
Abstract
Background
More than 75 common variant loci account for only a portion of the heritability for Alzheimer’s disease (AD). A more complete understanding of the genetic basis of AD can be deduced by exploring associations with AD-related endophenotypes.
Methods
We conducted genome-wide scans for cognitive domain performance using harmonized and co-calibrated scores derived by confirmatory factor analyses for executive function, language, and memory. We analyzed 103,796 longitudinal observations from 23,066 members of community-based (FHS, ACT, and ROSMAP) and clinic-based (ADRCs and ADNI) cohorts using generalized linear mixed models including terms for SNP, age, SNP × age interaction, sex, education, and five ancestry principal components. Significance was determined based on a joint test of the SNP’s main effect and interaction with age. Results across datasets were combined using inverse-variance meta-analysis. Genome-wide tests of pleiotropy for each domain pair as the outcome were performed using PLACO software.
Results
Individual domain and pleiotropy analyses revealed genome-wide significant (GWS) associations with five established loci for AD and AD-related disorders (BIN1, CR1, GRN, MS4A6A, and APOE) and eight novel loci. ULK2 was associated with executive function in the community-based cohorts (rs157405, P = 2.19 × 10–9). GWS associations for language were identified with CDK14 in the clinic-based cohorts (rs705353, P = 1.73 × 10–8) and LINC02712 in the total sample (rs145012974, P = 3.66 × 10–8). GRN (rs5848, P = 4.21 × 10–8) and PURG (rs117523305, P = 1.73 × 10–8) were associated with memory in the total and community-based cohorts, respectively. GWS pleiotropy was observed for language and memory with LOC107984373 (rs73005629, P = 3.12 × 10–8) in the clinic-based cohorts, and with NCALD (rs56162098, P = 1.23 × 10–9) and PTPRD (rs145989094, P = 8.34 × 10–9) in the community-based cohorts. GWS pleiotropy was also found for executive function and memory with OSGIN1 (rs12447050, P = 4.09 × 10–8) and PTPRD (rs145989094, P = 3.85 × 10–8) in the community-based cohorts. Functional studies have previously linked AD to ULK2, NCALD, and PTPRD.
Conclusion
Our results provide some insight into biological pathways underlying processes leading to domain-specific cognitive impairment and AD, as well as a conduit toward a syndrome-specific precision medicine approach to AD. Increasing the number of participants with harmonized cognitive domain scores will enhance the discovery of additional genetic factors of cognitive decline leading to AD and related dementias.
Similar content being viewed by others
Background
Late-onset Alzheimer’s disease (AD) occurring after age 65 is the most common type of dementia and is highly heritable, estimated at 60% to 80% [1]. Common single-nucleotide polymorphisms (SNPs) explain 24% to 33% of the total phenotypic variance of AD [2,3,4], of which up to 6% is accounted for by APOE [3]. More than 75 loci affecting AD risk have been identified in several large-scale genome-wide association studies (GWAS) [5,6,7,8,9,10], but much of the underlying genetic architecture of AD remains unknown [11].
Although GWAS conducted in larger samples will undoubtedly reveal additional AD loci, understanding of the genetic influence on AD risk can be improved by examining the association with endophenotypes that potentially highlight specific pathways underlying the complex disease phenotype. Previously, numerous genetic associations have been identified for AD-related endophenotypes, such as cognitive performance [12,13,14,15], brain imaging traits [16,17,18], neuropathological traits [19,20,21], and biomarkers measured in cerebrospinal fluid [22,23,24]. GWAS have found several loci for general cognitive ability [13, 25], but most findings for specific cognitive domains are not genome-wide significant (GWS), inconsistent, and rarely replicated in independent datasets, perhaps because of the variability in neuropsychological (NP) tests administered across cohorts [26,27,28,29]. To address this concern, Mukherjee and colleagues applied confirmatory factor analysis models to co-calibrate and harmonize composite scores for several cognitive domains. The scores obtained are on the same scale, making them comparable to each other regardless of the NP protocol [15, 30].
Here, we conducted a GWAS for cognitive scores of three domains derived from longitudinal, prospectively collected NP tests administered to participants of several large cohort studies. The statistical power for detecting associations with endophenotypes can be increased by studying outcomes of multiple correlated traits under a model of pleiotropy—a phenomenon where a single gene or variant affects multiple phenotypes [31,32,33,34]. This approach has successfully identified novel associations for neuropathological processes in AD [35,36,37]. Because measures of cognitive performance are highly heritable and correlated with each other [38,39,40], they are well suited as outcomes for cross-phenotype genetic association studies. Therefore, we also tested pleiotropy models for each pair of the three cognitive domains to identify novel loci which may be involved in AD.
Methods
Participants
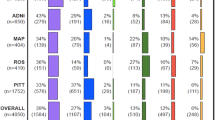
This study included non-Hispanic white participants of the Framingham Heart Study (FHS), National Institute on Aging sponsored Alzheimer’s Disease Research Centers whose phenotypic information was assembled and curated by the National Alzheimer’s Coordinating Center (NACC), the Adult Changes in Thought (ACT) Study, the Alzheimer’s Disease Neuroimaging Initiative (ADNI), and the Religious Orders Study/Rush Memory and Aging Project (ROSMAP). Briefly, FHS is a long-running multi-generation community-based study of cardiovascular disease and other age-related disorders [41,42,43], including cognitive decline and dementia [44, 45]. ACT and ROSMAP are also community-based cohorts that recruited unrelated cognitively normal participants who are followed longitudinally for cognitive disorders [46, 47]. Participants of NACC and ADNI were clinically ascertained for AD research and were cognitively normal or met the criteria for mild cognitive impairment (MCI) or AD at the time of enrollment [48,49,50,51,52]. Extensive cognitive testing of participants of all is conducted at all visits. Details regarding the ascertainment, evaluation, and diagnosis of members of these five cohorts were reported elsewhere [45,46,47,48, 52].
Cognitive domain scores
Scores for executive function, language, and memory domains were derived as previously described [15, 30, 53]. Scores are co-calibrated to put them on the same scale regardless of the cognitive battery administered. Briefly, an expert panel of neuropsychologists (EHT, AJS) and a behavioral neurologist (JBM) assigned each NP test item to one of the three domains. Confirmatory factor analysis in Mplus [54] was used for co-calibration. Cognitive data from the most recent visit were first used to derive scores for each domain, with each domain modeled separately. Test items administered in multiple cohorts functioned as anchors for co-calibration. Parameters for anchor items were forced to be the same across studies to put scores across studies on the same metric. Within study, multiple models (including single factor and bifactor models) were considered with the choice of model determined based on a combination of model fit and concordance with neuropsychological theory. Next, each study’s item parameters from calibration of data at the last visit were fixed and used to obtain scores for each person at each time point. Co-calibrated cognitive scores with a standard error (SE) > 0.6 or derived solely from the mini-mental state examination (MMSE), which has a ceiling effect, were excluded. Time points less than age 60 were only available for FHS and were excluded due to concern that cognitive performance under age 60 may have a different genetic architecture that was only being captured in a single study.
Genotype data processing
We obtained genome-wide SNP data that were processed and imputed using the Trans-Omics for Precision Medicine (TOPMed) reference panel and aligned to Genome Research Consortium human build 38 (GRCh38) [10, 55]. Variants with poor imputation quality (r2 < 0.3), minor allele frequencies (MAF) < 0.01, call rates < 95%, and Hardy-Weinberg Equilibrium (HWE) test p-value < 1 × 10–6 were excluded, and approximately nine million variants remained for each cohort after quality control (QC). Principal components (PCs) of population structure were generated for individuals within each cohort using the set of post-QC variants that were pruned on the basis of a linkage disequilibrium (LD) threshold of 0.1 using the R package GENESIS [56]. Measures of relatedness, kinship coefficients for family-based samples and empirical identity by descent (IBD) in the other samples, were estimated using established procedures [57,58,59].
Genetic and phenotypic correlation estimation
Genetic correlations between each pair of cognitive domain scores (executive function, language, and memory) were estimated in each cohort using GREML [60, 61]. The kinship matrix derived from self-reported FHS pedigrees was incorporated in the estimates using the kinship2 package [62]. We used the empirical genetic relationship matrix (GRM) to account for relatedness among individuals in the other cohorts. To concurrently investigate SNP associations with both performance at the median age and change in cognitive function over time in each domain, we applied a joint test of the marginal genetic effects and gene × age interaction together in a generalized linear mixed model framework with a random slope and intercept as implemented in the mixed‐model association test for gene-environment interactions (MAGEE) R package [63, 64]. Models included terms for SNP, the interaction between SNP and age, and covariates for age, sex, educational level (less than high school, high school, some college, or college graduate), and the first five PCs represented as follows:
where αA, αB, and αC indicate the effects of age, sex, and educational level, respectively; βG is the main SNP effect; γX represents the SNP × age interaction effect; αi is the effect of the ith PC (i between 1 and 5); and r is a random intercept. We subtracted the median age for all observations for all individuals in the dataset from the individual’s age at each exam in order to center age because the intercept will refer to the mean outcome value when an individual’s baseline age is equal to the mean age at baseline in each dataset. Models also incorporated the GRM as a random effect.
Cross-trait analyses
We performed cross-traits LD score regression [65] to estimate genetic correlations across general cognitive function, cognitive domain scores, and neuropsychiatric disorders. We used GWAS summary statistics for general cognitive function (n = 300,486) [13], cognitive factor scores (n = 23,066) from the current study, neuropsychiatric disorders—AD, bipolar disorder, schizophrenia (n = 420,531), and depression (n = 370,457)—from the Pan-UK Biobank, and LD scores derived from the 1000 Genomes Project (phase 3) European samples. We only included 4,815,014 variants with imputation quality r2 > 0.6 and MAF > 0.01 in cross-trait analyses.
Genetic association analyses
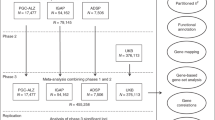
Analyses were performed in each dataset separately, and the GWAS results were combined across datasets by meta-analyses. To correct systematic inflation in a joint test of the SNP’s main and interaction effects [66, 67], we applied the joint meta-analysis method [68] which considers the covariance between the main and interaction effects, and the inverse variance weighted approach in METAL [69]. Meta-analyses were performed for each cognitive domain in the total sample, clinic-based cohorts (NACC and ADNI), and community-based cohorts (FHS, ACT, and ROSMAP) separately (Fig. S1). Results for the clinic- and community-based cohorts were considered separately because some associations might be unique to one of the cohort groups due to disparity in age or proportion of participants with AD. The genomic inflation factor (λ) was calculated for each GWAS and applied to adjust p-values for each test. A GWS threshold was set at P = 5 × 10–8.
Genome-wide pleiotropy analyses
We conducted a pleiotropy GWAS for each pair of cognitive domains in the total sample, clinic-based cohorts, and community-based cohorts using the pooled GWAS results from the joint meta-analysis (Fig. S1) and the R package PLACO [70, 71]. Because rejecting the global null hypothesis that neither phenotype is associated does not specifically imply the existence of pleiotropy, PLACO tests the composite null hypothesis that no more than one phenotype is associated with a variant. Thus, rejecting the composite null hypothesis implies that both phenotypes are associated with the variant, i.e., pleiotropy. This approach uses the product of the Z-statistics as the test statistic for the association of a given variant with each individual trait. The null distribution of the test statistic takes the form of a mixture distribution that allows for the variant to be associated with none or only one of the traits. Variants with squared Z-scores > 80 for one trait were removed because they could cause spurious pleiotropic signals [72, 73]. Because correlations between the Z-statistics for the association between a variant and the two traits can result in inflated type I errors [74], we adjusted for the Pearson correlation for variants with no effect (P > 1 × 10–4) as suggested by the developers of the method.
Pathway enrichment analyses
We performed several pathway analyses, each of which was seeded with genes containing variants associated with a single cognitive domain or pleiotropy for paired domains (P < 1 × 10–4) in the respective GWAS, using the Ingenuity Pathway Analysis software (QIAGEN Inc.) [75]. Enrichment p-values for each canonical pathway were adjusted for a false discovery rate (FDR) using the Benjamini-Hochberg method [76], and an FDR-adjusted P-value threshold was set at 0.001 to account for the 18 separate pathway analyses (six single and paired domains multiplied by three sample strata).
Results
Cognitive domains are phenotypically and genetically correlated
Compared to the community-based cohorts, the clinic-based cohorts had more males, participants who were younger and better educated, and higher proportion of participants who were diagnosed with MCI or AD (Table 1). Even though the mean and median ages at the last visits of individuals in the clinic-based cohorts were slightly lower than those in the community-based cohorts, scores for executive function and memory were significantly lower (P < 0.001), and the language score was significantly higher (P < 0.001) in the clinic-based cohorts. Phenotypic and genetic correlations for each pair of factor scores in each dataset were moderate to high (phenotypic r = 0.56–0.86, genetic r = 0.57–0.72) (Table S1). Most phenotypic and genetic correlations were higher for ROSMAP compared to the other cohorts. Cross-trait analyses revealed that factor scores for all three cognitive domains are significantly genetically correlated with general cognitive function (0.51 ≤ r ≤ 0.77) (Table S2). Although none of the traits were significantly correlated with AD or other psychiatric disorders, the language domain score was moderately associated with depression (r = 0.60, P = 0.11) and the memory domain score was strongly associated with AD (r = 0.90, P = 0.40). Lack of significance for these results may be due to insufficient power for genetic correlations with dichotomous outcomes.
GWAS identifies multiple established AD and novel loci associated with individual cognitive domains
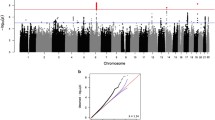
There was little evidence of genomic inflation (λ = 1.006–1.023) in the GWAS for each cognitive domain and strata of the sample (Figs. S2, S3 and S4). GWS associations were observed for many SNPs in the APOE region for all traits (Table S3). We also identified associations with several other established AD loci (Table 2). BIN1 SNP rs6733839 was associated with language (PJoint = 2.70 × 10–8) and memory (PJoint = 2.37 × 10–9) in the total sample and with both language (PJoint = 1.98 × 10–9) and memory (PJoint = 1.60 × 10–8) in the clinic-based cohorts. The significant joint effect of rs6733839 is due primarily to the SNP’s main effect rather than its interaction with age and is supported by multiple adjacent variants (Figs. S5 and S6). GWS associations for memory were also observed with CR1 SNP rs1752684 (PJoint = 8.85 × 10–9) and MS4A6A SNP rs7232 (PJoint = 3.97 × 10–8) in the clinic-based cohorts, findings which were supported by adjacent SNPs (Fig. S7). Similar to BIN1, results of the joint test of the main and interaction effects for rs1752684 and rs7232 reflect the SNPs’ main effects. GWS associations were also detected with SNPs in four additional loci, ULK2 (rs157405, PJoint = 2.19 × 10–9) with executive function in the community-based cohorts, CDK14 (rs705353, PJoint = 1.73 × 10–8) with language in the clinic-based cohorts, PURG (rs117523305, PJoint = 1.73 × 10–8) with memory in the community-based cohorts, and LINC02712 (rs145012974, PJoint = 3.66 × 10–8) with language in the total sample (Fig. 1). We also identified a GWS association of memory with GRN (rs5848, PJoint = 4.21 × 10–8) in the total sample (Fig. 1). Unlike the associations with the other known AD loci, the interactions of the ULK2 (PG×Age = 7.65 × 10–7), CDK14 (PG×Age = 2.54 × 10–9), PURG (PG×Age = 1.41 × 10–8), LINC02712 (PG×Age = 7.69 × 10–9), and GRN (PG×Age = 1.07 × 10–6) SNPs with age accounted for the significant joint test findings (Table 2).

Locus Zoom plots showing the association of SNPs in the regions of novel loci with cognitive domains. The SNP with the lowest p-value at each locus is indicated with a purple diamond. Computed estimates of linkage disequilibrium (r2) of SNPs in the region with top-ranked SNP are color-coded according to the key. Vertical blue lines indicate locations of high recombination rates. Locations of genes in the region are shown below the diagram. a Association of rs157405 with executive function in the community-based cohorts. b Association of rs705353 with language in the clinic-based cohorts. c Association of rs117523305 with memory in the community-based cohorts. d Association of rs145012974 with language in the total sample. e Association of rs5848 with memory in the total sample
The APOE region comprised many variants significantly associated with all the cognitive domains in all cohort groupings (Table S3). The high LD between these variants suggests that there are not multiple independent association signals, a conclusion supported by evidence that no genes in this region other than the APOE account for the observed association with AD risk or onset age [77]. Focusing on the APOE SNPs encoding the ε4 (rs429358) and ε2 (rs7412) alleles, the ε4 SNP was significantly associated with lower (worse) scores for all cognitive domains in an age-dependent manner, based on the negative sign of βG×Age. Notably, the magnitude of the effect of the ε4 SNP on memory was approximately 1.7 times greater than on executive function or language in the total sample at the median age (Table 2). In the same sample, the effect of interaction between ε4 and age was 1.5–1.9 times larger for memory compared to executive function or language. Conversely, the ε2 SNP was significantly associated with higher (better) cognitive domain scores in the clinic-based cohorts at the median age with some limited age-dependent effect (Table 2).
Numerous highly suggestive associations (P < 1 × 10–6), including several that were nearly GWS (P < 1 × 10–7), were found for individual cognitive domains with other loci (Table S3). Notably, language was associated with 18 ADCY2 SNPs in the community-based cohorts (top SNP: rs7734697, PJoint = 6.34 × 10–8) and with 19 DAPK2 SNPs in the clinic-based cohorts (top SNP: rs112972763, PJoint = 7.76 × 10–8). Six PLXDC2 SNPs were associated with executive function in the total sample (top SNP: rs7083449, PJoint = 7.02 × 10–8), and most of the evidence was derived from the community-based cohorts.
Genome-wide pleiotropy analysis identifies the association of cognitive domains with the progranulin gene and four novel loci
GWAS for the three pairs of cognitive domains collectively identified GWS evidence of association with SNPs in five independent loci (Table 3, Table S4) with little evidence of genomic inflation in the total sample or separately within the clinic-based and community-based cohorts (λ = 0.964–0.994, Figs. S8, S9 and S10). Consistent with the findings from analyses of individual cognitive domains, GWS evidence of pleiotropy was found for the association of the APOE region SNPs with all cognitive domain pairs in the total sample and the clinic-based and community-based cohorts (Table S4). GWS pleiotropy was also observed with rs6733839, located between BIN1 and CYP27C1, in the total sample (PJoint = 9.01 × 10–12) and the clinic-based cohorts (PJoint = 6.85 × 10–10) for language and memory (Table 3). The association with rs6733839 was evident in the community-based cohorts (PJoint = 2.52 × 10–4), strengthened in the total sample (PJoint = 9.01 × 10–12), well supported by association with neighboring variants (Fig. S11), and attributable primarily to its main effect for each domain (Table 3).
In the clinic-based cohorts, there was GWS pleiotropy for language and memory with rs73005629 (PJoint = 3.12 × 10–8) located in an intergenic region on chromosome 4 (Table 3, Fig. 2). The joint effect of rs73005629 on language (PJoint = 4.66 × 10–7) and memory (PJoint = 1.47 × 10–7) was equally attributable to its main and interaction effects. There was no evidence of pleiotropy for rs73005629 in the community-based cohorts. Conversely, significant pleiotropy for the same domain pair was observed with NCALD SNP rs56162098 (PJoint = 1.23 × 10–9) and PTPRD SNP rs145989094 (PJoint = 8.34 × 10–9) in the community-based cohorts (Table 3). The association with rs56162098 was not evident in the clinic-based cohorts but was supported by the association with neighboring variants (Fig. 2). The same PTPRD SNP was also pleiotropic for executive function and memory (PJoint = 3.85 × 10–8), but this association is not supported by findings in the clinic-based cohorts (Table 3) or neighboring SNPs (Fig. 2).

Locus Zoom plots showing genome-wide significant pleiotropy for SNPs in the regions of novel loci. The SNP with the lowest p-value at each locus is indicated with a purple diamond. Computed estimates of linkage disequilibrium (r2) of SNPs in the region with top-ranked SNP are color-coded according to the key. Vertical blue lines indicate locations of high recombination rates. Locations of genes in the region are shown below the diagram. a Association of rs12447050 with executive function and memory in the community-based cohorts. b Association of rs56162098 with language and memory in the community-based cohorts. c Association of rs145989094 with executive function and memory in the community-based cohorts. d Association of rs145989094 with language and memory in the community-based cohorts. e Association of rs73005629 with language and memory in the clinic-based cohorts
We also identified significant pleiotropy in the community-based cohorts for executive function and memory with rs12447050, located 5.5 kb upstream from OSGIN1 (PJoint = 4.09 × 10–8). This association was comparably supported by each domain and the SNP’s main effect and interaction with age (Table 3), as well as by neighboring SNPs (Fig. 2). There was no evidence of association with the individual domains or in the pleiotropy model in the clinic-based cohorts. However, the magnitude of effect for rs12447050 and its interaction with age in each domain, as well as the significance levels for the main, interaction, and joint pleiotropy tests in the community-based cohorts and the total sample, were nearly identical (Table 3).
Highly suggestive pleiotropy was observed in the community-based cohorts with two SNPs (rs7081658 and rs7070729) located in the USP6NL/ECHDC3 region, an established AD risk locus, for executive function and language (PJoint = 3.54 × 10–7 and PJoint = 8.76 × 10–8, respectively) and for executive function and memory (PJoint = 2.11 × 10–7 and PJoint = 5.09 × 10–8, respectively); rs7070729 was also pleiotropic for language and memory (PJoint = 7.76 × 10–7) (Table S4). There was also suggestive pleiotropy for executive function and language with two SNPs in the AD risk locus WWOX (rs13329990, PJoint = 8.45 × 10–7; rs11862902, PJoint = 9.60 × 10–7).
Pathways involved in neuronal development or signaling, vascular and endocrine systems are related to cognitive domain performance
A total of 28 canonical pathways were significantly enriched for loci associated with pleiotropy for paired domains (Table 4), noting that none of these pathways were specific to the clinic-based cohorts, and no significant pathways were identified in analyses seeded with top-ranked genes in the GWAS for individual cognitive domains. The evidence for approximately 60% (17/28) of these pathways was derived from analyses of the community-based cohorts only. The top-ranked pathway, synaptogenesis signaling, was significantly enriched for genes that emerged from pleiotropy analysis for all three pairs of cognitive domains. Several pathways are related to neuronal development or signaling (e.g., synaptogenesis signaling, synaptic long-term depression, endocannabinoid neuronal synapse, netrin signaling, GABA and glutamate receptor signaling, and calcium signaling), AD-associated vascular risk factors (e.g., type II diabetes and maturity onset diabetes of young signaling, insulin secretion signaling, dilated cardiomyopathy and cardiac hypertrophy signaling, and nitric oxide signaling in the cardiovascular system), and the endocrine system (e.g., G protein-coupled receptor-mediated nutrient sensing in enteroendocrine cells, insulin, corticotropin-releasing hormone, gonadotropin-releasing hormone, androgen, and oxytocin signaling). Details for suggestive pathways (FDR-adjusted P < 0.05) and the number of seed genes selected from each GWAS and pleiotropy analysis are summarized in Tables S5 and S6, respectively.
Discussion
Genome-wide scans for performance measures in three cognitive domains in two large clinically ascertained and three community-based cohorts revealed GWS associations with four well-established AD loci (BIN1, CR1, MS4A6A, and APOE) and eight loci not previously genetically linked to AD or cognitive decline (ULK2, CDK14, PURG, LINC02712, LOC107984373, NCALD, PTPRD, and OSGIN1), as well as with GRN which has been associated with AD and several other dementing illnesses [7, 10, 78,79,80]. These findings were based on analyses that leveraged data obtained from one or more cognitive examinations, considered cognitive performance changes over time, and examined genetic effects on individual or pairs of domains. In comparison to previous GWAS of cognitive performance, which were limited to the availability of data for particular NP tests and focused primarily on clinic-based or community-based samples [26,27,28,29], our study utilized harmonized measures that enabled pooling data obtained using multiple NP protocols and considered associations that may be common or unique to differentially ascertained samples.
To our knowledge, this is the first genome-wide pleiotropy study using harmonized cognitive domain scores. Compared to previous conventional GWAS or pleiotropy studies of individual cognitive traits [26,27,28,29], our genetic analysis of harmonized cognitive scores allows combining results from studies using different NP protocols and permits greater opportunities for replication and meta-analyses. This approach has been successfully used in a variety of studies of cognitive aging [81,82,83,84]. A recent study of five preclinical AD cohorts conducted a factor analysis on three domains—general cognitive performance, episodic memory, and executive function—and established a common algorithm for classifying MCI progression across the heterogeneously evaluated samples [85]. Cognitive factor scores derived in an identical fashion as those used in this study have been utilized for a variety of investigations of AD subgroups [81], which linked cognition to imaging [83], neuropathology [84], and genetics [15]. Similarly, they have been used in genetic studies of cognitive resilience to AD [82, 86].
We identified three novel loci that have functional relevance to processes implicated in AD. Dysfunction of the protein encoded by ULK2, unc51 like autophagy activating kinase 2, has been suggested to cause multiple diseases. ULK2 SNPs have been associated with schizophrenia [87], and a ULK2 circular RNA is expressed more than tenfold in a vascular dementia rat model [88]. Lee and colleagues recently demonstrated that amyloid-β 42 oligomer-mediated loss of excitatory synapses in cortical neurons and hippocampal CA1 neurons requires AMPK-mediated activation of ULK2-dependent mitophagy [89]. PTPRD, protein tyrosine phosphatase receptor type D, was previously reported to be associated with AD susceptibility [90]. A recent study identified a significant association of PTPRD with the accumulation of neurofibrillary tangles that was independent of amyloid-β pathology [20]. NCALD encodes a member of the neuronal calcium sensor family of calcium-binding proteins, which mediates signal transduction in response to calcium in neurons. NCALD is downregulated in the AD brain and may play a protective role in hippocampal CA1 and CA3 regions [91, 92]. This observation is consistent with a finding from a study of differentially expressed proteins in rats fed a high-fat diet suggesting that the memory-impairing effects of diet-induced obesity might potentially be mediated by down-regulated NCALD within the hippocampus [93].
We also identified a GWS signal in GRN, the gene that encodes the anti-inflammatory and neurotrophic factor progranulin (PGRN) [94]. GRN mutations are a well-established cause of frontotemporal lobar degeneration (FTLD). More than 60 disease-causing GRN mutations have been identified, accounting for 20% to 25% of familial FTLD cases and about 10% of all FTLD cases [95]. The most significantly associated GRN SNP in our study, rs5848, was found in the 3’-untranslated region, which is predicted to be a microRNA binding site. Rs5848 is the GRN variant most frequently associated with FTLD and is associated with a reduction in PGRN in plasma and cerebrospinal fluid [96, 97]. In addition to FTLD, several studies have shown an association between clinical AD and the rs5848 T allele, which we found to be linked to lower memory performance in both clinic- and community-based cohorts [98]. A recent large GWAS meta-analysis found a GWS association of AD risk with rs5848-T [10]. A recent study examining neuropathological AD correlates showed that rs5848 T allele carriers had a higher frequency of hippocampal sclerosis and TDP-43 deposits, significantly increased tau pathology burden, but showed no specific association with β-amyloid load or AD neuropathological diagnosis [99]. Interestingly, our finding was exclusive to the memory domain, which is affected in early stages of AD, but also commonly affected in hippocampal sclerosis and limbic-predominant age-related TDP-43 encephalopathy (LATE) [100, 101]. Effects were driven by both the SNP’s main and SNP × age interaction effects. This finding provides additional evidence that variation in GRN may be related to neurodegeneration more broadly and that restoring PGRN levels may be an effective way to prevent and treat dementia [102].
Highly suggestive pleiotropy (P < 1 × 10–7) was also found with other established AD risk loci, including USP6NL/ECHDC3 for all three paired cognitive domains and WWOX for executive function and language. ECHDC3, enoyl-CoA hydratase domain containing 3, was previously reported to be associated with AD [7, 10]. ADCY2, adenylate cyclase 2, was reported to be associated with AD-related changes in hippocampal gene expression [103, 104], as well as AD-associated structural changes detected by brain imaging [105]. A recent GWAS reported the association of DAPK2, death associated protein kinase 2, with amyloid deposition in the brain [106], a finding consistent with studies showing that DAPK1 promotes APP phosphorylation and amyloidogenic processing [107]. PLXDC2, plexin domain containing 2, is upregulated with increasing β-amyloid plaque load or Braak stages [108].
There are no established links of OSGIN1, CDK14, and PURG to AD. The product encoded by OSGIN1 is an oxidative stress response protein that regulates cell death and appears to be a key regulator of both inflammatory and anti-inflammatory molecules [109, 110]. CDK14 encodes a protein kinase whose expression is more than two-fold higher in the brain than in any other tissue. However, it has been linked to cancer in various tissues, primarily outside of the brain. Although the function of PURG is unknown, a SNP in this gene showed significant associations in GWAS of cognitive performance and intelligence [111, 112]. The biological significance of the pleiotropic association of memory and language with a chromosome 4 variant located about 170 kb from LOC107984373, which encodes a long non-protein coding RNA, is also puzzling at this time.
Bioinformatic analyses of the top-ranked genes emerging from GWAS implicated several biological pathways related to neuronal development and signaling, AD-associated vascular risk factors, and endocrine pathways. Notably, all of the significant pathways were identified from analyses of findings from the pleiotropy GWAS analyses, especially those supported by the community-based cohorts. Because pathways were constructed using information from well-established metabolic and cell signaling pathways, they tend to reflect more common or shared mechanisms rather than particular or trait-specific mechanisms. Therefore, pleiotropic loci affecting multiple cognitive domains may be more suitable as seed genes for canonical pathways than loci associated with a single domain. Indeed, in the analyses of community-based cohorts, the numbers of seed genes from pleiotropy GWAS were 1.5–1.7 times larger than those from GWAS of individual cognitive domains (Table S6). Considering that AD pathology results in progressive dysfunction in several cognitive domains over time, the majority of our findings, which emerged from analyses of the community-based rather than the clinic-based cohorts, may represent pathways underlying cognitive processes related to AD progression rather than AD risk.
Interestingly, associations with several well-established AD loci, including BIN1, CR1, and MS4A6A, were observed only in the clinic-based cohorts. Lack of replication in the community-based cohorts might be due to the relative paucity of AD cases and the higher likelihood of mixed pathologies. Conversely, the associations with the known AD locus USP6NL/ECHDC3 and novel loci, including ULK2, NCALD, PTPRD, ADCY2, and OSGIN1, were observed only in the community-based cohorts. Lack of replication in the clinic-based cohorts may indicate that these loci are associated with normal age-related cognitive changes rather than an AD process. However, this explanation seems less likely given their previous association with AD risk (USP6NL/ECHDC3) or functional relevance to processes implicated in AD and/or their association with other AD-related endophenotypes (NCALD, ULK2 and PTPRD). Alternatively, community-based cohort-specific findings may indicate that the effects of these genes are age-dependent or detectable when tracked over time. This idea is supported by the observation of the highly significant SNP × age interaction term for these loci, which for ULK2, PTPRD, ADCY2, and OSGIN1 were responsible for the significant joint effect to a much greater extent than the SNP’s main effect.
We employed a joint test that combines the main genetic effects and SNP × age interaction together to increase our power to detect genetic associations. Nonetheless, interpreting a joint test can be challenging, requiring examination of effect estimates for both the main and interaction terms. The contribution of the SNP’s main effect to the joint association for some findings, including the well-established AD loci and several novel ones (e.g., ULK2, CDK14, PURG, LINC02712, and GRN), was much stronger than its interaction with age. This may reflect that these loci are associated with the development rather than the progression of AD. This aligns with the fact that these loci were initially identified using a case-control design. In contrast, particularly for the more novel loci, the contribution of the SNP × age interaction to the joint association was stronger than the main effect. This may reflect that these loci are associated with the progression rather than the development of AD and could explain why they have not been identified previously, as few genetic studies of AD have utilized a longitudinal design.
Of note, all of the GWS pleiotropic associations and half of the GWS single-domain associations involved the memory domain. This is not surprising as prominent memory impairment is the most common cognitive feature in AD. Nonetheless, prominent impairment in other cognitive domains occurs in a reasonable number of AD cases. Those specific loci were implicated for particular cognitive domains may provide syndrome-specific therapeutic targets with an eye toward a precision medicine approach to AD.
Our results also highlight that the biology underlying cognitive performance in older individuals is complex and likely a function of multiple processes including lifelong ability, neurodegeneration and resilience to neurodegeneration. Genetic architecture may be influencing cognition through each of these processes. Without a measure of underlying pathology, disentangling the mechanism by which genes are affecting cognition is difficult. This point applies to both the current study and the large AD GWAS in which most participants received a diagnosis based only upon assessment of cognition in life. Our finding that AD risk is strongly genetically correlated with the factor score for memory but not executive function or language might provide some insight into these processes. GWAS findings for the individual cognitive domains showing that memory was associated only with established AD risk genes. However, all of the novel associations identified in the pleiotropy analysis included memory as part of the paired outcome. These genetic association patterns might argue that our phenotypes for executive function and language could reflect decline from AD (rather than development of AD), underlying cognitive ability and/or cognitive resilience. Future studies that utilize both measures of cognition and underlying pathology will be needed to better disentangle the genetic architecture underlying these different processes that influence cognition.
This study has several limitations. Although we included several large cohorts whose cognitive test data were co-calibrated and harmonized with each other, the sample size was small compared to the previous GWAS of AD risk. In addition, there was a reduction in power for tests of marginal genetic effects or SNP × age interactions because a large portion of the subjects had only one visit (FHS-21.5%, NACC-21.1%, ACT-9.9%, ADNI-13.3%, and ROSMAP-6.0%). The interpretation of our findings based on the joint effect of the SNP and SNP × age interaction is complicated because the identified loci could imply several meanings to cognitive domain functions or AD. Those findings may reflect genetic associations with the development or progression of AD or both, but additional work is needed to address this issue confidently. Further, our model assumes linearity in the cognitive trajectories, but cognitive trajectories at different disease stages may be non-linear. The observed associations with known AD loci provide validation for our modeling approach. Our results are not adjusted for the number of genome-wide scans performed, but the analyses for each cognitive domain and paired cognitive domain are testing separate hypotheses. Correction for analyses conducted separately in the clinic- and community-based cohorts would raise the significance threshold to 2.5 × 10–8, which would render associations for MS4A6A, LINC02712, GRN, LOC107984373, PTPRD, and OSGIN1 as borderline GWS. Another concern is the lack of replication which will require the availability of co-calibrated longitudinally obtained cognitive data from independent samples which are informative for AD. Ongoing phenotype harmonization efforts of the Alzheimer’s Disease Genetics Consortium, Alzheimer’s Disease Sequencing Project, and other studies will likely yield the data necessary for replication testing. Because some of the identified loci have no obvious connection to AD or cognition, further research is required to determine their mechanistic pathways. Finally, datasets from other population groups containing cognitive domain factor scores for adequately powered samples will be needed to extend our findings which were derived from non-Hispanic whites only.
Conclusion
Our results provide some insight into biological pathways underlying processes leading to domain-specific cognitive impairment and AD. The findings may provide a conduit toward a syndrome-specific precision medicine approach to AD. Increasing the number of datasets by harmonizing measures of cognitive performance in other cohorts, as applied in this study, would likely enhance the discovery of additional genetic factors of cognitive decline leading to AD and related dementias.
Availability of data and materials
Genetic and phenotype data for the ACT, ADNI, NACC, and ROSMAP GWAS datasets are available through the National Institute on Aging Genetics of Alzheimer Disease Data Storage site (https://www.niagads.org/). Framingham Heart Study data are available in dbGaP and/or through application to the FHS Research Committee (https://www.framinghamheartstudy.org/fhs-for-researchers/). GWAS summary statistics are available in NIAGADS.
Abbreviations
- ACT:
-
Adult Changes in Thought
- AD:
-
Alzheimer’s disease
- ADNI:
-
Alzheimer’s Disease Neuroimaging Initiative
- FDR:
-
False discovery rate
- FHS:
-
Framingham Heart Study
- FTLD:
-
Frontotemporal lobar degeneration
- GRM:
-
Genetic relationship matrix
- GWAS:
-
Genome-wide association study
- GWS:
-
Genome-wide significant
- HWE:
-
Hardy-Weinberg equilibrium
- IBD:
-
Identical by descent
- LATE:
-
Limbic-predominant age-related TDP-43 encephalopathy
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- MAGEE:
-
Mixed‐model association test for gene-environment interactions
- MCI:
-
Mild cognitive impairment
- MMSE:
-
Mini-mental State Examination
- NACC:
-
National Alzheimer’s Coordinating Center
- NP:
-
Neuropsychological
- PC:
-
Principal component
- PRGN:
-
Progranulin
- QC:
-
Quality control
- ROSMAP:
-
Religious Order Study and Rush Memory Aging Project
- SE:
-
Standard error
- SNP:
-
Single nucleotide polymorphism
- TOPMed:
-
Trans-Omics for Precision Medicine
References
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–74.
Lee SH, Harold D, Nyholt DR, Consortium AN, International Endogene C, Genetic, Environmental Risk for Alzheimer’s disease C, Goddard ME, Zondervan KT, Williams J, et al. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum Mol Genet. 2013;22:832–41.
Ridge PG, Mukherjee S, Crane PK, Kauwe JS. Alzheimer’s Disease Genetics C: Alzheimer’s disease: analyzing the missing heritability. PLoS ONE. 2013;8:e79771.
Brainstorm C, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, Escott-Price V, Falcone GJ, Gormley P, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360(6395):eaap8757.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hagg S, Athanasiu L, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414–30.
Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, Rongve A, Borte S, Winsvold BS, Drange OK, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53:1276–82.
Bellenguez C, Kucukali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54:412–36.
Holland D, Frei O, Desikan R, Fan CC, Shadrin AA, Smeland OB, Andreassen OA, Dale AM. The genetic architecture of human complex phenotypes is modulated by linkage disequilibrium and heterozygosity. Genetics. 2021;217(3):iyaa046.
Rietveld CA, Esko T, Davies G, Pers TH, Turley P, Benyamin B, Chabris CF, Emilsson V, Johnson AD, Lee JJ, et al. Common genetic variants associated with cognitive performance identified using the proxy-phenotype method. Proc Natl Acad Sci U S A. 2014;111:13790–4.
Davies G, Lam M, Harris SE, Trampush JW, Luciano M, Hill WD, Hagenaars SP, Ritchie SJ, Marioni RE, Fawns-Ritchie C, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun. 2018;9:2098.
Sherva R, Gross A, Mukherjee S, Koesterer R, Amouyel P, Bellenguez C, Dufouil C, Bennett DA, Chibnik L, Cruchaga C, et al. Genome-wide association study of rate of cognitive decline in Alzheimer’s disease patients identifies novel genes and pathways. Alzheimers Dement. 2020;16:1134–45.
Mukherjee S, Mez J, Trittschuh EH, Saykin AJ, Gibbons LE, Fardo DW, Wessels M, Bauman J, Moore M, Choi SE, et al. Genetic data and cognitively defined late-onset Alzheimer’s disease subgroups. Mol Psychiatry. 2020;25:2942–51.
Xu Z, Wu C, Pan W, Alzheimer’s Disease Neuroimaging I. Imaging-wide association study: integrating imaging endophenotypes in GWAS. Neuroimage. 2017;159:159–69.
Knutson KA, Deng Y, Pan W. Implicating causal brain imaging endophenotypes in Alzheimer’s disease using multivariable IWAS and GWAS summary data. Neuroimage. 2020;223:117347.
Knutson KA, Pan W. Integrating brain imaging endophenotypes with GWAS for Alzheimer’s disease. Quant Biol. 2021;9:185–200.
Chung J, Wang X, Maruyama T, Ma Y, Zhang X, Mez J, Sherva R, Takeyama H, Alzheimer’s Disease Neuroimaging I, Lunetta KL, et al. Genome-wide association study of Alzheimer’s disease endophenotypes at prediagnosis stages. Alzheimers Dement. 2018;14:623–33.
Chibnik LB, White CC, Mukherjee S, Raj T, Yu L, Larson EB, Montine TJ, Keene CD, Sonnen J, Schneider JA, et al. Susceptibility to neurofibrillary tangles: role of the PTPRD locus and limited pleiotropy with other neuropathologies. Mol Psychiatry. 2018;23:1521–9.
Tasaki S, Gaiteri C, Mostafavi S, De Jager PL, Bennett DA. The molecular and neuropathological consequences of genetic risk for Alzheimer’s dementia. Front Neurosci. 2018;12:699.
Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, Mayo K, Bertelsen S, Hinrichs A, Alzheimer’s Disease Neuroimaging I, Fagan AM, et al. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet. 2012;21:4558–71.
Deming Y, Xia J, Cai Y, Lord J, Holmans P, Bertelsen S, Holtzman D, Morris JC, Bales K, Pickering EH, et al. A potential endophenotype for Alzheimer’s disease: cerebrospinal fluid clusterin. Neurobiol Aging. 2016;37:208.e201-208.e209.
Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, Carrell D, Cai Y, Fernandez MV, Budde J, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017;133:839–56.
Trampush JW, Yang ML, Yu J, Knowles E, Davies G, Liewald DC, Starr JM, Djurovic S, Melle I, Sundet K, et al. GWAS meta-analysis reveals novel loci and genetic correlates for general cognitive function: a report from the COGENT consortium. Mol Psychiatry. 2017;22:336–45.
Debette S, Ibrahim Verbaas CA, Bressler J, Schuur M, Smith A, Bis JC, Davies G, Wolf C, Gudnason V, Chibnik LB, et al. Genome-wide studies of verbal declarative memory in nondemented older people: the Cohorts for Heart and Aging Research in Genomic Epidemiology consortium. Biol Psychiatry. 2015;77:749–63.
Ibrahim-Verbaas CA, Bressler J, Debette S, Schuur M, Smith AV, Bis JC, Davies G, Trompet S, Smith JA, Wolf C, et al. GWAS for executive function and processing speed suggests involvement of the CADM2 gene. Mol Psychiatry. 2016;21:189–97.
Davies G, Marioni RE, Liewald DC, Hill WD, Hagenaars SP, Harris SE, Ritchie SJ, Luciano M, Fawns-Ritchie C, Lyall D, et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N=112 151). Mol Psychiatry. 2016;21:758–67.
Homann J, Osburg T, Ohlei O, Dobricic V, Deecke L, Bos I, Vandenberghe R, Gabel S, Scheltens P, Teunissen CE, et al. Genome-wide association study of Alzheimer’s disease brain imaging biomarkers and neuropsychological phenotypes in the European Medical Information Framework for Alzheimer’s Disease Multimodal Biomarker Discovery dataset. Front Aging Neurosci. 2022;14:840651.
Mukherjee S, Choi SE, Lee ML, Scollard P, Trittschuh EH, Mez J, et al. Cognitive domain harmonization and cocalibration in studies of older adults. Neuropsychology. 2023;37:409–23.
Stearns FW. One hundred years of pleiotropy: a retrospective. Genetics. 2010;186:767–73.
Paaby AB, Rockman MV. The many faces of pleiotropy. Trends Genet. 2013;29:66–73.
Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 2013;14:483–95.
van Rheenen W, Peyrot WJ, Schork AJ, Lee SH, Wray NR. Genetic correlations of polygenic disease traits: from theory to practice. Nat Rev Genet. 2019;20:567–81.
Jun G, Moncaster JA, Koutras C, Seshadri S, Buros J, McKee AC, Levesque G, Wolf PA, St George-Hyslop P, Goldstein LE, Farrer LA. delta-Catenin is genetically and biologically associated with cortical cataract and future Alzheimer-related structural and functional brain changes. PLoS ONE. 2012;7:e43728.
Chung J, Zhang X, Allen M, Wang X, Ma Y, Beecham G, Montine TJ, Younkin SG, Dickson DW, Golde TE, et al. Genome-wide pleiotropy analysis of neuropathological traits related to Alzheimer’s disease. Alzheimers Res Ther. 2018;10:22.
Bone WP, Siewert KM, Jha A, Klarin D, Damrauer SM, Program VAMV, Chang KM, Tsao PS, Assimes TL, Ritchie MD, Voight BF. Multi-trait association studies discover pleiotropic loci between Alzheimer’s disease and cardiometabolic traits. Alzheimers Res Ther. 2021;13:34.
Harris SE, Deary IJ. The genetics of cognitive ability and cognitive ageing in healthy older people. Trends Cogn Sci. 2011;15:388–94.
Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, Posthuma D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702–9.
Plomin R, Deary IJ. Genetics and intelligence differences: five special findings. Mol Psychiatry. 2015;20:98–108.
Dawber TR, Kannel WB. The Framingham study. An epidemiological approach to coronary heart disease. Circulation. 1966;34:553–5.
Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham offspring study. Design and preliminary data. Prev Med. 1975;4:518–25.
Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, D’Agostino RB Sr, Fox CS, Larson MG, Murabito JM, et al. The third generation cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol. 2007;165:1328–35.
Seshadri S, Wolf PA, Beiser A, Au R, McNulty K, White R, D’Agostino RB. Lifetime risk of dementia and Alzheimer’s disease. The impact of mortality on risk estimates in the Framingham study. Neurology. 1997;49:1498–504.
Seshadri S, Beiser A, Au R, Wolf PA, Evans DA, Wilson RS, Petersen RC, Knopman DS, Rocca WA, Kawas CH, et al. Operationalizing diagnostic criteria for Alzheimer’s disease and other age-related cognitive impairment-part 2. Alzheimers Dement. 2011;7:35–52.
Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, van Belle G, Jolley L, Larson EB. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–46.
Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimers Dis. 2018;64:S161–89.
Beekly DL, Ramos EM, van Belle G, Deitrich W, Clark AD, Jacka ME, Kukull WA, Centers NI-AsD. The National Alzheimer’s Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18:270–7.
Beekly DL, Ramos EM, Lee WW, Deitrich WD, Jacka ME, Wu J, Hubbard JL, Koepsell TD, Morris JC, Kukull WA, Centers NIAAsD. The National Alzheimer’s Coordinating Center (NACC) database: the uniform data set. Alzheimer Dis Assoc Disord. 2007;21:249–58.
Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack C, Jagust W, Trojanowski JQ, Toga AW, Beckett L. The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clin N Am. 2005;15:869–77 (xi-xii).
Weiner MW, Aisen PS, Jack CR Jr, Jagust WJ, Trojanowski JQ, Shaw L, Saykin AJ, Morris JC, Cairns N, Beckett LA, et al. The Alzheimer’s disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6:202-211.e207.
Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR Jr, Jagust WJ, Shaw LM, Toga AW, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–9.
Scollard P, Choi S-E, Lee ML, Mukherjee S, Trittschuh E, Sanders RE, et al. Ceiling effects and differential measurement precision across calibrated cognitive scores in the Framingham study. Neuropsychology. 2023;37:383–97.
Muthén LK, Muthén BO. Mplus: statistical analysis with latent variables: user’s guide (version 8). Los Angeles: Muthén & Muthén; 2017.
Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–9.
Gogarten SM, Sofer T, Chen H, Yu C, Brody JA, Thornton TA, Rice KM, Conomos MP. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics. 2019;35:5346–8.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–73.
Conomos MP, Miller MB, Thornton TA. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet Epidemiol. 2015;39:276–93.
Conomos MP, Reiner AP, Weir BS, Thornton TA. Model-free estimation of recent genetic relatedness. Am J Hum Genet. 2016;98:127–48.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28:2540–2.
Sinnwell JP, Therneau TM, Schaid DJ. The kinship2 R package for pedigree data. Hum Hered. 2014;78:91–3.
Chen H, Wang C, Conomos MP, Stilp AM, Li Z, Sofer T, Szpiro AA, Chen W, Brehm JM, Celedon JC, et al. Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. Am J Hum Genet. 2016;98:653–66.
Wang X, Lim E, Liu CT, Sung YJ, Rao DC, Morrison AC, Boerwinkle E, Manning AK, Chen H. Efficient gene-environment interaction tests for large biobank-scale sequencing studies. Genet Epidemiol. 2020;44:908–23.
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, ReproGen C, Psychiatric Genomics C, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control C, Duncan L, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41.
Voorman A, Lumley T, McKnight B, Rice K. Behavior of QQ-plots and genomic control in studies of gene-environment interaction. PLoS ONE. 2011;6:e19416.
Almli LM, Duncan R, Feng H, Ghosh D, Binder EB, Bradley B, Ressler KJ, Conneely KN, Epstein MP. Correcting systematic inflation in genetic association tests that consider interaction effects: application to a genome-wide association study of posttraumatic stress disorder. JAMA Psychiat. 2014;71:1392–9.
Manning AK, LaValley M, Liu CT, Rice K, An P, Liu Y, Miljkovic I, Rasmussen-Torvik L, Harris TB, Province MA, et al. Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP x environment regression coefficients. Genet Epidemiol. 2011;35:11–8.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Ray D, Chatterjee N. A powerful method for pleiotropic analysis under composite null hypothesis identifies novel shared loci between Type 2 Diabetes and Prostate Cancer. PLoS Genet. 2020;16:e1009218.
Ray D, Venkataraghavan S, Zhang W, Leslie EJ, Hetmanski JB, Weinberg SM, Murray JC, Marazita ML, Ruczinski I, Taub MA, Beaty TH. Pleiotropy method reveals genetic overlap between orofacial clefts at multiple novel loci from GWAS of multi-ethnic trios. PLoS Genet. 2021;17:e1009584.
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics C, Patterson N, Daly MJ, Price AL, Neale BM. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5.
Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, Hemani G, Tansey K, Laurin C, Early G, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2017;33:272–9.
Lin DY, Sullivan PF. Meta-analysis of genome-wide association studies with overlapping subjects. Am J Hum Genet. 2009;85:862–72.
Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–30.
Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300.
Jun G, Vardarajan BN, Buros J, Yu CE, Hawk MV, Dombroski BA, Crane PK, Larson EB, Alzheimer’s Disease Genetics C, Mayeux R, et al. Comprehensive search for Alzheimer disease susceptibility loci in the APOE region. Arch Neurol. 2012;69:1270–9.
Goedert M, Spillantini MG. Frontotemporal lobar degeneration through loss of progranulin function. Brain. 2006;129:2808–10.
Eriksen JL, Mackenzie IR. Progranulin: normal function and role in neurodegeneration. J Neurochem. 2008;104:287–97.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–102.
Crane PK, Trittschuh E, Mukherjee S, Saykin AJ, Sanders RE, Larson EB, McCurry SM, McCormick W, Bowen JD, Grabowski T, et al. Incidence of cognitively defined late-onset Alzheimer’s dementia subgroups from a prospective cohort study. Alzheimers Dement. 2017;13:1307–16.
Dumitrescu L, Mahoney ER, Mukherjee S, Lee ML, Bush WS, Engelman CD, Lu Q, Fardo DW, Trittschuh EH, Mez J, et al. Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain. 2020;143:2561–75.
Groot C, Grothe MJ, Mukherjee S, Jelistratova I, Jansen I, van Loenhoud AC, Risacher SL, Saykin AJ, Mac Donald CL, Mez J, et al. Differential patterns of gray matter volumes and associated gene expression profiles in cognitively-defined Alzheimer’s disease subgroups. Neuroimage Clin. 2021;30:102660.
Uretsky M, Gibbons LE, Mukherjee S, Trittschuh EH, Fardo DW, Boyle PA, Keene CD, Saykin AJ, Crane PK, Schneider JA, Mez J. Longitudinal cognitive performance of Alzheimer’s disease neuropathological subtypes. Alzheimers Dement (N Y). 2021;7:e12201.
Gross AL, Hassenstab JJ, Johnson SC, Clark LR, Resnick SM, Kitner-Triolo M, Masters CL, Maruff P, Morris JC, Soldan A, et al. A classification algorithm for predicting progression from normal cognition to mild cognitive impairment across five cohorts: the preclinical AD consortium. Alzheimers Dement (Amst). 2017;8:147–55.
Eissman JM, Dumitrescu L, Mahoney ER, Smith AN, Mukherjee S, Lee ML, Scollard P, Choi SE, Bush WS, Engelman CD, et al. Sex differences in the genetic architecture of cognitive resilience to Alzheimer’s disease. Brain. 2022;145:2541–54.
Kang WS, Lee SM, Hwang D, Park HJ, Kim JW. Association between Unc-51-like autophagy activating kinase 2 gene polymorphisms and schizophrenia in the Korean population. Medicine (Baltimore). 2022;101:e28745.
Huang Y, Liao X, Luo J, Liu H, Zhong S, Chen J. Expression of circular RNAs in the vascular dementia rats. Neurosci Lett. 2020;735:135087.
Lee A, Kondapalli C, Virga DM, Lewis TL Jr, Koo SY, Ashok A, Mairet-Coello G, Herzig S, Foretz M, Viollet B, et al. Abeta42 oligomers trigger synaptic loss through CAMKK2-AMPK-dependent effectors coordinating mitochondrial fission and mitophagy. Nat Commun. 2022;13:4444.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41.
Shimohama S, Chachin M, Taniguchi T, Hidaka H, Kimura J. Changes of neurocalcin, a calcium-binding protein, in the brain of patients with Alzheimer’s disease. Brain Res. 1996;716:233–6.
Miller JA, Woltjer RL, Goodenbour JM, Horvath S, Geschwind DH. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 2013;5:48.
Ma WW, Ding BJ, Yuan LH, Zhao L, Yu HL, Xi YD, Xiao R. Neurocalcin-delta: a potential memory-related factor in hippocampus of obese rats induced by high-fat diet. Afr Health Sci. 2017;17:1211–21.
Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den Bosch L, Robberecht W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41.
Mackenzie IR, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. 2016;138(Suppl 1):54–70.
Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, Finch N, Rutherford NJ, Crook RJ, Josephs KA, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17:3631–42.
Nicholson AM, Finch NA, Thomas CS, Wojtas A, Rutherford NJ, Mielke MM, Roberts RO, Boeve BF, Knopman DS, Petersen RC, Rademakers R. Progranulin protein levels are differently regulated in plasma and CSF. Neurology. 2014;82:1871–8.
Xu HM, Tan L, Wan Y, Tan MS, Zhang W, Zheng ZJ, Kong LL, Wang ZX, Jiang T, Tan L, Yu JT. PGRN is associated with late-onset Alzheimer’s disease: a case-control replication study and meta-analysis. Mol Neurobiol. 2017;54:1187–95.
Vardarajan BN, Reyes-Dumeyer D, Piriz AL, Lantigua RA, Medrano M, Rivera D, Jimenez-Velazquez IZ, Martin E, Pericak-Vance MA, Bush W, et al. Progranulin mutations in clinical and neuropathological Alzheimer’s disease. Alzheimers Dement. 2022;18:2458–67.
Nag S, Yu L, Wilson RS, Chen EY, Bennett DA, Schneider JA. TDP-43 pathology and memory impairment in elders without pathologic diagnoses of AD or FTLD. Neurology. 2017;88:653–60.
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503–27.
Rhinn H, Tatton N, McCaughey S, Kurnellas M, Rosenthal A. Progranulin as a therapeutic target in neurodegenerative diseases. Trends Pharmacol Sci. 2022;43:641–52.
Gomez Ravetti M, Rosso OA, Berretta R, Moscato P. Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus’ gene expression profiles in Alzheimer’s disease. PLoS ONE. 2010;5:e10153.
Silver M, Janousova E, Hua X, Thompson PM, Montana G, Alzheimer’s Disease Neuroimaging I. Identification of gene pathways implicated in Alzheimer’s disease using longitudinal imaging phenotypes with sparse regression. Neuroimage. 2012;63:1681–94.
Vounou M, Janousova E, Wolz R, Stein JL, Thompson PM, Rueckert D, Montana G, Alzheimer’s Disease Neuroimaging I. Sparse reduced-rank regression detects genetic associations with voxel-wise longitudinal phenotypes in Alzheimer’s disease. Neuroimage. 2012;60:700–16.
Yan Q, Nho K, Del-Aguila JL, Wang X, Risacher SL, Fan KH, Snitz BE, Aizenstein HJ, Mathis CA, Lopez OL, et al. Genome-wide association study of brain amyloid deposition as measured by Pittsburgh Compound-B (PiB)-PET imaging. Mol Psychiatry. 2021;26:309–21.
Kim BM, You MH, Chen CH, Suh J, Tanzi RE, Ho Lee T. Inhibition of death-associated protein kinase 1 attenuates the phosphorylation and amyloidogenic processing of amyloid precursor protein. Hum Mol Genet. 2016;25:2498–513.
Wirz KT, Bossers K, Stargardt A, Kamphuis W, Swaab DF, Hol EM, Verhaagen J. Cortical beta amyloid protein triggers an immune response, but no synaptic changes in the APPswe/PS1dE9 Alzheimer’s disease mouse model. Neurobiol Aging. 2013;34:1328–42.
Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–92.
Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, Yamamoto M. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624.
Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, Nguyen-Viet TA, Bowers P, Sidorenko J, Karlsson Linner R, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50:1112–21.
Hill WD, Marioni RE, Maghzian O, Ritchie SJ, Hagenaars SP, McIntosh AM, Gale CR, Davies G, Deary IJ. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol Psychiatry. 2019;24:169–81.
Acknowledgements
Biological samples and associated phenotypic data used in primary data analyses were stored at study investigator institutions and at the National Cell Repository for Alzheimer’s Disease (NCRAD, U24-AG021886) at Indiana University funded by NIA. Associated phenotypic data used in primary and secondary data analyses were provided by the study investigators, the NIA funded Alzheimer’s Disease Centers (ADCs), the National Alzheimer’s Coordinating Center (NACC, U01-AG016976); and the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS, U24-AG041689) at the University of Pennsylvania, funded by NIA. Phenotypic data were harmonized by the ADSP Phenotype Harmonization Consortium (ADSP-PHC, U24-AG074855, U01-AG068057 and R01-AG059716). This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Library of Medicine. Contributors to the genetic and phenotypic data included the study investigators on projects that were individually funded by NIA and other NIH institutes, and by private US organizations, foreign governmental organizations, or nongovernmental organizations.
We also acknowledge the following investigators who assembled and characterized participants of cohorts included in this study.
Adult Changes in Thought: James D. Bowen, Paul K. Crane, Gail P. Jarvik, C. Dirk Keene, Eric B. Larson, W. William Lee, Wayne C. McCormick, Susan M. McCurry, Shubhabrata Mukherjee, Katie Rose Richmire
Chicago Health and Aging Project: Philip L. De Jager, Denis A. Evans.
Estudio Familiar de la Influencia Genetica en Alzheimer: Sandra Barral, Rafael Lantigua, Richard Mayeux, Martin Medrano, Dolly Reyes-Dumeyer, Badri Vardarajan.
Framingham Heart Study: Ting Fang Alvin Ang, Hugo J. Aparicio, Rhoda Au, Sanford Auerbach, Alexa S. Beiser, Anita DeStefano, Sherral Devine, Lindsay A. Farrer, Jesse Mez, Jose Raphael Romero, Sudha Seshadri.
Genetic Differences: Duane Beekly, James Bowen, Walter A. Kukull, Eric B. Larson, Wayne McCormick, Gerard D. Schellenberg, Linda Teri.
Mayo Clinic: Minerva M. Carrasquillo, Dennis W. Dickson, Nilufer Ertekin-Taner, Neill R. Graff-Radford, Joseph E. Parisi, Ronald C. Petersen, Steven G. Younkin.
Mayo PD: Gary W. Beecham, Dennis W. Dickson, Ranjan Duara, Nilufer Ertekin-Taner, Tatiana M. Foroud, Neill R. Graff-Radford, Richard B. Lipton, Joseph E. Parisi, Ronald C. Petersen, Bill Scott, Jeffery M. Vance.
Memory and Aging Project: David A. Bennett, Philip L. De Jager.
Multi-Institutional Research in Alzheimer’s Genetic Epidemiology Study: Sanford Auerbach, Helan Chui, Jaeyoon Chung, L. Adrienne Cupples, Charles DeCarli, Ranjan Duara, Martin Farlow, Lindsay A. Farrer, Robert Friedland, Rodney C.P. Go, Robert C. Green, Patrick Griffith, John Growdon, Gyungah R. Jun, Walter Kukull, Alexander Kurz, Mark Logue, Kathryn L. Lunetta, Thomas Obisesan, Helen Petrovitch, Marwan Sabbagh, A. Dessa Sadovnick, Magda Tsolaki.
National Cell Repository for Alzheimer’s Disease: Kelley M. Faber, Tatiana M. Foroud.
National Institute on Aging (NIA) Late Onset Alzheimer’s Disease Family Study: David A. Bennett, Sarah Bertelsen, Thomas D. Bird, Bradley F. Boeve, Carlos Cruchaga, Kelley Faber, Martin Farlow, Tatiana M. Foroud, Alison M. Goate, Neill R. Graff-Radford, Richard Mayeux, Ruth Ottman, Dolly Reyes-Dumeyer, Roger Rosenberg, Daniel Schaid, Robert A. Sweet, Giuseppe Tosto, Debby Tsuang, Badri Vardarajan.
NIA Alzheimer Disease Centers: Erin Abner, Marilyn S. Albert, Roger L. Albin, Liana G. Apostolova, Sanjay Asthana, Craig S. Atwood, Lisa L. Barnes, Thomas G. Beach, David A. Bennett, Eileen H. Bigio, Thomas D. Bird, Deborah Blacker, Adam Boxer, James B. Brewer, James R. Burke, Jeffrey M. Burns, Joseph D. Buxbaum, Nigel J. Cairns, Chuanhai Cao, Cynthia M. Carlsson, Richard J. Caselli, Helena C. Chui, Carlos Cruchaga, Mony de Leon, Charles DeCarli, Malcolm Dick, Dennis W. Dickson, Nilufer Ertekin-Taner, David W. Fardo, Martin R. Farlow, Lindsay A. Farrer, Steven Ferris, Tatiana M. Foroud, Matthew P. Frosch, Douglas R. Galasko, Marla Gearing, David S. Geldmacher, Daniel H. Geschwind, Bernardino Ghetti, Carey Gleason, Alison M. Goate, Teresa Gomez-Isla, Thomas Grabowski, Neill R. Graff-Radford, John H. Growdon, Lawrence S. Honig, Ryan M. Huebinger, Matthew J. Huentelman, Christine M. Hulette, Bradley T. Hyman, Suman Jayadev, Lee-Way Jin, Sterling Johnson, M. Ilyas Kamboh, Anna Karydas, Jeffrey A. Kaye, C. Dirk Keene, Ronald Kim, Neil W. Kowall, Joel H. Kramer, Frank M. LaFerla, James J. Lah, Allan I. Levey, Ge Li, Andrew P. Lieberman, Oscar L. Lopez, Constantine G. Lyketsos, Daniel C. Marson, Ann C. McKee, Marsel Mesulam, Jesse Mez, Bruce L. Miller, Carol A. Miller, Abhay Moghekar, John C. Morris, John M. Olichney, Joseph E. Parisi, Henry L. Paulson, Elaine Peskind, Ronald C. Petersen, Aimee Pierce, Wayne W. Poon, Luigi Puglielli, Joseph F. Quinn, Ashok Raj, Murray Raskind, Eric M. Reiman, Barry Reisberg, Robert A. Rissman, Erik D. Roberson, Howard J. Rosen, Roger N. Rosenberg, Martin Sadowski, Mark A. Sager, David P. Salmon, Mary Sano, Andrew J. Saykin, Julie A. Schneider, Lon S. Schneider, William W. Seeley, Scott Small, Amanda G. Smith, Robert A. Stern, Russell H. Swerdlow, Rudolph E. Tanzi, Sarah E. Tomaszewski Farias, John Q. Trojanowski, Juan C. Troncoso, Debby W. Tsuang, Vivianna M. Van Deerlin, Linda J. Van Eldik, Harry V. Vinters, Jean Paul Vonsattel, Jen Chyong Wang, Sandra Weintraub, Kathleen A. Welsh-Bohmer, Shawn Westaway, Thomas S. Wingo, Thomas Wisniewski, David A. Wolk, Randall L. Woltjer, Steven G. Younkin, Lei Yu, Chang-En Yu.
Religious Orders Study: David A. Bennett, Philip L. De Jager.
Texas Alzheimer’s Research and Care Consortium: Perrie Adams, Alyssa Aguirre, Lisa Alvarez, Gayle Ayres, Robert C. Barber, John Bertelson, Sarah Brisebois, Scott Chasse, Munro Culum, Eveleen Darby, John C. DeToledo, Thomas J. Fairchild, James R. Hall, John Hart, Michelle Hernandez, Ryan Huebinger, Leigh Johnson, Kim Johnson, Aisha Khaleeq, Janice Knebl, Laura J. Lacritz, Douglas Mains, Paul Massman, Trung Nguyen, Sid O’Bryant, Marcia Ory, Raymond Palmer, Valory Pavlik, David Paydarfar, Victoria Perez, Marsha Polk, Mary Quiceno, Joan S. Reisch, Monica Rodriguear, Roger Rosenberg, Donald R. Royall, Janet Smith, Alan Stevens, Jeffrey L. Tilson, April Wiechmann, Kirk C. Wilhelmsen, Benjamin Williams, Henrick Wilms, Martin Woon.
University of Miami: Larry D. Adams, Gary W. Beecham, Regina M. Carney, Katrina Celis, Michael L. Cuccaro, Kara L. Hamilton-Nelson, James Jaworski, Brian W. Kunkle, Eden R. Martin, Margaret A. Pericak-Vance, Farid Rajabli, Michael Schmidt, Jeffery M Vance.
University of Toronto: Ekaterina Rogaeva, Peter St. George-Hyslop.
University of Washington Families: Thomas D. Bird, Olena Korvatska, Wendy Raskind, Chang-En Yu.
Vanderbilt University: John H. Dougherty, Harry E. Gwirtsman, Jonathan L. Haines, Angela Jefferson.
Washington Heights-Inwood Columbia Aging Project: Adam Brickman, Rafael Lantigua, Jennifer Manly, Richard Mayeux, Christiane Reitz, Nicole Schupf, Yaakov Stern, Giuseppe Tosto, Badri Vardarajan.
Funding
This work was supported in part by NIH grants U19-AG068753, R01-AG048927, U54-AG052427, U01-AG058654, U01-AG032984, RF1-AG057519, U01-AG062602, P30-AG072878, U24-AG074855, U01-AG068057, and R01-AG059716.
Author information
Authors and Affiliations
Contributions
MK, RS, KLL, JBM, and LAF planned and designed the study. SM, LEG, PS, ML, S-EC, BK, CN, PKC, and JBM derived the cognitive domain factor scores. EHT, LCD, AD, TJH, MLC, and AJS harmonized phenotypic data for the ACT, ADNI, ROSMAP, and NACC datasets. TFAA, SAD, RA, and JBM collected and harmonized FHS data. WAK assembled and harmonized ADRC phenotypic data into NACC. DAB collected and harmonized ROSMAP phenotype data. MK performed the statistical analyses. KLL, JBM, and LAF supervised the analyses. MK and LAF wrote the manuscript. All authors edited the manuscript. L-SW, RPM, JLH, MAP-V, GDS, RA, and LAF obtained funding for this study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was performed in accordance with the Declaration of Helsinki and approved by the Boston University Institutional Review Board (Protocol # H-28168). Informed consent was obtained from all subjects and/or their legal guardians.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Schemes for combining results from GWAS and pleiotropy analyses across datasets. Fig. S2. Manhattan and QQ plots for GWAS of individual cognitive domain scores in the total sample. Fig. S3. Manhattan and QQ plots for GWAS of individual cognitive domain scores in the clinic-based cohorts. Fig. S4. Manhattan and QQ plots for GWAS of individual cognitive domain scores in the community-based cohorts. Fig. S5. Locus Zoom plots showing association of SNPs in the BIN1 region with language. Fig. S6. Locus Zoom plots showing association of SNPs in the BIN1 region with memory. Fig. S7. Locus Zoom plots showing association of SNPs in theCR1 andMS4A6A regions with memory in the clinic-based cohorts. Fig. S8. Manhattan and QQ plots for pleiotropy GWAS in pairs of cognitive domain scores in the total sample. Fig. S9. Manhattan and QQ plots for pleiotropy GWAS in pairs of cognitive domain scores in the clinic-based cohorts. Fig. S10. Manhattan and QQ plots for pleiotropy GWAS in pairs of cognitive domain scores in the community-based cohorts. Fig. S11. Locus Zoom plots showing pleiotropy of SNPs in the BIN1 region with language and memory.
Additional file 2: Supplementary Table 1.
Correlations among cognitive domain scores by cohort. Supplementary Table 2. Genetic correlations of cognitive domain scores with general cognitive function and neuropsychiatric disorders. Supplementary Table 3. Genome-wide significant and suggestive associations for cognitive domain scores in the total sample, clinic-based cohorts, and community-based cohorts. Supplementary Table 4. Genome-wide significant and suggestive pleiotropy for each pair of cognitive domains in the total sample, clinic-based cohorts, and community-based cohorts. Supplementary Table 5. Canonical pathways significantly enriched for top-ranked GWAS genes with corrected FDR < 0.05. Supplementary Table 6. Number of seed genes from individual GWAS and pleiotropy analyses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kang, M., Ang, T.F.A., Devine, S.A. et al. A genome-wide search for pleiotropy in more than 100,000 harmonized longitudinal cognitive domain scores. Mol Neurodegeneration 18, 40 (2023). https://doi.org/10.1186/s13024-023-00633-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00633-4