
Abstract
Background
Quick genetic diagnosis of a patient with congenital heart disease (CHD) is quite important for proper health care and management. Copy number variations (CNV), chromosomal imbalances and rearrangements have been frequently associated with CHD. Previously, due to limitations of microscope based standard karyotyping techniques copious CNVs and submicroscopic imbalances could not be detected in numerous CHD patients. The aim of our study is to identify cytogenetic abnormalities among the selected CHD cases (n = 17) of the cohort using high density oligo arrays.
Results
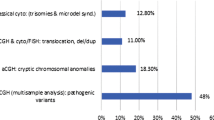
Our screening study indicated that six patients (~35%) have various cytogenetic abnormalities. Among the patients, only patient 2 had a duplication whereas the rest carried various deletions. The patients 1, 4 and 6 have only single large deletions throughout their genome; a 3.2 Mb deletion on chromosome 7, a 3.35 Mb deletion on chromosome 3, and a 2.78 Mb a deletion on chromosome 2, respectively. Patients 3 and 5 have two deletions on different chromosomes. Patient 3 has deletions on chromosome 2 (2q24.1; 249 kb) and 16 (16q22.2; 1.8 Mb). Patient 4 has a 3.35 Mb an interstitial deletion on chromosome 3 (3q13.2q13.31).
Based on our search on the latest available literature, our study is the first inclusive array CGH evaluation on Saudi cohort of CHD patients.
Conclusions
This study emphasizes the importance of the arrays in genetic diagnosis of CHD. Based on our results the high resolution arrays should be utilized as first-tier diagnostic tool in clinical care as suggested before by others. Moreover, previously evaluated negative CHD cases (based on standard karyotyping methods) should be re-examined by microarray based cytogenetic methods.
Similar content being viewed by others


Background
Congenital heart disease (CHD) is the most common anomaly affecting newborns and also leading cause of mortality and morbidity among neonates [1,2,3,4]. This group of disorders is predicted to have an incidence rate of 8–9 in every 1000 live birth [4, 5] and leads to ~10% of spontaneous miscarriages [5]. Despite the still largely ambiguous pathophysiology of CHD, genetic factors were found to contribute to the etiology in many cases. In addition, numerous incidents of CHD were found to have chromosomal abnormalities; particularly among cases with associated multiple organ malformations, developmental delays, and growth abnormalities [6, 7]. Interestingly enough such cases are prone to harbor morbidities of additional chromosomal syndromes such as Williams-Beuren and DiGeorge or even monogenic hereditary disorders such as Noonan [6].
Advances in molecular and cytogenetic techniques in the recent years gave rise to tools of higher sensitivity such as single nucleotide polymorphism (SNP) based microarrays [8], array comparative genomic hybridization (aCGH) platforms [9,10,11] and nextgen sequencing [12] techniques, which are enabling the detection of chromosomal aberrations and sub-microscopic copy number variations (CNVs) on an unprecedented resolution that was not possible with standard and high-resolution karyotyping techniques. This facilitated the discovery of novel pathogenic copy number variations, genes and mutations, and the establishment of genotype-phenotype correlations for various diseases [10, 13,14,15] including heart defects [16]. The dense coverage of the microarray probes can also be quite helpful in refining breakpoints of novel genomic imbalances as well as further characterization and fine mapping of already known gains and losses in different human chromosomes [15, 17].
It has been well-established that standard microscope based chromosome analysis misses quite many gains and losses due to its low resolution. Hence, aCGH and/or similar array platforms have been proposed to be utilized as a first-tier diagnostic tool for various disorders including autism, intellectual disability and more recently for newborn screenings of CHD patients [18,19,20,21]. In this study we screened Saudi CHD patients using high density oligo arrays to identify likely chromosomal imbalances.
Methods
Patients
We ascertained 223 patients inflicted with one or more of the following clinical problems: autism spectrum disorder, intellectual disability, heart defects, developmental delay, language delay, and dysmorphic features of unknown origin evaluated at the Kind Faisal Specialist Hospital and Research Center using the institutionally approved IRB protocols (RAC# 2040042, 2,030,046, 2,120,022, 2,080,032). Before the sample collection, the patients and parents were signed the written informed consents. All the patients were clinically examined and underwent a consistent study protocol for with perinatal history, and neurological assessment. The patients also underwent aCGH testing as a first-tier approach and then tested with one of the followings; FISH, standard cytogenetics, and targeted sequencing.
DNA isolation
Blood samples were collected from all participants. DNA was isolated using PureGene DNA Purification Kit (Gentra Systems, Inc. Minneapolis, MN, US).
Affymetrix microarrays and analysis
Affymetrix’s Cytogenetics Whole-Genome 2.7 M arrays (Affymetrix Inc., Santa Clara, CA, US) and CytoScan HD arrays were used in the study. Both assays have over 2 million probes that interrogate polymorphic and nonpolymorphic genomic sequences. The assay preparation, scanning, image processing, genotyping, and preliminary data analysis were all done according to manufacturer’s protocols and guidelines. CNV detection was done using Affymetrix’s in-house developed software called “Chromosome Analysis Software” otherwise known as ChAS using the software’s default detection settings for high resolution. Previously reported benign CNVs were excluded from the analysis.
Cytogenetic banding analysis
The microscope based standard karyotype analysis was performed onTrypsin-Wright (GTW) banded metaphase spreads (at least 20 metaphases were analyzed and 2 were karyotyped using cultured peripheral blood lymphocytes according to standard protocols. Karyotypes were interpreted according to the International System for Human Cytogenetic Nomenclature.
Array CGH
A custom designed oligonucleotide microarray assay from Agilent (Agilent Technologies, Santa Clara, CA, USA) was utilized for CNV assessment [22]. The assay was developed and tested through an academic laboratory consortium [22]. Human male or female DNA (Promega Corp., Nepean, Canada) was used as a reference control. After DNA GC check, good quality DNA was digested, labelled, and then hybridized onto the custom arrays. Then, the slides were washed and scanned with either Agilent DNA Microarray Scanner (Agilent Technologies) or GenePix 4000B (Molecular Devices, Sunnyvale, CA). The images were processed using Agilent Feature Extraction software (v10.0), and transferred to Agilent Genomic Workbench software for data analysis, CNV visualization, and detection. During the analysis, softwares defaults were not changed and the analysis was based on human genome build hg19/GRCh37. All the assays protocols were performed according to the manufacturer’s instructions (Agilent Inc.).
Fish
Confirmatory Fluorescence in situ hybridization (FISH) analysis was performed using p-arm and q-arm specific probes for the all chromosomes using standard protocols (Abbott Laboratories, Abbott Parl, IL, USA). In the absence of ready probes, custom FISH probes were designed and used. The experiments were carried out standard protocols.
Results
Case 1
The patient is now a 13 year old male who was initially admitted to the hospital for percutaneous valve implantation. He was born with pulmonary atresia with ventricular septal defect (VSD) that was completely repaired with RV to PA conduit in July 2003. He did not suffer any significant cardiac complications, and the follow-up examination reported no cardiac symptoms but the patient had speech delays. Cardiovascular examination showed S1 and S2 were normal with systolic murmur in the left lower sternal border. Echocardiogram revealed normal biventricular systolic function. Right-sided ventricular chambers were mild to moderately dilated but the dilatation has improved post-PPVI. Respiratory and gastrointestinal examinations were insignificant. The patient had speech and developmental delays and brain malformation was suspected. He is mentally retarded. An MRI examination revealed a nonspecific right frontal centrum semiovale hyperintense lesion.
Molecular cytogenetic analysis indicated that he has a novel 3.2 Mb deletion extending on 7q33-q34 (Fig. 1). The deletion begins at 137,917,363 bp genomic position and ends at 141,131,675 bp. The deleted region contains 44 genes including 12 uncharacterized genes, 20 pseudogenes, and one miRNA according to Mapviewer Human Annotation Release 107.

The visual diagram is adopted from Chromosome Analysis Suite (Affymetrix Inc.,). From right to left the diagram presents copy number coordinates, the patient’s probe distribution, paternal and maternal probes distributions, OMIM genes, miRNAs, all SNP and copy number probes in the region, and chromosomal coordinates. The patient has 3214 kb deletion (presented in blue color) while father and mother are normal
Case 2
The patient is a 13-years old female referred to our center for evaluation of dysmorphic features and congenital heart disease. She was born at term with uneventful prenatal and peripartum period. She was noticed after birth to have hypoplastic right thumb. Her parents, who are not related, have 4 other normal children. When she was initially evaluated in our center at the age of 6 and ½ years, her examination showed a head circumference of 45 cm (4.7 SD below the mean), a weight of 14.6 kg (4 SD below the mean) and a height of 100.5 cm (3 SD below the mean). She had upslanting palpebral fissures, bulbous nose, malformed right ear, retrognathia, low posterior hairline, webbed neck, and widely spaced nipples. The chest and abdominal examination was unremarkable. The cardiovascular examination revealed normal first heart sound, fixed split of second heart sound, and a systolic murmur grade 3/6 over the left upper sternal border. She had normal tone, power and deep tendon reflexes. The musculoskeletal examination revealed right thumb hypoplasia with absent thenar muscles, absent extensor pollicis longus, and thumb extensors. There was significant instability of the metacarpophalangeal joints of the right thumb. Skeletal survey revealed ankylosis between C3, C4 and C5 spine. The thoraacolumbar spine and the long tubular bones of both upper limbs were osteopenic. The right fifth metacarpal bone was short with hypoplasia of the first right metacarpal bone. The epiphysis of the first metacarpal bone was absent bilaterally. There was mild bilateral subluxation of the hip joints. The tibia and fibula were normal bilaterally. Hallux valgus at the interphalangeal joint was seen bilaterally. There was coning of the epiphysis of the second to fourth toes bilaterally. Echocardiogram revealed large secundum atrial septal defect measuring 12 mm with left to right shunt. Ultrasound of abdomen and pelvis indicated that the left kidney was rotated and ectopic lying down into the pelvis.
As part of routine diagnostic procedures a high-resolution GTW-banding study was carried on the patient’s sample. No gross abnormality was detected. Then, an aCGH experiment was performed as a further clinical screening and indicated an interstitial duplication pointed by 33 oligonucleotide probes on 5q35.2-q35.3. An interphase FISH using a probe (CTD-2301A4) within the duplicated interval re-confirmed the findings. To better characterize the duplication a high-resolution array (Cytogenetics Whole-Genome 2.7 M) from Affymetrix Inc. (Affymetrix Inc., San Paolo, CA, US) was utilized to further delineate the gain missed during the initial standard microscope based-karyotyping. This particular chip array utilizes 2.7 million markers including 400,000 SNP probes that provide whole-genome coverage with the one of the highest density coverage among the present platforms. Based on Affymetrix’s cytogenetic assay results, the duplication extends from 175,349,728 to 177,347,753 bp (hg19) and comprises 58 genes targeted by more than 600 SNP and copy number (CN) probes covering approximately 2.0 Mb (Fig. 2). In comparison to the previously published duplications this duplication seems novel and does not share breakpoints with the compared cases.

The diagram presents interrogated region, results of the patient’s and parental samples. Copy number status is given next to each tested sample. The patient a duplication comprising more than 50 genes and expanding on approximately 2 Mb region on chromosome 5q35.3. Apparently, parental samples do not carry the gain indicating de novo status of the duplication. The visuals are adopted from Chromosome Analysis Suite (Affymetrix Inc.)
Case 3
A 5- year-old Saudi male, the first child of non-consanguineous healthy parents, was born at term following in-vitro fertilization (IVF) pregnancy via cesarean section. His birth weight was 3.5 Kg. He was admitted to the neonatal intensive care unit for 1 week because of Jaundice, treated with phototherapy, and was discovered to have congenital heart disease (patent ductus arteriosus [PDA]). Since early infancy, he was noted to have slow psychomotor development, sitting at 10 months and starting to walk independently at 2 years of age. He had significant delay in initiation of language which he developed. At age 1 month, he was admitted to the hospital with febrile illness and treatment as a case of sepsis. When he was 2 years of age, he underwent surgery to place testes (bilateral orchidopexy). There is no previous history of convulsion; however, recently he developed one episode of unprovoked convulsion with semiology of cyanosis and jerky movements of the limbs. Mother had history of two abortions following IVF pregnancy, and there is no family history of epilepsy or neurological problems. Examination at the age of 4 ½ years revealed no dysmorphic features and no neurocutaneuos marks apart from a single hypo pigmented patch at the right forearm. His growth parameters were: weight 18.2 Kg (75th centile), height 110 cm (75th centile), and head circumference 50 cm (50th centile). Vision and hearing were normal. Cardiovascular examination showed apex beat in the fifth intercostal space within the midclavicular line. There were no thrills, no left parasternal heave or palpable P2. Auscultation revealed no murmurs. Neurological examination revealed no gross abnormalities. Laboratory investigations (including complete blood count [CBC], renal function tests, bone profile, liver function tests [LFT], thyroid function tests, and serum lactate and ammonia) were all normal. Brain magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) were unremarkable. Electroencephalography (EEG) showed normal findings. Recently, the patient had psychometry with IQ score of 57. He was also evaluated by a cardiologist and echocardiogram revealed a small PDA (1.5 mm) with good cardiac functions.
Whole genome screening of chromosomal aberrations using Affymetrix’s cytogenetic microarrays revealed presence of two large hemizygous deletions at chromosomes 2 (2q24.1 Size: 249 kb) and 16 (16q22.2 Size:1819 kb). Genomic locations of the deletions are chromosome 2, positions: 159,140,953–159,390,141 bp, and chromosome 16 positions: 71,589,375–73,408,685 according hg19 (Figs. 3 and 4). The deleted region in chromosome 2 contains 2 genes and chromosome 16 comprises 18 genes, 5 of which are OMIM-annotated and associated with Tyrosinemia type II (OMIM#: 613,018), Anhaptoglobinemia (OMIM#: 140,100), Hypohaptoglobinemia (OMIM#: 140,100), Prostate cancer susceptibility (OMIM#: 104,155). The patient’s mother was also tested and found negative for above mentioned deletions. Unfortunately, paternal DNA sample was not available for testing and we were unable to recruit the father for further investigation.

Microarray results are displayed for chromosome 2q23.3–24.3 bands. A deletion is seen on 2q24.1 cytoband expanding over more than 249 kb genomic region

Microarray result displays for chromosome 16. The second deletion on the patient was observed on chromosome 16q22.2–22.3 bands. The deletion is over 1800 kb detected by more than 1450 molecular markers
Case 4
This is a 7-year-old boy who was the product of full normal spontaneous vaginal delivery with a birthweight of 3.5 kg. He was well until the age of 8 months when he was noticed to have flexion swinging movements in the hands and wrists that were spontaneous and exaggerated by irritability. There were no other abnormal movements or seizures. He was delayed in attaining milestones with severely impaired cognitive, linguistics and social skills. He was also diagnosed with atrial septal defect. His parents are first-cousins. They have 2 other children who are alive and well. There was no family history of a similar disease. On examination, his head circumference was around 50th percentile, weight was at the 97th percentile, and height was just above the 97th percentile. He had low-set ears and prominent philtrum. His tone, power and reflexes were normal. There were no neurocutaneous manifestations. Skeletal survey, brain MRI and ultrasound of abdomen were normal.
Molecular cytogenetic studies identified a 3.35 Mb interstitial deletion on the long arm of chromosome 3 from 112,146,815 bp to 115,496,750 bp (3q13.2q13.31) (hg19). This region contains over 27 genes, one of which is an annotated OMIM disease gene (DRD3). All remaining regions did not show any significant DNA copy number gains or losses.
Case 5
This is a three-year-old girl who was a product of an in vitro fertilization delivered at 34 weeks of gestation to consanguineous parents with negative family history. She was noticed to have dysmorphic features with low set ears, depressed nasal bridge, and long philtrum. Ophthalmological examination revealed left choroidal coloboma involving the macula and optic disc. Echocardiogram showed large atrial septal defect, large, perimembranous ventricular septal defect, hypoplastic right upper pulmonary vein. She had surgical intervention with ASD and VSD closure and repair of upper pulmonary vein stenosis. She has had global developmental delay and growth failure with all the parameters below the 5th percentile (head circumference − 2.7 SD, weight -6SD, height -4SD). Brain MRI and ultrasound abdomen were normal.
Based on 20 metaphase cells, standard g-banding karyotyping at 425 band resolution indicated an apparently balanced translocation between the q-arm of chromosome 10 and the q arm of chromosome 12 (46,XX,t(10;12)(q22;q22)) was found in all the cells; however, loss of chromosomal material cannot be ruled out based on the low band resolution seen. Although this translocation could be de-novo most likely one of the parents is a carrier for the translocation. Follow-up aCGH study indicated presence of two deletions; one on chromosome 12 (12p12.1p11.21; 25,320,816–31,285,151; ~5.96 Mb) and the other on the chromosome 16 (16p11.2; 29,567,295–30,321,320; ~754 Kb), (hg19). The deletion on chromosome 16 is paternally inherited. The larger deletion (chromosome 12) observed in this patient is not present in either parent and therefore appears to be a de novo event.
Case 6
This is a 4-year-old female who was the product of full normal spontaneous vaginal delivery with a birthweight of 1.45. She stayed in the NICU for 2 months and was diagnosed with perimembranous ventricular septal defect. She was noticed to have global developmental delay and poor growth. There was no history of seizures. The parents are not consanguineous. There is no family history of a similar problem. Her physical examination was notable for microcephaly (7 SD below the mean). Her weight was -5SD and height was -3SD. She had low anterior hair line, squint, broad nasal bridge, short philtrum, and micrognathia. The muscle tone was mildly increased in the upper and lower extremities. Ultrasound of abdomen showed mild right upper pole caliectasis.
Molecular cytogenetic analysis revealed presence of 2.78 Mb interstitial deletion in the short arm of chromosome 2 (extending between 59,170,950 bp and 61, 946, 784 bp over chromosome 2p16.1 deletion syndrome region) (Fig. 5). Although the region is large in size, it is relatively gene-poor region with a total of only 27 genes, six of which are OMIM-annotated. Among these PEX13 is known to associate with Zellweger syndrome.

A deletion on chromosome 2p16.1-p15 is presented in the diagram. The deletion comprises several genes and ~2776 kb genomic region, and detected by 2780 SNP and CN probes
Discussion
Case 1
Deletions in the 7q33-q34 region are rarely reported in the scientific literature. Reported deletions in this region are mostly associated with developmental delay, intellectual disability, microcephaly, and significant morphological and developmental phenotypes. The deleted region in this case contains 50 genes including the BRAF; the mutation of which is known to be associated with cardiofaciocutaneous (CFC) syndrome [23], a disease characterized by heart defects, mental retardation and a distinctive facial appearance. BRAF encodes for the BRAF protein, which is involved in the MAP kinase/ERK signalling pathway; an important pathway that implicates various cell processes including growth, differentiation, proliferation, senescence and apoptosis [24]. Mutations in BRAF disrupt the regulation of MAP kinase/ERK pathway and can lead to a range of complications including various types of cancers as well as developmental disorders such as Noonan syndrome (NS), Costello syndrome, LEOPARD syndrome, and Cardiofaciocutaneous syndrome (CFC). Interestingly, only one of the previously described cases shared a deletion in the genomic region constituting the BRAF gene [25]. This makes it a likely candidate to explain the clinical features in these cases.
Case 2
Chromosome 5q35.2-q35.3 deletions are well-known mainly due to Sotos syndrome. Altogether, these genomic alterations reach to a significant number [26,27,28]. Compared to deletions [27, 29,30,31,32,33] duplications in the region are rare and not well-characterized [34,35,36,37]. Moreover, there is no well-established genotype-phenotype correlation for these gains currently since they are in variable sizes and lack precise breakpoints. Interestingly among these cases only singleton have been reported to have Sotos syndrome-like symptom [38]. The rest of cases have different phenotypic findings mostly in the form of developmental delay and short stature. Among these cases, two duplications exceed nearly twice the size of the rest of the gains located on the 5q35.2-q35.3 region [34, 38]. In the present study we describe a patient with a duplication leading to congenital heart disease, cervical ankylosis, and thumb hypoplasia in addition to microcephaly, short stature, and various dysmorphic features. Intriguingly, among the duplication carrying patients, beside our case, there are only three patients who have heart defects [38, 39].
In their study Jamsheer et al. [38] pointed out likely involvement of MSX2 in radial agenesis as well as complex heart defect, and FGFR4 as causative factor of limb formation. Although FGFR4 is shared by both gains (ours and that of Jamsheer et al. [38]), MSX2 is located outside the boundaries of our duplication. Deletions of both genes, NSD1 and FGFR4, were previously reported with congenital heart anomalies [40]. However, interestingly, FGFR4 is not a shared gene between all four cases having heart defects. In other words it is not in the shared region of the patients reported in Rosenfeld et al.’s study [39]. Hence, involvement of this gene in the reported heart defects is less likely. Relatedly Rosenfeld et al. raised the likely contribution of another candidate gene PDLIM7 which is shared among all the cases with the heart defect including ours according to recent human assembly (hg38, 39]. PDLIM7 is a scaffold protein that regulates Tbx5 which has critical roles in heart and limb development. Moreover, suppressed expression of Pdlim7in zebrafish led to the development of heart abnormalities in the animals.
Case 3
The deletion is large (1.8 Mb) and comprises 18 genes (TAT, MARVELD3, PHLPP2, SNORA70D, AP1G1, SNORD71, ATXN1L, ZNF821, IST1, PKD1L3, DHODH, HP, HPR, TXNL4B, DHX38, PMFBP1, ZFHX3, HCCAT5, C16orf47) located in 16q22.1-q24 cytogenetic region. Such deletions are frequently seen among breast cancer patients [41]. Moreover, two different cases having cytogenetic abnormalities in the long arm of chromosome 16, were previously described with ventricular septal defect [42, 43]. However, these abnormalities are not overlapping with 16q22.2 cytogenetic band.
The partial deletion of 16q is implicated in the rare 16q22 deletion syndrome (OMIM #614541) characterized by failure to thrive, growth retardation, dysmorphic facial features, and hypotonia. DHODH is one of the implicated genes that encodes for dihydroorotate dehydrogenase which catalyzes the oxidation of dihydroorotate to orotate, thereby facilitating the biosynthesis of pyrimidine blocks. Moreover, the mutations in DHODH lead to Miller syndrome, also known as postaxial acrofacial dysostosis [44]. Interestingly, it was also found that DHODH is involved in the transcriptional elongation of BRAF [45] that is a well-known oncogene, a member of the Raf kinase family, and an important molecule for RAS/MAPK signaling pathway. Mutations in this gene cause different hereditary disorders such as cardiofaciocutaneous syndrome, multiple lentigines syndrome, and Noonan syndrome as well as the development of birth defects.
Small deletions in the 2q24.1q24.2 region are quite rare [46]. A female patient was screened with SNP arrays and found to carry de novo deletion of 2q24.1q24.2 region. The patient had mental retardation and generalized hypotonia but lacking any cardiovascular problem [46]. The deleted region on chromosome 2 in our patient harbors two genes: CCDC148 and PKP4. PKP4 has been speculated to be a modifier gene for LMNA in which a splicing mutation caused sudden death, ventricular arrhythmia, cardiomyopathy, and heart failure in a 63-year-old male with a family history of individuals (>10) with similar problems [47].
It is also noteworthy to mention that paternal DNA sample was not available for cytogenetic testing. Hence, we were unable to confirm the de novo status of the deletions in our patient.
Case 4
Our molecular cytogenetic studies identified an interstitial microdeletion on 3q13.2q13.31 cytobands. Such deletions are rare [48] and only few cases have been reported by now. There are more cases of larger deletions in the region (3q11q23) with a range of various phenotypic features such as developmental delay, facial dysmorphisms, and musculoskeletal abnormalities [49,50,51,52,53,54]. A recent study summarized most of these cases excluding few recently reported patients [55,56,57]. The study narrowed down all the deletions to a shared region that harbors expectedly significantly lesser genes than those of the larger region from the previous 3q deletion studies [58]. In this study the smallest deletion was nearly 0.6 Megabases of size and located on 3q13.31. This region contains over 27 genes, one of which is an annotated OMIM disease gene (DRD3). Genotype-phenotype comparison of more than 20 patients shared 3q13.31 deletion and all shared some common phenotypic features developmental delay, muscular hypotonia, a high arched palate, and recognizable facial features including short philtrum and protruding lips. Heart related abnormalities were not among the listed characteristics. The authors speculated that developmental delay seen in these patients is related to DRD3 and ZBTB20. Intriguingly none of these cases of 3q13.31 deletion have heart related abnormality. Moreover, considering that the parents are closely related, the phenotype is likely to originate from the consanginuity. However, this needs further investigations.
Case 5
Chromosome 12p (12p12.1p11.21) and chromosome 16p (16p11.2) deletions are not commonly co-occurring. There are reports for deletions for 12p and 16 p regions [59, 60]. There is only a single report of an interesting patient, who harbors two-hits, maternally inherited 16p13.11-p12.3 duplication and a de novo 12p12.1 deletion [61] whereas our patient has a paternally inherited 16p11.2 deletion and a de novo 12p12.1p11.21 deletion. However, considering the gain type and affected cytogenetic bands our patient is unique and will add to literature of two-hits patients. Deletions of 16p11.2 have been associated with the highly variable phenotype ranging from intellectual disability autism and congenital abnormalities to mildly affected or unaffected cases [62]. In such cases a child may be on one and of the conical spectrum. A recent comprehensive study revisits deletions and duplications in this region in 246 patients. The interesting difference between carriers of the 16p11.2 deletions and duplications is the frequent encounter of macrocephaly among the deletion cases [63]. However, the larger deletion (Chromosome 12) observed in this patient is not present in either parents and therefore appears to be a de novo event as such it is likely to be a significant contributor to the patient’s phenotype. Chromosome 12p12.1 deletions harboring SOX5 have been previously reported [64,65,66]. Among these few patients were reported to have heart related problems such as ventricular septal defect, slight arrhythmia, secundumatrial septal defect, and atrioventricular canal [66]. Phenotypic consequences of these patients were linked to SOX5 haploinsufficiency.
Case 6
2p16.1p15 deletion harbors 27 genes including PEX13. While compound heterozygous and homozygous mutations in PEX13 are associated with Zellweger syndrome (Type PDB11A (OMIM#614883). Haploinsufficiency of this gene due to a heterozygous deletion has not been reported to be a cause of the disease. A PEX13 sequence based mutation on the non-deleted chromosome could conceivably give rise to a clinical phenotype that differs from Zellweger syndrome and sequencing of this gene is considered. However, the plasma very long chain fatty acids assay was normal in the patient; hence, the sequencing of PEX13 was not performed. Until recently, microdeletions of 2p15–16.1 were identified in 15 patients with a recognizable syndrome of dysmorphic features, microcephaly and intellectual disability [67] in addition to the patients deposited to the public databases such as DECIPHER and ISCA. Among the cases, no patient has a report of heart related defect. Hence, the relationship of this deletion to our patient’s phenotype needs further delineation. Moreover, parental studies would be useful in order to determine whether this alteration represents a familial variant or a de novo change. De novo changes are more likely to be clinically significant.
Conclusions
In conclusion, we present the first chromosomal imbalances associated with congenital heart abnormalities among Saudi patients. Such information, combined with further delineation of similar cases and relatedly collection of Saudi Specific CNVs, will allow better understanding of the pathobiology as well as management of the CHD patients in Saudi Arabia.
References
Bernier PL, Stefanescu A, Samoukovic G, Tchervenkov CI. The challenge of congenital heart disease worldwide: epidemiologic and demographic facts. Semin Thorac Cardiovasc Surg Pediatr Card Surg Ann. 2010;13(1):26–34.
Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890–900.
Dolk H, Loane M, Garne E, European Surveillance of Congenital Anomalies Working G. Congenital heart defects in Europe: prevalence and perinatal mortality, 2000 to 2005. Circulation. 2011;123(8):841–9.
van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58(21):2241–7.
Yan Y, Wu Q, Zhang L, Wang X, Dan S, Deng D, Sun L, Yao L, Ma Y, Wang L. Detection of submicroscopic chromosomal aberrations by array-based comparative genomic hybridization in fetuses with congenital heart disease. Ultrasound Obstet Gynecol. 2014;43(4):404–12.
Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association congenital cardiac defects committee, council on cardiovascular disease in the young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):3015–38.
Thienpont B, Mertens L, de Ravel T, Eyskens B, Boshoff D, Maas N, Fryns JP, Gewillig M, Vermeesch JR, Devriendt K. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J. 2007;28(22):2778–84.
Colak D, Al-Dhalaan H, Nester M, Albakheet A, Al-Younes B, Al-Hassnan Z, Al-Dosari M, Chedrawi A, Al-Owain M, Abudheim N, et al. Genomic and transcriptomic analyses distinguish classic Rett and Rett-like syndrome and reveals shared altered pathways. Genomics. 2011;97(1):19–28.
Kaya N, Al-Owain M, Albakheet A, Colak D, Al-Odaib A, Imtiaz F, Coskun S, Al-Sayed M, Al-Hassnan Z, Al-Zaidan H, et al. Array comparative genomic hybridization (aCGH) reveals the largest novel deletion in PCCA found in a Saudi family with propionic acidemia. Eur J Med Genet. 2008;51(6):558–65.
Kaya N, Colak D, Albakheet A, Al-Owain M, Abu-Dheim N, Al-Younes B, Al-Zahrani J, Mukaddes NM, Dervent A, Al-Dosari N, et al. A novel X-linked disorder with developmental delay and autistic features. Ann Neurol. 2012;71(4):498–508.
Kaya N, Imtiaz F, Colak D, Al-Sayed M, Al-Odaib A, Al-Zahrani F, Al-Mubarak BR, Al-Owain M, Al-Dhalaan H, Chedrawi A, et al. Genome-wide gene expression profiling and mutation analysis of Saudi patients with Canavan disease. Genet Med. 2008;10(9):675–84.
Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, Hanscom C, Pillalamarri V, Seabra CM, Abbott MA, et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet. 2016;
Bonnet C, Andrieux J, Beri-Dexheimer M, Leheup B, Boute O, Manouvrier S, Delobel B, Copin H, Receveur A, Mathieu M, et al. Microdeletion at chromosome 4q21 defines a new emerging syndrome with marked growth restriction, mental retardation and absent or severely delayed speech. J Med Genet. 2010;47(6):377–84.
Shaffer LG, Theisen A, Bejjani BA, Ballif BC, Aylsworth AS, Lim C, McDonald M, Ellison JW, Kostiner D, Saitta S, et al. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9(9):607–16.
Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40(3):322–8.
Ballif BC, Theisen A, Rosenfeld JA, Traylor RN, Gastier-Foster J, Thrush DL, Astbury C, Bartholomew D, McBride KL, Pyatt RE, et al. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am J Hum Genet. 2010;86(3):454–61.
Rosenfeld JA, Stephens LE, Coppinger J, Ballif BC, Hoo JJ, French BN, Banks VC, Smith WE, Manchester D, Tsai AC, et al. Deletions flanked by breakpoints 3 and 4 on 15q13 may contribute to abnormal phenotypes. Eur J Hum Genet. 2011;19(5):547–54.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–64.
Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, Filippi T, Carey JC. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. 2013;17(6):589–99.
Connor JA, Hinton RB, Miller EM, Sund KL, Ruschman JG, Ware SM. Genetic testing practices in infants with congenital heart disease. Congenit Heart Dis. 2014;9(2):158–67.
Bachman KK, DeWard SJ, Chrysostomou C, Munoz R, Madan-Khetarpal S. Array CGH as a first-tier test for neonates with congenital heart disease. Cardiol Young. 2015;25(1):115–22.
Baldwin EL, Lee JY, Blake DM, Bunke BP, Alexander CR, Kogan AL, Ledbetter DH, Martin CL. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet Med. 2008;10(6):415–29.
Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38(3):294–6.
Hussain MR, Baig M, Mohamoud HS, Ulhaq Z, Hoessli DC, Khogeer GS, Al-Sayed RR, Al-Aama JY. BRAF gene: from human cancers to developmental syndromes. Saudi J Biol Sci. 2015;22(4):359–73.
Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13(9):777–84.
Dikow N, Maas B, Gaspar H, Kreiss-Nachtsheim M, Engels H, Kuechler A, Garbes L, Netzer C, Neuhann TM, Koehler U, et al. The phenotypic spectrum of duplication 5q35.2-q35.3 encompassing NSD1: is it really a reversed Sotos syndrome? Am J Med Genet A. 2013;161(9):2158–66.
Kurotaki N, Harada N, Shimokawa O, Miyake N, Kawame H, Uetake K, Makita Y, Kondoh T, Ogata T, Hasegawa T, et al. Fifty microdeletions among 112 cases of Sotos syndrome: low copy repeats possibly mediate the common deletion. Hum Mutat. 2003;22(5):378–87.
Mochizuki J, Saitsu H, Mizuguchi T, Nishimura A, Visser R, Kurotaki N, Miyake N, Unno N, Matsumoto N. Alu-related 5q35 microdeletions in Sotos syndrome. Clin Genet. 2008;74(4):384–91.
Fickie MR, Lapunzina P, Gentile JK, Tolkoff-Rubin N, Kroshinsky D, Galan E, Gean E, Martorell L, Romanelli V, Toral JF, et al. Adults with Sotos syndrome: review of 21 adults with molecularly confirmed NSD1 alterations, including a detailed case report of the oldest person. Am J Med Genet A. 2011;155A(9):2105–11.
Hoglund P, Kurotaki N, Kytola S, Miyake N, Somer M, Matsumoto N. Familial Sotos syndrome is caused by a novel 1 bp deletion of the NSD1 gene. J Med Genet. 2003;40(1):51–4.
Miyake N, Kurotaki N, Sugawara H, Shimokawa O, Harada N, Kondoh T, Tsukahara M, Ishikiriyama S, Sonoda T, Miyoshi Y, et al. Preferential paternal origin of microdeletions caused by prezygotic chromosome or chromatid rearrangements in Sotos syndrome. Am J Hum Genet. 2003;72(5):1331–7.
Peredo J, Quintero-Rivera F, Bradley JP, Tu M, Dipple KM. Cleft lip and palate in a patient with 5q35.2-q35.3 microdeletion: the importance of chromosomal microarray testing in the craniofacial clinic. Cleft Palate Craniofac J. 2013;50(5):618–22.
Sohn Y, Lee C, Ko J, Yang JA, Yun JN, Jung EJ, Jin HS, Park SJ, Jeong S. Clinical and genetic spectrum of 18 unrelated Korean patients with Sotos syndrome: frequent 5q35 microdeletion and identification of four novel NSD1 mutations. J Hum Genet. 2013;58(2):73–7.
Chen CP, Lin SP, Lin CC, Chen YJ, Chern SR, Li YC, Hsieh LJ, Lee CC, Pan CW, Wang W. Molecular cytogenetic analysis of de novo dup(5)(q35.2q35.3) and review of the literature of pure partial trisomy 5q. Am J Med Genet A. 2006;140(14):1594–600.
Franco LM, de Ravel T, Graham BH, Frenkel SM, Van Driessche J, Stankiewicz P, Lupski JR, Vermeesch JR, Cheung SW. A syndrome of short stature, microcephaly and speech delay is associated with duplications reciprocal to the common Sotos syndrome deletion. Eur J Hum Genet. 2010;18(2):258–61.
Kasnauskiene J, Cimbalistiene L, Ciuladaite Z, Preiksaitiene E, Kucinskiene ZA, Hettinger JA, Sismani C, Patsalis PC, Kucinskas V. De novo 5q35.5 duplication with clinical presentation of Sotos syndrome. Am J Med Genet A. 2011;155A(10):2501–7.
Kirchhoff M, Bisgaard AM, Bryndorf T, Gerdes T. MLPA analysis for a panel of syndromes with mental retardation reveals imbalances in 5.8% of patients with mental retardation and dysmorphic features, including duplications of the Sotos syndrome and Williams-Beuren syndrome regions. Eur J Med Genet. 2007;50(1):33–42.
Jamsheer A, Sowinska A, Simon D, Jamsheer-Bratkowska M, Trzeciak T, Latos-Bielenska A. Bilateral radial agenesis with absent thumbs, complex heart defect, short stature, and facial dysmorphism in a patient with pure distal microduplication of 5q35.2-5q35.3. BMC Med Genet. 2013;14:13.
Rosenfeld JA, Kim KH, Angle B, Troxell R, Gorski JL, Westemeyer M, Frydman M, Senturias Y, Earl D, Torchia B, et al. Further evidence of contrasting phenotypes caused by reciprocal deletions and duplications: duplication of NSD1 causes growth retardation and Microcephaly. Mol Syndromol. 2013;3(6):247–54.
Fagali C, Kok F, Nicola P, Kim C, Bertola D, Albano L, Koiffmann CP. MLPA analysis in 30 Sotos syndrome patients revealed one total NSD1 deletion and two partial deletions not previously reported. Eur J Med Genet. 2009;52(5):333–6.
Driouch K, Dorion-Bonnet F, Briffod M, Champeme MH, Longy M, Lidereau R. Loss of heterozygosity on chromosome arm 16q in breast cancer metastases. Genes Chromosom Cancer. 1997;19(3):185–91.
Fryns JP, Melchoir S, Jaeken J, van den Berghe H. Partial monosomy of the long arm of chromosome 16 in a malformed newborn: karyotype 46,XX,del(16)(q21). Hum Genet. 1977;38(3):343–6.
Stratakis CA, Lafferty A, Taymans SE, Gafni RI, Meck JM, Blancato J. Anisomastia associated with interstitial duplication of chromosome 16, mental retardation, obesity, dysmorphic facies, and digital anomalies: molecular mapping of a new syndrome by fluorescent in situ hybridization and microsatellites to 16q13 (D16S419-D16S503). J Clin Endocrinol Metab. 2000;85(9):3396–401.
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–5.
White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471(7339):518–22.
Palumbo O, Palumbo P, Palladino T, Stallone R, Zelante L, Carella M. A novel deletion in 2q24.1q24.2 in a girl with mental retardation and generalized hypotonia: a case report. Mol Cytogenet. 2012;5(1):1.
Zaragoza MV, Fung L, Jensen E, Oh F, Cung K, LA MC, Tran CK, Hoang V, Hakim SA, Grosberg A. Exome sequencing identifies a novel LMNA splice-site mutation and Multigenic Heterozygosity of potential modifiers in a family with sick sinus syndrome, dilated Cardiomyopathy, and sudden cardiac death. PLoS One. 2016;11(5):e0155421.
Lowther C, Costain G, Melvin R, Stavropoulos DJ, Lionel AC, Marshall CR, Scherer SW, Bassett AS. Adult expression of a 3q13.31 microdeletion. Mol Cytogenet. 2014;7(1):23.
Jenkins MB, Stang HJ, Davis E, Boyd L. Deletion of the proximal long arm of chromosome 3 in an infant with features of turner syndrome. Ann Genet. 1985;28(1):42–4.
Okada N, Hasegawa T, Osawa M, Fukuyama Y. A case of de novo interstitial deletion 3q. J Med Genet. 1987;24(5):305–8.
Fujita H, Meng J, Kawamura M, Tozuka N, Ishii F, Tanaka N. Boy with a chromosome del (3)(q12q23) and blepharophimosis syndrome. Am J Med Genet. 1992;44(4):434–6.
Genuardi M, Calvieri F, Tozzi C, Coslovi R, Neri G. A new case of interstitial deletion of chromosome 3q, del(3q)(q13.12q21.3), with agenesis of the corpus callosum. Clin Dysmorphol. 1994;3(4):292–6.
Mackie Ogilvie C, Rooney SC, Hodgson SV, Berry AC. Deletion of chromosome 3q proximal region gives rise to a variable phenotype. Clin Genet. 1998;53(3):220–2.
Lawson-Yuen A, Berend SA, Soul JS, Irons M. Patient with novel interstitial deletion of chromosome 3q13.1q13.3 and agenesis of the corpus callosum. Clin Dysmorphol. 2006;15(4):217–20.
Gimelli S, Leoni M, Di Rocco M, Caridi G, Porta S, Cuoco C, Gimelli G, Tassano E. A rare 3q13.31 microdeletion including GAP43 and LSAMP genes. Mol Cytogenet. 2013;6(1):52.
Vuillaume ML, Delrue MA, Naudion S, Toutain J, Fergelot P, Arveiler B, Lacombe D, Rooryck C. Expanding the clinical phenotype at the 3q13.31 locus with a new case of microdeletion and first characterization of the reciprocal duplication. Mol Genet Metab. 2013;110(1–2):90–7.
Herve B, Fauvert D, Dard R, Roume J, Cognard S, Goidin D, Lozach F, Molina-Gomes D, Vialard F. The emerging microduplication 3q13.31: expanding the genotype-phenotype correlations of the reciprocal microdeletion 3q13.31 syndrome. Eur J Med Genet. 2016;59(9):463–9.
Molin AM, Andrieux J, Koolen DA, Malan V, Carella M, Colleaux L, Cormier-Daire V, David A, de Leeuw N, Delobel B, et al. A novel microdeletion syndrome at 3q13.31 characterised by developmental delay, postnatal overgrowth, hypoplastic male genitals, and characteristic facial features. J Med Genet. 2012;49(2):104–9.
Leyser M, Dias BL, Coelho AL, Vasconcelos M, Nascimento OJ. 12p deletion spectrum syndrome: a new case report reinforces the evidence regarding the potential relationship to autism spectrum disorder and related developmental impairments. Mol Cytogenet. 2016;9:75.
Iourov IY, Vorsanova SG, Kurinnaia OS, Zelenova MA, Silvanovich AP, Yurov YB. Molecular karyotyping by array CGH in a Russian cohort of children with intellectual disability, autism, epilepsy and congenital anomalies. Mol Cytogenet. 2012;5(1):46.
Quintela I, Barros F, Lago-Leston R, Castro-Gago M, Carracedo A, Eiris J. A maternally inherited 16p13.11-p12.3 duplication concomitant with a de novo SOX5 deletion in a male patient with global developmental delay, disruptive and obsessive behaviors and minor dysmorphic features. Am J Med Genet A. 2015;167(6):1315–22.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, TH V, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–46.
Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, Green Snyder L, D'Angelo D, Chen Q, Chung WK, et al. 16p11.2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohort. Am J Med Genet A. 2016;170(11):2943–55.
Lee RW, Bodurtha J, Cohen J, Fatemi A, Batista D. Deletion 12p12 involving SOX5 in two children with developmental delay and dysmorphic features. Pediatr Neurol. 2013;48(4):317–20.
Schanze I, Schanze D, Bacino CA, Douzgou S, Kerr B, Zenker M. Haploinsufficiency of SOX5, a member of the SOX (SRY-related HMG-box) family of transcription factors is a cause of intellectual disability. Eur J Med Genet. 2013;56(2):108–13.
Lamb AN, Rosenfeld JA, Neill NJ, Talkowski ME, Blumenthal I, Girirajan S, Keelean-Fuller D, Fan Z, Pouncey J, Stevens C, et al. Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum Mutat. 2012;33(4):728–40.
Balci TB, Sawyer SL, Davila J, Humphreys P, Dyment DA. Brain malformations in a patient with deletion 2p16.1: a refinement of the phenotype to BCL11A. Eur J Med Genet. 2015;58(6–7):351–4.
Acknowledgements
We gratefully thank to the patients and the parents for their kind participation to the study. We also thank KFSHRC Core Facilities at Genetics Department, Research Advisory Council Committees and Purchasing Departments, especially Mr. Faisal Al-Otaibi and his team, for facilitating and expediting our requests.
Funding
This study was supported by King Faisal Specialist Hospital and Research center’s seed grants (KFSHRC-RAC# 2120022) and funds King Abdulaziz City for Science and Technology (KACST# 11-MED1439–20, KACST#14-MED2007–20, KACST#14-BIO2221–20, and KACST#11-BIO2072–20) and King Salman Center for Disability Research (Project#: 02-R-0029-NE-02-AU-1). M. A. S. was supported by the Deanship of Scientific Research, King Saud University, Riyadh, Saudi Arabia through research group no RGP-VPP- 301.
Availability of data and materials
Data deposition and sharing are not applicable for the study.
Declaration
We have obtained consents from the studied patients for the study. The patients were evaluated at the Kind Faisal Specialist Hospital and Research Center using the institutionally approved IRB protocols (RAC# 2040042, 2030046, 2120022, 2080032).
Web resources
http://dgvbeta.tcag.ca/gb2/gbrowse/dgv2_hg19/
Author information
Authors and Affiliations
Contributions
NK conceived and designed the experiments, drafted manuscript, reviewed the data analyses. DC involved in experimental design, reviewed the data analyses, involved in drafting the manuscript, ZA involved in experimental designed, reviewed the charts, evaluated the patients, undertook patient care and management, collected clinical data, delineated the patients’ phenotype and drafted the manuscript. WA involved in drafting the manuscript, analyzed the data. FA, RA, AA performed the experiments. OMM reviewed the charts and involved in drafting and revising the manuscript. ZS, SW involved in performing experiments, MAS, MA, SMH, MAJ reviewed the charts, evaluated the patients, undertook patient care and management, and collected clinical data, OA, BL, BA involved in data analyses, read and revise the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patients were ascertained under Kind Faisal Specialist Hospital and Research Center’s institutionally approved IRB protocols (KFSHRC’s Research Advisory Council Committees including Basic Research Committee and Research Ethics Committee: RAC# 2040042, 2030046, 2120022, 2080032). Before the sample collection, the patients and/or parents (legal guardians) signed the written informed consents.
Consent for publication
Informed written consents were obtained from the patients. See ethics approval. Copy of the signed consent forms is available upon request.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Al-Hassnan, Z.N., Albawardi, W., Almutairi, F. et al. Identification of novel genomic imbalances in Saudi patients with congenital heart disease. Mol Cytogenet 11, 9 (2018). https://doi.org/10.1186/s13039-018-0356-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-018-0356-6