
Abstract
Background
Deletion or duplication on the distal portion of the long arm of chromosome 1 result in complex and highly variable clinical phenotype including.
intellectual disability and autism.
Case presentation
We report on a patient with intellectual disability and a 763.3 Kb duplication on 1q43 that includes only CHRM3, which was detected by next generation sequencing (NGS). The patient presented with intellectual disability, developmental delay, autistic behavior, limited or no speech, social withdrawal, self-injurious, feeding difficulties, strabismus, short stature, hand anomalie, and no seizures, anxiety, or mood swings, and clinodactyly.
Conclusions
We propose that CHRM3 is the critical gene responsible for the common characteristics in the cases with 1q43 duplication and deletion.
Similar content being viewed by others

Introduction
The clinical phenotype associated with chromosome 1q43-q44 deletion and duplication is characterized by substantial intellectual disability, limited or no speech, and craniofacial dysmorphology, hand and foot anomalies, congenital heart, genital malformation, and central nervous system (CNS) abnormalities, including microcephaly, seizures, and agenesis of the corpus callosum [1]. The clinical phenotype of the terminal region of chromosome 1 deletion and duplication reported by references is highly variable, due to the size of the segment and the number of genes involved [2,3,4,5,6,7,8,9,10,11]. Accordingly, qualification and quantification of the critical region/regions associated with these particular characteristics would provide more evidence in genotype-phenotype correlation.
Here, we report the first case of a 763.3 kb duplication at chromosome 1q43 involving only CHRM3 which encodes for the M3-muscarinic receptor, who has intellectual disability, developmental delay, autistic behavior, short stature, strabismus, hand anomalies, limited or no speech, feeding difficulties, social withdrawal, self-injurious, and a tendency toward self-injurious stereotypic behavior. The clinical features of this patient with pure 1q43 duplication are different from a previously reported case of a ~ 3½ year-old boy with a pure 473 kb deletion at 1q43 comprising only CHRM3, who has no short stature and hand anomalies [12]. We propose that CHRM3 is the critical gene responsible for the common characteristics in the cases with 1q43 duplication and deletion.
Materials and methods
Case presentation
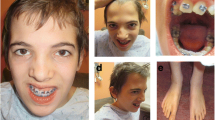
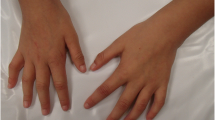
The patient, a boy, was born at term via uncomplicated spontaneous vaginal delivery to an 24-year-old gravida at 38 weeks of gestation. His birth weight was 3.5 kg. Prenatal course had no preeclampsia; neonatal history was benign. Both parents had no history of neurological disease and developmental delays. At ~ 12 months, his parents became concerned for delays in language skills. At 3 yrs., he was given a diagnosis of autism disorder by pediatric evaluation. His past medical history is significant for strabismus, short stature and hand anomalie (Fig. 1). He is impulsive, hyperactive and inattentive in terms of behavior, and has severely limited social skills.

The figure shows some clinical features like strabismus, short stature, and hand anomalie
Next generation sequencing (NGS) analysis
DNA was extracted from peripheral blood at 2018 January using QIAamp DNA Blood Mini Kit (Qiagen, Germany), and was quantified by using Qubit dsDNA BR Assay Kit and Qubit 2.0 Fluorometer (Life Technologies, USA). KAPA HTP Library Preparation Kit Illumina® Platforms (Kapa Biosystems, USA) was utilized for highthroughput library construction for Illumina sequencing, starting from fragmented, double-stranded DNA (dsDNA) according to manufacturer’s instructions. Reads of the sequencing run for patient, patient’s father and mother were 8.5 M, 9.2 M, 9.8 M, respectively. After quality control, coverage percentages of ROI were calculated using the filtered reads at 0.20X, 0.22X, 0.23X for patient, patient’s father and mother, respectively. Genome Analyzer Toolkit (GATK) was used for base quality score recalibration, and single-nucleotide variants (SNV) discovery across was performed for all three samples. The whole-exome enrichment and sequencing (WES) was determined based on the Agilent SureSelect Human All Exon platform (Agilent Technologies), which targets ~ 50 Mb of the human exonic regions. The SureSelect system uses ~ 120-base RNA probes to capture known coding DNA sequences (CDS) from the NCBI Consensus CDS Database as well as other major RNA coding sequence databases, such as Sanger miRBase.
Results
As a part of intellectual disability and autism workup, the patient undergoes genetic testing. NGS analysis showed a single duplication of 763.3 Kb at 1q43 region (239,049,010-239,812,310, GRCh37, NCBI) (Figs. 2 and 3), including only CHRM3. The schematic of chromosome 1q43 region is shown in Fig. 2. The clinical manifestations of the case with a pure 473 kb deletion at 1q43 comprising only CHRM3 reported by Petersen et al. [12], along with the present case are presented in Table 1. The inheritance of duplication in our case could not be observed in parental samples. MR images of the patient’s head showed normal structures (Fig. 4). CHRM3 gene was determined using whole-exome enrichment and sequencing. And the single-nucleotide polymorphism (SNPs) were not found in CHRM3 (Fig. 5).

Overview of chromosome 1 with a 763.3 kb duplication of 1q43 region involving CHRM3. The region of interest has been magnified. The inheritance of duplication in our case could not be observed in parental samples

Genome maps of patient, patient’s father and mother

MRI of the head showed normal structures. a Axial FLAIR image at the level of the lateral ventricles. b Diffusion Tensor Imaging (DTI) image. c, d DTI color maps

Whole-exome sequencing. The single-nucleotide polymorphism (SNPs) were not found in CHRM3
Discussion
We report the first case of a 763.3 kb duplication at chromosome 1q43 involving only CHRM3 (Acetylcholine receptor, muscarinic, 3; OMIM: 118494) which encodes for the M3-muscarinic acetylcholine receptor. M3-muscarinic acetylcholine receptor is the predominant muscarinic subtype mediating acetylcholine-induced airway smooth muscle contraction, one of 5 subtypes (M1-M5) of muscarinic receptors, found throughout the CNS and peripheral tissue. The clinical features of this patient with pure 1q43 duplication are different from a previously reported case of a ~ 3½ year-old boy with a pure 473 kb deletion at 1q43 comprising only CHRM3 [12] (Table 1). Both children exhibit substantial intellectual disability, developmental delay, autistic behavior, limited or no speech, social withdrawal, self-injurious, feeding difficulties, strabismus, and no seizures, anxiety, mood swings, and clinodactyly. However, the patient in our report has short stature and hand anomalie, which were not observed in the patient with 473 kb deletion including CHRM3 reported by Petersen et al. [12]. The deletion in the patient reported by Petersen et al. includes a 380 kb upstream region and the first four exons of CHRM3, whereas in our patient with a 763.3 kb duplication at 1q43 includes 343 kb in CHRM3 which involves the first seven exons of CHRM3 [13]. The entire cDNA encoding the m3 receptor comprise 4559 bp in 8 exons, exon 1 includes a cluster of transcriptional start sites, exons 2, 4, 6 and 7 are subject to alternative splicing [13]. The difference in clinical features might because of the various length of CHRM3 gene involved. As short stature was observed in a previously reported case of a 7 year-old autistc boy with a 911 kb deletion at 1q43 encompassing all 8 exons of CHRM3 and two other genes: RPS7P5 and FMN2 [14]. Moreover, compared with 1q43-q44 deletion, hand abnormalities appear to be more associated with 1q43-q44 duplication [15,16,17,18,19,20,21,22,23]. However, abnormal hand was not observed in a report that a deletion and a duplication of approximately 6.0 Mb at 1q43-q44 involving CHRM3 in the proband and her younger brother [15]. Compared with 1q43-q44 deletion, the clinical manifestations of the patient with 1q43-q44 duplication appear to be mild and mainly include macrocephaly, mental retardation and mild malformation.
The muscarinic acetylcholine receptors belong to the family of G protein-coupled receptors that regulate numerous fundamental functions of the central and peripheral nervous systems. The muscarinic acetylcholine receptors consist of five distinct subtypes (M1, M2, M3, M4, M5) which encoded by the genes CHRM1, CHRM2, CHRM3, CHRM4 and CHRM5. M1, M3 and M5 receptors tend to couple to G proteins of the Gq/11 family, whereas M2 and M4 receptors preferentially signal through the Gi/o family of G proteins [24]. The M3 muscarinic acetylcholine receptor (M3R) is distributed dominantly in the central and peripheral nervous systems, cardiac muscles, smooth muscles, and exocrine glands, plays a central role in many fundamental functions of the CNS and other human physiology, including regulating heart rate, smooth muscle contraction, and glandular secretion [25]. In CNS, M3-muscarinic receptors have been specifically related to behavior modulation, for example, M3-muscarinic receptor activation could stimulation dopamine release and enhances signal transduction in the hippocampus (related to memory acquisition), striatal nucleus (related to reward and conditioning), nucleus accumbens (related to addiction and pleasure) and amygdala (related to fear conditioning and risk taking). The KO mice for the CHRM3 gene were found deficits in tasks involving fear conditioning, learning and memory [26]. Furthermore, CHRM3 expression was different in the 12 different brain regions. The brain stem, cerebellum, and olfactory region showed decreased expression patterns. As 8 exons of CHRM3 gene were derived from three distinct promoter regions (T1: L1HS, T2, T4: original, T3, T3–1:THE1C) [27], these transposable element-derived transcripts showed tissue-specific expression patterns. One retroelement could be cleaved by other retroelement integration events in different tissues. Thus, the various exons of CHRM3 gene involved might account for the differences in clinical features between our patient with 1q43 duplication and the case with 1q43 deletion. Much about the specific role of CHRM3 in human CNS pathophysiology remains to be elucidated, identification of well-detailed genotype-phenotype correlations in this case as well as others of 1q43 duplication and deletion would provide more possible links between M3-muscarinic receptor pathophysiology and the neurocognitive phenotype. For example, it was reported that SNPs rs6700381 in CHRM3 gene in 1q43 was associate with its role in multiple brain functional connectivities [28]. In this case, SNPs were not found in CHRM3. Therefore, a duplication of 763.3 Kb at 1q43 region might result in abnormal protein structure of CHRM3, which might account for the clinical features.
Conclusions
We report the first case of a 763.3 kb duplication at chromosome 1q43 involving only CHRM3. In view of these common characteristics in our patient and the case with 1q43 deletion, we propose CHRM3 as the critical gene responsible for intellectual disability, developmental delay, autistic behavior, limited or no speech, social withdrawal, self-injurious, feeding difficulties, strabismus, and other overlapping phenotype in our patient as well as other patients with 1q43 deletions and duplication including CHRM3.
Abbreviations
- CNS:
-
Central nervous system
- M3R:
-
M3 muscarinic acetylcholine receptor
- NGS:
-
Next generation sequencing
References
Ballif BC, Rosenfeld JA, Traylor R, Theisen A, Bader PI, Ladda RL, Sell SL, Steinraths M, Surti U, McGuire M, et al. High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet. 2012;131(1):145–56.
Meinecke P, Vogtel D. A specific syndrome due to deletion of the distal long arm of chromosome 1. Am J Med Genet. 1987;28(2):371–6.
Halal F, Vekemans M, Kaplan P, Zeesman S. Distal deletion of chromosome 1q in an adult. Am J Med Genet. 1990;35(3):379–82.
Rotmensch S, Liberati M, Luo JS, Tallini G, Mahoney MJ, Hobbins JC. Prenatal diagnosis of a fetus with terminal deletion of chromosome 1 (q41). Prenat Diagn. 1991;11(11):867–73.
Dallapiccola B, Ferranti G, Pachi A. Prenatal diagnosis of terminal deletion 1 (q42). Prenat Diagn. 1992;12(10):853.
Ioan DM, Maximilian C, Kleczkowska A, Fryns JP. Distal deletion of the long arm of chromosome number 1 (q43-->qter) associated with severe mental retardation and a nonspecific dysmorphic syndrome. Ann Genet. 1992;35(3):167–9.
Schefels J, Keller-Rottger E, Esser KJ. Terminal deletion of chromosome 1--a recognizable condition. Eur J Pediatr. 1996;155(8):720.
Gentile M, Di Carlo A, Volpe P, Pansini A, Nanna P, Valenzano MC, Buonadonna AL. FISH and cytogenetic characterization of a terminal chromosome 1q deletion: clinical case report and phenotypic implications. Am J Med Genet A. 2003;117A(3):251–4.
van Bever Y, Rooms L, Laridon A, Reyniers E, van Luijk R, Scheers S, Wauters J, Kooy RF. Clinical report of a pure subtelomeric 1qter deletion in a boy with mental retardation and multiple anomalies adds further evidence for a specific phenotype. Am J Med Genet A. 2005;135(1):91–5.
Hill AD, Chang BS, Hill RS, Garraway LA, Bodell A, Sellers WR, Walsh CA. A 2-Mb critical region implicated in the microcephaly associated with terminal 1q deletion syndrome. Am J Med Genet A. 2007;143A(15):1692–8.
Hiraki Y, Okamoto N, Ida T, Nakata Y, Kamada M, Kanemura Y, Yamasaki M, Fujita H, Nishimura G, Kato M, et al. Two new cases of pure 1q terminal deletion presenting with brain malformations. Am J Med Genet A. 2008;146A(10):1241–7.
Petersen AK, Ahmad A, Shafiq M, Brown-Kipphut B, Fong CT, Anwar Iqbal M. Deletion 1q43 encompassing only CHRM3 in a patient with autistic disorder. European journal of medical genetics. 2013;56(2):118–22.
Forsythe SM, Kogut PC, McConville JF, Fu Y, McCauley JA, Halayko AJ, Liu HW, Kao A, Fernandes DJ, Bellam S, et al. Structure and transcription of the human m3 muscarinic receptor gene. Am J Respir Cell Mol Biol. 2002;26(3):298–305.
Perrone MD, Rocca MS, Bruno I, Faletra F, Pecile V, Gasparini P. De novo 911 kb interstitial deletion on chromosome 1q43 in a boy with mental retardation and short stature. Eur J Med Genet. 2012;55(2):117–9.
Luo A, Cheng D, Yuan S, Li H, Du J, Zhang Y, Yang C, Lin G, Zhang W, Tan YQ. Maternal interchromosomal insertional translocation leading to 1q43-q44 deletion and duplication in two siblings. Mol Cytogenet. 2018;11:24.
Hemming IA, Forrest AR, Shipman P, Woodward KJ, Walsh P, Ravine DG, Heng JI. Reinforcing the association between distal 1q CNVs and structural brain disorder: a case of a complex 1q43-q44 CNV and a review of the literature. Am J Med Genet B Neuropsychiatr Genet. 2016;171B(3):458–67.
Wang D, Zeesman S, Tarnopolsky MA, Nowaczyk MJ. Duplication of AKT3 as a cause of macrocephaly in duplication 1q43q44. Am J Med Genet A. 2013;161A(8):2016–9.
Flatz S, Fonatsch C. Partial trisomy 1q due to tandem duplication. Clin Genet. 1979;15(6):541–2.
Clark BJ, Lowther GW, Lee WR. Congenital ocular defects associated with an abnormality of the human chromosome 1: trisomy 1q32-qter. J Pediatr Ophthalmol Strabismus. 1994;31(1):41–5.
Duba HC, Erdel M, Loffler J, Bereuther L, Fischer H, Utermann B, Utermann G. Detection of a de novo duplication of 1q32-qter by fluorescence in situ hybridisation in a boy with multiple malformations: further delineation of the trisomy 1q syndrome. J Med Genet. 1997;34(4):309–13.
Cocce MC, Villa O, Obregon MG, Salido M, Barreiro C, Sole F, Gallego MS. Duplication dup (1)(q41q44) defined by fluorescence in situ hybridization: delineation of the 'trisomy 1q42-->qter syndrome. Cytogenetic and genome research. 2007;118(1):84–6.
Kulikowski LD, Bellucco FT, Nogueira SI, Christofolini DM, Smith Mde A, de Mello CB, Brunoni D, Melaragno MI. Pure duplication 1q41-qter: further delineation of trisomy 1q syndromes. Am J Med Genet A. 2008;146A(20):2663–7.
Nowaczyk MJ, Bayani J, Freeman V, Watts J, Squire J, Xu J. De novo 1q32q44 duplication and distal 1q trisomy syndrome. Am J Med Genet A. 2003;120A(2):229–33.
Zhang W, Yamada M, Gomeza J, Basile AS, Wess J. Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1-M5 muscarinic receptor knock-out mice. J Neurosci. 2002;22(15):6347–52.
Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482(7386):552–6.
Poulin B, Butcher A, McWilliams P, Bourgognon JM, Pawlak R, Kong KC, Bottrill A, Mistry S, Wess J, Rosethorne EM, et al. The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/arrestin-dependent manner. Proc Natl Acad Sci U S A. 2010;107(20):9440–5.
Huh JW, Kim YH, Lee SR, Kim H, Kim DS, Kim HS, Kang HS, Chang KT. Gain of new exons and promoters by lineage-specific transposable elements-integration and conservation event on CHRM3 gene. Molecules and cells. 2009;28(2):111–7.
Wang Q, Cheng W, Li M, Ren H, Hu X, Deng W, Ma X, Zhao L, Wang Y, Xiang B, et al. The CHRM3 gene is implicated in abnormal thalamo-orbital frontal cortex functional connectivity in first-episode treatment-naive patients with schizophrenia. Psychol Med. 2016;46(7):1523–34.
Acknowledgements
We would like to thank the patients and their family members who kindly agreed to participate in this study.
Funding
This study was supported by grant from the Natural Science Foundation of Inner Mongolia, China (No. 2014MS0815).
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
XC, RH, YL contributed to design the study, collected the literature data and wrote a part of the manuscript. XC, QY performed genetic studies on the patients, contributed to design the study, collected the literature data and wrote a part of the manuscript. JL, JY, HX performed clinical evaluation of the patients. GZ, YP, XL performed genetic studies on the patients. XC, RH, YL contributed to design the study, wrote a part of the article and critically read the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this case report and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cheng, X., Yang, Q., Liu, J. et al. Constitutional 763.3 Kb chromosome 1q43 duplication encompassing only CHRM3 gene identified by next generation sequencing (NGS) in a child with intellectual disability. Mol Cytogenet 12, 16 (2019). https://doi.org/10.1186/s13039-019-0427-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-019-0427-3