
Abstract
Background
Alzheimer’s disease is characterized by aggregated β-amyloid and tau proteins, but the clinical presentations and patterns of brain atrophy vary substantially. A part of this heterogeneity may be linked to the risk allele APOE ε4. The spread of tau pathology is related to atrophy and cognitive decline, but little data exist on the effects of APOE ε4 on tau. The objective of this preliminary study was therefore to test if tau load and brain structure differ by APOE ε4 in Alzheimer’s disease.
Methods
Sixty-five β-amyloid-positive patients at the prodromal and dementia stages of Alzheimer’s disease were enrolled, including APOE ε4-positive (n = 46) and APOE ε4-negative (n = 19) patients. 18F-AV-1451 positron emission tomography was used to measure tau and brain magnetic resonance imaging (MRI) was used to measure cortical thickness.
Results
Compared with their APOE ε4-positive counterparts, APOE ε4-negative patients had greater tau load and reduced cortical thickness, with the most pronounced effects for both in the parietal cortex. Relative to the overall cortical tau load, APOE ε4-positive patients had greater tau load in the entorhinal cortex. APOE ε4-positive patients also had slightly greater cortical β-amyloid load. There was an interaction between APOE ε4 and 18F-AV-1451 on cortical thickness, with greater effects of 18F-AV-1451 on cortical thickness in APOE ε4-negative patients. APOE ε4 and 18F-AV-1451 were independent predictors of cognition, but showed distinct associations with different cognitive tests.
Conclusions
APOE genotype may be associated with differences in pathways in Alzheimer’s disease, potentially through differential development and spread of tau, as well as through effects on cognitive outcomes involving non-tau-related mechanisms.
Similar content being viewed by others


Background
Most Alzheimer’s disease (AD) patients have an amnestic-predominant cognitive impairment profile, while others have more prominent executive, language, or visuospatial deficits [1, 2]. The variations in presentations may be associated with specific patterns of neurodegeneration or tau pathology [3,4,5,6], but the reason why the regional involvement of neurodegeneration and tau varies is largely unclear [7]. One possibility is that the apolipoprotein (APOE) ε4 allele contributes to the variability. APOE ε4 is a major genetic risk factor for sporadic AD, and is present in 50–70% of patients [8,9,10]. However, roughly a third of all AD patients develop the disease without carrying an APOE ε4 allele. APOE ε4-negative patients are characterized by relatively more nonamnestic deficits and anatomically by greater frontoparietal atrophy, while APOE ε4-positive patients predominantly demonstrate memory impairment and temporal lobe atrophy [11,12,13,14,15]. Given the intimate link between tau pathology and neurodegeneration in neuroimaging [3, 16,17,18,19], neuropathology [20], and cell and animal studies [21], it is possible that APOE ε4 status affects the spread of tau in AD and subsequent brain atrophy, but only few studies have explored this [22,23,24]. We tested if APOE ε4 was associated with regional and global differences in 18F-AV-1451 in prodromal AD and AD dementia patients, and if APOE ε4 status and 18F-AV-1451 interacted to predict atrophy and cognition. We hypothesized that APOE ε4-positive patients would have more tau pathology and atrophy in the temporal lobe, while APOE ε4-negative patients would be more affected by tau and atrophy in other brain regions, in particular the frontal and parietal areas.
Methods
Participants
All participants were recruited from the Swedish BioFINDER study. Inclusion and exclusion criteria have been described previously [25]. We included prodromal AD (amyloid-β (Aβ)-positive mild cognitive impairment [MCI]) [26] and mild-to-moderate (Aβ-positive) AD dementia patients who were all assessed by physicians with expertise in dementia. Aβ-positivity was defined by cerebrospinal fluid (CSF) Aβ42 in all cases except one for whom 18F-flutemetamol positron emission tomography (PET) was used. The inclusion criteria for MCI were: referred to a memory clinic due to possible cognitive impairment; objective impairment in one or more cognitive domains; preservation of independence in functional abilities; and not fulfilling criteria for any dementia disorder. All dementia patients met the NIA-AA criteria for dementia due to AD [27]. The exclusion criteria were: cognitive impairment that without doubt could be explained by another condition other than prodromal AD or AD dementia; significant systemic illness making it difficult to participate; and significant alcohol or drug abuse.
Cognitive measures
Cognitive measures included Mini-Mental State Examination (MMSE) [28] for global cognition, the immediate and delayed conditions of the 10-word list recall tests from the Alzheimer’s Disease Assessment Scale (ADAS)-cognitive subscale [29] for memory, Trail Making Test-A (TMT-A) [30], and A Quick Test of cognitive speed—Color & Form subtest (AQT-CF) [31] for attention and processing speed, and category fluency [30] for language and semantic memory (animal fluency) and executive function (letter S fluency). Data were missing for MMSE in one subject, for ADAS immediate recall in six subjects, for ADAS delayed recall in seven subjects, for TMT-A in nine subjects, for letter S fluency and animal fluency in 12 subjects, and for AQT-CF in 14 subjects. The reason for missing cognitive data was either refusal or cognitive inability to perform the task. We kept all eligible subjects for this analysis, even if they lacked some cognitive data, since exclusion based on missing data would have increased the risk of a selection bias.
CSF biomarkers
Lumbar CSF sampling was performed following the Alzheimer’s Association Flow Chart [32]. Samples were stored in 1-ml polypropylene tubes at −80 °C until analysis. Enzyme-linked immunosorbent assays (ELISAs) were used for CSF Aβ42, total (T)-tau, and phosphorylated (P)-tau (ADx/Euroimmun AG, Lübeck, Germany, and Innotest, Fujirebio, Ghent, Belgium). All analyses were performed by board-certified laboratory technicians who were blinded for clinical data and diagnoses. Aβ-positivity was defined as CSF Aβ42 < 647 ng/L [33]. For one subject who refused to undergo lumbar puncture we determined Aβ-positivity by 18F-flutemetamol PET (see below for details).
Magnetic resonance imaging (MRI)
T1-weighted MRI was performed on a 3-T MR scanner in all subjects (Siemens Tim Trio 3 T, Siemens Medical Solutions, Erlangen, Germany), producing a high resolution anatomical MP-RAGE image (TR = 1950 ms, TE = 3.4 ms, 1 mm isotropic voxels, and 178 slices). Cortical reconstruction and volumetric segmentation were performed with FreeSurfer (v5.3) (http://surfer.nmr.mgh.harvard.edu/) using an in-house developed image analysis pipeline. The MP-RAGE images underwent correction for intensity homogeneity [34], removal of nonbrain tissue, and segmentation into gray matter (GM) and white matter (WM) with intensity gradient and connectivity among voxels [35,36,37,38]. Cortical thickness was measured as the distance from the GM/WM boundary to the corresponding pial surface [36]. Reconstructed datasets were visually inspected for accuracy, and segmentation errors were corrected. The surface area-weighted average cortical thickness was calculated for seven FreeSurfer-based metaregions: lateral parietal, medial parietal, lateral temporal, medial temporal, frontal, occipital, and whole brain cortex (Additional file 1: Table S1). We also calculated the ratio between entorhinal cortical thickness and overall cortical thickness in accordance with Whitwell et al. [24] (E/C ratio, the overall cortical region included the whole cortex except for entorhinal and the inferior temporal gyri, since tau accumulation there may be closely linked to entorhinal tau).
Automated segmentation of white matter lesions (WML) was performed using the lesion prediction algorithm as implemented in the LST toolbox (www.statistical-modelling.de/lst.html) for SPM. This algorithm consists of a binary classifier in the form of a logistic regression model; as covariates, a lesion belief map [39] is used as well as a spatial covariate that takes into account voxel-specific changes in lesion probability. Parameters of this model fit are used to segment lesions by providing an estimate for the lesion probability for each voxel.
18F-AV-1451 tau PET imaging and processing
Tau PET was performed in all subjects as described previously [40]. Briefly, 18F-AV-1451 was synthesized at Skåne University Hospital, Lund [41], and PET scans were performed on a GE Discovery 690 PET scanner (General Electric Medical Systems). FreeSurfer parcellation in the MR space of the anatomical scan was applied to processed, coregistered and time-averaged PET images to extract regional uptake values. 18F-AV-1451 standardized uptake value ratio (SUVr) images were based on mean uptake over 80–100 min postinjection normalized to uptake in a GM-masked inferior cerebellum reference region. The signal was not corrected for partial volume effects.
The same FreeSurfer metaregions as for MRI, including the E/C ratio, were calculated for 18F-AV-1451 (volume weighted; Additional file 1: Table S1). We excluded the hippocampus from the medial temporal metaregion because of its susceptibility to spill-over effects from the anatomically proximate choroid plexus [40].
18F-flutemetamol PET imaging
Fibrillary brain Aβ was quantified in a subgroup (n = 54) using 18F-flutemetamol PET. PET/computed tomography (CT) scanning was conducted at two sites using the same type of scanner, a Philips Gemini TF 16. PET sum images from 90 to 110 min postinjection were generated for the average uptake. FreeSurfer parcellation in the MR space of the anatomical scan was applied to the processed images. The SUVr images were normalized to the mean uptake in a composite region consisting of cerebellar white matter, brainstem and cerebral white matter. We used the same FreeSurfer metaregions (volume weighted) as for 18F-AV-1451 and additionally assessed the striatum.
18F-flutemetamol PET SUVr in a composite neocortical region-of-interest indicated Aβ-positivity in one subject who refused to undergo lumbar puncture [42].
Statistical analysis
Statistical analyses were performed with R (v. 3.3.2, The R Foundation for Statistical Computing). The relationships between demographics and APOE ε4 were evaluated with Fisher’s exact test and Wilcoxon-Mann-Whitney rank sum test.
We used linear regression to test effects of APOE ε4 status on 18F-AV-1451, 18F-flutemetamol, cortical thickness, and interactions of APOE ε4 status and 18F-AV-1451 to predict cortical thickness and cognition. Covariates included age, sex, education, CSF Aβ42, WML, and CSF P-tau.
In secondary analyses, we compared models for APOE ε4 and 18F-AV-1451 to predict cognition, with and without adjusting for each other, to test if they provided independent information. We adjusted for brain MRI measures to test if effects of APOE ε4 and 18F-AV-1451 on cognition were independent of atrophy. Akaike Information Criterion (AIC) was used for model comparisons. Statistical significance was determined at P < 0.05.
Primary research question
Does regional cortical thickness and 18F-AV-1451 PET uptake differ by APOE ε4 status in AD patients at the prodromal and dementia stage of the disease?
Results
We included 46 APOE ε4-positive and 19 APOE ε4-negative AD patients, with no significant group differences regarding age, sex, education, WML, hippocampal volume, global cognition (MMSE), or CSF Aβ42 (Table 1). APOE ε4-negative patients had higher CSF T-tau (P = 0.035) and P-tau (P = 0.012), better memory function (the ADAS delayed recall, P = 0.018), and worse executive function (TMT-A, P = 0.021). Fifty-four patients (37 APOE ε4-positive and 17 APOE ε4-negative) had 18F-flutemetamol PET data. The APOE ε4-positive patients had slightly greater Aβ load (Additional file 1: Figure S1).
Associations for APOE ε4 with 18F-AV-1451 tau PET and cortical thickness
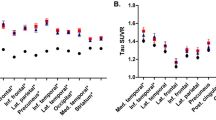
APOE ε4-negative patients had greater 18F-AV-1451 uptake in lateral parietal, medial parietal, occipital, and whole brain cortical areas compared with APOE ε4-positive patients, while APOE ε4-positive patients had greater 18F-AV-1451 E/C ratio uptake (Fig. 1). All differences remained significant when adjusting for CSF Aβ42 or WML, and differences in the medial parietal cortex also remained significant when adjusting for age and sex (Table 2 and Additional file 1: Table S2). When adjusting for CSF P-tau, effects remained significant in the medial parietal cortex, with trends for the lateral parietal cortex and E/C ratio (Additional file 1: Table S2). APOE ε4-negative patients also had thinner cortex in lateral and medial parietal areas compared with APOE ε4-positive patients (Fig. 2 and Table 2).


In a secondary analysis, we tested effects in the AD dementia subgroup only (15 APOE ε4-negative and 28 APOE ε4-positive patients) to better account for differences in disease severity. APOE ε4 status was not related to global cognition (MMSE (mean ± SD) 20.9 ± 5.7 vs. 20.8 ± 4.3, P = 0.93) or age (68.9 ± 8.1 vs. 72.1 ± 7.9, P = 0.18) in these patients, but APOE ε4-negative patients still had increased 18F-AV-1451 in the lateral and medial parietal and whole brain cortical areas (Additional file 1: Figure S2) and had thinner parietal cortices (Additional file 1: Figure S3).
Interactions of APOE ε4 and 18F-AV-1451 tau PET to predict cortical thickness
Next, we tested models with interactions for APOE ε4 and 18F-AV-1451 to predict cortical thickness (Fig. 3). There were significant interactions in the lateral parietal, medial parietal, and frontal regions, and in whole brain cortex (P < 0.05), indicating that the effects of 18F-AV-1451 on cortical thickness were more pronounced in APOE ε4-negative than in APOE ε4-positive patients. The main effects of 18F-AV-1451 on cortical thickness were significant in APOE ε4-negative patients in all regions (except for the E/C ratio), but only in lateral temporal and lateral parietal regions in APOE ε4-positive patients.

Associations between 18F-AV-1451 and cortical thickness in regions and in whole brain cortex. Models included the interaction between 18F-AV-1451 and APOE ε4 status. P values are shown for the individual APOE ε4 groups, and for the interaction effect. See Additional file 1 (Table S1) for definitions of regions. All models were adjusted for age and sex. SUVr, standardized uptake value ratio
Effects of APOE ε4 and 18F-AV-1451 PET on cognition
There were no significant interactions between APOE ε4 and (regional or global) 18F-AV-1451 to predict cognitive scores. We therefore only present models without interaction terms. APOE ε4 was a significant predictor of worse ADAS delayed recall, and better TMT-A, while greater (whole cortical) 18F-AV-1451 signal was a significant predictor of worse animal fluency and (at trend level, P = 0.054) worse MMSE (Table 3). We next included both APOE ε4 and 18F-AV-1451 as predictors, which had minor effects on their respective separate estimates. The effect of 18F-AV-1451 on MMSE was β = −3.838 (P = 0.054) before and β = −4.024 (P = 0.046) after adjustment for APOE ε4, a difference of 4.9%. Similarly, the effect of APOE ε4 positivity on ADAS delayed recall was β = 2.124 (P = 0.01) before and β = 2.170 (P = 0.01) after adjustment for 18F-AV-1451, a difference of 2.2%. R2 for the models was only minimally affected by combining the modalities, and comparisons of AIC never favored the combination of APOE ε4 and 18F-AV-1451 over the best individual model (Table 3). Taken together, this suggests that APOE ε4 and 18F-AV-1451 had independent effects on the cognitive measures.
These models used global cortical 18F-AV-1451, but the results for regional 18F-AV-1451 were very similar (data not shown). For TMT-A, the effect of 18F-AV-1451 was significant in occipital, lateral parietal, and lateral temporal regions, corresponding to the trend for global 18F-AV-1451 (Table 3).
Effects of APOE ε4 and 18F-AV-1451 PET on cognition, adjusted for cortical thickness
For cognitive tests that were associated with APOE ε4 (ADAS delayed recall and TMT-A) or 18F-AV-1451 (MMSE and animal fluency), we performed exploratory analyses with additional adjustments for brain structure.
The effects of 18F-AV-1451 on cognition were reduced when adjusting for cortical thickness. The association with MMSE was eliminated (P = 0.25) by adjusting for global cortical thickness (which had an independent significant effect; β = 12.1, P = 0.015). The effect on animal fluency was attenuated by adjusting for global cortical thickness (P = 0.055) and eliminated (P = 0.18) by adjusting for medial temporal lobe thickness (which had an independent significant effect; β = 6.81, P = 0.027).
The effects of APOE ε4 in this exploratory analysis were largely independent from cortical thickness. The effect on ADAS delayed recall was not affected by adjusting for global cortical thickness, and only marginally affected (β = 1.74, P = 0.024) by adjusting for medial temporal lobe cortical thickness (which had an independent significant effect; β = −2.97, P = 0.022). The effect on ADAS delayed recall also remained (β = 1.94, P = 0.013) when adjusting for hippocampal volume (which was not significantly associated with memory in this combined model; β = −0.0013, P = 0.079). For TMT-A, the effect was not affected by adjusting for global cortical thickness, and only marginally affected (β = −55.2, P = 0.0094) when adjusting for lateral temporal thickness (which had an independent significant effect; β = −118, P = 0.027).
In summary, the effects of 18F-AV-1451 on cognition largely depended on cortical thickness, while the effects of APOE ε4 on cognition were largely independent of cortical thickness and hippocampal volume.
Associations between 18F-AV-1451 and demographic factors
Lower age was correlated with greater 18F-AV-1451 uptake. This was independent of APOE ε4 status, except in the medial temporal lobe where lower age was associated with 18F-AV-1451 uptake in APOE ε4-positive but not in APOE ε4-negative patients (Fig. 4). There were no significant associations between sex and 18F-AV-1451, and no interactions between sex and APOE ε4 status to predict 18F-AV-1451 (data not shown).

APOE ε4, age and 18F-AV-1451 PET. Regional and whole brain cortical 18F-AV-1451 PET in AD patients. Models included the interaction between age and APOE ε4 status. P values are shown for the individual APOE ε4 groups, and for the interaction effect. See Additional file 1 (Table S1) for definitions of regions. All models were adjusted for sex. SUVr, standardized uptake value ratio
Associations between 18F-flutemetamol and 18F-AV-1451
There were no significant associations between 18F-flutemetamol and 18F-AV-1451,
in either APOE ε4-positive or APOE ε4-negative patients (Additional file 1: Figure S4).
Discussion
APOE ε4-negative AD patients had increased 18F-AV-1451 (tau) uptake and reduced cortical thickness compared with APOE ε4-positive patients, particularly in the parietal cortex. In contrast, APOE ε4-positive patients had slightly higher uptake of 18F-AV-1451 in the entorhinal cortex relative to the whole cortex, and also had higher cortical 18F-flutemetamol (Aβ) uptake. Regional associations between 18F-AV-1451 and cortical thickness depended on APOE ε4 status, as greater 18F-AV-1451 uptake was associated with cortical thinning mainly in APOE ε4-negative patients. Both 18F-AV-1451 and APOE ε4 were independently associated with cognition, but with different cognitive tests. These differences were found despite that age, sex, and global cognitive impairment not differing by APOE ε4 status. Taken together, these results suggest that APOE ε4 status influences differences in disease pathways, both through differential development and spread of tau pathology, and through effects on cognitive outcomes that may involve non-tau-related mechanisms. The proportion of APOE ε4-negative patients was 29%. This is in line with previous estimates of APOE ε4 prevalence in people with biomarker evidence of Aβ pathology, where APOE ε4 positivity is especially common in cohorts from northern Europe [43]. However, even at this relatively low frequency, the number of AD patients who are APOE ε4 negative will still be considerable in the population as a whole.
Our first main finding was that APOE ε4-negative patients had increased global 18F-AV-1451 uptake, with significant increases in the lateral parietal, medial parietal, and occipital areas. The APOE ε4-negative patients also had increased CSF T-tau and P-tau, which agrees with previous comparisons between 18F-AV-1451 and CSF tau [42, 44]. In contrast, APOE ε4-positive patients had higher relative 18F-AV-1451 uptake in the entorhinal cortex compared with the remaining cortex. Few studies have tested effects of APOE ε4 on tau PET imaging, but our results are consistent with a study in 62 (typical and atypical) AD patients, where the absence of APOE ε4 was associated with a greater 18F-AV-1451 signal in the neocortex and less signal in the entorhinal cortex (using a similar E/C ratio as here) [24]. A smaller study in 20 (mostly atypical) AD patients found that APOE ε4 was associated with greater 18F-AV-1451 signal in the bilateral medial temporal and right temporoparietal cortex [3]. Also, few studies have tested the effects of APOE ε4 on CSF tau measures when restricting the analysis to people with biomarker evidence of Aβ pathology. Altmann et al. found increased CSF tau levels in APOE ε4-positive controls and MCI patients [45], but since that study also included people without biomarkers supporting Aβ positivity the results are difficult to compare with our study.
Our second main finding was that APOE ε4-negative patients also had reduced thickness in the lateral and parietal areas (but there were no APOE ε4-dependent differences in hippocampal volumes). This partly agrees with results that APOE ε4-negative AD patients have less temporal lobe atrophy [12] and hypometabolism [46] but more frontoparietal atrophy [11, 13] compared with APOE ε4-positive patients. Furthermore, 18F-AV-1451 and APOE ε4 status interacted to predict cortical thickness, with stronger effects of 18F-AV-1451 on atrophy in APOE ε4-negative patients. The interaction was mainly present in the parietal and frontal lobes.
Our results support a hypothetical model in which APOE ε4-dependent differences in patterns of atrophy are at least partly related to differences in the spread of tau pathology. One intriguing possibility is that the cortex, with frontoparietal regions in particular, is more vulnerable to tau in patients who develop AD despite lacking APOE ε4. The reason for this increased vulnerability is not clear. We considered the possibility that Aβ pathology could contribute to the differences in atrophy and tau, but this is unlikely since the APOE ε4-positive patients had slightly greater Aβ load than the APOE ε4-negative patients. Previous studies have found divergent results for associations between APOE ε4 and Aβ in AD, with either no effect of APOE ε4 on Aβ pathology [47], less Aβ pathology [46, 48], or more Aβ pathology [49] in APOE ε4-positive AD. Potentially, patients who develop AD despite lacking APOE ε4 have other genetic abnormalities which put them at risk for degeneration [50]. Another possibility is that patients who develop AD and have an APOE ε4 allele may have partly non-tau-dependent cortical atrophy, perhaps due to impaired neuronal repair in the presence of APOE ε4 [51]. This may fit with the attenuated association between 18F-AV-1451 uptake and cortical thickness in APOE ε4-positive patients in this study.
The remaining part of our study was on cognition. Due to the relatively small sample size and the inherent variability of cognitive measures, these results should be interpreted with caution and be regarded as preliminary. However, we reproduced known APOE ε4-dependent profiles with relatively preserved memory function and more attention/processing speed impairment in APOE ε4-negative AD [11,12,13,14]. The fact that the associations between APOE ε4 status and delayed recall memory (worse in APOE ε4-positive) and TMT-A (worse in APOE ε4-negative) remained significant when adjusting for 18F-AV-1451 or brain atrophy suggests that the relationship between APOE ε4 status and cognition at least partly depends on non-tau-related mechanisms. In contrast, the associations between 18F-AV-1451 and cognitive tests were attenuated by adjusting for brain atrophy, which supports the hypothesis that tau accumulation leads to cognitive decline partly through atrophy [5].
Strengths of the study include the multimodal dataset, which combines brain MRI, tau and amyloid PET, CSF biomarkers, genetics, and cognitive data. Furthermore, the patients were recruited at a secondary referral clinic, which may make the sample more representative of the general AD population than samples recruited at more specialized centers. Another strength is that the APOE ε4 groups were matched for age, sex, education, presence of dementia, and overall cognitive impairment (measured by MMSE). One limitation is the cross-sectional design. Longitudinal analyses are required to understand causal relationships between tau, atrophy, and cognition. For example, we cannot exclude the possibility of reverse causality for some of the associations, i.e., that APOE ε4-negative patients had premorbid thinner cortices (especially in frontoparietal regions) which predisposed them to tau accumulation. In line with this, a study on neonates found that APOE ε4 carriers had reductions in volumes in temporal regions, but greater volumes in the parietal, frontal, and occipital cortex compared with APOE ε4-negative neonates, which suggests that some APOE ε4-dependent differences in brain structure (and perhaps vulnerability to tau) may already be present at birth [52]. In the future, it will be informative to extend the current study with longitudinal data.
Conclusions
APOE ε4 status is associated with heterogeneity not only in clinical presentation and atrophy patterns in AD, but also in the accumulation of tau pathology. APOE ε4-negative patients may be more likely to deposit tau and have more atrophy in the parietal lobes compared with APOE ε4-positive AD, which predispose them to a nonamnestic clinical phenotype.
References
Stopford CL, Snowden JS, Thompson JC, Neary D. Variability in cognitive presentation of Alzheimer’s disease. Cortex. 2008;44:185–95.
Scheltens P, Blennow K, Breteler MMB, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet Lond Engl. 2016;388:505–17.
Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain J Neurol. 2016;139:1551–67.
Zhang X, Mormino EC, Sun N, Sperling RA, Sabuncu MR, Yeo BTT, et al. Bayesian model reveals latent atrophy factors with dissociable cognitive trajectories in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2016;113:E6535–44.
Bejanin A, Schonhaut DR, La Joie R, Kramer JH, Baker SL, Sosa N, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain J Neurol. 2017;140:3286–300.
Risacher SL, Anderson WH, Charil A, Castelluccio PF, Shcherbinin S, Saykin AJ, et al. Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology. 2017;89:2176–86.
Cummings JL. Cognitive and behavioral heterogeneity in Alzheimer’s disease: seeking the neurobiological basis. Neurobiol Aging. 2000;21:845–61.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3.
Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–9.
Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology. 2012;38:1–17.
Wolk DA, Dickerson BC. Alzheimer’s Disease Neuroimaging Initiative. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107:10256–61.
Pievani M, Galluzzi S, Thompson PM, Rasser PE, Bonetti M, Frisoni GB. APOE4 is associated with greater atrophy of the hippocampal formation in Alzheimer’s disease. NeuroImage. 2011;55:909–19.
van der Flier WM, Pijnenburg YA, Fox NC, Scheltens P. Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE ɛ4 allele. Lancet Neurol. 2011;10:280–8.
Ossenkoppele R, Cohn-Sheehy BI, La Joie R, Vogel JW, Möller C, Lehmann M, et al. Atrophy patterns in early clinical stages across distinct phenotypes of Alzheimer’s disease. Hum Brain Mapp. 2015;36:4421–37.
Whitwell JL, Dickson DW, Murray ME, Weigand SD, Tosakulwong N, Senjem ML, et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer’s disease: a case-control study. Lancet Neurol. 2012;11:868–77.
Hanseeuw BJ, Betensky RA, Schultz AP, Papp KV, Mormino EC, Sepulcre J, et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann Neurol. 2017;81:583–96.
LaPoint MR, Chhatwal JP, Sepulcre J, Johnson KA, Sperling RA, Schultz AP. The association between tau PET and retrospective cortical thinning in clinically normal elderly. NeuroImage. 2017;157:612–22.
Iaccarino L, Tammewar G, Ayakta N, Baker SL, Bejanin A, Boxer AL, et al. Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer’s disease. Neuro Image Clin. 2018;17:452–64.
Nasrallah IM, Chen YJ, Hsieh M-K, Phillips JS, Ternes K, Stockbower GE, et al. 18 F-Flortaucipir PET/MRI correlations in nonamnestic and amnestic variants of Alzheimer disease. J Nucl Med Off Publ Soc Nucl Med. 2018;59:299–306.
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81.
Spires-Jones TL, Hyman BT. The intersection of amyloid Beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82:756–71.
Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA. Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol Aging. 2016;37:19–25.
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–7.
Whitwell JL, Graff-Radford J, Tosakulwong N, Weigand SD, Machulda M, Senjem ML, et al. [18 F]AV-1451 clustering of entorhinal and cortical uptake in Alzheimer’s disease. Ann Neurol. 2018;83:248–57.
Palmqvist S, Zetterberg H, Mattsson N, Johansson P, Alzheimer’s Disease Neuroimaging Initiative, Minthon L, et al. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology. 2015;85:1240–9.
Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–94.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc. 2011;7:263–9.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98.
Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141:1356–64.
Delis DC, Kaplan E, Kramer JH. Delis-Kaplan executive function system TM: Examiner’s manual. San Antonio: The Psychological Corporation, A Harcourt Assessment Company; 2001.
Wiig EH, Annas P, Basun H, Andreasen N, Lannfelt L, Zetterberg H, et al. The stability of AQT processing speed, ADAS-Cog and MMSE during acetylcholinesterase inhibitor treatment in Alzheimer’s disease. Acta Neurol Scand. 2010;121:186–93.
Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–44.
Palmqvist S, Zetterberg H, Blennow K, Vestberg S, Andreasson U, Brooks DJ, et al. Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid β-amyloid 42: a cross-validation study against amyloid positron emission tomography. JAMA Neurol. 2014;71:1282–9.
Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging. 1998;17:87–97.
Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I Segmentation and surface reconstruction. Neuroimage. 1999;9:179–94.
Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97:11050–5.
Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–55.
Fischl B, Salat DH, van der Kouwe AJW, Makris N, Ségonne F, Quinn BT, et al. Sequence-independent segmentation of magnetic resonance images. NeuroImage. 2004;23(Suppl 1):S69–84.
Schmidt P, Gaser C, Arsic M, Buck D, Förschler A, Berthele A, et al. An automated tool for detection of FLAIR-hyperintense white-matter lesions in multiple sclerosis. NeuroImage. 2012;59:3774–83.
Smith R, Schain M, Nilsson C, Strandberg O, Olsson T, Hägerström D, et al. Increased basal ganglia binding of (18) F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord Off J Mov Disord Soc. 2017;32:108–14.
Hahn A, Schain M, Erlandsson M, Sjölin P, James GM, Strandberg OT, et al. Modeling strategies for quantification of in vivo 18F-AV-1451 binding in patients with tau pathology. J Nucl Med Off Publ Soc Nucl Med. 2017;58:623–31.
Mattsson N, Smith R, Strandberg O, Palmqvist S, Schöll M, Insel PS, et al. Comparing 18F-AV-1451 with CSF t-tau and p-tau for diagnosis of Alzheimer disease. Neurology. 2018;90:e388–95.
Mattsson N, Groot C, Jansen WJ, Landau SM, Villemagne VL, Engelborghs S, et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc. 2018;14:913–24.
La Joie R, Bejanin A, Fagan AM, Ayakta N, Baker SL, Bourakova V, et al. Associations between [18F]AV1451 tau PET and CSF measures of tau pathology in a clinical sample. Neurology. 2018;90:e282–90.
Altmann A, Tian L, Henderson VW, Greicius MD. Sex modifies the APOE-related risk of developing Alzheimer’s disease. Ann Neurol. 2014;75:563–73.
Lehmann M, Ghosh PM, Madison C, Karydas A, Coppola G, O’Neil JP, et al. Greater medial temporal hypometabolism and lower cortical amyloid burden in ApoE4-positive AD patients. J Neurol Neurosurg Psychiatry. 2014;85:266–73.
Landen M, Thorsell A, Wallin A, Blennow K. The apolipoprotein E allele epsilon 4 does not correlate with the number of senile plaques or neurofibrillary tangles in patients with Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1996;61:352–6.
Ossenkoppele R, van der Flier WM, Zwan MD, Adriaanse SF, Boellaard R, Windhorst AD, et al. Differential effect of APOE genotype on amyloid load and glucose metabolism in AD dementia. Neurology. 2013;80:359–65.
Drzezga A, Grimmer T, Henriksen G, Mühlau M, Perneczky R, Miederer I, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72:1487–94.
Jiang S, Tang L, Zhao N, Yang W, Qiu Y, Chen H-Z. A systems view of the differences between APOE ε4 carriers and non-carriers in Alzheimer’s disease. Front Aging Neurosci. 2016;8:171.
Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms, and therapy. Nat Rev Neurol. 2013;9:106–18.
Knickmeyer RC, Wang J, Zhu H, Geng X, Woolson S, Hamer RM, et al. Common variants in psychiatric risk genes predict brain structure at birth. Cereb Cortex N Y N 1991. 2014;24:1230–46.
Acknowledgements
Doses of 18F-flutemetamol injection were sponsored by GE Healthcare, and the precursor of 18F-AV1451 was provided by AVID radiopharmaceuticals.
Funding
This study was supported by the European Research Council, the Swedish Research Council, the Marianne and Marcus Wallenberg foundation, the Strategic Research Area MultiPark (Multidisciplinary Research in Parkinson’s disease) at Lund University, the Swedish Brain Foundation, the Skåne University Hospital Foundation, the Swedish Alzheimer Association, and the Swedish federal government under the ALF agreement.
Availability of data and materials
The dataset used during the current study is available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
Drafting the manuscript: NM and RO. Revising the manuscript for content: all authors. Study concept or design: NM, RO, and OH. Analysis or interpretation of data: NM, RO, and OH. Acquisition of data: SP, RS, ES, OS, TO, and JJ. Statistical analysis: NM and RO. Study supervision or coordination: OH. Obtaining funding: NM and OH. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants gave written informed consent to participate in the study. Ethical approval was given by the Ethical Committee of Lund University, Lund, Sweden, and all the methods were carried out in accordance with the approved guidelines. 18F-AV-1451 PET imaging approval was obtained from the Swedish Medicines and Products Agency and the local Radiation Safety Committee at Skåne University Hospital, Sweden.
Consent for publication
Not applicable.
Competing interests
OH has acquired research support (for the institution) from Roche, GE Healthcare, Biogen, AVID Radiopharmaceuticals, Fujirebio, and Euroimmun. In the past 2 years, he has received consultancy/speaker fees (paid to the institution) from Lilly, Roche, and Fujirebio. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Supplementary material. (DOCX 286 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mattsson, N., Ossenkoppele, R., Smith, R. et al. Greater tau load and reduced cortical thickness in APOE ε4-negative Alzheimer’s disease: a cohort study. Alz Res Therapy 10, 77 (2018). https://doi.org/10.1186/s13195-018-0403-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-018-0403-x