
Abstract
Purpose
XX male syndrome also known as De la Chapelle syndrome/Testicular Disorder of Sex Development (DSD) is a rare genetic abnormality, identified by a partial or complete mismatch between phenotypic and genotypic gender of an individual. The present study describes the pertinent clinical, biochemical, cytogenetics, and molecular findings in four phenotypically normal males, presented with gonadal dysgenesis and hypergonadotrophic hypogonadism.
Method
Clinical characteristics and biochemical parameters in four patients were assessed. Further, chromosomal analysis has been performed using conventional karyotyping. FISH and Y chromosome microdeletion assays were carried out to confirm the presence of male-specific genes followed by microarray analysis.
Result
Chromosomal analysis revealed a 46,XX karyotype, FISH showed the presence of 2 normal X chromosomes along with translocation of the SRY gene on the short (p) arm of one of the X chromosome. Molecular analysis for Y chromosome microdeletion revealed the presence of the SRY gene with a complete absence of azoospermic factor regions (AZFa, AZFb, and AZFc) on the long (q) arm of the Y chromosome. Chromosomal microarray revealed no significant copy number variation.
Conclusions
The peculiar translocation of the SRY gene in 46,XX males strongly supports the inclusion of cytogenetic testing for establishing diagnosis and genetic counseling for infertility and/or hormonal imbalances in individuals. The present study provides insight into the cascade of events triggered by the SRY gene in the XX genome, which reinforces the differentiation towards the formation of testes while actively inhibiting ovarian development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sex development and determination in humans is an intricate process, orchestrated by a cascade of molecular events. The Y chromosome is one of the most important factors for the gender assignment of a fetus. Universally, 46,XX embryos develop as females and 46,XY as males. However, genomic alterations result in atypical sexual development, ranging from full sex reversal to the presence of both ovarian and testicular tissues. Hence individuals with 46,XY genotype may develop as phenotypically females, while those with 46,XX genotype having SRY gene presence/absence may be phenotypically males. 46,XX testicular disorder of sex development is a peculiar abnormality of sex determination with an occurrence of 1 in 20,000 and is characterized by a variable degree of mismatch between the genotype and phenotype of the affected individual [1]. It is characterized into three groups; individuals with male external genitalia, individuals with atypical genitalia, and individuals with male external genitalia with signs of hypovirilization (e.g., hypospadias) and in adulthood, typically small testicular volume. The most common phenotype in testicular DSD patients at birth is the one with male external genitalia. Sometimes, they are also reported to have rudimentary testis, hyalinization of the seminiferous tubules, primary hypogonadism, azoospermia, gynecomastia, hypospadias, and/or infertility [2].
The sex-determining region located on the Y chromosome (SRY) encodes a testis-determining factor (TDF) that promotes male development by initiating the undifferentiated gonadal tissue of an embryo to form testis [3]. SRY gene is a genetic switch controlling male development. Therefore, 46,XX males can be divided into SRY-positive and SRY-negative based on the occurrence of the SRY gene [4]. Approximately, 90% of SRY-positive 46,XX males exhibit normal male genitalia at birth, whereas, the remaining 10% have atypical features, like hypospadias [5]. This disorder is caused by the translocation of the SRY gene during the spermatogenesis process. The testicular disorder in 46,XX males with the presence of the SRY gene is almost never inherited as it is genetic and usually associated with infertility [6]. There are various reasons for the male phenotype in XX male syndrome. One of them could be due to an erroneous exchange of varying levels of Y chromosome regions, including the SRY gene, translocating most often to the short (p) arm of chromosome X, resulting in a 46,XX karyotype with a male phenotype. The other causes could be due to a mutation in an unknown autosomal or X-linked gene of the testis-determining pathway or any cryptic Y chromosome mosaicism [7]. SRY-positive 46,XX males present with clinically variable features. These individuals frequently present with appropriate male external genitalia in the prepubertal stage while showing post-pubertal signs of hypovirilization (infertility and small testicular volume).
Y chromosomal microdeletion contributes significantly to spermatogenetic impairment in XX males due to deletions in the Azoospermia factor (AZF) region which can be further divided into AZFa, AZFb, AZFc, and AZFd regions. Deletions of the entire AZFa or AZFb regions is associated with azoospermia, however, AZFc deletions result in variable phenotype ranging from azoospermia to mild oligozoospermia [8]. The phenotypes of 46,XX males are determined by the sex determination cascade encompassing SRY, DAX1, GFG9, WT1, SF1, SOX3, and SOX9 genes [9]. The present study emphasizes the genotype-phenotype correlation in SRY-positive 46,XX individuals resulting in male phenotype, abnormal hormone levels, and eventually infertility. We have analyzed clinical characteristics, biochemical parameters, chromosomal karyotype, and related genes in four individuals presented with hypogonadotropic hypogonadism.
Materials and Methods
Patient information
From 2019 to 2022, four males of Indian origin having a history of gonadal dysgenesis and hypogonadotropic hypogonadism were referred for routine hormonal and cytogenetic testing. Physical, biochemical, and routine clinical examinations are listed in Table 1. The ethics committee of the reference laboratory approved the study. In view of a retrospective observational study and to maintain the anonymity of data, a waiver for an informed consent form was obtained.
Conventional chromosomal investigations
Cytogenetic analysis was performed on the peripheral blood lymphocytes of four patients. The samples were cultured in complete RPMI 1640 medium for 72 h, followed by arrest of the metaphases using colcemid (50 μg/ml) Metaphase chromosome spreads were stained using GTG-Banding at a band level of 500–550 following standard protocol [10]. At least 30 metaphases per patient were analyzed as per routine chromosomal analysis using Cytovision 7.0 image analysis software (Leica Biosystems, Germany). Chromosomal abnormalities were designated following the International System for Human Cytogenetic Nomenclature, 2020 [10].
Fluorescence in situ hybridization analysis (FISH)
FISH analysis was performed on the metaphases spread on slides to ascertain the presence of the SRY gene [11]. FISH probes, X centromere (CEP X), and SRY-locus specific (LSI SRY) (Vysis Inc., USA) were targeted on metaphases, as well as, interphase nuclei according to the manufacturer’s protocol. FISH microscopic examination was assessed employing an Olympus BX63 microscope (Olympus, Japan)
Y chromosome microdeletion assay
Genomic DNA was extracted from peripheral blood leukocytes of patients using a Genomic DNA Isolation Kit (PUREGENE, USA) as per the manufacturer’s instructions. The genomic DNA concentration was determined at 260 nm using a spectrophotometer. The patient’s DNA samples were considered for Y microdeletions assay using a Promega Y chromosome microdeletion kit (version 2.0). Y chromosome microdeletion assay was performed by setting up a multiplex polymerase chain reaction to detect deletions in AZF regions and the SRY gene. The set of STS tagged sites for the diagnosis of microdeletion of AZFa, AZFb, and AZFc regions included: AZFa: sY81, sY84, sY86, sY182; AZFb: sY121, sY133, sY124, sY127, sY128, sY130, sY134, sYPR3; AZFc: sY157, sY254, sY255, sY145, sY152, sY242, sY208; SRY: sY14 and ZFX/ZFY. PCR-generated amplicons were then electrophoresed on 4% agarose gel.
Chromosomal single nucleotide polymorphism (SNP) array
The genomic DNA of patients was subjected to microarray analysis using the Affymetrix Cytoscan 750K assay (Affymetrix, USA) as per the manufacturer’s instructions. Genomic DNA was denatured, subjected to restriction digestion, and amplified by PCR technique. The amplicons were purified, fragmented, and end-labeled with biotin followed by hybridization as per the standard protocol [10]. Copy number variation was analyzed by Chromosome Analysis Suite software (ChAS) v4.2 (Affymetrix, Inc.) by mapping it against the reference genome, GRCh37 (Hg19), to identify the chromosome positions. ChAS software enables the detection of the chromosomal imbalance with the respective clinical phenotype by collecting and overlapping the generated data against different public repositories namely Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER), a database of genomic variants (DGVa) and Online Mendelian Inheritance in Man (OMIM) database.
Results
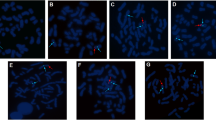
Biochemical parameters revealed that all four individuals had normal prolactin and estradiol levels, higher FSH and LH levels, and lower testosterone levels (Table 1). Conventional chromosome analysis showed the presence of an abnormal chromosomal complement with 2 normal X chromosomes and no Y chromosome in all subjects, suggesting a 46,XX karyotype in phenotypically male individuals. Case 1 showed a normal female chromosome complement along with the presence of heterochromatin on the long arm of chromosome 9 (46,XX,9qh+) (heterochromatic region is not considered abnormal). Cases 2, 3, and 4 had a normal female, 46,XX karyotype (Fig. 1). The FISH analysis confirmed the presence of two copies of X chromosomes, along with the SRY gene translocated on the distal end of the short (p) arm of one of the X chromosomes, in all four cases (Fig. 2). Molecular analysis specific to Y chromosome microdeletion revealed the presence of the SRY gene, with complete deletion of the AZF regions (Table 2 and Fig. 3). Microarray analysis performed on cases 1 and 2 showed similar abnormal molecular karyotypes. A mosaic gain of ~ 7.5 Mb was noted in the short arm of chromosome X-arr[GRCh37]Xp22.33p22.31(168,552_7,716,350)x2~3. A microduplication of ~ 908 kb was noted in the short arm of chromosome X-Xp22.33(1,832,913_2,740,608)x3. A gain of ~ 3.4 Mb was observed in the short arm of chromosome Y-Yp11.31p11.2(2,650,425_6,114,035)x1(Data not shown). Microarray analysis revealed the presence of SRY gene along with other OMIM significant genes which did not have any clinical implications on the present condition of the patients studied (data not shown).

Conventional karyotyping of G-banded chromosomes. G-banded chromosomal analysis revealed a female chromosome complement for all the cases. a Case 1 depicts a karyotype; 46,XX,9qh+, the heterochromatin on the long arm of chromosome 9 is observed in all cells. b–d Cases 2, 3, and 4 show a karyotype, 46,XX in all the cells examined

Fluorescent in situ hybridization (FISH) on metaphase chromosomes with SRY-locus specific (orange)/CEP X (green) probes: a–d G-banded metaphase spread and FISH image using targeted probe showing normal X chromosomes (green signal for centromeric DXZ1 locus) and SRY gene (orange) translocated to the distal end of the short (p) arm of one of the X chromosome in all the 4 cases studied. Yellow arrows indicate the SRY signal and green arrows indicate the X centromere

Y chromosome microdeletion analysis. The amplification products from a, b, c, d, and e reactions. Lanes: 1 Marker; 2, male genomic DNA control; 3-6, cases 1–4 sample DNA (containing deletions). Bands generated with male genomic DNA control are compared to those generated with a patient sample (cases 1–4) containing Y chromosome microdeletions. The marker (M) lanes contain 50 bp DNA Step Ladder. The sole presence of the SRY gene in the cases studied is highlighted with a yellow box
Discussion
46,XX male syndrome is an unusual genetic sex reversal condition associated with a male phenotype in an individual with a 46,XX karyotype. The testis determining factor encoded by the SRY gene located on the distal end of the Y chromosome is one of the main determining factors of male sex determination of the growing fetus [12]. The genetics of SRY-positive 46,XX male syndrome is due to unequal crossing over between pseudoautosomal regions of sex chromosomes during paternal gametogenesis. Thus, the abnormal X chromosomes in 46,XX SRY-positive patients could be a de novo event originating during paternal meiosis I in spermatogenesis. SRY-positive 46,XX males are diagnosed by a multidisciplinary approach involving the evaluation of clinical features, biochemical parameters, and cytogenetics testing. During the adolescence stage, a majority of the 46,XX males have normal levels of testosterone and free testosterone which gradually decrease during adulthood resulting in hypogonadotropic hypogonadism [13]. Our patients were reported to have reduced testicular volume and presented with hypogonadotropic hypogonadism. Our observations correlated with that of the existing literature as the levels of testosterone were found to be decreased in all four patients studied [13]. The elevated levels of FSH and LH support the notion that though the majority of 46,XX SRY-positive males are born with normal male external genitalia they will, at least in adulthood, show signs of hypovirilization (typically reduced testicular volume) and impaired spermatogenesis. The patients in the present study were less than 173 cm in height (the average height of Asian males) which is similar to the normal female’s height and shorter than men with Klinefelter syndrome (KS) [14].
These patients present with infertility due to the deletion of AZF regions, involved in regulating normal spermatogenesis [15]. The Y-chromosome microdeletion analysis in all four patients showed a complete lack of the AZF regions on the Y chromosome. The infertility is probably due to the deletion of AZFa, AZFb, and AZFc regions since the complete mechanism and genes involved in spermatogenesis are still unknown. Studies have reported that testicular biopsies performed on 46,XX males showed the absence of spermatogenic cells with the presence of only Leydig and Sertoli cells [16]. The presence of rudimentary testes in XX males is supported by the absence of germ cells [17]. In some studies conducted on gonadal histological samples of children, immature germ cells were found. The gametogenesis could, therefore, not be completely absent but rather defective and mostly unable to proceed into the later maturation phases [18, 19]. Additionally, a study has reported neurocognitive impairment in 46,XX males with AZF deletion [20]. In our study, microarray analysis of cases 1 and 2 showed duplication of Xp22.33 region with the presence of an OMIM significant gene namely NLGN4X known to be associated with intellectual disability [21]. Microarray findings were considered benign as they did not correlate with the present clinical condition of the subjects. About 30% of cases have been reported in neoplastic transformation (gonadoblastoma) of dysgenetic gonads, particularly upon detection of Y chromosome material [22]. Gonadectomy /gonadal biopsy in the nonfunctioning gonads might reduce the chances of developing malignancy, albeit cosmetic surgery and testosterone replacement therapy should be proposed for patients with gynecomastia and hypogonadism, respectively [23]. Periodic self-assessment and timely gonadal ultrasound imaging should be encouraged as part of cancer surveillance. Androgen replacement psychosocial support and reconstructive surgery could aid in the management of 46,XX males.
Conclusion
46,XX testicular disorder is a rare sex chromosome abnormality, bearing clinical and reproductive consequences for the patients. If left undiagnosed and untreated, the patient may develop testicular failure and therefore testosterone deficiency. A multidisciplinary approach is imperative to diagnose, manage, and prognosticate patients with XX syndrome. For cases presenting with hypogonadotropic hypogonadism, azoospermia, and infertility, cytogenetics analysis is recommended to rule out the differential diagnosis of this rare syndrome, including Klinefelter’s syndrome, or any mosaic variations of disorders of sex development. Our study provides evidence that conventional cytogenetics in conjunction with PCR-based DNA diagnostic methodology plays an indispensable role in establishing diagnosis of sex disorders in individuals and is also important in identifying patients with a higher risk of infertility.
Availability of data and materials
The datasets analyzed during the current study are available upon request.
Abbreviations
- DSD:
-
Disorder of sex development
- AZF:
-
Azoospermic factor regions
- TDF:
-
Testis determining factor
- SRY :
-
Sex-determining region Y gene
References
Chen T, et al. Clinical and genetic analysis in males with 46,XX disorders of sex development: a reproductive centre experience of 144 cases. Andrologia. 2019;51(4):e13232.
Grigorescu-Sido A, et al. Three new 46,XX male patients: a clinical, cytogenetic and molecular analysis. J Pediatr Endocrinol Metab. 2005;18(2):197–203.
Anık A, Çatlı G, Abacı A, Böber E. 46,XX male disorder of sexual development:a case report. J Clin Res Pediatr Endocrinol. 2013;5(4):258–60.
Jain M, Veeramohan V, Chaudhary I, Halder A. The sertoli cell only syndrome and glaucoma in a sex - determining region Y (SRY) positive XX infertile male. J Clin Diagn Res. 2013;7(7):1457–9.
Terribile M, et al. 46,XX Testicular Disorder of Sex Development (DSD): A Case Report and Systematic Review. Medicina (Kaunas). 2019;55(7):371.
Frühmesser A, Kotzot D. Chromosomal variants in klinefelter syndrome. Sex Dev. 2011;5(3):109–23.
Acién P, Acién M. Disorders of sex development: classification, review, and impact on fertility. J Clin Med. 2020;9(11):3555.
Krausz C, Riera-Escamilla A. Genetics of male infertility. Nat Rev Urol. 2018;15(6):369–84.
Ono M, Harley VR. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol. 2013;9(2):79–91.
Zhu Y, Hu L, Cao D, Ou X, Jiang M. Chromosomal microarray analysis of infertile men with azoospermia factor microdeletions. Gene. 2020;20(735):144389.
Mishra SR, et al. Complex rearrangement in acute myeloid leukemia M2 with RUNX1/RUNX1T1 fusion involving chromosomes 8, 17 and 21. Mol Cytogenet. 2021;14(1):28.
Rizvi AA. 46, XX man with SRY gene translocation: cytogenetic characteristics, clinical features and management. Am J Med Sci. 2008;335(4):307–9.
Wang T, Liu JH, Yang J, Chen J, Ye ZQ. 46, XX male sex reversal syndrome: a case report and review of the genetic basis. Andrologia. 2009;41(1):59–62.
Poplinski A, Wieacker P, Kliesch S, Gromoll J. Severe XIST hypomethylation clearly distinguishes (SRY+) 46,XX-maleness from Klinefelter syndrome. Eur J Endocrinol. 2010;162(1):169–75.
Li TF, et al. 46,XX testicular disorder of sexual development with SRY-negative caused by some unidentified mechanisms: a case report and review of the literature. BMC Urol. 2014;14:104.
Majzoub A, Arafa M, Starks C, Elbardisi H, Al Said S, Sabanegh E. 46 XX karyotype during male fertility evaluation; case series and literature review. Asian J Androl. 2017;19(2):168–72.
Lapoirie M, Dijoud F, Lejeune H, Plotton I. Effect of androgens on Sertoli cell maturation in human testis from birth to puberty. Basic Clin Androl. 2021;31(1):31.
Boucekkine C, et al. Clinical and anatomical spectrum in XX sex reversed patients. Relationship to the presence of Y specific DNA-sequences. Clin Endocrinol (Oxf). 1994;40(6):733–42.
Costanzo M, et al. Gonadal tumor development in 46,XX disorders of gonadal development. Eur J Endocrinol. 2022;187(3):451–62.
Chen T, et al. Possible misdiagnosis of 46,XX testicular disorders of sex development in infertile males. Int J Med Sci. 2020;17(9):1136–41.
Laumonnier F, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74(3):552–7.
Dicken BJ, et al. Gonadal dysgenesis is associated with worse outcomes in patients with ovarian nondysgerminomatous tumors: a report of the Children's Oncology Group AGCT 0132 study. Pediatr Blood Cancer. 2018;65(4):10.
Surampudi PN, Wang C, Swerdloff R. Hypogonadism in the aging male diagnosis, potential benefits, and risks of testosterone replacement therapy. Int J Endocrinol. 2012;2012:625434.
Acknowledgements
The authors are grateful to the patients and their family members for supporting the case study. We would also like to thank the management of Dr. Lal Path Labs for providing excellent diagnostic facilities.
Funding
None.
Author information
Authors and Affiliations
Contributions
LR, SP, RK, GS, and MS performed the cytogenetics studies and contributed to data collection. LR interpreted the data and drafted the manuscript. SJ performed the FISH analysis. RN, VL, and VKT revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Dr. Lal Pathlabs Ltd. (EC/NEW/INST/2021/1702) on October 10, 2022. Informed and written consent was obtained.
Consent for publication
Informed and written consent for the publication was obtained from the participants/guardian.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rawal, L., Prabhash, S., Kumar, R. et al. 46,XX males with SRY gene translocation: cytogenetics and molecular characterization. J Rare Dis 3, 1 (2024). https://doi.org/10.1007/s44162-023-00025-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44162-023-00025-8