
Abstract
Background
APOE variants are strongly associated with abnormal amyloid aggregation and additional direct effects of APOE on tau aggregation are reported in animal and human cell models. The degree to which these effects are present in humans when individuals are clinically unimpaired (CU) but have abnormal amyloid (Aβ+) remains unclear.
Methods
We analyzed data from CU individuals in the Anti-Amyloid Treatment in Asymptomatic AD (A4) and Longitudinal Evaluation of Amyloid Risk and Neurodegeneration (LEARN) studies. Amyloid PET data were available for 4486 participants (3163 Aβ-, 1323 Aβ+) and tau PET data were available for a subset of 447 participants (55 Aβ-, 392 Aβ+). Linear models examined APOE (number of e2 and e4 alleles) associations with global amyloid and regional tau burden in medial temporal lobe (entorhinal, amygdala) and early neocortical regions (inferior temporal, inferior parietal, precuneus). Consistency of APOE4 effects on regional tau were examined in 220 Aβ + CU and mild cognitive impairment (MCI) participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI).
Results
APOE2 and APOE4 were associated with lower and higher amyloid positivity rates, respectively. Among Aβ+ CU, e2 and e4 were associated with reduced (−12 centiloids per allele) and greater (+15 centiloids per allele) continuous amyloid burden, respectively. APOE2 was associated with reduced regional tau in all regions (-0.05 to -0.09 SUVR per allele), whereas APOE4 was associated with greater regional tau (+0.02 to +0.07 SUVR per allele). APOE differences were confirmed by contrasting e3/e3 with e2/e3 and e3/e4. Mediation analyses among Aβ+ s showed that direct effects of e2 on regional tau were present in medial temporal lobe and early neocortical regions, beyond an indirect pathway mediated by continuous amyloid burden. For e4, direct effects on regional tau were only significant in medial temporal lobe. The magnitude of protective e2 effects on regional tau was consistent across brain regions, whereas detrimental e4 effects were greatest in medial temporal lobe. APOE4 patterns were confirmed in Aβ+ ADNI participants.
Conclusions
APOE influences early regional tau PET burden, above and beyond effects related to cross-sectional amyloid PET burden. Therapeutic strategies targeting underlying mechanisms related to APOE may modify tau accumulation among Aβ+ individuals.
Similar content being viewed by others
Background
APOE4 is the strongest genetic predictor of sporadic Alzheimer’s disease (AD) dementia [1,2,3,4] and is consistently associated with risk of abnormal amyloid during the stages preceding dementia onset [5,6,7]. APOE influences amyloid accumulation through interactions with production, fibrillization, and clearance mechanisms, as well as interactions between APOE, amyloid, tau, neuroinflammation, and neuronal structure and function [8,9,10]. On average, APOE4 carriers typically reach the threshold for abnormal amyloid (Aβ+) approximately 10–15 years before e3/3 carriers, whereas amyloid positivity is delayed by approximately 4 years in e2 carriers compared to e3/e3 carriers [11].
Tau abnormalities are a stronger predictor of concurrent cognitive ability and future cognitive impairment than Aβ status alone [12,13,14]. Although both amyloid and tau abnormalities are strongly related such that elevated tau is very uncommon among Aβ- individuals, there is substantial variability in the magnitude of tau PET uptake within Aβ+ individuals [12, 15,16,17,18,19,20], highlighting that additional factors beyond amyloid contribute to downstream tau accumulation. Interestingly, there is growing evidence suggesting that APOE may directly impact variability in tau accumulation. For instance, tau PET studies in Aβ+ patients across the AD clinical spectrum have shown a more medial temporal lobe (MTL)-dominant pattern of tau pathology in e4 carriers compared to non-carriers [16, 21] even after adjusting for global amyloid burden [22, 23]. This suggests that APOE genotype may influence disease progression even after amyloid positivity is reached. This may also partially explain why in early clinical trials for AD dementia patients, which did not yet routinely assess amyloid status, e4 carriers declined more rapidly than non-e4 carriers [24].
However, analyses examining APOE effects on tau PET specifically among clinically unimpaired (CU) participants have found mixed results. For instance, Lowe and colleagues [25] reported no effect of e4 on tau, and Ramanan and colleagues [26] found similar null effects after controlling for global levels of amyloid. In contrast, Ossenkoppele and colleagues [27] found a significant effect of e4 on entorhinal tau and Ghisays and colleagues [28] reported that e4 modified the association between age and entorhinal tau. Thus, the extent to which APOE4 influences early regional tau burden in Aβ+ CU remains unclear [29]. Additionally, little has been established regarding whether APOE2 is associated with reduced tau burden in Aβ+ individuals. A primary barrier to examining e2 effects is that e2 carriership is uncommon in Aβ+ CU individuals. Within Aβ+ CU individuals, approximately 4–8% will be e2 carriers whereas around 50% will be e4 carriers [29,30,31]. One study examining CU combined with mild cognitive impairment (MCI) showed comparable levels of regional tau burden and tau accumulation over time in 45 e2 carriers compared to 257 e3/e3 carriers [32], though only 15 of the 45 e2 carriers were Aβ+. Post-mortem work examining the full disease spectrum from CU to AD dementia has shown mixed results, with one study of 411 e2 carriers showing no effect on Braak staging after controlling for neuritic amyloid plaque severity [33], and another study of 163 e2 carriers showing fewer tau tangles in the Aβ+ e2 group compared to Aβ+ e2 non-carriers [34]. To date, it is likely that studies focused on CU cohorts have been underpowered to determine whether e2 effects influence tau accumulation among Aβ+ .
The overall goal of the present study was to establish whether APOE genotype influences early tau burden in preclinical AD beyond effects attributable to amyloid burden. We leveraged the Anti-Amyloid Treatment in Asymptomatic AD (A4) and Longitudinal Evaluation of Amyloid Risk and Neurodegeneration (LEARN) amyloid PET screening dataset of 4486 CU [30, 35] along with the tau PET substudy [29, 30] that included 392 Aβ+ CU individuals with tau PET. APOE4 effects were additionally confirmed in 220 Aβ+ CU and MCI participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). We hypothesized that APOE genotype would influence regional tau PET values in the MTL and that these effects would be present after controlling for continuous amyloid burden.
Methods
The A4 Study is a secondary prevention trial that focused on participants with preclinical AD (i.e., Aβ+ CU) [30]. The LEARN study is a companion to the A4 study focused on Aβ- individuals. After initial telephone and in-clinic screenings to determine study eligibility including cognitive status, 4486 participants underwent amyloid PET scanning to allow for identification of Aβ+ individuals prior to treatment randomization. A subset enriched for amyloid positivity also underwent tau PET scanning prior to treatment randomization. All A4/LEARN participants completed written informed consent before participation. The ADNI is a public-private partnership launched in 2003 with the primary goal of testing whether serial neuroimaging and biological markers, and clinical and neuropsychological assessments can be combined to measure the progression of MCI and early AD. All ADNI participants provided written informed consent in compliance with local IRBs. For up-to-date information, see www.adni-info.org.
In the A4/LEARN dataset, all participants included in this study (Table 1 and Table S1) were 65–85 years old, CU (Clinical Dementia Rating score = 0, Mini-Mental State Examination score = 25–30, and Logical Memory Delayed Recall score = 6–18), and had completed amyloid PET scans ([18F]-florbetapir). A subset of these participants also completed tau PET scans ([18F]-flortaucipir). Amyloid status was determined using a hybrid quantitative and qualitative method established by the A4/LEARN study team [30]. Global standardized uptake value ratios (SUVRs) for amyloid PET (reference region: whole cerebellum) and regional SUVRs for tau PET (reference region: cerebellar gray) were extracted using previously published pipelines [29]. Briefly, amyloid and tau PET data were processed with in-house scripts using FSL, SPM, and FreeSurfer. Five-minute amyloid and tau PET frames corresponding to 50–70 min and 90–110 min post-injection, respectively, were realigned and summed. Each participant’s MRI data (including aparc+aseg FreeSurfer labels) were coregistered to the summed PET data. Mean uptake values for each aparc+aseg FreeSurfer region were extracted. Cortical amyloid SUVR was calculated following ADNI procedures (see “Florbetapir (AV45) processing methods” from http://adni.loni.usc.edu) and regional tau analyses focused on entorhinal, amygdala, inferior temporal, inferior parietal, and precuneus regions as defined by FreeSurfer.
In the ADNI dataset, only participants with a tau PET scan ([18F]-flortaucipir), an amyloid positive PET scan ([18F]-florbetaben (FBB) or [18F]-florbetapir (FBP)), available APOE status, and a diagnosis of CU or MCI within 1 year of the tau PET scan were included (Table 2). Amyloid PET SUVRs and cutoffs for amyloid status (reference region: whole cerebellum) as well as regional tau PET (reference region: inferior cerebellum) were downloaded from the ADNI LONI website.
For both A4/LEARN and ADNI amyloid data, amyloid SUVRs were converted to the common Centiloid (CL) scale using previously published equations [36] (Fig. S1). This conversion allows for direct comparison of data collected from different tracers and from different studies. ADNI analyses combined [18F]-florbetaben and [18F]-florbetapir ligands using CL values.
Statistical analysis
Data were analyzed using R v4.1.2. Analyses primarily focused on the A4/LEARN dataset given the substantially larger sample of Aβ+ CU individuals with tau PET. First, to examine the association between APOE and amyloid burden, logistic regression models using the questionr package to obtain odds ratios examined the effects of e2 and e4 dosage on dichotomous amyloid status (e.g., e3/e3 carriers were coded with an e2 dosage of 0 and an e4 dosage of 0; e3/e4 carriers were coded with an e2 dosage of 0 and an e4 dosage of 1; e2/e4 carriers were coded with an e2 dosage of 1 and an e4 dosage of 1). Within Aβ+s, linear regression models also examined the effects of e2 and e4 dosage on continuous amyloid burden. Models were repeated to include age*APOE interactions and sex*APOE interactions. Second, across Aβ-s and Aβ+s, linear regression models examined the effects of continuous amyloid on regional tau. Third, linear regression models examined the effects of e2 and e4 dosage on regional tau within Aβ+s only. Primary analyses included all genotypes. Additionally, we conducted a series of sensitivity analyses to ensure that the APOE effects on regional tau were not primarily driven by the presence of uncommon genotypes or our allele dosage modeling approach: (1) linear regression models with e2 and e4 dosage were repeated after excluding e2/e2 and e4/e4 homozygotes, (2) linear regression models with e2 and e4 dosage were repeated after excluding e2/e4 carriers, (3) instead of modeling e2 and e4 dosage, linear regression models contrasted common APOE genotype groups (e2/e3 vs. e3/e3, e3/e4 vs. e3/e3). Finally, mediation models using the lavaan package examined whether continuous amyloid mediated the association between APOE dosage and regional tau in Aβ+s. Bootstrapping procedures (n = 10,000) were used to determine significance of effects in mediation models as well as to compare e2 and e4 effect sizes. All models controlled for mean centered age (71.3 years) and sex.
Analyses were repeated in Aβ+ CU and MCI participants from ADNI. Given the small number of Aβ+ e2 individuals in ADNI (n = 7 CU and n = 6 MCI), ADNI was only used to examine effects of e4. Mediation models examining whether continuous amyloid mediated the association between e4 dosage and regional tau were repeated in 220 Aβ+ CU and MCI participants. These models also controlled for mean centered age (75.9 years) and sex.
Results
APOE predicts amyloid status
APOE genotype was significantly related to amyloid status after accounting for age and sex. As expected, e2 dosage was associated with amyloid negativity and e4 dosage was associated with amyloid positivity (Table 3A). The impact of age on amyloid positivity was reduced in e2 carriers and the protective effects of e2 on amyloid positivity was not observed in males (Table 3A and Fig. 1).

APOE interactions with age and sex. The top row shows associations between age and probability of Aβ+ status as a function of APOE carriership. The top left panel depicts reduced effects of age on Aβ+ status in e2 carriers. The top right panel shows increasing associations between age and Aβ+ status across all levels of e4 carriership. The bottom row shows associations between sex and probability of Aβ+ status as a function of APOE carriership. The bottom left panel depicts protective effects of e2 in females. The bottom right panel shows no significant difference between sexes in probability of Aβ+ status across all levels of e4 carriership
Global amyloid burden associations with regional tau
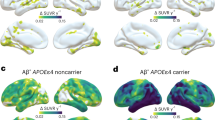
Among Aβ+s with tau PET data, APOE genotype was significantly associated with continuous amyloid burden but interactions with age and sex were not significant (Table 3B). Higher continuous amyloid burden was associated with greater regional tau levels across all MTL and early neocortical tau regions (Fig. 2 and Table S2). In general, Aβ- CU individuals showed little evidence of tau elevations. Although the Aβ+ group showed elevated regional tau SUVRs, a wide range was present, with some Aβ+ individuals showing evidence of tau elevations and others showing levels comparable to the Aβ- group (Fig. 2). Overall, among the Aβ+ group, age, sex, and continuous amyloid burden explained 7–15% of the total variance of regional tau (Table S2).

Association between amyloid and tau burden in (A) Aβ- and Aβ+ participants and in (B) only Aβ+ participants. Plotted tau SUVRs and amyloid centiloids (CLs) are residualized by age and sex
APOE associations with regional tau
Given the minimal levels of tau in Aβ-s combined with the small sample size of this group (n = 55), we next examined the influence of APOE on regional tau in Aβ+s only (n = 392). APOE e2 and e4 were significantly associated with reduced and increased tau, respectively, in MTL and early neocortical regions (Table 4). APOE2 effects on reduced tau in amygdala and precuneus were strongest at older ages (Table S3). Results were similar after excluding the single e2/e2 Aβ+ participant and the 25 e4/e4 Aβ+ homozygotes (Table S4) as well as after excluding 13 e2/e4 Aβ+ carriers (Table S5).
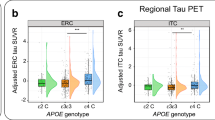
APOE differences among Aβ+ s were further confirmed by directly contrasting e2/3 (n = 17) and e3/4 (n = 182) against the e3/3 group (n = 147). APOE e2/e3 individuals had significantly lower tau SUVRs than e3/e3 individuals in entorhinal cortex, amygdala, inferior temporal, and precuneus (0.05–0.09 SUVRs), whereas e3/e4 individuals had significantly higher tau SUVRs than e3/e3 individuals in all examined MTL and early neocortical regions (0.03–0.08 SUVRs; Fig. 3 and Table S6).

Regional tau differences between Aβ+ APOE e2/e3, e3/e3, and e3/e4 groups. Plotted tau SUVRs are residualized by age and sex. Note: * p < 0.05, **p < 0.01, ***p < 0.001
Direct effects of APOE on regional tau in Aβ+ CU
Because APOE genotype remains a significant predictor variable of continuous amyloid burden even within Aβ+ s (Fig. 2 and Table S2), we conducted a series of mediation models to determine whether global amyloid burden mediates the association between APOE and regional tau in Aβ+ CU individuals (Fig. 4A; Fig. S2A). Continuous amyloid burden accounted for 14–27% of the effect of e2 on regional tau and 21–68% of the effect of e4 on regional tau. For e2, partial mediation was found in both MTL and early neocortical regions with significant direct effects remaining between e2 and regional tau across all regions. In contrast, partial mediation effects were found for e4 in MTL regions only; direct effects of e4 on tau in early neocortical regions were not significant.

Mediating effects of amyloid (centiloids) on APOE and regional tau SUVRs. (A) Stacked bar plots depict the total effect, as well as indirect and direct subcomponents, extracted from mediation models examining the effects of APOE on regional tau among Aβ+ clinically unimpaired (CU) individuals from A4. A comparison of e2 and e4 direct effect sizes are also shown. Error bars reflect standard error. (B) Stacked bar plots depict the total effect, as well as indirect and direct subcomponents, extracted from mediation models examining the effects of APOE e4 on regional tau among Aβ+ CU and mild cognitive impairment (MCI) individuals from ADNI. Note: * p < 0.05, **p < 0.01, ***p < 0.001
The e2 and e4 effect sizes were compared within and across tau regions using bootstrapping procedures (Fig. 4A). The direct effect of e4 on regional tau SUVRs was significantly stronger in the amygdala in comparison to neocortical regions. The strength of direct effects of e2 in MTL were qualitatively larger but not significantly different than those observed in neocortical regions, with the exception of stronger e2 effects in amygdala than inferior parietal tau. Within each tau region, e2 direct effects were qualitatively stronger than e4 direct effects, though these differences were not significant except in precuneus.
Amyloid mediates the association between APOE4 and tau in Aβ+ CU and MCI
Mediation analyses examining effects of e4 were conducted in 220 Aβ+ CU and MCI individuals from ADNI (Fig. 4B). APOE2 effects were not examined given the small sample size of only 13 Aβ+ e2 carriers (9 e2/e3 and 4 e2/e4) in this dataset. This analysis showed that e4 effects on regional tau were 2–3 times larger than e4 effects from the A4/LEARN analyses, presumably because the ADNI cohort included Aβ+ MCI and therefore a broader range of regional tau values. Among Aβ+ CU and MCI, continuous amyloid burden accounted for 40–51% of the variance of the effect of e4 on MTL tau and both indirect and direct effects were significant. In neocortical regions, continuous amyloid burden fully accounted for the variance of the effect of e4, and only indirect pathways through continuous amyloid were significant (i.e., no significant direct effect of e4 on neocortical tau remained). Thus, e4 effects on regional tau were consistent across A4/LEARN and ADNI analyses, with both cohorts showing significant direct effects between e4 and MTL tau PET, and no significant direct effects of e4 on neocortical regions after accounting for continuous amyloid PET burden. All effects were similar when only including the 162 participants who had tau PET and amyloid PET scans within 2 years of each other (Fig. S3).
Discussion
In a large cohort of Aβ+ CU from the A4/LEARN study, we found that APOE genotype is associated with both global amyloid PET burden as well as regional tau PET in MTL and early neocortical regions. Cross-sectional examinations of APOE, amyloid, and tau demonstrated both amyloid-mediated and direct effects of e2 and e4 on tau within the MTL. For tau in early neocortical regions, both amyloid-mediated and direct effects were present for e2 whereas only amyloid-mediated effects were present for e4. Our findings provide evidence that APOE genotype influences early tau burden in preclinical AD beyond effects that are attributable to amyloid burden as measured with PET. This work highlights that although amyloid positivity is a key driver of downstream tau accumulation, APOE genotype additionally influences disease progression through mechanisms directly related to tau accumulation.
APOE4 is involved in amyloid production, formation of amyloid plaques and APOE/amyloid complexes, cellular clearance of amyloid, amyloid clearance through the blood-brain barrier, and proteolytic degradation [8, 9, 37]. Given the numerous mechanisms of e4 influencing amyloid, it is no surprise that amyloid PET studies consistently demonstrate that e4 is associated with an earlier onset of amyloid accumulation [38] as well as increased amyloid deposition rate and burden [6, 7, 39, 40]. Our results are consistent with these studies as we show that e4 dosage is associated with increased risk of amyloid positivity as well as continuous amyloid burden even after amyloid positivity is reached. Specifically, each e4 and e2 allele was associated with +15 and −12 centiloids, respectively, in Aβ+ CU. Given these effects, it is critical to account for continuous amyloid burden in analyses examining the impact of APOE on regional tau burden [41,42,43,44].
Our finding that the effects of e4 on amyloid burden are greater than the protective effects of e2 for amyloid positivity is consistent with mechanistic work that shows more pathways linking e4 to amyloid processing than e2 [9, 37]. For instance, whereas both e4 and e2 have been associated with amyloid plaques and amyloid clearance [45], e4 may additionally affect extracellular APOE/amyloid complexes [46], cellular clearance of amyloid [47, 48], and proteolytic degradation of monomeric amyloid [49]. Overall, our analyses of the full A4/LEARN screening dataset of 4486 CU confirmed the expected effects of e4 and e2 on both overall amyloid positivity and continuous amyloid burden within the Aβ+ CU group.
Our main finding was that both e4 and e2 influence early regional tau levels among Aβ+ individuals. Across two datasets (i.e., 392 Aβ+ CU from A4/LEARN; 220 Aβ+ CU and MCI from ADNI), we showed that e4 was associated with higher MTL tau PET burden, an effect that was only partially mediated by continuous amyloid burden. Interestingly, the overall pattern of significant direct effects of e4 on MTL accounting for ~50% of the total effect was consistent between Aβ+ CU data from A4/LEARN and combined Aβ+ CU and MCI data from ADNI. One difference between the A4/LEARN and ADNI results was that the overall magnitude of the total e4 effect on regional tau in MTL and neocortical regions was 2–3 times stronger in ADNI (increase of 0.039–0.152 SUVR per allele) than A4/LEARN (increase in 0.019–0.073 SUVR per allele). The stronger total effects in ADNI can be explained by the inclusion of Aβ+ MCI in the ADNI analyses, who are known to have a greater range of tau PET values than Aβ+ CU [27, 50]. Despite differing magnitudes, the fact that the proportion of e4 direct and indirect effects remained consistent in both cohorts provides further support for an association between e4 on tau during the pre-dementia stages of disease. Potential mechanisms may involve mislocalization of tau from the axon to the soma and dendrites, promotion of aberrant hyperphosphorylation, acceleration of spread from diseased to healthy neurons, enhancement of aggregation of p-tau into insoluble neurofibrillary tangles, and disruption of tau clearance due to e4 [9, 51].
In addition to e4 effects on regional tau, we also found that e2 was associated with reduced tau levels in both MTL and neocortical regions. More than 75% of the total effect of e2 on tau in MTL and cortex was direct (i.e., not accounted for by continuous amyloid burden). Our findings are consistent with animal models and iPSC-derived human brain cell cultures that have identified direct associations between APOE and tau accumulation [51,52,53,54,55]. APOE2 in particular may be protective against AD partially through lipid and metabolic mechanisms that lead to fewer tau tangles [44]. It is likely that other non-amyloid factors have a similar impact on disease progression, accelerating progression in some Aβ+ individuals but allowing for resilience to downstream changes in other Aβ+ individuals [56]. Given the link between elevated tau burden and risk of clinical progression from CU to MCI [14], the identification of factors that influence tau burden among Aβ+s will improve individual level prediction and shed insight into mechanisms of disease risk and resilience.
Our results also show that e4 and e2 differ in their impact on tau spatial patterns. Specifically, e4 was only linked to regional effects within the MTL whereas the effect of e2 was consistent across MTL and neocortical regions. Our findings linking e4 specifically to MTL tau is consistent with several studies combining participants across the AD clinical spectrum [16, 21,22,23]. Mattsson and colleagues [21] reported that in AD dementia patients, e4 carriers were similar to non-carriers in the magnitude of regional tau, but e4 carriers had greater entorhinal cortex uptake relative to neocortical uptake quantified using a ratio. APOE4 is known to lead to blood-brain barrier dysfunction [57,58,59,60] and regional susceptibility to blood-brain barrier breakdown [61] may explain the MTL dominant pattern of tau in e4 carriers. While blood-brain barrier dysfunction contributes to cognitive decline independent of AD pathology [62], there are also inflammatory and immune-related pathways linking e4 to Aβ and tau [63, 64]. Recent human work linking blood-brain barrier dysfunction to APOE4 status showed selective blood-brain barrier dysfunction in the MTL [62], suggesting that the MTL in particular may have increased susceptibility to e4-related blood-brain barrier breakdown in aging. Our results extend these findings to provide further support for a selective effect of e4 on the MTL and suggest that this effect emerges during the preclinical stage of AD.
In A4/LEARN, we found that e2 was associated with a global reduction in tau burden across the MTL as well as all three neocortical regions. Given this more global pattern, it may be fruitful for future studies to explore e2 associations with cerebrospinal fluid or plasma biomarkers. To our knowledge, this is the first study to show a global effect of e2 on regional tau levels in preclinical AD. In vivo PET imaging and postmortem findings have provided evidence for regional variation in tau pathology that was associated with factors aside from amyloid such as differing levels of APOE expression, Related Orphan Receptor B positive neurons, homeostatic astrocytes, and inflammatory microglia function [65]. Although our effects are predominantly independent of continuous amyloid burden as measured by PET, the e2 effect we observed may still be mediated by amyloid-related processes. Given that e2 has been associated with slower longitudinal amyloid accumulation over time [6], it is possible that slower rates of accumulation are also associated with less downstream tau accumulation. For instance, a slower rate of abnormal amyloid deposition may afford greater opportunities for compensatory mechanisms and/or a more effective response to neuronal injury.
A recent case report highlighted a PSEN1-E280A individual with two copies of the Apoe Christchurch mutation, low to intermediate levels of tau burden, extremely high levels of amyloid, and preserved cognition for nearly 30 years beyond the typical age of AD symptoms for the PSEN1-E280A kindred [65]. Although the Apoe Christchurch mutation is only a candidate protective variant in this case, this mutation is thought to act through similar mechanisms to e2. Both the Apoe Christchurch mutation and e2 homozygosity are associated with Type III hyperlipoproteinemia [66] and lipid metabolism could potentially be an APOE-related protective mechanisms against significant tau accumulation. Overall, e2 seems to exert additional protection against AD dementia risk via pathways directly related to tau accumulation, although future work is needed to identify exact underlying mechanisms.
Although we showed that both e4 and e2 influence regional tau levels, differences in the overall pattern of these findings highlight that underlying mechanistic pathways may differ across APOE genotypes. While e2, e3, and e4 show a stepwise association with risk of clinical AD dementia as well as overall amyloid plaque burden [33, 48, 67], the impact of e2 on Aβ-related and Aβ-independent mechanisms of AD are less established than e4 effects [37]. This lack of understanding may be because e2 is less common than e4 in the general population. The expected frequency of e2 and e4 is approximately 8 and 13%, respectively, in individuals with European ancestry [68, 69], but e2 is especially underrepresented among Aβ+ CU (4–8% [29,30,31]) and AD dementia patients (approximately 11% [70]). Importantly, the rarity of e2 carriers among Aβ+ individuals limits the ability to evaluate e2-related mechanisms that influence disease progression above and beyond amyloid abnormalities in humans, such as downstream tau accumulation. This was evident in our analyses with ADNI, where the presence of only 13 e2 Aβ+ individuals precluded analysis (however, see previous work in ADNI that found no effect between e2 and tau PET when examining e2 effects in a combined group of Aβ- and Aβ+ CU and MCI [32]). The large sample size of the A4/LEARN CU dataset provided the opportunity to evaluate 31 Aβ+ e2 carriers that also have tau PET (1 e2/e2, 17 e2/e3, 13 e2/e4), which is still a small sample but nevertheless is larger than previous work. Given limited sample sizes for each e2 genotype, our general approach was to model e2 allele count, and additionally confirm results by directly contrasting e2/e3 and e3/e3 groups in sensitivity analyses. Future work that combines data across cohorts may provide one strategy to validate our A4/LEARN e2 findings and further characterize the impact of e2 on non-amyloid pathways.
There are several limitations to consider. First, only cross-sectional amyloid and cross-sectional tau data were examined. Because APOE genotype has been shown to influence rates of amyloid accumulation over time [6], it is possible that differences in tau burden relate to amyloid rate rather than APOE genotype. Similarly, it has been suggested that tau pathology begins to accelerate, particularly in neocortex, at some point after an individual becomes amyloid positive [15, 71]. Because APOE4 and APOE2 carriers tend to become amyloid positive at earlier and later ages, respectively, compared to APOE3 carriers [2, 72], it is possible that e2 carriers have been amyloid positive for a shorter period of time and that reduced tau reflects this shorter period rather than a direct effect on tau. Longitudinal data and/or modeling of amyloid duration [73,74,75,76] will be helpful for understanding the contribution of amyloid rate and duration. Second, although this is the largest cohort of preclinical AD participants to date, the limited number of Aβ+ e2 carriers that underwent tau PET reduces the power and reliability of our findings. This may be especially true for examinations of hypothesized age and sex interactions, which requires further subgrouping within the sample of 31 Aβ+ CU e2 carriers. However, autopsy and cohort studies have shown that e2 carriers are less likely to ever be Aβ+. Third, amyloid PET is unable to distinguish between amyloid plaques relevant to AD and amyloid buildup in arteries indicative of cerebral amyloid angiopathy (CAA), and there are known APOE genotype differences in CAA [77, 78]. Thus, if amyloid PET is capturing CAA-relevant amyloid in the A4/LEARN and ADNI samples, our analyses may not accurately capture the magnitude of amyloid-mediated effects of APOE on tau. Relatedly, amyloid PET tracers bind to amyloid plaques whereas [18F]-flortaucipir binds to neuropil threads, ghost tangles, and neuritic plaques [79]. There may be additional links between APOE and other types of amyloid and tau deposits, such as oligomers, that are not captured by PET and thus not assessed in this study. Finally, because we only examined Aβ+ individuals, we are unable to determine whether direct APOE effects on tau only emerge after amyloid positivity has been reached or whether the same direct APOE effects on tau would be present in the absence of amyloid. Although elevated tau PET values are sparse among Aβ- individuals, these elevations have been associated with reduced cognitive performance and greater atrophy [80]. Thus, it is possible that APOE exerts effects even among Aβ- individuals, warranting future larger studies that can determine whether effects of APOE on tau pathology are dependent on the presence of abnormal amyloid.
Conclusions
Our results suggest that APOE genotype influences regional tau PET burden in the early stages of AD pathological accumulation. E4 was specifically associated with elevations in the MTL, whereas protective e2 effects were present in the MTL and neocortex. Given our findings for both amyloid-mediated and direct effects of APOE on tau, gene therapies [81,82,83] that leverage protective e2 mechanisms may have a global protective impact on limiting amyloid and tau across the brain. Our results also suggest that targeting amyloid removal in e4 carriers at the preclinical stage may not be enough for impacting downstream tau accumulation given that amyloid burden only explained 22–39% of the total effect of e4 on regional tau in the MTL. APOE effects on tau accumulation in Aβ+ CU highlight that across individuals, there are variable rates of progression throughout the AD cascade, as opposed to a uniform canonical pathway that follows from amyloid positivity. Treatments may need to target different mechanisms depending on disease stage, and these treatments may vary as a function of APOE genotype. For example, whereas anti-amyloid treatments may be effective among e3 and e2 Aβ+ individuals, e4 carriers may only benefit from anti-amyloid treatments before amyloid positivity is reached (and may require combination therapies targeting both amyloid and tau after amyloid positivity). Understanding APOE mechanisms on tau may inform the development of anti-tau treatments and more broadly, therapeutic strategies targeting underlying mechanisms related to APOE may be effective in preventing amyloid-mediated and direct effects of APOE on tau accumulation.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available through LONI as well as request to the corresponding author.
Abbreviations
- A4:
-
Anti-Amyloid Treatment in Asymptomatic AD
- AD:
-
Alzheimer’s disease
- APOE :
-
Apolipoprotein
- ADNI:
-
Alzheimer’s Disease Neuroimaging Initiative
- CAA:
-
Cerebral amyloid angiopathy
- CL:
-
Centiloid
- CU:
-
Clinically unimpaired
- FBB:
-
Florbetaben
- FBP:
-
Florbetapir
- LEARN:
-
Longitudinal Evaluation of Amyloid Risk and Neurodegeneration
- MCI:
-
Mild cognitive impairment
- MTL:
-
Medial temporal lobe
- PET:
-
Positron emission tomography
- SUVR:
-
Standardized uptake value ratio
References
Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–4.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3.
Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. 2016;41:200.e13–20.
Ge T, Sabuncu MR, Smoller JW, Sperling RA, Mormino EC. Dissociable influences of APOE ε4 and polygenic risk of AD dementia on amyloid and cognition. Neurology. 2018;90:e1605–12.
Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BNM, et al. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA. 2015;313:1939–49.
Lim YY, Mormino EC. Alzheimer’s Disease Neuroimaging Initiative. APOE genotype and early β-amyloid accumulation in older adults without dementia. Neurology. 2017;89:1028–34.
Toledo JB, Habes M, Sotiras A, Bjerke M, Fan Y, Weiner MW, et al. APOE effect on amyloid-β PET spatial distribution, deposition rate, and cut-points. J Alzheimers Dis JAD. 2019;69:783–93.
Liu C-C, Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Koutsodendris N, Nelson MR, Rao A, Huang Y. Apolipoprotein E and Alzheimer’s disease: findings, hypotheses, and potential mechanisms. Annu Rev Pathol. 2022;17:73–99.
Fernández-Calle R, Konings SC, Frontiñán-Rubio J, García-Revilla J, Camprubí-Ferrer L, Svensson M, et al. APOE in the Bullseye of neurodegenerative diseases: impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol Neurodegener. 2022;17:62.
Lopresti BJ, Campbell EM, Yu Z, Anderson SJ, Cohen AD, Minhas DS, et al. Influence of apolipoprotein-E genotype on brain amyloid load and longitudinal trajectories. Neurobiol Aging. 2020;94:111–20.
Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, et al. Association of Amyloid and tau with Cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76:915–24.
Ossenkoppele R, Smith R, Mattsson-Carlgren N, Groot C, Leuzy A, Strandberg O, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head-to-head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78:961–71.
Strikwerda-Brown C, Hobbs DA, Gonneaud J, St-Onge F, Binette AP, Ozlen H, et al. Association of elevated amyloid and tau positron emission tomography signal with near-term development of alzheimer disease symptoms in older adults without cognitive impairment. JAMA Neurol. 2022;79:975–85.
Jack CR, Wiste HJ, Botha H, Weigand SD, Therneau TM, Knopman DS, et al. The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain J Neurol. 2019;142:3230–42.
La Joie R, Visani AV, Lesman-Segev OH, Baker SL, Edwards L, Iaccarino L, et al. Association of APOE4 and clinical variability in Alzheimer disease with the pattern of tau- and amyloid-PET. Neurology. 2021;96:e650–61.
Buckley RF, Mormino EC, Rabin JS, Hohman TJ, Landau S, Hanseeuw BJ, et al. Sex differences in the Association of Global Amyloid and Regional tau Deposition Measured by positron emission tomography in clinically Normal older adults. JAMA Neurol. 2019;76:542–51.
Betthauser TJ, Koscik RL, Jonaitis EM, Allison SL, Cody KA, Erickson CM, et al. Amyloid and tau imaging biomarkers explain cognitive decline from late middle-age. Brain J Neurol. 2020;143:320–35.
Lowe VJ, Bruinsma TJ, Wiste HJ, Min H-K, Weigand SD, Fang P, et al. Cross-sectional associations of tau-PET signal with cognition in cognitively unimpaired adults. Neurology. 2019;93:e29–39.
Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, et al. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. NeuroImage. 2017;157:448–63.
Mattsson N, Ossenkoppele R, Smith R, Strandberg O, Ohlsson T, Jögi J, et al. Greater tau load and reduced cortical thickness in APOE ε4-negative Alzheimer’s disease: a cohort study. Alzheimers Res Ther. 2018;10:77.
Therriault J, Benedet AL, Pascoal TA, Mathotaarachchi S, Chamoun M, Savard M, et al. Association of Apolipoprotein E ε4 with medial temporal tau independent of amyloid-β. JAMA Neurol. 2020;77:470–9.
Baek MS, Cho H, Lee HS, Lee JH, Ryu YH, Lyoo CH. Effect of APOE ε4 genotype on amyloid-β and tau accumulation in Alzheimer’s disease. Alzheimers Res Ther. 2020;12:140.
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–33.
Lowe VJ, Bruinsma TJ, Min H-K, Lundt ES, Fang P, Senjem ML, et al. Elevated medial temporal lobe and pervasive brain tau-PET signal in normal participants. Alzheimers Dement Diagn Assess Dis Monit. 2018;10:210–6.
Ramanan VK, Castillo AM, Knopman DS, Graff-Radford J, Lowe VJ, Petersen RC, et al. Association of Apolipoprotein E ɛ4, educational level, and sex with tau deposition and tau-mediated metabolic dysfunction in older adults. JAMA Netw Open. 2019;2:e1913909.
Ossenkoppele R, Leuzy A, Cho H, Sudre CH, Strandberg O, Smith R, et al. The impact of demographic, clinical, genetic, and imaging variables on tau PET status. Eur J Nucl Med Mol Imaging. 2021;48:2245–58.
Ghisays V, Goradia DD, Protas H, Bauer RJ, Devadas V, Tariot PN, et al. Brain imaging measurements of fibrillar amyloid-β burden, paired helical filament tau burden, and atrophy in cognitively unimpaired persons with two, one, and no copies of the APOE ε4 allele. Alzheimers Dement. 2020;16:598-609.
Young CB, Winer JR, Younes K, Cody KA, Betthauser TJ, Johnson SC, et al. Divergent cortical tau positron emission tomography patterns among patients with preclinical Alzheimer disease. JAMA Neurol. 2022;79(6):592–603.
Sperling RA, Donohue MC, Raman R, Sun C-K, Yaari R, Holdridge K, et al. Association of Factors with Elevated Amyloid Burden in clinically Normal older individuals. JAMA Neurol. 2020;77:735–45.
Mattsson N, Groot C, Jansen WJ, Landau SM, Villemagne VL, Engelborghs S, et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc. 2018;14:913–24.
Salvadó G, Grothe MJ, Groot C, Moscoso A, Schöll M, Gispert JD, et al. Differential associations of APOE-ε2 and APOE-ε4 alleles with PET-measured amyloid-β and tau deposition in older individuals without dementia. Eur J Nucl Med Mol Imaging. 2021;48:2212–24.
Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11:667.
Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA. Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol Aging. 2016;37:19–25.
Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6:228fs13.
Royse SK, Minhas DS, Lopresti BJ, Murphy A, Ward T, Koeppe RA, et al. Validation of amyloid PET positivity thresholds in centiloids: a multisite PET study approach. Alzheimers Res Ther. 2021;13:99.
Li Z, Shue F, Zhao N, Shinohara M, Bu G. APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Mol Neurodegener. 2020;15:63.
Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a Meta-analysis. JAMA. 2015;313:1924–38.
Mishra S, Blazey TM, Holtzman DM, Cruchaga C, Su Y, Morris JC, et al. Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE ε4 genotype. Brain J Neurol. 2018;141:1828–39.
Insel PS, Hansson O, Mattsson-Carlgren N. Association between apolipoprotein E ε2 vs ε4, age, and β-amyloid in adults without cognitive impairment. JAMA Neurol. 2021;78:229–35.
Gavett BE, John SE, Gurnani AS, Bussell CA, Saurman JL. The role of Alzheimer’s and cerebrovascular pathology in mediating the effects of age, race, and apolipoprotein E genotype on dementia severity in pathologically-confirmed Alzheimer’s disease. J Alzheimers Dis JAD. 2016;49:531–45.
Bennett DA, Schneider JA, Wilson RS, Bienias JL, Berry-Kravis E, Arnold SE. Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J Neurol Neurosurg Psychiatry. 2005;76:1194–9.
Koffie RM, Hashimoto T, Tai H-C, Kay KR, Serrano-Pozo A, Joyner D, et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain J Neurol. 2012;135:2155–68.
Serrano-Pozo A, Qian J, Monsell SE, Betensky RA, Hyman BT. APOEε2 is associated with milder clinical and pathological Alzheimer disease. Ann Neurol. 2015;77:917–29.
Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110:E1807–16.
Tai LM, Bilousova T, Jungbauer L, Roeske SK, Youmans KL, Yu C, et al. Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J Biol Chem. 2013;288:5914–26.
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–13.
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3:89ra57.
Jiang Q, Lee CYD, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, et al. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–93.
Young CB, Landau SM, Harrison TM, Poston KL, Mormino EC. Influence of common reference regions on regional tau patterns in cross-sectional and longitudinal [18F]-AV-1451 PET data. NeuroImage. 2021;243:118553.
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–7.
Huynh T-PV, Wang C, Tran AC, Tabor GT, Mahan TE, Francis CM, et al. Lack of hepatic apoE does not influence early Aβ deposition: observations from a new APOE knock-in model. Mol Neurodegener. 2019;14:37.
Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98:1141–1154.e7.
Wang C, Najm R, Xu Q, Jeong D-E, Walker D, Balestra ME, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24:647–57.
Zhao Z, Song H, Zhang R, Pang K, Liu C, Song H, et al. Dynamic spatiotemporal beams that combine two independent and controllable orbital-angular-momenta using multiple optical-frequency-comb lines. Nat Commun. 2020;11:4099.
Frisoni GB, Altomare D, Thal DR, Ribaldi F, van der Kant R, Ossenkoppele R, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci. 2022;23:53–66.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–96.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease Meta analysis consortium. JAMA. 1997;278:1349–56.
Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2016;36:216–27.
Hultman K, Strickland S, Norris EH. The APOE ɛ4/ɛ4 genotype potentiates vascular fibrin (ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2013;33:1251–8.
Iadecola C. Dangerous leaks: blood-brain barrier woes in the aging Hippocampus. Neuron. 2015;85:231–3.
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature. 2020;581:71–6.
Panitch R, Hu J, Xia W, Bennett DA, Stein TD, Farrer LA, et al. Blood and brain transcriptome analysis reveals APOE genotype-mediated and immune-related pathways involved in Alzheimer disease. Alzheimers Res Ther. 2022;14:30.
Riphagen JM, Ramakers IHGM, Freeze WM, Pagen LHG, Hanseeuw BJ, Verbeek MM, et al. Linking APOE-ε4, blood-brain barrier dysfunction, and inflammation to Alzheimer’s pathology. Neurobiol Aging. 2020;85:96–103.
Sepulveda-Falla D, Sanchez JS, Almeida MC, Boassa D, Acosta-Uribe J, Vila-Castelar C, et al. Distinct tau neuropathology and cellular profiles of an APOE3 Christchurch homozygote protected against autosomal dominant Alzheimer’s dementia. Acta Neuropathol (Berl). 2022;144:589–601.
Bu G. APOE targeting strategy in Alzheimer’s disease: lessons learned from protective variants. Mol Neurodegener. 2022;17:51.
Bennett DA, De Jager PL, Leurgans SE, Schneider JA. Neuropathologic intermediate phenotypes enhance association to Alzheimer susceptibility alleles. Neurology. 2009;72:1495–503.
Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002;155:487–95.
Kern S, Mehlig K, Kern J, Zetterberg H, Thelle D, Skoog I, et al. The distribution of apolipoprotein E genotype over the adult lifespan and in relation to country of birth. Am J Epidemiol. 2015;181:214–7.
van der Lee SJ, Wolters FJ, Ikram MK, Hofman A, Ikram MA, Amin N, et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol. 2018;17:434–44.
van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21:21–35.
Noguchi S, Murakami K, Yamada N, Payami H, Kaye J, Heston LL, et al. Apolipoprotein E genotype and Alzheimer’s disease. Lancet. 1993;342:737–8.
Betthauser TJ, Koscik RL, Jonaitis EM, Van Hulle CA, Basche KE, Kohli A, et al. Amyloid time: quantifying the onset of abnormal biomarkers and cognitive impairment along the Alzheimer’s disease continuum. Alzheimers Dement. 2021;17:e056269.
Koscik RL, Betthauser TJ, Jonaitis EM, Allison SL, Clark LR, Hermann BP, et al. Amyloid duration is associated with preclinical cognitive decline and tau PET. Alzheimers Dement Diagn Assess Dis Monit. 2020;12:e12007.
Insel PS, Donohue MC, Berron D, Hansson O, Mattsson-Carlgren N. Time between milestone events in the Alzheimer’s disease amyloid cascade. NeuroImage. 2021;227:117676.
Schindler SE, Li Y, Buckles VD, Gordon BA, Benzinger TLS, Wang G, et al. Predicting symptom onset in sporadic Alzheimer disease with amyloid PET. Neurology. 2021;97:e1823–34.
Yu L, Boyle PA, Nag S, Leurgans S, Buchman AS, Wilson RS, et al. APOE and cerebral amyloid angiopathy in community-dwelling older persons. Neurobiol Aging. 2015;36:2946–53.
Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, et al. APOE-ε2 and APOE-ε4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol. 2013;72:708–15.
Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Mol Neurodegener. 2017;12:19.
Yoon B, Guo T, Provost K, Korman D, Ward TJ, Landau SM, et al. Abnormal tau in amyloid PET negative individuals. Neurobiol Aging. 2022;109:125–34.
Lexeo Therapeutics, Alzheimer’s Drug Discovery Foundation, Weill Medical College of Cornell University. Gene Therapy for APOE4 Homozygote of Alzheimer’s Disease. Available from: https://clinicaltrials.gov/ct2/show/NCT03634007.
Yamazaki Y, Painter MM, Bu G, Kanekiyo T. Apolipoprotein E as a therapeutic target in Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs. 2016;30:773–89.
dos Santos RB, Kanekiyo T, Singh J. ApoE-2 brain-targeted gene therapy through transferrin and Penetratin tagged liposomal nanoparticles. Pharm Res. 2019;36:161.
Acknowledgements
Data used in this article were obtained from the Anti-Amyloid Treatment in Asymptomatic AD (A4) Study screening database. As such, the investigators in the A4 Study Team contributed to the data collection, but did not participate in the analysis or writing of this report. The full listing of the A4 Study team investigators can be found at: https://a4study.org/a4-study-team/. The A4 Study is a secondary prevention trial in preclinical Alzheimer’s disease, aiming to slow cognitive decline associated with brain amyloid accumulation in clinically normal older individuals. The A4 Study is funded by a public-private-philanthropic partnership, including funding from the National Institutes of Health-National Institute on Aging, Eli Lilly and Company, Alzheimer’s Association, Accelerating Medicines Partnership, GHR Foundation, an anonymous foundation and additional private donors, with in-kind support from Avid and Cogstate. The companion observational Longitudinal Evaluation of Amyloid Risk and Neurodegeneration (LEARN) Study is funded by the Alzheimer’s Association and GHR Foundation. The A4 and LEARN Studies are led by Dr. Reisa Sperling at Brigham and Women’s Hospital, Harvard Medical School and Dr. Paul Aisen at the Alzheimer’s Therapeutic Research Institute (ATRI), University of Southern California. The A4 and LEARN Studies are coordinated by ATRI at the University of Southern California, and the data are made available through the Laboratory for Neuro Imaging at the University of Southern California. The participants screening for the A4 Study provided permission to share their de-identified data in order to advance the quest to find a successful treatment for Alzheimer’s disease. We would like to acknowledge the dedication of all the participants, the site personnel, and all of the partnership team members who continue to make the A4 and LEARN Studies possible.
Data used in preparation of thisAQ4 article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at https://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Funding
This study was supported by grants K99AG071837 (CBY), K99AG075238 (MEB), U24AG074855 (ECM), P30AG066515 (ECM, KLP, MEB), R01NS115114 (KLP, ECM), K01AG051718 (ECM), P01AG036694 (RAS, KAJ), and P50AG005134 (RAS) from the National Institute of Health; and AARFD-21-849349 (CBY) and AARF-20-683984 (MEB) from the Alzheimer’s Association.
Author information
Authors and Affiliations
Consortia
Contributions
CBY, RAS, KAJ, and ECM made substantial contributions to the conception and design of the work. RAS and KAJ acquired the data. CBY, EJ, GK, and ECM analyzed the data. All authors interpreted the data and have drafted or substantively revised the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All A4 participants completed written informed consent before participation. All ADNI participants provided written informed consent in compliance with local IRBs.
Consent for publication
Not applicable.
Competing interests
KLP received consulting fees from CuraSen Therapeutics Inc. ECM is a paid consultant for Roche, Genentech, and Eli Lilly. All other authors have no disclosures relevant to the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Methods. Description of how amyloid SUVRs are converted to centiloids. Table S1. Demographic information for the 392 Aβ+ CU individuals with tau PET imaging from A4/LEARN. Table S2. Association between amyloid burden (centiloids) and regional tau SUVRs in (A) Aβ- and Aβ+ participants and in (B) only Aβ+ participants from A4/LEARN. Unstandardized beta values (SE), p-values, and model R2 values are listed. Each column represents a separate regression model. Table S3.APOE associations with regional tau SUVRs in 392 Aβ+s with APOE*Age and APOE*Sex interactions included in the model. Unstandardized beta values (SE) and p-values are listed. Each column represents a separate regression model. Table S4. (A) APOE associations with regional tau SUVRs in 359 Aβ+ individuals (B) with APOE*age and APOE*sex interactions after excluding APOE homozygotes (1 e2/e2 participant and 25 e4/e4 participants). Unstandardized beta values (SE) and p-values are listed. Each column represents a separate regression model. Table S5. (A) APOE associations with regional tau SUVRs in 372 Aβ+ individuals (B) with APOE*age and APOE*sex interactions after excluding 13 e2/e4 participants. Unstandardized beta values (SE) and p-values are listed. Each column represents a separate regression model. Table S6. Comparisons of regional tau SUVRs between APOE (A) e2/e3 and e3/e3 groups as well as (B) e3/e4 and e3/e3 groups after adjusting for age and sex. Unstandardized beta values (SE) and p-values are listed. Each column represents a separate regression model. Figure S1. Conversion of global SUVRs to centiloids (CL). (A) Conversion of florbetapir (FBP) global amyloid SUVRs to CLs for A4/LEARN data. (B) Conversion of FBP and florbetaben (FBB) global amyloid SUVRs to CLs for ADNI data. (C) CL distributions across A4/LEARN FBP, ADNI FBP, and ADNI FBB data. Figure S2. Mediation models examining direct and indirect effects of APOE on regional tau SUVRs with continuous amyloid (centiloids) as a mediator in (A) Aβ+ clinically unimpaired (CU) individuals from A4 and (B) Aβ+ CU and mild cognitive impairment (MCI) individuals from ADNI. Both e4 and e2 effects were examined in A4, but only e4 effects were examined in ADNI due to the small sample size of e2 Aβ+ participants in ADNI (n = 13). Unstandardized betas are listed. Note: * p < 0.05, ** p < 0.01, *** p < 0.001. Figure S3. Mediation models examining the effects of APOE4 on regional tau SUVRs with continuous amyloid (centiloids) as a mediator among 162 Aβ+ CU and mild cognitive impairment (MCI) participants who had tau and amyloid PET scans within 2 years of each other. Results are consistent with the effects in the larger ADNI sample. Note: * p < 0.05, ** p < 0.01, *** p < 0.001.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Young, C.B., Johns, E., Kennedy, G. et al. APOE effects on regional tau in preclinical Alzheimer’s disease. Mol Neurodegeneration 18, 1 (2023). https://doi.org/10.1186/s13024-022-00590-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-022-00590-4