (E14.5 Ctrl: N = 3, Pdk1 cKO: N = 3, P = 0.00045; E18.5 Ctrl: N = 3, Pdk1 cKO: N = 3, P = 0.00016, Multiple t-test, with alpha = 0.05/2 = 0.025). (q-r’) Immunofluorescence for PROX1 showed that the number of PROX1+ cortical interneurons was decreased in the Pdk1 cKO cortex at E18.5. (s) Statistical analysis of PROX1+ interneurons (Ctrl: N = 3; Pdk1 cKO: N = 3; P = 0.0018, Student’s t-test, with alpha = 0.05). The data are presented as the mean ± SEM. Scale bar, 100 μm
Results
Loss of Pdk1 results in a severe reduction in cortical interneurons at postnatal stages
As a number of AGC protein kinase family members, PDK1 has been reported to be ubiquitously expressed [26]. To investigate its role in the development of cortical interneurons, the Pdk1fl/fl line was crossed with Dlx5/6-Cre-IRES-EGFP mice [38] to conditionally delete Pdk1 in the subpallium. Quantitative real-time PCR showed an approximate 80% deletion efficiency in the GE (Fig. 1a). As for western blot, the PDK1 antibody detected total three including one stronger and two weaker bands between the range of 55kD and 70kD. The stronger band showed a significant reduction in the Pdk1 cKO subpallium compared to control (Fig. 1b). We have tested several other commercial PDK1 antibodies but found they do not work well especially at developmental stages with nonspecific staining in Pdk1 cKO tissues. Here considering the Dlx5/6-Cre is a powerful tool to delete genes in SVZ progenitors in the ventral telencephalon [14], and the Pdk1fl/fl line has been employed to disrupt Pdk1 in several studies [30,31,32], combined with the result of Quantitative real-time PCR, Pdk1 could be considered to be efficiently deleted. Most Pdk1 cKO mice died shortly after birth, with a very small number surviving to P15. We next quantified the total number of GFP+ cells, which represented the cortical interneurons, in the postnatal somatosensory cortex. As shown in Fig. 1c-d’, a large number of GFP+ interneurons populated to the cortex in control mice, while the number was significantly decreased in the Pdk1 cKO cortex. Statistical analysis showed an approximately 28.2% reduction in GFP+ interneurons in the Pdk1 cKO cortex compared to the control cortex (Fig. 1e).
To further examine the changes in distinct subgroups of cortical interneurons, we performed in situ hybridization to detect Somatostatin (SST)- and parvalbumin (PV)-expressing cells, which represented MGE-derived interneurons, and immunostaining for PROX1, a specific marker of CGE-derived interneurons. At P6, many SST+ interneurons were observed in the control cortex, which exhibited a superficial layerless/deep-layermore distribution pattern (Fig. 1f’). In the Pdk1 cKO cortex, this distribution pattern seemed unchanged; however, the number of SST+ interneurons was obviously decreased ((Fig. 1g’). There was a 32% decrease compared to the control cortex (Fig. 1h). Furthermore, we observed a 67% decrease in the number of PV+ interneurons at P15 (Fig. 1i-k). As shown by PROX1 immunostaining, the control cortex displayed a superficial layermore/deep-layerless distribution pattern of CGE-derived interneurons that was consistent with previously reported results. In the Pdk1 cKO cortex, only a few PROX1+ interneurons were detected. The percentage was reduced to 36.2% compared to that of control mice (Fig. 1l-n). These results suggested that the number of cortical interneurons, including both MGE- and CGE-derived interneurons, was significantly decreased after Pdk1 ablation.
Embryonic cell loss in the cortex with unchanged tangential migration after Pdk1 deletion
The migration of cortical interneurons is initiated as early as E12.5. Once exiting the cell cycle, newborn cortical interneurons tangentially migrate towards the dorsal cortex [43]. Considering the important role of PDK1 in the migration of cortical excitatory neurons and cancer cells, we next examined whether the reduction of cortical interneurons in the Pdk1 cKO cortex was caused by defects of tangential migration. At E12.5, a small portion of GFP+ cortical interneurons in Dlx5/6-Cre-EGFP; Pdk1fl/+ control mice had already migrated across the pallial-subpallial boundary (PSB) and entered the dorsal cortex (Fig. 2a). The same result was observed in Pdk1 cKO mice, and the distribution patterns and the numbers of GFP+ were comparable (Fig. 2b and t). Similar to the results observed at E12.5, more GFP+ cortical interneurons migrated into the developing cortical plate, and no significant differences were observed at E13.5 (Fig. 2c-d’ and 2 T). These results indicated that the number of newborn cortical interneurons was not altered at early stages. From E14.5 onwards, we observed that interneurons that in the cortical plate and in the migratory routes were gradually decreased (Fig. 2e-j’). At E14.5, this reduction seemed subtle (Fig. 2e and f), while at E16.5, the reduction became more obvious (Fig. 2g and h). Till E18.5 the number of cortical interneurons was significantly decreased (Fig. 2i-j’ and t). To further address whether Pdk1 deletion had effects on tangential migration ability, we quantified the dorsal/ventral ratio of the number of GFP+ Interneurons at E13.5 and E18.5, respectively. No significant differences were detected between Pdk1 cKO and control mice (Fig. 2c-d” and i-j” and u). Combined with unchanged numbers of GFP+ interneurons in the dorsal cortex at early stages, these results indicated that Pdk1 deletion had no effects on tangential migration ability.
To further address whether Pdk1 deletion affects distinct subtypes of cortical interneurons derived from the MGE and CGE, respectively, we performed in situ hybridization to detect SST and immunostaining to detect PROX1. As shown in Fig. 2k-p’, the number of MGE-derived SST+ interneurons in the Pdk1 cKO subpallium was obviously decreased from E14.5 onwards. At E18.5, the number of SST+ interneurons was further reduced in the Pdk1 cKO cortex (Fig. 2o-p’ and v). Notably, no SST+ interneurons accumulated in the ventral telencephalon (Fig. 2k-p), further demonstrating that Pdk1 is not required for the tangential migration. Next, we determined the number of CGE-derived PROX1+ interneurons in the cortical plate at E18.5, and similar to SST+ interneurons, a significant decrease was observed (Fig. 2q-r’ and s). Taken together, these results show that cortical interneuron loss occurred at early embryonic stages but Pdk1 ablation had no obvious effects on the tangential migration.
The progenitor Pool in the ventral telencephalon was not altered
Previously, we have showed that loss of PDK1 leads to a remarkable expansion of progenitor pools in the dorsal cortex and that PDK1 controls the transition from apical progenitors to basal progenitors during cortical excitatory neurogenesis [31]. To elucidate whether PDK1 plays a similar role in the development of interneurons, we examined progenitor pools in the ventral telencephalon. The transcription factor Ascl1 is highly expressed in a subset of progenitors in the VZ and gradually down-regulated in the SVZ in the developing ventral telencephalon [44]. Loss of Pdk1 has been shown to lead to a decreased level of Ascl1 and cause an abnormal differentiation of interneurons [36]. Ascl1 mutant mice exhibit a severe loss of progenitors, particularly in the MGE [45]. Here, no obvious differences in progenitors were observed in Pdk1 cKO mice at E13.5 (Fig. 3a and b). Similar results were obtained at E14.5 (Fig. 3c and d) and E16.5 (Fig. 3e and f). These results indicated that loss of Pdk1 had no effects on the ASCL1 + progenitor pool in the ventral telencephalon.

The progenitor pool in the ventral telencephalon was not altered. (a-f) Immunofluorescence for ASCL1 in coronal sections showed that the ASCL1+ progenitor pool in the VZ was unaffected from E12.5 to E16.5. (g-l) Immunofluorescence for OLIG2 showed a comparable number of OLIG2+ progenitors between control and Pdk1 cKO mice from E12.5 to E16.5. (m-r) In situ hybridization for CyclinD2 showed that the SVZ progenitor pool in the Pdk1 cKO subpallium was normal from E12.5 to E16.5. Scale bar, 100 μm
Olig2 is expressed in most progenitors in the VZ and particularly strongly expressed in the MGE [46, 47]. OLIG2-expressing progenitors give rise to both cortical interneurons and oligodendrocytes [47, 48]. At E13.5, there were no significant differences in the GE between Pdk1 cKO and control mice (Fig. 3g and h). Similarly, the staining was comparable at E14.5 (Fig. 3i and j) and E16.5 (Fig. 3k and l), suggesting an unchanged VZ progenitor pool.
Next, we examined the SVZ progenitor pool. Previous studies have demonstrated that CyclinD2 is strongly expressed in intermediate progenitor cells in the SVZ of the ventral telencephalon [49, 50]. CyclinD2 promotes transit-amplifying divisions in the SVZ and ensures the proper output of at least a subset of PV+ interneurons [51]. We did not find significant differences in CyclinD2-expressing progenitors in Pdk1 cKO mice (Fig. 3m-r), suggesting that the intermediate progenitor pool was normal. Taken together, these results show that in contrast to the role of PDK1 in the dorsal cortex, loss of Pdk1 had no effects on progenitor pools in the developing ventral telencephalon.
Normal cell proliferation in the ventral telencephalon of Pdk1 cKO mice
PDK1 is highly expressed in cancer cells and promotes oncogenesis by regulating proliferation and survival [34]. Inhibition of the PDK1/AKT/GSK3 pathway causes cell proliferation defects by inducing cell cycle arrest (at G0/G1 and G2/M phases) [52,53,54]. We next evaluated whether cell proliferation contributes to the reduction in cortical interneurons observed in Pdk1 cKO mice. The generation of MGE-derived SST+ interneurons peaks at E12.5, with PV+ interneurons peaking at E14.5 [55,56,57], while GCE-derived interneuron production appears to peaks at E16.5 [13, 58]. Thus, three time points, E12.5, E14.5, and E16.5, were chosen to assess cell proliferation. Immunostaining for phospho-histone-3 (PH3) was carried out to quantify progenitors in M phase of the cell cycle. As shown in Fig. 4a and b, at E12.5 PH3+ progenitors were distributed in the VZ and SVZ of the GE, and no obvious differences were observed. Cell counting showed that total numbers of PH3+ cells in the VZ and SVZ of the MGE were comparable (Fig. 4s). Similar results were obtained at E14.5 (Fig. 4c-d, and s) and E16.5 (Fig. 4e-f’, and s). We also measured progenitors in S phase by acute BrdU labeling. No significant differences were detected at E12.5 (Fig. 4g-h, and t), E14.5 (Fig. 4i-j, and t), or E16.5 (Fig. 4k-l’, and t). To further confirm the proliferation of progenitors, we next performed immunostaining of anti- Ki67. Consistent with the results of PH3 staining and BrdU-labeling, no obvious differences were identified between control and Pdk1 cKO mice (Fig. 4m-r’ and u). Taken together, these results indicated that a lack of Pdk1 has no effects on the proliferation of progenitors in the ventral telencephalon during cortical interneuron neurogenesis.

Normal cell proliferation in the Pdk1 cKO subpallium. (a-f′) Immunofluorescence for PH3 showed that the number of PH3-labeled M phase cells was unaffected in the MGE of Pdk1 cKO mice compared with control at E12.5, E14.5, and E16.5. (s) Statistical analysis of PH3+ cells (E12.5 Pdk1 cKO: N = 3, Ctrl: N = 3, P = 0.7922; E14.5 Pdk1 cKO: N = 3, Ctrl: N = 3, P > 0.9999; E16.5 Pdk1 cKO: N = 3, Ctrl: N = 3, P = 0.5767, Multiple t-test, with alpha = 0.05/3 = 0.0167). (g-l’) Immunofluorescence for BrdU showed that the number of S phase cells within the Pdk1 cKO MGE was similar to that in the control MGE at E12.5, E14.5, and E16.5. (t) Statistical analysis of BrdU+ cells (E12.5 Pdk1 cKO: N = 3, Ctrl: N = 3, P = 0.9502; E14.5 Pdk1 cKO: N = 3, Ctrl: N = 3, P = 0.9265; E16.5 Pdk1 cKO: N = 5, Ctrl: N = 5, P = 0.7189, Multiple t-test, with alpha = 0.05/3 = 0.0167). (m-r’) Immunofluorescence for Ki67 revealed comparable numbers of Ki67+ cells between Pdk1 cKO and control mice at E12.5, E14.5, and E16.5. (u) Statistical analysis of Ki67+ cells (E12.5 Pdk1 cKO: N = 3, Ctrl: N = 3, P = 0.9954; E14.5 Pdk1 cKO: N = 4, Ctrl: N = 4, P = 0.8033; E16.5 Pdk1 cKO: N = 6, Ctrl: N = 6, P = 0.9003, Multiple t-test, with alpha = 0.05/3 = 0.0167). The data are presented as the mean ± SEM. Scale bar, 100 μm
PDK1 regulates the survival of the developing cortical interneurons
The unchanged tangential migration and cell proliferation in Pdk1 cKO mice led us to further explore whether the reduction in cortical interneurons was due to increased apoptosis. To examine this hypothesis, we performed immunostaining for the cleaved (active) form of Caspase-3. At E12.5 in both control and Pdk1 cKO mice, only a few Caspase-3+ cells were distributed in the GE (Fig. 5a-b’). Statistical analysis showed no remarkable differences (Fig. 5i), suggesting that cell apoptosis was not increased at this time point, coincident with the unchanged number of GFP+ interneurons at E12.5 (Fig. 2a-b and s). In contrast, we observed a significant increase in apoptosis in the GE at E14.5 (Fig. 5c-d’). Compared with control mice, the number of Caspase-3+ cells was increased by 297.3% (Fig. 5i). Similarly, at E16.5, the number of Caspase-3+ cells was significantly increased as well (Fig. 5e-f’ and 5I). We also observed a remarkable increase in cell apoptosis in the postnatal neocortex (Fig. 5g-h’ and i). These results thus demonstrated that Pdk1 ablation resulted in severely increased apoptosis and subsequently led to a reduction in cortical interneurons. PDK1 plays a critical role in regulating the survival of cortical interneurons. Since Pdk1 was deleted in the SVZ progenitors in the ventral telencephalon in this study, the possibility that apoptotic cells includes a very small number of progenitors in the SVZ can’t be excluded.

The loss of Pdk1 resulted in increased cell apoptosis. (a-b′) Immunofluorescence showing that the number of Caspase-3+ cells was no differences in the subpallium of Pdk1 cKO and control mice at E12.5. (c-d’) The number of Caspase-3+ cells was significantly increased in the Pdk1 cKO subpallium compared with control at E14.5. (e-f′) Increased number of Caspase-3+ cells in the Pdk1 cKO subpallium compared to control at E16.5. (g-h′) Immunofluorescence for Caspase-3 showed that the number of Caspase-3+ cells was increased in the cortex of Pdk1 cKO mice compared with control mice at P6. (i) Statistical analysis of Caspase-3+ cells (E12.5 Pdk1 cKO: N = 3; Ctrl: N = 3; P = 0.9231; E14.5 Pdk1 cKO: N = 3; Ctrl: N = 3; P = 0.0048; E16.5 Pdk1 cKO: N = 3; Ctrl: N = 3; P = 0.0114; P6 Pdk1 cKO: N = 3; Ctrl: N = 3; P = 0.00004, Multiple t-test, with alpha = 0.05/4 = 0.0125). The data are presented as the mean ± SEM. Scale bar, 100 μm
Decreased activity of AKT-GSK3β signaling pathway after Pdk1 deletion
The PDK1-AKT pathway is important for cell survival [59,60,61,62,63]. Previous studies have demonstrated that the phosphorylation of AKT at Thr308 by PDK1 is essential for its activation [64, 65]. Loss of Pdk1 results in decreased AKT activity, consequently leading to an increased apoptosis of excitatory neurons at postnatal rather than embryonic stages [30]. Here, we first examined total AKT level in the ventral telencephalon and observed no significant differences at E16.5 (Fig. 6a and b); however, the phosphorylation level of AKT at Thr308 (p-AKTThr308) was significantly decreased (Fig. 6c and d), reflecting a decreased activity after Pdk1 ablation. In contrast, the relative level of AKT phosphorylation at Ser473 (p-AKTSer473) was increased (Fig. 6e and f), indicating a compensatory effect for the decreased level of p-AKTThr308. Studies have shown that active GSK3β plays a proapoptosis role [52, 54, 65,66,67], and that GSK3β can be inactivated when it is phosphorylated at Ser9 by AKT [68, 69]. We found that although the total level of GSK3β was unchanged (Fig. 6g and h), the level of GSK3β phosphorylated at Ser9 (p-GSK3βSer9) was significantly decreased (Fig. 6i and j), consistent with the decrease of p-AKTThr308. Phosphatase and tensin homolog (PTEN) serves as an inhibitor of the PI3K-PDK1-AKT signaling [70, 71]. It has been reported that the loss of Pten results in the preferential apoptosis of SST+ interneurons [8]. However, we did not detect significant changes in PTEN after Pdk1 ablation (Fig. 6k and l). In summary, these results indicated that the loss of Pdk1 resulted in a decreased AKT activity accompanied by a decreased level of inactivated GSK3βSer9, ultimately leading to an increase in total GSK3β activity. Thus, the AKT-GSK3β signaling pathway may be involved in the regulation of the survival of cortical interneurons.

Decreased activity of AKT-GSK3β signaling pathway after Pdk1 deletion. Western blotting analysis of the AKT-GSK3β signaling pathway in the subpallium at E16.5. (a) The relative expression levels of total AKT were no differences between Pdk1 cKO and control mice. (b) Statistical analysis of total AKT protein expression levels (Pdk1 cKO: N = 4; Ctrl: N = 4; P = 0.1186, Student’s t-test, with alpha = 0.05). (c) The relative expression levels of p-AKTThr308 was significantly reduced. (d) Statistical analysis of p-AKTThr308 protein expression levels (Pdk1 cKO: N = 5; Ctrl: N = 3; P = 0.0027, Student’s t-test, with alpha = 0.05). (e) While p-AKTSer473 was obviously increased in the Pdk1 cKO subpallium compare to control. (f) Statistical analysis of p-AKTSer473 protein expression levels (Pdk1 cKO: N = 4; Ctrl: N = 4; P = 0.0153, Student’s t-test, with alpha = 0.05). (g) There were no significant differences in the relative expression levels of total GSK3β between Pdk1 cKO and control mice. (h) Statistical analysis of total GSK3β protein expression levels (Pdk1 cKO: N = 4; Ctrl: N = 4; P = 0.5900, Student’s t-test, with alpha = 0.05). (i) The relative expression levels of p-GSK3βSer9 was significantly decreased in the Pdk1 cKO subpallium compared to control. (j) Statistical analysis of p-GSK3βSer9 protein expression levels (Pdk1 cKO: N = 4; Ctrl: N = 4; P = 0.0273, Student’s t-test, with alpha = 0.05). (k) There were no significant differences in the relative expression levels of PTEN between Pdk1 cKO and control mice. (l) Statistical analysis of PTEN protein expression levels (Pdk1 cKO: N = 4; Ctrl: N = 4; P = 0.8366, Student’s t-test, with alpha = 0.05). GAPDH was used as an internal control
Discussion
The PDK1-AKT signaling pathway regulates a variety of biological processes, including cell proliferation, differentiation, cell survival, and cell metabolism, in many tissues [72]. By inactivating GSK3β through phosphorylation that subsequently stabilizing the protein level of CyclinD1, PDK1-AKT pathway regulates cell proliferation in mouse fibroblasts [72, 73]. In the olfactory bulb PDK1 promotes stem cells to differentiate into neurons and astrocytes [74]. In Drosophila PDK1-AKT pathway modulates the function of multiple regulatory proteins, such as S6K, resulting in stimulation of glucose uptake and storage, and moreover controls protein synthesis and storage of amino acids [21, 75, 76]. Our previous studies have shown that PDK1 plays essential roles in the development of the neocortex and the dentate gyrus. In the dorsal cortex, PDK1 regulates the transitional period between apical progenitors and basal progenitors through mediating asymmetric division, further controlling neuronal output [31]. In the developing dentate gyrus, PDK1 is required to maintain a balance between cell proliferation and neurogenesis [32]. PDK1 is also involved in the radial migration of cortical projection neurons [29]. Previously, it was shown that PDK1 promotes the differentiation of progenitors into GABAergic interneurons rather than excitatory neurons [36]. However, the detailed function of PDK1 during interneuron development remains unclear. In this study, we report that PDK1 is required for the survival of cortical interneurons but has no obvious effects on cell proliferation and tangential migration. Deletion of Pdk1 in the subpallium leads to decreased AKT signaling activity, which may be involved in the survival of the developing cortical interneurons.
PDK1 serves as an antiapoptotic factor in a variety of cell types, such as cancer cells, endothelial cells, and skeletal muscle cells [28, 61, 62, 77,78,79,80,81]. Recently, it was reported that the ablation of Pdk1 in the dorsal telencephalon leads to the increased apoptosis of cortical excitatory neurons at postnatal rather than embryonic stages [30, 31]. Here, we found that during cortical interneuron development apoptosis occurred in the embryonic stages and lasted to the postnatal stages, resulting in a dramatical reduction in cortical interneurons, including MGE-derived SST+ and PV+ interneurons and the CGE-derived PROX1+ subtypes. We further demonstrated that AKT activity was significantly decreased, as reflected by the level of p-AKTThr308. Subsequently, the activity of GSK3β, a key downstream effector of the PDK1-AKT signaling pathway, was increased, as reflected by the decreased phosphorylation of GSK3β at Ser9. Studies have demonstrated that increased GSK3β activity enhances apoptosis [54, 66, 67]. Thus, the AKT-GSK3β signaling pathway may be involved in the regulation of the survival of interneurons.
By inhibiting the PI3K-PDK1-AKT signaling pathway, PTEN promotes apoptosis [82,83,84,85,86]. Previous studies have demonstrated that loss of Pten leads to the preferential apoptosis of SST+ interneurons, producing an abnormal PV+ interneuron/SST+ interneuron ratio in the cortex [8]. Here, we did not detect any changes in PTEN expression in the Pdk1 cKO mice. This result indicated that PTEN is not involved in the increased apoptosis of cortical interneurons.
Cortical interneurons originate in the GE [10, 11, 13, 87,88,89,90]. Inhibition of the PDK1/AKT/GSK3 pathway reduces cell proliferation defects by inducing cell cycle arrest [52,53,54]. Pdk1 ablation leads to expansion of the progenitor pool in the dorsal cortex [31]. In this study, no alteration in progenitor pools was observed. Immunostaining to detect ASCL1 and OLIG2 showed comparable ASCL1 and OLIG2 expression patterns in control and the Pdk1 cKO mice, and the numbers of progenitors in the VZ and SVZ were similar. In situ analysis of CyclinD2 further revealed an unaltered SVZ progenitor pool. Our data suggested that PDK1 is not involved in the transition of apical progenitors to basal progenitors in the ventral telencephalon. Moreover, BrdU labeling and immunostaining to detect PH3 and Ki67 demonstrated that the loss of Pdk1 had no effects on cell proliferation. Gotoh et al. have shown that deletion of Pdk1 in VZ progenitors achieved by Nestin-Cre mediated recombination leads to abnormal differentiation of interneurons alone with a decreased level of Ascl1 [36], here we did not observed alteration of Ascl1 expression when Pdk1 was deleted in SVZ progenitors, suggesting PDK1 may play different roles in distinct progenitor pools. Future studies will explore molecular mechanisms underlying the function of PDK1 in distinct progenitor populations. Previously studies have demonstrated that PDK1 regulates the radial migration of excitatory neurons by controlling the microtubule stabilization [29]. Here, we found that the tangential migration of cortical interneurons was normal after Pdk1 disruption. These results suggest that the roles of PDK1 in the excitatory neurons and interneurons are quite different. Future studies will explore more detailed mechanisms underlying PDK1-mediated regulation of cell migration, proliferation and survival in neurodevelopment.
Availability of data and materials
The data generated or analyzed during this study are included in this published article.
References
Whittington MA, Traub RD. Interneuron diversity series: inhibitory interneurons and network oscillations in vitro. Trends Neurosci. 2003;26(12):676–82.
Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5(10):793–807.
Moore CI, Carlen M, Knoblich U, Cardin JA. Neocortical interneurons: from diversity, strength. Cell. 2010;142(2):189–93.
Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Developmental neurobiology. 2011;71(1):45–61.
Tuncdemir SN, Fishell G. Batista-Brito R: miRNAs are essential for the survival and maturation of cortical interneurons. Cereb Cortex. 2015;25(7):1842–57.
Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2(5):255–67.
Marin O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13(2):107–20.
Vogt D, Cho KK, Lee AT, Sohal VS, Rubenstein JL. The parvalbumin/somatostatin ratio is increased in Pten mutant mice and by human PTEN ASD alleles. Cell Rep. 2015;11(6):944–56.
Denaxa M, Neves G, Rabinowitz A, Kemlo S, Liodis P, Burrone J, Pachnis V. Modulation of apoptosis controls inhibitory interneuron number in the cortex. Cell Rep. 2018;22(7):1710–21.
Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA. Origins of cortical interneuron subtypes. J Neurosci. 2004;24(11):2612–22.
Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci. 2006;7(9):687–96.
Gelman DM, Martini FJ, Nobrega-Pereira S, Pierani A, Kessaris N, Marin O. The embryonic preoptic area is a novel source of cortical GABAergic interneurons. J Neurosci. 2009;29(29):9380–9.
Miyoshi G, Hjerling-Leffler J, Karayannis T, Sousa VH, Butt SJ, Battiste J, Johnson JE, Machold RP, Fishell G. Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J Neurosci. 2010;30(5):1582–94.
Yang Y, Shen W, Ni Y, Su Y, Yang Z, Zhao C. Impaired interneuron development after Foxg1 disruption. Cereb Cortex. 2017;27(1):793–808.
Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128(3):353–63.
Jimenez D, Lopez-Mascaraque LM, Valverde F, De Carlos JA. Tangential migration in neocortical development. Dev Biol. 2002;244(1):155–69.
Ang ES Jr, Haydar TF, Gluncic V, Rakic P. Four-dimensional migratory coordinates of GABAergic interneurons in the developing mouse cortex. J Neurosci. 2003;23(13):5805–15.
Martini FJ, Valiente M, Lopez Bendito G, Szabo G, Moya F, Valdeolmillos M, Marin O. Biased selection of leading process branches mediates chemotaxis during tangential neuronal migration. Development. 2009;136(1):41–50.
Yoshida S, Sakagami H, Owada Y, Kokubun S, Kondo H. Localization of PDK-1 mRNA in the brain of developing and adult rats. Tohoku J Exp Med. 1999;187(3):249–55.
Kohn M, Hameister H, Vogel M, Kehrer-Sawatzki H. Expression pattern of the Rsk2, Rsk4 and Pdk1 genes during murine embryogenesis. Gene expression patterns : GEP. 2003;3(2):173–7.
Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15(2):161–70.
Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11(1):9–22.
Polleux F, Whitford KL, Dijkhuizen PA, Vitalis T, Ghosh A. Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development. 2002;129(13):3147–60.
Onishi K, Higuchi M, Asakura T, Masuyama N, Gotoh Y. The PI3K-Akt pathway promotes microtubule stabilization in migrating fibroblasts. Genes to cells : devoted to molecular & cellular mechanisms. 2007;12(4):535–46.
Morgan-Smith M, Wu Y, Zhu X, Pringle J, Snider WD. GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. eLife. 2014;3:e02663.
Gagliardi PA, di Blasio L, Primo L. PDK1: a signaling hub for cell migration and tumor invasion. Biochim Biophys Acta. 2015;1856(2):178–88.
Di Blasio L, Gagliardi PA, Puliafito A, Primo L: Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a Key Regulator of Cell Migration and Cancer Dissemination. Cancers 2017, 9(3).
Wang W, Sun X, Hu T, Wang L, Dong S, Gu J, Chu Y, Wang X, Li Y, Ru Y, et al. PDK1 regulates definitive HSCs via the FOXO pathway during murine fetal liver hematopoiesis. Stem Cell Res. 2018;30:192–200.
Itoh Y, Higuchi M, Oishi K, Kishi Y, Okazaki T, Sakai H, Miyata T, Nakajima K, Gotoh Y. PDK1-Akt pathway regulates radial neuronal migration and microtubules in the developing mouse neocortex. Proc Natl Acad Sci U S A. 2016;113(21):E2955–64.
Xu C, Yu L, Hou J, Jackson RJ, Wang H, Huang C, Liu T, Wang Q, Zou X, Morris RG, et al. Conditional deletion of PDK1 in the forebrain causes neuron loss and increased apoptosis during cortical development. Front Cell Neurosci. 2017;11:330.
Han X, Wei Y, Wu X, Gao J, Yang Z, Zhao C. PDK1 regulates transition period of apical progenitors to basal progenitors by controlling asymmetric cell division. Cereb Cortex. 2019.
Xu M, Han X, Liu R, Li Y, Qi C, Yang Z, Zhao C, Gao J. PDK1 deficit impairs the development of the dentate Gyrus in mice. Cereb Cortex. 2019;29(3):1185–98.
Lin HJ, Hsieh FC, Song H, Lin J. Elevated phosphorylation and activation of PDK-1/AKT pathway in human breast cancer. Br J Cancer. 2005;93(12):1372–81.
Fyffe C, Falasca M. 3-Phosphoinositide-dependent protein kinase-1 as an emerging target in the management of breast cancer. Cancer Manag Res. 2013;5:271–80.
Kim EJ, Kim GT, Kim BM, Lim EG, Kim SY, Kim YM. Apoptosis-induced effects of extract from Artemisia annua Linne by modulating PTEN/p53/PDK1/Akt/ signal pathways through PTEN/p53-independent manner in HCT116 colon cancer cells. BMC Complement Altern Med. 2017;17(1):236.
Oishi K, Watatani K, Itoh Y, Okano H, Guillemot F, Nakajima K, Gotoh Y. Selective induction of neocortical GABAergic neurons by the PDK1-Akt pathway through activation of Mash1. Proc Natl Acad Sci U S A. 2009;106(31):13064–9.
Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002;21(14):3728–38.
Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23(1):167–74.
Shen W, Ba R, Su Y, Ni Y, Chen D, Xie W, Pleasure SJ, Zhao C. Foxg1 regulates the postnatal development of cortical interneurons. Cereb Cortex. 2019;29(4):1547–60.
Tian C, Gong Y, Yang Y, Shen W, Wang K, Liu J, Xu B, Zhao J, Zhao C. Foxg1 has an essential role in postnatal development of the dentate gyrus. J Neurosci. 2012;32(9):2931–49.
Liu B, Zhou K, Wu X, Zhao C. Foxg1 deletion impairs the development of the epithalamus. Molecular brain. 2018;11(1):5.
Wu X, Gu X, Han X, Du A, Jiang Y, Zhang X, Wang Y, Cao G, Zhao C. A novel function for Foxm1 in interkinetic nuclear migration in the developing telencephalon and anxiety-related behavior. J Neurosci. 2014;34(4):1510–22.
Marin O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2(11):780–90.
Porteus MH, Bulfone A, Liu JK, Puelles L, Lo LC, Rubenstein JL. DLX-2, MASH-1, and MAP-2 expression and bromodeoxyuridine incorporation define molecularly distinct cell populations in the embryonic mouse forebrain. J Neurosci. 1994;14(11 Pt 1):6370–83.
Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126(3):525–34.
Takebayashi H, Yoshida S, Sugimori M, Kosako H, Kominami R, Nakafuku M, Nabeshima Y. Dynamic expression of basic helix-loop-helix Olig family members: implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mech Dev. 2000;99(1–2):143–8.
Alzu'bi A, Lindsay S, Kerwin J, Looi SJ, Khalil F, Clowry GJ. Distinct cortical and sub-cortical neurogenic domains for GABAergic interneuron precursor transcription factors NKX2.1, OLIG2 and COUP-TFII in early fetal human telencephalon. Brain Struct Funct. 2017;222(5):2309–28.
Miyoshi G, Butt SJ, Takebayashi H, Fishell G. Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J Neurosci. 2007;27(29):7786–98.
Glickstein SB, Alexander S, Ross ME. Differences in cyclin D2 and D1 protein expression distinguish forebrain progenitor subsets. Cereb Cortex. 2007;17(3):632–42.
Glickstein SB, Monaghan JA, Koeller HB, Jones TK, Ross ME. Cyclin D2 is critical for intermediate progenitor cell proliferation in the embryonic cortex. J Neurosci. 2009;29(30):9614–24.
Glickstein SB, Moore H, Slowinska B, Racchumi J, Suh M, Chuhma N, Ross ME. Selective cortical interneuron and GABA deficits in cyclin D2-null mice. Development. 2007;134(22):4083–93.
Kurosu T, Nagao T, Wu N, Oshikawa G, Miura O. Inhibition of the PI3K/Akt/GSK3 pathway downstream of BCR/ABL, Jak2-V617F, or FLT3-ITD downregulates DNA damage-induced Chk1 activation as well as G2/M arrest and prominently enhances induction of apoptosis. PLoS One. 2013;8(11):e79478.
Venigalla RK, McGuire VA, Clarke R, Patterson-Kane JC, Najafov A, Toth R, McCarthy PC, Simeons F, Stojanovski L, Arthur JS. PDK1 regulates VDJ recombination, cell-cycle exit and survival during B-cell development. EMBO J. 2013;32(7):1008–22.
Du J, Zhu X, Guo R, Xu Z, Cheng FF, Liu Q, Yang F, Guan L, Liu Y, Lin J. Autophagy induces G0/G1 arrest and apoptosis in menstrual blood-derived endometrial stem cells via GSK3-beta/beta-catenin pathway. Stem Cell Res Ther. 2018;9(1):330.
Butt SJ, Fuccillo M, Nery S, Noctor S, Kriegstein A, Corbin JG, Fishell G. The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron. 2005;48(4):591–604.
Inan M, Welagen J, Anderson SA. Spatial and temporal bias in the mitotic origins of somatostatin- and parvalbumin-expressing interneuron subgroups and the chandelier subtype in the medial ganglionic eminence. Cereb Cortex. 2012;22(4):820–7.
Bandler RC, Mayer C, Fishell G. Cortical interneuron specification: the juncture of genes, time and geometry. Curr Opin Neurobiol. 2017;42:17–24.
Nery S, Fishell G, Corbin JG. The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat Neurosci. 2002;5(12):1279–87.
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278(5338):687–9.
Vivanco I, Chen ZC, Tanos B, Oldrini B, Hsieh WY, Yannuzzi N, Campos C, Mellinghoff IK. A kinase-independent function of AKT promotes cancer cell survival. eLife. 2014;3.
Mao D, Sun X. Reactivation of the PI3K/Akt signaling pathway by the Bisperoxovanadium compound bpV (pic) attenuates photoreceptor apoptosis in experimental retinal detachment. Invest Ophthalmol Vis Sci. 2015;56(9):5519–32.
Zhou BH, Tan PP, Jia LS, Zhao WP, Wang JC, Wang HW. PI3K/AKT signaling pathway involvement in fluoride-induced apoptosis in C2C12cells. Chemosphere. 2018;199:297–302.
Liu AH, Chu M, Wang YP. Up-regulation of Trem2 inhibits hippocampal neuronal apoptosis and alleviates oxidative stress in epilepsy via the PI3K/Akt pathway in mice. Neurosci Bull. 2019;35(3):471–85.
Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR. The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Current biology : CB. 2000;10(8):439–48.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74.
Gomez-Sintes R, Hernandez F, Bortolozzi A, Artigas F, Avila J, Zaratin P, Gotteland JP, Lucas JJ. Neuronal apoptosis and reversible motor deficit in dominant-negative GSK-3 conditional transgenic mice. EMBO J. 2007;26(11):2743–54.
Zhang Y, Zhu C, Sun B, Lv J, Liu Z, Liu S, Li H. Integrated high throughput analysis identifies GSK3 as a crucial determinant of p53-mediated apoptosis in lung Cancer cells. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;42(3):1177–91.
Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11(8):539–51.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381–405.
Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19(2):231–4.
Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int J Biochem Cell Biol. 2009;41(4):757–61.
Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2(10):760–8.
Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12(22):3499–511.
Otaegi G, Yusta-Boyo MJ, Vergano-Vera E, Mendez-Gomez HR, Carrera AC, Abad JL, Gonzalez M, de la Rosa EJ, Vicario-Abejon C, de Pablo F. Modulation of the PI 3-kinase-Akt signalling pathway by IGF-I and PTEN regulates the differentiation of neural stem/precursor cells. J Cell Sci. 2006;119(Pt 13):2739–48.
Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114(Pt 16):2903–10.
Volarevic S, Thomas G. Role of S6 phosphorylation and S6 kinase in cell growth. Prog Nucleic Acid Res Mol Biol. 2001;65:101–27.
Zhong Y, Huang Y, Cao J, Lu X, Feng M, Shen G, Shen A, Yu X. Increase in phosphorylation of PDK1 and cell survival after acute spinal cord injury. J Neurol Sci. 2012;320(1–2):38–44.
Chiarini A, Liu D, Armato U, Dal Pra I. Bcl10 crucially nucleates the pro-apoptotic complexes comprising PDK1, PKCzeta and caspase-3 at the nuclear envelope of etoposide-treated human cervical carcinoma C4-I cells. Int J Mol Med. 2015;36(3):845–56.
Park H, Lee S, Shrestha P, Kim J, Park JA, Ko Y, Ban YH, Park DY, Ha SJ, Koh GY, et al. AMIGO2, a novel membrane anchor of PDK1, controls cell survival and angiogenesis via Akt activation. J Cell Biol. 2015;211(3):619–37.
Peng Y, Qiu L, Xu D, Zhang L, Yu H, Ding Y, Deng L, Lin J. M4IDP, a zoledronic acid derivative, induces G1 arrest, apoptosis and autophagy in HCT116 colon carcinoma cells via blocking PI3K/Akt/mTOR pathway. Life Sci. 2017;185:63–72.
Wiedenhoft H, Hayashi L, Coffin AB. PI3K and inhibitor of apoptosis proteins modulate gentamicin- induced hair cell death in the Zebrafish lateral line. Front Cell Neurosci. 2017;11:326.
Dupont J, Renou JP, Shani M, Hennighausen L, LeRoith D. PTEN overexpression suppresses proliferation and differentiation and enhances apoptosis of the mouse mammary epithelium. J Clin Invest. 2002;110(6):815–25.
Li L, Liu F, Salmonsen RA, Turner TK, Litofsky NS, Di Cristofano A, Pandolfi PP, Jones SN, Recht LD, Ross AH. PTEN in neural precursor cells: regulation of migration, apoptosis, and proliferation. Mol Cell Neurosci. 2002;20(1):21–9.
Wan X, Yokoyama Y, Shinohara A, Takahashi Y, Tamaya T. PTEN augments staurosporine-induced apoptosis in PTEN-null Ishikawa cells by downregulating PI3K/Akt signaling pathway. Cell Death Differ. 2002;9(4):414–20.
Choy MS, Bay BH, Cheng HC, Cheung NS. PTEN is recruited to specific microdomains of the plasma membrane during lactacystin-induced neuronal apoptosis. Neurosci Lett. 2006;405(1–2):120–5.
Zhu Y, Hoell P, Ahlemeyer B, Krieglstein J. PTEN: a crucial mediator of mitochondria-dependent apoptosis. Apoptosis : an international journal on programmed cell death. 2006;11(2):197–207.
Wonders C, Anderson SA. Cortical interneurons and their origins. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2005;11(3):199–205.
Sultan KT, Shi W, Shi SH. Clonal origins of neocortical interneurons. Curr Opin Neurobiol. 2014;26:125–31.
Crandall SR, Connors BW. Diverse ensembles of inhibitory interneurons. Neuron. 2016;90(1):4–6.
Mi D, Li Z, Lim L, Li M, Moissidis M, Yang Y, Gao T, Hu TX, Pratt T, Price DJ, et al. Early emergence of cortical interneuron diversity in the mouse embryo. Science. 2018;360(6384):81–5.
Acknowledgments
We thank Yiquan Wei and Li Liu for their assistance in the laboratory and with animal care and other members of the laboratory for discussion.
Funding
This work was supported by grants 31930045, 81870899 from the National Natural Science Foundation of China, and 2016YFA0501001 from the National Key R&D Program of China to C.Z.
Author information
Authors and Affiliations
Contributions
C.Z. conceptualized the study. Y.W., X.H., and C.Z. designed the experiments; Y.W. performed the immunostaining, in situ hybridization, and western blotting. Y.W., X.H., and C.Z. analyzed the data. C.Z. and Y.W. wrote the paper. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All animals were housed in the animal facility of Southeast University, and all experimental procedures followed the guidance approved by Southeast University.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Statistical results of raw data.
Additional file 2.
Raw data supplementary materials
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wei, Y., Han, X. & Zhao, C. PDK1 regulates the survival of the developing cortical interneurons. Mol Brain 13, 65 (2020). https://doi.org/10.1186/s13041-020-00604-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-020-00604-6