
Abstract
Background
Individuals with X chromosomal translocations, variable phenotypes, and a high risk of live birth defects are of interest for scientific study. These characteristics are related to differential breakpoints and various types of chromosomal abnormalities. To investigate the effects of X chromosome translocation on clinical phenotype, a retrospective analysis of clinical data for patients with X chromosome translocation was conducted. Karyotype analysis plus endocrine evaluation was utilized for all the patients. Additional semen analysis and Y chromosome microdeletions were assessed in male patients.
Results
X chromosome translocations were detected in ten cases, including seven females and three males. Infantile uterus and no ovaries were detected in case 1 (FSH: 114 IU/L, LH: 30.90 mIU/mL, E2: < 5.00 pg/ml), and the karyotype was confirmed as 46,X,t(X;22)(q25;q11.2) in case 1. Infantile uterus and small ovaries were both visible in two cases (FSH: 34.80 IU/L, LH: 17.06 mIU/mL, E2: 15.37 pg/ml in case 2; FISH: 6.60 IU/L, LH: 1.69 mIU/mL, E2: 23.70 pg/ml in case 3). The karyotype was detected as 46,X,t(X;8)(q13;q11.2) in case 2 and 46,X,der(X)t(X;5)(q21;q31) in case 3. Normal reproductive hormone levels and fertility abilities were found for cases 4, 6 and 7. The karyotype were detected as 46,X,t(X;5)(p22.3;q22) in case 4 and 46,X,der(X)t(X;Y)(p22.3;q11.2) in cases 6 and 7. These patients exhibited unremarkable clinical manifestations but experienced a history of abnormal chromosomal pregnancy. Normal phenotype and a complex reciprocal translocation as 46,X,t(X;14;4)(q24;q22;q33) were observed in case 5 with a history of spontaneous abortions. In the three male patients, multiple semen analyses confirmed the absence of sperm. Y chromosome microdeletion and hormonal analyses were normal. The karyotypes were detected as 46,Y,t(X;8)(q26;q22), 46,Y,t(X;1)(q26;q23), 46,Y,t(X;3)(q26;p24), respectively.
Conclusions
Our study provides insights into individuals with X chromosome translocations. The clinical phenotypes are variable and unpredictable due to differences in breakpoints and X chromosome inactivation (XCI) patterns. Our results suggest that physicians should focus on the characteristics of the X chromosome translocations and provide personalized clinical evaluations in genetic counselling.
Similar content being viewed by others


Background
Reciprocal translocations are common chromosomal abnormalities that are reported to occur in approximately 0.2% of humans and 2.1% of couples who experience spontaneous abortion [1, 2]. X chromosome translocations are extremely rare and are estimated to occur in 1:30,000 live births [3]. With balanced chromosomal structural rearrangements of no gain or loss of genetic material, the individuals are expected to be phenotypically normal but at a high risk for infertility, spontaneous abortion, embryo failure, and birth defects due to cytogenetically unbalanced pregnancies. This imbalance consists of partial monosomies and/or trisomies that may affect the chromosomal segments involved in the translocations [4, 5]. In contrast to autosomal translocations, X chromosome translocations are quite rare structural abnormalities of chromosomes. The clinical phenotype associated with X chromosome translocation is variable. In practice, certain individuals exhibit unremarkable clinical manifestations. Patients with X-autosome translocations are likely to experience short stature, mental retardation, germinal aplasia and X-linked disorders due to X chromosome inactivation [6,7,8]. With X–Y translocations females are phenotypically normal or short stature according to the breakpoint of the X chromosome, and anomalies of gonadal development tend to occur in males [9]. Additionally, X chromosome translocation patients have a higher rate of offspring with X chromosome aneuploidy than patients with autosomal translocation for X chromosome inactivation [10, 11]. This study aimed to investigate laboratory data and phenotypes in X chromosome translocations to provide meaningful clinical evidence for genetic counselling and reproductive risk assessment.
Results
Between January 2021 and February 2023, 30,815 patients visited the Medical Genetics Center of Jiangxi Maternal and Child Health Hospital for clinical genetic consultation. Ethical approval was obtained from the Ethics Committee of Jiangxi Maternal and Child Health Hospital. Among the total study population (N = 30,815), females (N = 16,543) including infertility, history of spontaneous abortion, history of abnormal pregnancy, amenorrhea, and males (N = 14,272) including infertility underwent karyotyping. Of these, ten cases of X chromosome translocations were detected. In cases 1, 2 and 3, secondary amenorrhea, infantile or small uterus, and absent or small ovaries were observed. In cases 4 and 6, an abnormal chromosomal pregnancy was shown. In cases 5 and 7, there was a spontaneous abortion history. Cases 8, 9, and 10 involved three men with infertility due to azoospermia (Table 1).
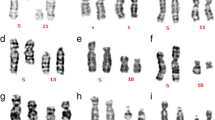
Anti-Müllerian hormone (AMH) < 0.01 ng/ml was observed in cases 1, 2 and 3 with secondary amenorrhea. Transabdominal ultrasound revealed an infantile uterus and small ovaries in cases 2 and 3 (Follicle-stimulating hormon (FSH): 34.80 IU/L, Luteinizing hormone (LH): 17.06 mIU/mL, Oestradiol (E2): 15.37 pg/ml; FISH: 6.60 IU/L, LH: 1.69 mIU/mL, and E2: 23.70 pg/ml) compared to no ovaries in case 1 (FSH: 114 IU/L, LH: 30.90 mIU/mL, E2: < 5.00 pg/ml). A significant correlation was observed between AMH levels and ovarian insufficiency. The karyotypes were detected as 46,X,t(X;22)(q25;q11.2) for case 1, 46,X,t(X;8)(q13;q11.2) for case 2, and 46,X,der(X)t(X;5)(q21;q31) for case 3 (Fig. 1). According to karyotype, the breakpoints located on the long arm of the X chromosome (Xq25 for case 1, Xq13 for case 2, and Xq21 for case 3) correlated with low AMH levels and ovarian insufficiency. Balanced reciprocal translocations between X chromosome and autosomes were observed in cases 1 and 2, and imbalanced translocation was observed in case 3.

A The karyotype was 46,X,t(X;22)(q25;q11.2) for case 1. B The karyotype was 46,X,t(X;8)(q13;q11.2) for case 2. C The karyotype was 46,X,der(X)t(X;5)(q21;q31) for case 3
Additionally, Cases 4 and 6 involved normal hormonal levels and fertility abilitiy but with a history of abnormal chromosomal pregnancy. The karyotypes were 46,X,t(X;5)(p22.3;q22) and 46,X,der(X)t(X;Y)(p22.3;q11.2). In case 4, ultrasonography at 20 weeks of gestation indicated that the foetus had tetralogy of Fallot, an intense left heart spot, a single umbilical artery, and small kidneys. Prenatal diagnosis from the amniotic fluid cells suggested that the karyotype of the foetus was 47,XN, +der(X)t(X;5)(p22.3;q32)mat with duplication of Xp22.3-qter and 5q32-qter as confirmed by chromosomal microarray (Fig. 2). The 5q35-qter duplication syndrome presents a phenotype of mental and growth retardation, microcephaly, cardiac defects, pulmonary hypertension, genital defects, brachydactyly, congenital hipdysplasia, dental caries and eczema. The foetus in case 4 also exhibited abdominal, limb, and cardiac malformations. Imbalanced translocations between X and Y were present in cases 6 and 7. The G-banding karyotype was established as 46,X,der(X)t(X;Y)(p22.3;q11.2) and C-banding karyotype revealed heterochromatin ligated to Xp22.3 (Fig. 3). In case 6, karyotype and copy number variation sequencing (CNV-seq) of amniofluid cells revealed 46,Y,der(X)t(X;Y)mat for the male foetus inheriting der(X) from the mother. The foetus had short limbs due to SHOX haploinsufficiency associated with Leri-Weill dyschondrosteosis (LWD). In case 5 which involved spontaneous abortion 2 times, normal hormone levels were detected and complex reciprocal translocation among chromosomes X, 14, and 4 was identified by karyotyping (Table 2).

A The karyotype was 46,X,t(X;5)(p22.3;q22) for case 4. B The karyotype was 47,XN, +der(X)t(X;5)(p22.3;q32)mat for the foetus of case 4. C The karyotype was 46,X,t(X;14;4)(q24;q22;q33) for case 5

A The karyotype was 46,X,der(X)t(X;Y)(p22.3;q11.2) for case 6. B Heterochromatin visualized and ligated to Xp22.3 in the C-band for case 6. C The karyotype was 46,X,der(X)t(X;Y)(p22.3;q11.2) for case 7. D CNV-seq revealed seq[GRCh37]Xp22.33p22.31(990,745–6426956) × 1 chrX:g.990745-6426956del chrY: 16,207,792–28819361 for case 6. E CNV-seq revealed seq[GRCh37]del(X)(p22.33p22.33) chrX:g.287328-2757513del seq[GRCh37]dup(Y)(q11.21q12) chrY:g.14567885-28819361dup for the foetus of case 7
Cases 8, 9 and 10 involved male patients admitted to our hospital for primary infertility. All of these patients underwent karyotype and semen analysis. The karyotypes were detected as 46,Y,t(X;8)(q26;q22), 46,Y,t(X;1)(q26;q23), and 46,Y,t(X;3)(q26;p24) (Fig. 4). The breakpoints were primarily located at Xq26. Multiple semen analyses confirmed the absence of sperm, though endocrine evaluation was normal. Y chromosome microdeletion analysis revealed the presence of a sex-determining region on the Y chromosome and azoospermia factors a, b, and c (AZFa, AZFb, and AZFc) (Table 3).

A The karyotype was 46,Y,t(X;8)(q26;q22) for case 8. B The karyotype was 46,Y,t(X;1)(q26;q23) for case 9. C The karyotype was 46,Y,t(X;3)(q26;q24) for case 10. D PCR results of the Y chromosome deletions. The AZFa, AZFb and AZFc regions include: sY14 (SRY, blue in tube A), ZFX/ZFY (blue in tube B), sY84 (green in tube A), sY86 (green in tube B), sY127 (orange in tube A), sY134 (orange in tube B), sY254 (red in tube B), and sY255 (red in tube A). Reference value: Typical S-type amplification and curve with Ct < 32
Discussion
Human cells possess two sex chromosomes, with males carrying an X and a Y chromosome, and females carrying two X chromosomes. There are more genes on the X chromosome than the Y chromosome. Dosage compensation is achieved by silencing one X chromosome to achieve equal dosages of XX and XY [12]. In early embryogenesis, one of the two X chromosomes in each female embryo cell is randomly inactivated, known as the lyonization [13, 14]. In fact, only partial fractions on the inactivated X chromosome are deactivated and lack transcriptional activity, in other fractions certain gene sites undergo transcription and performed double-dose functions such as primary pseudoautosomal regions (PAR1), secondary pseudoautosomal regions (PAR2), with an X–Y shared fragment of approximately 4 Mb in length located at Xq21.3 [15, 16]. Transcriptional silencing is initiated by an X-chromosome inactivation centre (XIC) located at Xq13. Which X chromosome is inactivated is a random process [17]. If the derived/translocated X chromosome is inactivated, gene silencing spreads to the connected autosomal segment. This phenomenon may cause phenotypic abnormalities [18, 19]. Theoretically, normal phenotypes and gene functions can only be achieved by normal X-chromosome inactivation. Therefore, the difference between which X is inactivated may determine a normal or abnormal phenotype [12].
Here, we report ten cases of X chromosome translocation. An abnormal reproductive system was observed in cases 1, 2 and 3. Extremely low AMH levels indicate ovarian insufficiency. AMH has recently been considered for assessing ovarian reserve, given its high sensitivity and specificity in predicting ovarian response and good intercycle reliability. FSH increases much earlier and more sharply than LH. The FSH/LH ratio is an independent factor to predict poor ovarian response and is associated with poor outcomes in vitro fertilization (IVF) treatment [20]. Patients with the X chromosome translocation and premature ovarian insufficiency (POI) constitute an interesting study in regard to the location of breakpoints. The Xq critical region is known for its role in maintaining ovarian function and normal reproductive lifespan and is located ranging between Xq13 and Xq27 [21,22,23]. To investigate the effects of balanced X-autosome translocation resulting in POI, Di Battista fine-mapped breakpoints in six patients with POI and balanced X-autosome translocation and addressed gene expression and chromatin accessibility changes in four of them. The results revealed that translocation exerts a broad effect on chromatin structure, this study thus appears to support the hypothesis that positional effects are a causal mechanism for premature ovarian insufficiency associated with X-autosome translocation [24]. The reasonable prevalence of X chromosome structural anomalies and X-autosome translocations related to POI was calculated to range from 4.2 to 12.0% [25].
A significant association between X chromosome translocations and abnormal pregnancy has been documented. X-autosome translocations may occur without a significant detrimental phenotype due to normal X chromosome inactivation, however, the carriers will face increased risks of abnormal pregnancy such as recurrent spontaneous abortion, stillbirths, and congenital disabilities [26]. A female child carrying an unbalanced X-autosome translocation with 45,X,der(X)t(X;14)(q28;q11.2)mat presented poor growth and development. Diagnostic procedures for developmental delay included brain MRI scan showing hypoplasia of the corpus callosum with microcephaly and cerebral atrophy for the derived X chromosome inactivation. Her mother with 46,X,t(X;14)(q28;q11.2) was phenotypically normal [3]. A family with 4 women of 3 different generations carrying an unbalanced X chromosome translocation with 46,X,der(X)t(X;7)(q26;q35) has been reported. None of the carriers showed intellectual disability, and all of them had a very mild clinical presentation mainly characterized by gynaecological/hormonal issues and autoimmune disorders. The XCI pattern showed a skewed X inactivation pattern with a preferential activation of the normal X [27]. Unpredictable detrimental phenotypes associated with skewed XCI patterns may occur in female carriers, whereas azoospermia or severe oligozoospermia occurs in all male carriers [28, 29].
X–Y chromosome translocation is a rare event, and the major breakpoints are located at Xq22 and Yq11 [30,31,32]. The clinical phenotype is heterogeneously associated with the size of the Xp deletion and the genes involved. For the XCI pattern preferring to the derived X chromosome silencing females with t(X;Y) are generally phenotypically normal except for short stature when the short stature homeobox (SHOX) gene is absent [33]. Patients may have ichthyotic skin disease and Kallmann syndrome when the STS and KAL1 genes are deleted [34]. Intellectual disability is linked to loss of the VCX-A gene [34]. Additionally, other genes have been mapped to Xp22.3 including X-linked recessive chondrodysplasia punctata (CDPX) and ocular albinism type I (OA1) [35]. A familial maternally inherited X–Y chromosome translocation encompassing SHOX and ARSE was documented in two Moroccan siblings with sensorineural deafness. The mother was short in stature but had normal intelligence and no hearing loss [36]. Although this X–Y chromosome translocation was maternally derived, the mother's phenotype was mild and her sons had abnormalities [37,38,39].
Chromosomal abnormalities are the primary genetic factors that lead to azoospermia and male infertility. Even if the copy number of the cellular chromosome is balanced, almost all hemizygous males with X-autosomal translocations are infertile [40]. Choi reported a 26-year-old male seeking initial infertility evaluation. The detailed physical examination and laboratory tests were normal, with the exception of an abnormal karyotype with a reciprocal translocation at chromosomes X and 16. An open testicular biopsy demonstrated late maturation arrest at the spermatid stage without evidence of significant peritubular fibrosis or hyalinization on pathology, thus confirming significantly reduced reproductive potential [41]. With immature sperm present on testicular biopsy, carriers may be candidates for testicular excisional sperm extraction using intracytoplasmic sperm injection (ICSI) and in vitro fertilization (IVF) [29].
Conclusions
Our study provides insights into individuals with the X chromosome translocations. Clinical phenotypes are variable and unpredictable with regard to differences in breakpoints and XCI patterns, including phenotypically normal, ovarian insufficiency, infertility, a high risk of birth defects and other syndromes for related genes loss. The effects of different breakpoints and XCI patterns are worthy of further study. Our results suggest that physicians should focus on the characteristics of the X chromosome translocations and provide personalized clinical evaluations in genetic counselling.
Methods
This study was approved by the Institutional Research Ethics Committee of Jiangxi Maternal and Child Health Hospital. All patients agreed to participate in the study and provided written informed consent. A total of 30,815 patients were referred for karyotyping for genetic counselling at the medical genetic centre between January 2021 and February 2023. Indications for karyotype analysis included infertility, amenorrhea, history of spontaneous abortion, and history of abnormal pregnancy.
Karyotype analysis
Peripheral lymphocyte blood specimens were subjected to cell culture and harvest, and slides were prepared according to standard operation protocols. Sufficiently dried slides were treated with trypsin at 37 °C and Giemsa staining for G-bands [42]. If necessary, the slides were incubated in Ba(OH)2 at 50 °C for denaturation of chromosomes and then in 2× SSC at 60 °C for reannealing to promote the visualization of highly repetitive C-band regions [43]. Thirty cells were counted, and five cells were analysed by two physicians according to International System for Cytogenetic Nomenclature (ISCN 2020).
CNV-seq analysis
DNA was extracted from blood or amniotic fluid using a DNeasy Blood and Tissue Kit (Qiagen, Germany) and generated to an average fragment size of 200 bp. All samples passing quality control (> 500 ng; OD260/OD280 > 1.8; OD260/OD230 > 1.5) were prepared for library construction and sequenced using the MGISEQ-2000 platform. By using 100 kb as a basic unit of analysis, all data were aligned to the human reference genome (GRCh37) [44].
Endocrine evaluation
Blood samples were collected on Days 2–4 of the menstrual cycles and measured using an automatic chemiluminescence immunoassay instrument (Cobas E801, Roche). Levels of FSH, LH, E2, PROG, PRL, T and AMH were quantified according to the instruments of the corresponding kits (Roche, Germany) [20].
Semen analysis and Y chromosome microdeletions
Two separate semen samples were centrifuged at 3000 × g for 15 min and subjected to microscopic evaluation [45].
Whole blood genomic DNA was extracted using BloodGen Mini Kit (Tegen, Shanghai), and polymerase chain reaction amplification was performed using a PCR instrument (Roche LightCycler 480). Each set of PCRs was carried out via duplex PCR. The primers used included those targeting SRY (sY14), ZFX/ZFY, AZFa (sY84, sY86), AZFb (sY127, sY134), AZFc (sY254, sY255) [46].
Availability of data and materials
All data generated or analysed during this study are included in the article.
Abbreviations
- FSH:
-
Follicle-stimulating hormone
- LH:
-
Luteinizing hormone
- E2:
-
Oestradiol
- PROG:
-
Progesterone
- PRL:
-
Prolactin
- T:
-
Testosterone
- AMH:
-
Anti-Müllerian hormone
- POI:
-
Primary ovarian insufficiency
- CNV-seq:
-
Copy number variation sequencing
- AZF:
-
Azoospermia factor
- PCR:
-
Polymerase chain reaction
References
Liu Y, Kong XD, Wu QH, et al. Karyotype analysis in large-sample infertile couples living in Central China: a study of 14965 couples. J Assist Reprod Genet. 2013;30(4):547–53. https://doi.org/10.1007/s10815-013-9964-6.
Park SJ, Min JY, Kang JS, et al. Chromosomal abnormalities of 19,000 couples with recurrent spontaneous abortions: a multicenter study. Fertil Steril. 2022;117(5):1015–25. https://doi.org/10.1016/j.fertnstert.2022.01.011.
Gupta N, Goel H, Phadke SR. Unbalanced X; autosome translocation. Indian J Pediatr. 2006;73(9):840–2. https://doi.org/10.1007/BF02790399.
Madan K, Nieuwint AW, van Bever Y. Recombination in a balanced complex translocation of a mother leading to a balanced reciprocal translocation in the child. Review of 60 cases of balanced complex translocations. Hum Genet. 1997;99(6):806–15. https://doi.org/10.1007/s004390050453.
Tharapel AT, Tharapel SA, Bannerman RM. Recurrent pregnancy losses and parental chromosome abnormalities: a review. Br J Obstet Gynaecol. 1985;92(9):899–914. https://doi.org/10.1111/j.1471-0528.1985.tb03069.x.
Watanabe T, Ishibashi M, Suganuma R, et al. Mild phenotypes associated with an unbalanced X-autosome translocation, 46, X, der(X)t(X;8)(q28;q13). Clin Case Rep. 2018;6(8):1561–4. https://doi.org/10.1002/ccr3.1596.
Giorda R, Bonaglia MC, Milani G, et al. Molecular and cytogenetic analysis of the spreading of X inactivation in a girl with microcephaly, mild dysmorphic features and t(X;5)(q22.1;q31.1). Eur J Hum Genet. 2008;16(8):897–905. https://doi.org/10.1038/ejhg.2008.28.
Gómez-Laguna L, Martínez-Herrera A, Reyes-de la Rosa ADP, et al. Nance-Horan syndrome in females due to a balanced X;1 translocation that disrupts the NHS gene: familial case report and review of the literature. Ophthalmic Genet. 2018;39(1):56–62. https://doi.org/10.1080/13816810.2017.1363245.
McElreavey K, Cortes LS. X-Y translocations and sex differentiation. Semin Reprod Med. 2001;19(2):133–9. https://doi.org/10.1055/s-2001-15393.
Lin L, Zhao C, Lv Y, et al. Clinical genetic analysis of an infant with 1q21.3-qter duplication and Xpter-p11 deletion caused by maternal balanced chromosomal translocation. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2022;39(5):514–7. https://doi.org/10.3760/cma.j.cn511374-20210918-007613.
Yuan S, Cheng D, Luo K, et al. Reproductive risks and preimplantation genetic testing intervention for X-autosome translocation carriers. Reprod Biomed Online. 2021;43(1):73–80. https://doi.org/10.1016/j.rbmo.2021.03.010.
Brockdorff N, Turner BM. Dosage compensation in mammals. Cold Spring Harb Perspect Biol. 2015;7(3): a019406. https://doi.org/10.1101/cshperspect.a019406.
Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372–3. https://doi.org/10.1038/190372a0.
Sun Z, Fan J, Wang Y. X-chromosome inactivation and related diseases. Genet Res (Camb). 2022;27(2022):1391807. https://doi.org/10.1155/2022/1391807.
Garieri M, Stamoulis G, Blanc X, et al. Extensive cellular heterogeneity of X inactivation revealed by single-cell allele-specific expression in human fibroblasts. Proc Natl Acad Sci USA. 2018;115(51):13015–20. https://doi.org/10.1073/pnas.1806811115.
San Roman AK, Skaletsky H, Godfrey AK, et al. The human Y and inactive X chromosomes similarly modulate autosomal gene expression. bioRxiv [Preprint]. 2023;doi https://doi.org/10.1101/2023.06.05.543763.
Furlan G, Galupa R. Mechanisms of choice in x-chromosome inactivation. Cells. 2022;11(3):535. https://doi.org/10.3390/cells11030535.
Sharp AJ, Spotswood HT, Robinson DO, et al. Molecular and cytogenetic analysis of the spreading of X inactivation in X;autosome translocations. Hum Mol Genet. 2002;11(25):3145–56. https://doi.org/10.1093/hmg/11.25.3145.
White WM, Willard HF, Van Dyke DL, et al. The spreading of X inactivation into autosomal material of an x;autosome translocation: evidence for a difference between autosomal and X-chromosomal DNA. Am J Hum Genet. 1998;63(1):20–8. https://doi.org/10.1086/301922.
Jiao X, Meng T, Zhai Y, et al. Ovarian reserve markers in premature ovarian insufficiency: within different clinical stages and different etiologies. Front Endocrinol (Lausanne). 2021;18(12): 601752. https://doi.org/10.3389/fendo.2021.601752.
Therman E, Laxova R, Susman B. The critical region on the human Xq. Hum Genet. 1990;85(5):455–61. https://doi.org/10.1007/BF00194216.
Fortuño C, Labarta E. Genetics of primary ovarian insufficiency: a review. J Assist Reprod Genet. 2014;31(12):1573–85. https://doi.org/10.1007/s10815-014-0342-9.
Rao Kandukuri L, Padmalatha V, Kanakavalli M, et al. Unique case reports associated with ovarian failure: necessity of two intact x chromosomes. Case Rep Genet. 2012;2012: 640563. https://doi.org/10.1155/2012/640563.
Di-Battista A, Favilla BP, Zamariolli M, et al. Premature ovarian insufficiency is associated with global alterations in the regulatory landscape and gene expression in balanced X-autosome translocations. Epigenetics Chromatin. 2023;16(1):19. https://doi.org/10.1186/s13072-023-00493-8.
Chen M, Jiang H, Zhang C. Selected genetic factors associated with primary ovarian insufficiency. Int J Mol Sci. 2023;24(5):4423. https://doi.org/10.3390/ijms24054423.
Migeon BR. X-linked diseases: susceptible females. Genet Med. 2020;22(7):1156–74. https://doi.org/10.1038/s41436-020-0779-4.
Ciaccio C, Redaelli S, Bentivegna A, Marelli S, Crosti F, Sala EM, Cavallari U. Unbalanced X;Autosome translocations may lead to mild phenotypes and are associated with autoimmune diseases. Cytogenet Genome Res. 2020;160(2):80–4. https://doi.org/10.1159/000506097.
Vianna EQ, Piergiorge RM, Gonçalves AP, et al. Understanding the landscape of X-linked variants causing intellectual disability in females through extreme X chromosome inactivation skewing. Mol Neurobiol. 2020;57(9):3671–84. https://doi.org/10.1007/s12035-020-01981-8.
Chamayou S, Sicali M, Lombardo D, et al. The decision on the embryo to transfer after preimplantation genetic diagnosis for X-autosome reciprocal translocation in male carrier. Mol Cytogenet. 2018;29(11):63. https://doi.org/10.1186/s13039-018-0409-x.
Frints SG, Fryns J, Lagae L, Syrrou M, Marynen P, Devriendt K. Xp22.3; Yq11.2 chromosome translocation and its clinical manifestations. Ann Genet. 2001;200144(2):71–6. https://doi.org/10.1016/s0003-3995(01)01071-1.
Yen PH, Tsai SP, Wenger SL, et al. X/Y translocations resulting from recombination between homologous sequences on Xp and Yq. Proc Natl Acad Sci USA. 1991;88(20):8944–8. https://doi.org/10.1073/pnas.88.20.8944.
Khudr G, Benirschke K. X-Y translocation. Am J Obstet Gynecol. 1973;116(4):584–5. https://doi.org/10.1016/0002-9378(73)90923-x.
Panasiuk B, Usinskiené R, Kostyk E, et al. Genetic counselling in carriers of reciprocal chromosomal translocations involving short arm of chromosome X. Ann Genet. 2004;47(1):11–28. https://doi.org/10.1016/j.anngen.2004.01.001.
Fukami M, Kirsch S, Schiller S, et al. A member of a gene family on Xp22.3, VCX-A, is deleted in patients with X-linked nonspecific mental retardation. Am J Hum Genet. 2000;67(3):563–73. https://doi.org/10.1086/303047.
Ballabio A, Andria G. Deletions and translocations involving the distal short arm of the human X chromosome: review and hypotheses. Hum Mol Genet. 1992;1(4):221–7. https://doi.org/10.1093/hmg/1.4.221.
Amasdl S, Smaili W, Natiq A, et al. Familial X/Y translocation encompassing ARSE in two moroccan siblings with sensorineural deafness. Cytogenet Genome Res. 2017;153(2):66–72. https://doi.org/10.1159/000485071.
Doherty MJ, Glass IA, Bennett CL, et al. An Xp; Yq translocation causing a novel contiguous gene syndrome in brothers with generalized epilepsy, ichthyosis, and attention deficits. Epilepsia. 2003;44(12):1529–35. https://doi.org/10.1111/j.0013-9580.2003.61702.x.
Liu S, Zheng J, Liu X, et al. Comprehensive analysis of three female patients with different types of X/Y translocations and literature review. Mol Cytogenet. 2023;16(1):7. https://doi.org/10.1186/s13039-023-00639-z.
Yin T, Wang Y, Wang Z, et al. Genetic study of a child carrying a maternally derived unbalanced 46, Y, der(X)t(X;Y)(p22;q11) chromosomal translocation. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2021;38(4):376–9. https://doi.org/10.3760/cma.j.cn511374-20200321-00188.
Li Y, Sha Y, Wei Z, et al. A familial analysis of two brothers with azoospermia caused by maternal 46, Y, t(X; 1) (q28; q21) chromosomal abnormality. Andrologia. 2021;53(1): e13867. https://doi.org/10.1111/and.13867.
Choi L, Levy G, Donlon T, et al. Azoospermia secondary to a novel X-autosomal reciprocal translocation: 46, Y, t(X:16)(p221:p112). Mil Med. 2020;185(9–10):e1860–3. https://doi.org/10.1093/milmed/usaa047.
Bangs CD, Donlon TA. Metaphase chromosome preparation from cultured peripheral blood cells. Curr Protoc Hum Genet. 2005. https://doi.org/10.1002/0471142905.hg0401s45.
Sumner AT. A simple technique for demonstrating centromeric heterochromatin. Exp Cell Res. 1972;75(1):304–6. https://doi.org/10.1016/0014-4827(72)90558-7.
Zheng Y, Zhu B, Tan J, et al. Experience of low-pass whole-genome sequencing-based copy number variant analysis: a survey of chinese tertiary hospitals. Diagnostics (Basel). 2022;12(5):1098. https://doi.org/10.3390/diagnostics12051098.
Björndahl L, Kirkman Brown J, other Editorial Board Members of the WHO Laboratory Manual for the Examination and Processing of Human Semen. The sixth edition of the WHO Laboratory Manual for the Examination and Processing of Human Semen: ensuring quality and standardization in basic examination of human ejaculates. Fertil Steril. 2022;117(2):246–251. doi: https://doi.org/10.1016/j.fertnstert.2021.12.012
Krausz C, Hoefsloot L, Simoni M, et al. EAA/EMQN best practice guidelines for molecular diagnosis of Y-chromosomal microdeletions: state-of-the-art 2013. Andrology. 2014;2(1):5–19. https://doi.org/10.1111/j.2047-2927.2013.00173.x.
Acknowledgements
We would like to thank all the patients and family members who participated in this study for their cooperation and patience.
Funding
Funding for this study was provided by the Science and Technology Project of the Health Commission of Jiangxi Province (202211059, 202130770), the Traditional Chinese Medicine of Administration of Jiangxi Province (2020B0343), and the Jiangxi Provincial Clinical Research Center for Birth Defects (20223BCG74002).
Author information
Authors and Affiliations
Contributions
The first draft of the manuscript was written by NH. Karyotype analysis was performed by NH, JZ, and WL. CNV-seq data analyses were performed by LL. Genetic counselling was performed by HY, and hormone analysis and semen analysis were performed by SD. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Jiangxi Maternal and Child Health Hospital. This was a retrospective study with no patient identifiers. All patients who participated in the study provided informed consent.
Consent for publication
All patients in this study provided their consent for publication.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, N., Zhou, J., Lu, W. et al. Characteristics and clinical evaluation of X chromosome translocations. Mol Cytogenet 16, 36 (2023). https://doi.org/10.1186/s13039-023-00669-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-023-00669-7