
Abstract
Background
Oxidative stress (OS) is an imbalance between oxidant and antioxidant species and, together with other numerous pathological mechanisms, leads to the degeneration and death of motor neurons (MNs) in amyotrophic lateral sclerosis (ALS).
Main body
Two of the main players in the molecular and cellular response to OS are NRF2, the transcription nuclear factor erythroid 2-related factor 2, and its principal negative regulator, KEAP1, Kelch-like ECH (erythroid cell-derived protein with CNC homology)-associated protein 1. Here we first provide an overview of the structural organization, regulation, and critical role of the KEAP1-NRF2 system in counteracting OS, with a focus on its alteration in ALS. We then examine several compounds capable of promoting NRF2 activity thereby inducing cytoprotective effects, and which are currently in different stages of clinical development for many pathologies, including neurodegenerative diseases.
Conclusions
Although challenges associated with some of these compounds remain, important advances have been made in the development of safer and more effective drugs that could actually represent a breakthrough for fatal degenerative diseases such as ALS.
Similar content being viewed by others


Background
Oxidative stress (OS) is a toxic cellular status due to the imbalance between oxidant and antioxidant mechanisms resulting from increased production of oxidants, a dysfunction in the antioxidant system, or both [1, 2]. OS can be triggered by free radicals or other oxidant species produced by either exogenous processes (environmental pollutants, drugs, viruses, bacteria, ionizing radiations, heavy metals, poor diet, and smoking) [3,4,5,6,7,8,9,10,11] or endogenous processes (such as immune cell activation, inflammation, ischemia, infection, cancer, and aging) [12,13,14,15,16,17,18,19].
Free radicals are molecules that are highly reactive to other cellular structures due to the presence of unpaired electrons [16, 20,21,22]. Oxygen (O2) is essential for life and plays a crucial role in many biological processes [23] and, given to its high oxidation-reduction (redox) potential, it is an ideal electron acceptor. However, its reactivity has a cost: O2 strips electrons from biological macromolecules causing the formation of reactive oxygen species (ROS), which induce intracellular damage [24]. Among them, the best known ROS in human cells include superoxide anion (O2•-), which is the precursor of most other ROS, hydroxyl radical (•OH), peroxyl radical (ROO•), hydroperoxyl radical (•HO2), and hydrogen peroxide (H2O2), although the last one is not technically a free radical because it does not have unpaired electrons, but it represents a source of them [25] (Fig. 1). Several sub-cellular compartments generating ROS have been identified, including cytosol, peroxisomes, plasma, and endoplasmic reticulum (ER) membranes [26, 27]. Nevertheless, under physiological conditions, in most tissues mitochondria are the major contributors to the cellular ROS production [28,29,30,31], mainly as a side effect of aerobic respiration via oxidative phosphorylation (OXPHOS) occurring on the inner membrane of the mitochondrion [32, 33], but also through other metabolic routes, such as pentose-phosphate pathway and glutathione (GSH) metabolism [34].

Redox imbalance in OS. An increased production of oxidants (ROS, RNS, and RSS) and a dysfunction, mostly a decrease, in the antioxidant system (Enzymatic, Non-enzymatic, and Dietary antioxidants) result in damage to cells and can contribute to the pathogenesis and progression of many neurodegenerative diseases, including ALS. ROS: reactive oxygen species, RNS: reactive nitrogen species, RSS: reactive sulfur species. •Refers to an unpaired electron; R and R' represent functional groups ≠ H
Although oxidative damage in biological systems has in the past been completely attributed to ROS, it is becoming increasingly clear that also reactive nitrogen species (RNS) and reactive sulfur species (RSS) are integral components of signal transduction processes in cells, and play important roles in OS generation [2, 25, 35]. The predominant RNS is nitric oxide radical (NO•) [36], formed in biological tissues from the oxidation of L-arginine to citrulline by nitric oxide synthase [37, 38], and precursor of other two RNS, the peroxynitrite (ONOO−), and the nitrogen dioxide radical (•NO2) (Fig. 1). The latter was once believed to predominantly be an environmental pollutant [39] but, it has been extensively demonstrated to be also produced endogenously by a subtype of white blood cells [40, 41]. The main sulfur-centered reactive species, RSS, are the products of oxidation of the thiol groups in proteins [2, 35, 42] (Fig. 1). RNS and RSS both contribute to fluctuations in redox homeostasis within the cells and, like ROS, are mainly produced within mitochondria [42,43,44,45]. Basal levels of ROS, RNS, and RSS are physiologically generated in cells and serve important regulatory and mediator functions. However, an uncontrolled increase in their concentrations leads to a chain of unwanted radical reactions that target the main biomolecules (DNA, RNA, lipids, and proteins) and alters key processes such as nucleic acid oxidation, lipid peroxidation, thereby contributing to the loss of cellular architecture, to impaired cell signalling and, ultimately, to cell death [25, 46].
Evidence suggests that OS is a common signature in neurodegenerative diseases such as Alzheimer’s disease (AD) [47,48,49], Huntington’s disease (HD) [50,51,52], Parkinson’s disease (PD) [53,54,55] and, in particular, it is part of the tightly connected and multi-factorial set of mechanisms involved in the pathogenesis and progression of Amyotrophic lateral sclerosis (ALS) [25, 56,57,58]. For counteracting OS effects, cells activate multiple antioxidant defense systems based on both enzymatic and non-enzymatic components. Enzymatic activities include superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase (CAT); some of the important non-enzymatic antioxidant molecules endogenously produced are lipoic acid, reduced GSH, L-arginine (L-Arg), and coenzyme Q10 [59,60,61,62]. Together with dietary antioxidants such as Vitamin C, Vitamin E, melatonin, and carotenoids [63, 64], these antioxidant systems serve as “free radical scavengers” by preventing, buffering, and repairing damages caused by OS, and enhancing the immune defense against degenerative diseases [65] (Fig. 1).
In that regard, the transcription factor NRF2 (nuclear factor erythroid 2-related factor 2) and its main negative regulator KEAP1 (Kelch-like ECH (erythroid cell-derived protein with CNC homology)-associated protein 1) represent a finely regulated antioxidant system, with a key role in the response to OS and maintenance of redox homeostasis. KEAP1 acts as an OS sensor to regulate NRF2 turnover within the cell/body, while NRF2 activation leads to the production of antioxidant and detoxifying substances that decrease OS [66,67,68].
This review aims to highlight the central role of OS in the pathophysiology of ALS, specifically focusing on the KEAP1-NRF2 system and its alteration in ALS, while discussing the potential applications of NRF2 activators in ALS therapy.
Main text
Role of oxidative stress in ALS
ALS is an adult-onset neurodegenerative disease, characterized by degeneration and loss of upper motor neurons (MNs) in the cerebral cortex and lower MNs in the brainstem and spinal cord [69], leading to progressive weakness of voluntary muscles, and death caused by a diaphragmatic failure within 2–5 years [70, 71]. Most of the cases of ALS are sporadic (sALS) with no genetic linkage, while around 10% are associated with familial forms (fALS), presenting mutations in over 20 genes. The most common of them include Cu2+/Zn2+ superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TARDBP), fused in Sarcoma (FUS), and chromosome 9 open reading frame 72 (C9orf72) genes, involved in protein homeostasis and RNA metabolism [72,73,74,75]. sALS and fALS’ progression is clinically and pathologically very similar, both characterized by the progressive accumulation of toxic misfolded protein aggregates within cells [76] and the significant loss of MNs [77]. To date, there is still no effective treatment for ALS, and the two currently available drugs, riluzole and edaravone, only extend survival of ALS patients by few months [78,79,80,81,82,83].
The precise regulation of ALS pathogenetic mechanisms is still elusive, despite the abundance of identified mechanisms mediating MN degeneration and death, including neuroinflammation, glutamate excitotoxicity, altered RNA metabolism, mitochondrial dysfunction, impaired cytoskeletal integrity, altered axonal transport dynamics, OS [71, 84, 85].
Given the complexity of the disease, it is therefore not surprising that dissecting cause and effect is not a simple matter. The role of OS in ALS pathology, whether as a primary cause of disease or a secondary consequence, is now well recognized and many preclinical and clinical studies have consistently demonstrated that the disease is characterized by high levels of OS markers able to induce cellular damage, impaired cell signalling, and finally cell death [25, 86]. So, it is likely that OS can be triggered by following an increase in the production of O2•- and NO• in MNs as well as in the central glia, starting from the presymptomatic phase of ALS. This unfavorable condition could also be facilitated by the early reduction of the GSH level in the various tissues of subjects affected by ALS [87]. However, as the main evidence comes from post mortem tissues, the temporal evolution of OS markers in ALS is not yet well defined.
Early evidence demonstrated that high levels of protein carbonyl groups, a general marker of OS, were present in both postmortem spinal cord [88, 89] and motor cortex [90] from sALS patients, compared to control samples. Likewise, increased 3-nitrotyrosine (3-NT) levels, a marker for peroxynitrite-mediated damage, were observed within spinal cord MNs [91]. High levels of lipid peroxidation-derived aldehydes, including 4-hydroxy-nonenal (4-HNE) and malondialdehyde (MDA) [92], protein glycoxidation markers such as pentosidine [93], and stress-induced DNA damage molecules, more specifically 8-hydroxy-2′-deoxyguanosine (8-OHdG) [94, 95], have also been revealed in the spinal cord from sALS patients. In addition, high levels of most of these OS biomarkers [95,96,97,98], together with high levels of ascorbate free radical [96], and two products of NO oxidation, such as nitrite and nitrate [99], were also detected in cerebrospinal fluid (CSF) samples from ALS patients, showing a positive correlation with 8-OHdG level and disease severity over the 9-month duration of the prospective study [95]. These findings were consistent with the hypothesis that oxidative pathology accompanies the neurodegenerative process in ALS and suggest that 8-OHdG could be a sensitive biomarker to be dynamically quantified during ALS to gather more information on the temporal evolution of OS and on disease progression.
While OS is recognized as an important feature in ALS, the role of impaired GSH metabolism is still debated. In two studies no detectable [100] or unchanged [101] activities of GSH peroxidase were found in spinal cord or cerebral cortex of ALS patients. Some other studies, on the contrary, have revealed a decrease in the content of antioxidant GSH. In particular, back in 1996 Przedborski and colleagues indicated that GSH peroxidase activity is reduced in a specific brain region affected in ALS, the precentral gyrus, suggesting that the observed deficit could be involved in the oxidative damage observed in sporadic ALS [102]. In another study they described a similar GSH reduction in red blood cells of sALS patients treated with insulin-like growth factor 1 (IGF-1), but probably only as a secondary cytotoxic effect due to the treatment with IGF-1 [103]. The oxidant-antioxidant imbalance in the erythrocytes of sALS patients was confirmed, more recently, in another study where GSH reductase and reduced GSH levels were also found to be significantly lower at 24 months from the onset of ALS, in patients who died shortly after [104]. In addition, a study performed along 72 months has shown a significant impairment of erythrocytes GSH peroxidase activity especially in subjects with a faster rate of disease progression [105]. Similarly, the first in vivo imaging studies in humans, confirmed an increase in OS and a simultaneous decrease in GSH levels in the motor cortex of patients with ALS (with an age of 65.2 ± 9.4 years, and a mean disease duration of 25.8 ± 17.1 months) compared to the controls [106, 107]. Furthermore, the fact that tracer retention in cortical regions increased with clinical disease severity, as estimated by the Revised ALS Functioning Rating Scale (ALSFRS-R), suggested that OS may be an important factor associated with the development of neurodegeneration in ALS patients [107]. Then in 2019, a meta-analysis study tried to clarify the OS marker profile in ALS patients: the authors demonstrated that 8-OHdG and MDA were significantly increased in the peripheral blood of ALS patients when compared with control subjects, while GSH levels were significantly reduced. However, due to the heterogeneity between studies and other clinical variables, more studies are needed to better understand how prooxidative imbalances contribute to the pathophysiology of ALS [108].
In line with patient studies, cellular and mouse models of ALS also showed high levels of ROS, along with elevated oxidation of membrane phospholipids (HNE, MDA), DNA (8-OHdG), and proteins (protein carbonyl groups, 3-NT) [94, 109,110,111]. Another preclinical study using presymptomatic and symptomatic SOD1G93A transgenic mice revealed an association between the accumulation of ceramides and cholesterol esters in the spinal cords of ALS patients and OS, thus highlighting the pivotal role of OS in inducing an alteration in the metabolism of sphingolipids metabolism which mediates MN death in ALS [112]. To further strengthen the contribution of OS in ALS, an increase in the level of several proteins oxidatively modified and consequently inactivated has been found in SOD1G93A transgenic mice and correlated with SOD1 mutation: such proteins are SOD1 itself, translationally controlled tumor protein (TCTP, which normally processes calcium binding activity and acts as a cytoprotective factor), and ubiquitin carboxyl-terminal hydrolase isoenzyme L1 (UCH-L1, which plays an important role in the ubiquitin-proteasome system, UPS). Both oxidatively modified SOD1 and UCH-L1 are involved in the formation of cytoplasmic inclusions in ALS mouse models and in ALS patients [113]. Taken together, these findings indicate a potential relationship between protein oxidation, Ca2+ regulation, and protein aggregation in ALS (Fig. 2).

OS markers in ALS. Increased levels of products of lipid peroxidation (and altered lipid metabolism), DNA oxidation, protein oxidation, as well as stress proteins, oxidatively modified proteins, and ROS have been found in ALS patients (blue points) and in SOD1G93A transgenic mice (yellow points); the presence of impaired antioxidant defense has been highlighted
Overall, considering the evidence reported here, the cellular antioxidant defense in ALS appears to be strongly diminished, with an imbalance towards high and persistent OS levels. Therefore, a better understanding of the dysregulation of the antioxidant KEAP1-NRF2 activity in ALS could help to develop novel therapeutic strategies able to halt disease progression, prolonging patients’ survival.
KEAP1-NRF2: the cellular response to OS
Structural insights
KEAP1 and NRF2 are two relevant players in the molecular and cellular response to OS [114, 115].
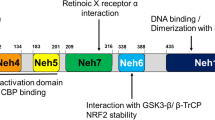
KEAP1 belongs to the BTB-Kelch family of proteins, known to be involved in ubiquitination [116, 117]. It was cloned for the first time using the Neh2 domain of NRF2 as bait in a yeast two-hybrid system by Yamamoto and coworkers [118]. It acts as a substrate linker for the interaction of Cullin 3 (CUL3)-dependent E3 ubiquitin ligase complex [119], with NRF2 leading to continuous ubiquitination and proteasomal degradation of NRF2 [120], thus representing the main negative regulator of NRF2 activity. Three major functional domains represent the essence of the KEAP1 protein structure: the N-terminal Broad complex/Tramtrack/Bric à brac (BTB) domain, the central intervening region (IVR) and the C-terminal Kelch/Double glycine repeat (DGR) domain [121, 122]. The BTB domain binds Cul3, contains the Cysteine151 residue (C151), which is one of the important cysteine residues in stress sensing, and mediates KEAP1 homodimerization [123,124,125]. The IVR domain contains a 3-box motif in its proximal part, which provides an additional CUL3 interaction site [116], and two critical cysteine residues, Cysteine273 (C273) and Cysteine288 (C288), that are important for the repression of NRF2 activity [126]; IVR connects the BTB domain with the Kelch/DGR domain. The latter domain is required for substrate capture and can bind separately to both DLG (Aspartic acid-Leucine-Glycine) and ETGE (Glutamic acid-Threonine-Glycine-Glutamic acid) motifs on the NRF2-ECH homology (Neh) 2 domain of NRF2 [127,128,129] (Fig. 3a).

Domain architecture of the human KEAP1 (a) and NRF2 (b) proteins. Functional domains, relevant motifs (3-box, DLG, ETGE, DSGIS, DSAPGS), and the most important cysteine residues (C151, C273, C288) involved in stress sensing are indicated. BTB: Broad complex/Tramtrack/Bric à Brac, IVR: intervening region, DGR: double glycine region, Neh1–7: NRF2-ECH homology 1–7
NRF2 is a member of the cap ‘n’ collar (CNC) subfamily of the basic region leucine zipper (bZIP) transcription factors, involved in multiple cellular processes [130,131,132,133,134,135]. Among them, its most prominent role is to induce the antioxidant responsive element (ARE)-mediated genes in response to OS, thereby ensuring cytoprotection [136]. In fact, NRF2 deletion causes an increase in OS and cellular death both in vitro and in vivo [137, 138]. The Nuclear factor erythroid 2 like 2 (NFE2L2) gene, encoding NRF2 protein, was cloned as a factor that binds to the β-globin promoter [139, 140]. Moving from the N-terminal to the C-terminal region, the protein structure includes seven highly conserved Neh domains [121, 122, 141, 142]. The Neh2 domain is a regulatory domain containing the DLG and ETGE binding motifs, through which NRF2 interacts with its major negative regulator, KEAP1, and is essential for the KEAP1-mediated degradation of the protein by the proteasome [128, 143,144,145]. The Neh4 and Neh5 domains are two important transactivation domains; they recruit cAMP response element-binding protein (CREB)-binding protein (CBP) or receptor-associated coactivator 3 (RAC3), to facilitate the transactivation of NRF2 target genes [146, 147]. The Neh7 domain mediates the binding to retinoid X receptor alpha (RXRα), another negative regulator of NRF2 [148]. The Neh6 domain, which is a serine-rich region, contributes to regulate NRF2 stability and mediates the interaction with a third negative regulator, β-transducin repeat-containing protein (β-TrCP), through its DSGIS (Aspartic acid-Serine-Glycine-Isoleucine-Serine) and DSAPGS (Aspartic acid-Serine-Alanine-Proline-Glycine-Serine) motifs not recognized by KEAP1. Phosphorylation of the DSGIS motif in NRF2 mediated by glycogen synthase kinase-3 beta (GSK-3β) promotes the binding of β-TrCP to NRF2 which is therefore marked for ubiquitination and then degraded by the proteasome [145, 149,150,151]. The Neh1 domain contains a bZIP binding motif responsible for the formation of a heterodimer with small musculoaponeurotic fibrosarcoma (sMaf) proteins and mediates the binding to ARE sequences, within the promoter of NRF2 target genes [152]. Finally, the C-terminal Neh3 domain is another transactivation domain that recruits chromo-ATPase/helicase DNA-binding protein 6 (CHD6), functioning as an NRF2 transcriptional coactivator [153] (Fig. 3b).
Spatiotemporal regulation of the KEAP1-NRF2 system
Under physiological conditions, KEAP1 is the main regulator of the NRF2 protein level (NRF2 has a half-life of about 20 min) [120, 145]. More specifically, KEAP1 homodimerizes [124], binds to the ETGE and DLG motifs on Neh2 domain of NRF2, and also acts as a substrate adaptor protein [154] for the KEAP1-NEDD8-CUL3-RBX1 E3 ubiquitin ligase complex [119, 120, 155]. CUL3 is linked to the ubiquitin-like protein NEDD8 [156] and represents a scaffolding protein between KEAP1 and RBX1. This conformation of KEAP1-NRF2 complex allows an adequate Lysine (Lys) rich α-helix orientation in the Neh2 domain for KEAP1-dependent polyubiquitination [157]. In addition, the UBX7-p97-UFD1/NPL4 complex, consisting of a ubiquitin-targeted ATP-dependent segregase, p97, and two heterodimeric cofactors, UBX7 and UFD1/NPL4, extracts ubiquitinated NRF2 from the KEAP1-NRF2 complex and transfers it to the 26S proteasome for its degradation [154] (Fig. 4a).

Schematic model of the KEAP1-NRF2 signalling pathway under basal conditions (a), OS (b), and after the recovery of cellular redox homeostasis (c). (a) Under basal conditions NRF2 is sequestered by the KEAP1-NEDD8-CUL3-RBX1 complex in the cytoplasm, transferring ubiquitin (Ub) proteins from E2 ligase to the Neh2 domain in NRF2. In addition, the UBX7-p97-UFD1/NPL4 complex interacts with ubiquitinated NRF2 and NEDD8-CUL3 complex and transfers NRF2 to 26S proteasome for its degradation. (b) OS causes the oxidation of cysteine residues in KEAP1, inducing a conformational change in its structure and preventing NRF2 ubiquitination. NRF2 is therefore stabilized and translocates to the nucleus, where it binds to sMaf proteins activating ARE-driven genes. (c) Upon the recovery of redox homeostasis, KEAP1 translocates into the nucleus and induces NRF2 nuclear export. In the cytosol, NRF2 is ubiquitinated and degraded, and its level returns to be physiologically low
In response to OS, different metabolites oxidize highly reactive cysteine residues in KEAP1, inducing a conformational change in the KEAP1-NEDD8-CUL3-RBX1-E3 ubiquitin ligase complex, with an improper orientation of Lys residues on NRF2 [122, 158]. This leads to impaired ubiquitination of NRF2, an increase of NRF2 protein levels [157], and a reduction of KEAP1 levels able to sequester de novo synthesized NRF2 [159]. NRF2 therefore accumulates and is free to translocate to the nucleus through the nuclear pore complex (NPC). This translocation is supported by Karyopherin α1 (importin α5), that functions as an adaptor molecule, and Karyopherin β1 (importin β1), the transport receptor that allows NRF2 translocation from the cytoplasm to the nuclear side of the NPC, due to Nuclear Localization Signal (NLS) sequences in NRF2 [160]. NRF2 then dimerizes with members of the sMaf proteins through its bZIP domains and binds to the ARE sequences, which are regulatory enhancers, within gene promoters [156, 161]. Classical NRF2 target genes encoding antioxidant and detoxifying enzymes are responsible for maintaining redox balance [162]. Some examples include: NADPH quinone dehydrogenase (NQO1), heme oxygenase 1 (HO1), the two subunits of glutamate-cysteine ligase (GCL), glutamate-cysteine ligase modifier (GCLM, the rate-limiting enzyme for GSH synthesis), SOD 1, CAT, sulfiredoxin, thioredoxin, and peroxiredoxin [68, 137, 162, 163]. In addition, NRF2 is capable of increasing the expression of many autophagy-related genes that present a common ARE sequence in their promoters, including Sequestosome-1 (SQSTM1) gene (encoding protein 62) [164], and of attenuating inflammation by blocking pro-inflammatory cytokine (IL-6 and IL-1β) transcription [132], thus highlighting its crucial contribution to different cellular pathways. Furthermore, NRF2 controls genes encoding enzymes governing different metabolic pathways, such as lipogenesis and lipid degradation, lipid transport and uptake, enzymes of the pentose phosphate pathway, and some enzymes of glycogen metabolism, like glycogen branching enzyme (GBE) and phosphorylase b kinase subunit A1 (PHKA1), recently linked to NRF2 signalling in the muscle [165] (Fig. 4b). After the recovery of cellular redox homeostasis, KEAP1 translocates into the nucleus through Karyopherin α6 (importin α7), inducing NRF2 nuclear export to the cytosol, in a CRM1-dependent process through Nuclear Export Signal (NES) sequences present in NRF2. Low basal level of NRF2 is therefore re-established by KEAP1-mediated ubiquitination and degradation, and the NRF2 signalling pathway is turned off [166,167,168] (Fig. 4c).
Regulatory mechanisms
Many cellular mechanisms and processes tightly modulate the activity of KEAP1-NRF2 system at multiple levels [169]. More specifically, a complex regulation at the DNA level (transcriptional control), at the mRNA level (post-transcriptional control), and at the protein level (protein stability, availability of binding partners, and post-translational control) is responsible for maintaining cellular redox homeostasis (Fig. 5). A better understanding of the detailed interactions between the factors involved will contribute to further elucidate the cell defense capability, which is impaired in neurodegenerative diseases such as ALS [67, 141, 170], and may reveal the path for identifying novel potential therapeutic targets.

Schematic overview of the cellular mechanisms and processes modulating the KEAP1-NRF2 system. Activators (upper panels) and inhibitors (bottom panels) tightly modulate KEAP1-NRF2 activity at the level of DNA, RNA, and protein: transcriptional regulation, post-transcriptional regulation, protein stability, availability of binding partners, and post-translational regulation are involved. Red arrows indicate the activating effect, while blue bars indicate an inhibitory effect
Activators
At the transcriptional level, NFE2L2 gene is induced by several transcription factors, including the aryl hydrocarbon receptor (AhR) in response to environmental pollutants like polycyclic aromatic hydrocarbons, breast cancer susceptibility gene 1 (BRCA1) in response to xenobiotic stress, and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in response to inflammatory stimuli. The first two bind to the xenobiotic response element (XRE) sequences, while the last binds to the NF-κB binding site in the NFE2L2 promoter, inducing its transcription [171,172,173,174], with a consequent increase of NFE2L2 mRNA available within the cell. To note, NRF2 regulates BRCA1 by the presence of an ARE sequence in BRCA1 promoter, creating a positive-feedback loop, with an increase of both transcripts [175]. Additionally, the neurogenic locus notch homolog protein 1 (NOTCH1) signalling pathway [133] and the phosphoinositide 3-kinase (PI3K)-protein kinase B (AKT) pathway [176, 177], have also been shown to augment NFE2L2 transcription, and consequently activate NRF2 signalling, thus maintaining redox balance.
At the post-transcriptional level, it has been demonstrated that the KEAP1-NRF2 system can be positively controlled by a microRNA (miRNA) able to target KEAP1 gene [178]. miRNAs are small (20–23-nucleotides), endogenous, single-stranded noncoding RNAs that regulate gene expression by pairing to sequence-specific binding sites in mRNAs to direct post-transcriptional repression and consequent inhibition of protein translation or degradation of the mRNA targets [179,180,181]. In particular, Eades’group in 2011 identified and validated for the first time in MDA-MB-231 and Hs578T breast cancer cells a miRNA, miR-200a, able to target the KEAP1 3′-untranslated region (3′-UTR), leading to KEAP1 mRNA degradation and, consequently, NRF2 stabilization and translocation to the nucleus [178]. MiR-7 also activates NRF2 by targeting KEAP1 expression in human SH-SY5Y neuroblastoma cells, resulting in increased levels of the reduced form of GSH, indicative of OS mitigation [182]. Another post-transcriptional mechanism that can regulate NRF2 is alternative splicing. In fact, it has been observed that aberrant NFE2L2 transcript variants missing exon 2, or exons 2 and 3, are present in lung cancers and head and neck cancers. The NRF2 protein isoforms encoded by these splice variants lack the KEAP1 interaction domain, thus preventing NRF2 degradation and resulting in protein stabilization and transcription of NRF2 target genes [183].
Like many other stress responsive transcription factors, NRF2 is mainly regulated at the level of protein stability by a variety of proteins; some of them interact directly with NRF2, while others interact indirectly with it and directly with KEAP1 [169]. More precisely, among them, the cyclin-dependent kinase inhibitor p21Cip1/WAF1, induced by p53 in response to OS and involved in the regulation of cell cycle arrest, replication, cellular proliferation, and apoptosis, competes with KEAP1 for the binding to NRF2 through the DLG motif, thus preventing NRF2 ubiquitination, and increasing its stabilization [184, 185]. BRCA1, which regulates cell cycle progression, DNA damage signalling and repair, and maintenance of genomic integrity, interacts with NRF2 either directly, through its ETGE motif, preventing KEAP1-dependent ubiquitination of NRF2 and protecting cells against OS, or indirectly, through the interaction with CBP or with c-Myc [186]. Other proteins that bind KEAP1 and interfere with KEAP1-NRF2 interaction, promoting NRF2 accumulation and function in the nucleus, include: phosphoglycerate mutase 5 (PGAM5, a serine/threonine phosphatase related to the phosphoglycerate mutase family), which has been reported to be associated with mitochondria, so its interaction with KEAP1 may be regulated by mitochondrial signalling processes [187, 188]; partner and localizer of BRCA2 (PALB2), which is a nuclear protein playing an important role in maintaining genome integrity [189]; dipeptidyl-peptidase 3 (DPP3), which is a zinc aminopeptidase involved in protein turnover [190, 191]; prothymosin alpha (ProTα, a nuclear protein involved in cellular proliferation and protection against apoptosis [192], whose interaction with KEAP1 has been found increased after cell treatment with the OS inducer diethyl maleate (DEM) [193, 194]. NRF2 activity is also increased by stress associated chemicals, such as NO, Zn2+, and alkenals (a class of carbonyls produced by lipid peroxidation) through their interaction with KEAP1 [195]. In any case, the most acknowledged cytoplasmic protein able to regulate NRF2 by promoting its stability and nuclear role is p62 [196]. It is a stress-inducible protein involved in multiple signalling pathways, such as autophagy, where it acts as a cargo receptor for the degradation of ubiquitinated proteins and mitochondria, thus preserving organisms from proteotoxic/oxidative stresses [196, 197]. p62 competes with NRF2 for binding to KEAP1, and this interaction induces a dependent autophagic degradation of KEAP1 and subsequent NRF2 stabilization and activation in both MEFs and HEK293 cells [198]. Interestingly, as for BRCA1, p62 expression is regulated by NRF2, due to the presence of ARE sequences in the p62 promoter, implying a positive-feedback loop [199].
NRF2 activity is also regulated by the availability of many binding partners, including a group of bZIP bi-directional transcription regulators named sMaf proteins (MafF, G and K) [200, 201] which, when bound to NRF2, under OS, favor its maintenance inside the nucleus thereby enhancing its activity [202].
In addition, at the post-translational level, many kinases and acetyl groups are critical for NRF2 subcellular localization and transcriptional activation. Among them, protein kinase C (PKC), a family of protein kinase enzymes involved in various cellular mechanisms, has been shown to phosphorylate NRF2 at Ser40 in its Neh2 domain, disrupting the association with KEAP1 and promoting its activation [203, 204]. Also, the casein kinase 2 (CK2), a highly conserved serine/threonine protein kinase with dual functionality in cellular growth/proliferation and suppression of apoptosis [205], can phosphorylate NRF2 in more than ten sites within Neh4 and Neh5 domains [206], leading to NRF2 translocation to the nucleus and its activation [207]. Alongside these, several studies focused on elucidating the role of mitogen-activated protein kinases (MAPKs) in NRF2 activation, suggested the involvement of c-jun N-terminal kinase 1/2 (JNK1/2) and extracellular signal-regulated kinase 1/2 (ERK1/2) in the NRF2 activation and nuclear accumulation [208, 209]. However, in a later study, it has been demonstrated that direct phosphorylation of NRF2 by MAPKs has a limited contribution in modulating NRF2 activity; rather, MAPKs appear to regulate the NRF2 signalling pathway mainly through indirect mechanisms [210]. Most recently, adenosine monophosphate (AMP)-activated protein kinase (AMPK), a Ser/Thr kinase involved in the cellular response to stressful stimuli [211], has been identified as a novel player in NRF2 activation through its phosphorylation [212]. AMPK phosphorylates NRF2 at the Ser550 residue located in the canonical NES sequence, facilitating NRF2 nuclear accumulation, probably by inhibiting nuclear export [212], and this effect is enhanced by GSK-3β inhibition [213]. Since it is well known that both ubiquitination and phosphorylation are involved in NRF2 regulation, it was no surprise that the acetylation of NRF2 also enhances its activity [214]. It has indeed been demonstrated that p300/CBP transcriptional co-activator proteins (two central players in coordinating and integrating multiple signal dependent events with the transcription apparatus [215, 216]) bind to NRF2 in response to OS induced by arsenite and acetylate several Lys residues within the Neh1, the DNA binding region of NRF2, stabilizing it, increasing protein abundance, and promoting NRF2 nuclear localization [146, 214, 217] (Fig. 5, upper panels).
Inhibitors
Pieces of evidence for the transcriptional regulation of NRF2 highlighted that modifications of the NFE2L2 promoter region such as hypermethylation or single nucleotide polymorphisms, are responsible for a decreased NRF2 expression; however, these findings need further investigation to be confirmed [218,219,220].
As already discussed above, at the post-transcriptional level, an important role in the regulation of the KEAP1-NRF2 system is played by miRNAs. Many studies revealed the existence of several miRNAs able to target NFE2L2, resulting in NFE2L2 mRNA degradation and inhibition of NRF2 protein translation, such as miR-144, miR-28, miR-93, miR-153, miR-27a, miR142-5p, and miR-34. miR-144 was the first miRNA identified as a NRF2 negative regulator in reticulocytes of patients with homozygous sickle cell disease (HbSS) [221]. Increased levels of miR-144 have been found in a subset of HbSS patients with typical severe anemia and reduced NRF2 protein levels, causing a reduction of GSH regeneration and impairing cell response to OS, thereby providing a possible mechanism for the increased anemia severity seen in these patients [221]. Soon after, a similar inverse association has been found between NRF2 and miR-28 in human MCF7 breast cancer cells, where ectopic expression of miR-28 alone was able to reduce NFE2L2 mRNA and protein levels, and, as an abnormal miR-28 expression has been seen in a variety of cancers, a role in this regard has been hypothesized [222]. miR-93 is another miRNA able to decrease NFE2L2 mRNA and protein levels, whose high expression has been associated with human breast carcinogenesis [223]. Subsequently, Mahimainathan’s group identified and validated for the first time a series of miRNAs, miR-153, miR27a, miR142-5p, and also miR-144 in neuronal cells, specifically in human SH-SY5Y neuroblastoma cells, which directly mediate repression of NFE2L2 in a KEAP1-independent manner [224]. The inhibitory effect on NRF2 activation by miR142-5p was also confirmed by Lv and colleagues using a cellular model of oxygen-glucose deprivation and reoxygenation-induced injury in hippocampal neurons in vitro [225]. Another miRNA shown to regulate Nrf2 is miR-34, able not only to target NFE2L2 but also downstream genes involved in cellular response to OS, such as microsomal glutathione S-transferase 1 (Mgst1) and Sirtuin 1 (Sirt1) genes in rat liver during aging [226].
In addition to KEAP1, which is the main negative regulator of NRF2 protein stability, many other factors which negatively regulate NRF2 have been discovered and characterized. One of them is GSK-3β, a Ser/Thr protein kinase involved in several metabolic processes, including glycogen metabolism and insulin signalling [213]. Cuadrado and colleagues first showed that GSK-3β inhibits NRF2 activity, but the identification of sites in NRF2 that are phosphorylated by GSK-3β was achieved several years later using two-dimensional electrophoresis and mass spectrometry: GSK-3β phosphorylates a group of Ser residues at the level of DSGIS motif in the Neh6 domain of NRF2, and this event represents the signal for the recognition by the adapter protein β-TrCP, that targets NRF2 for ubiquitination and proteasomal degradation in a KEAP1-independent manner [150, 151]. As discovered more recently by Zhang’s group, NRF2 can also be negatively regulated through the E3 ubiquitin-protein ligase synoviolin (HRD1), recently associated with the ER stress produced by misfolded protein accumulation. HRD1 interacts with the Neh4 and Neh5 domains of NRF2 and causes NRF2 ubiquitination and subsequent degradation through a KEAP1-independent mechanism, just like GSK-3β [227, 228]. Other negative modulators of NRF2 are: CR6-interacting Factor 1 (CRIF1), a protein previously known as a cell cycle regulator and transcription cofactor, which is able to negatively regulate NRF2 protein stability under both reducing and OS conditions through physical interaction with both N- and C-terminal regions of NRF2 [229]; seven in absentia homolog 2 (SIAH2), a key regulator, together with hypoxia-inducible factor 1 alpha (HIF-1α), of the cellular hypoxic responses under pathological conditions such as ischemia-reperfusion, whose association with NRF2 causes the degradation of NRF2 irrespective of its phosphorylation status [230]; and caveolin-1 (CAV1), a scaffold protein of caveolar membranes involved in signal transduction and in the uptake of lipophilic compounds, which directly interacts with NRF2, probably competing with KEAP1 for binding to NRF2, and subsequently suppresses its transcriptional activity [231].
Among the numerous partners that interact with NRF2 regulating its activity, two members of the CNC-bZIP protein family – BTB and CNC homolog 1 (BACH1) and BTB and CNC homolog 2 (BACH2) – are two negative regulators of NRF2: they compete with NRF2 for the binding to ARE sequences of target genes, thereby preventing the production of antioxidant enzymes and cellular defense against toxicity [232, 233].
At the post-translational level, just as CBP/p300-mediated acetylation of Lys within the Neh1 domain increasing NRF2-dependent transcription, SIRT1 mediates Lys deacetylation, antagonizing the NRF2 signal [234]. Interestingly, unlike other MAPKs, such as ERK1/2 and JNK1/2, it has been shown that p38 phosphorylates NRF2 and promotes its association with KEAP1, thereby preventing its nuclear translocation; this effect can be reversed by sulforaphane, a molecule with chemopreventive ability, and, in fact, represents a potential mechanism of action for sulforaphane-mediated induction of NRF2 [235]. Finally, the phosphorylation of tyrosine (Tyr) residues in NRF2 by another kinase, Fyn, present in the nucleus negatively regulates NRF2. More specifically, Fyn phosphorylates NRF2 at Tyr568 leading to nuclear export, ubiquitination, and degradation of NRF2 [236]. It has been demonstrated that GSK-3β acts upstream of Fyn, activating its phosphorylation and nuclear accumulation; in the nucleus Fyn itself can phosphorylate NRF2 and act as a negative regulator [237] (Fig. 5, bottom panels).
Alteration of KEAP1-NRF2 activity in ALS
The human brain is an organ with elevated demand of energy, relying on oxidative metabolism and high consumption of O2 to perform its functions [68]. In addition, non-neuronal cells residing in both the central nervous system (CNS) and peripheral nervous system (PNS) – called glial cells, and among them mainly astrocytes, microglia, and oligodendrocytes – provide essential metabolic and functional support to neurons, contributing to their plasticity [238,239,240,241]. Due to their high and variable metabolic and mitochondrial activity, both neurons and astrocytes are especially vulnerable to OS [242].
Therefore, it is essential to activate neuroprotective systems such as NRF2 and its downstream antioxidant signalling, able to counteract pathological insults from highly reactive oxidant species in the brain and limit the accumulation of oxidative damage [68].
Even if it is still unclear whether OS is a primary or a secondary cause of neurodegeneration in ALS, overwhelming evidence indicates the pathological role of OS, and alterations in KEAP1 and NRF2 expression and in KEAP1-NRF2 activity may explain the progressive MN degeneration and death associated with ALS [170]. Dysregulation of KEAP1-NRF2 activity has been observed in ALS cellular and animal models, and confirmed in human ALS tissue [67, 141, 243]. More specifically, NRF2 and KEAP1 have been analyzed in postmortem motor cortex and spinal cord specimens from ALS patients and a reduction in both NFE2L2 mRNA and NRF2 protein has been found in ALS patient tissues when compared to control ones. The level of KEAP1 mRNA was shown to be increased in the motor cortex but not in the spinal cord of ALS patients compared to controls; although no significant differences were seen at the protein level [244]. KEAP1 protein has also been shown to co-localise with intracellular inclusions in MNs of postmortem ALS spinal cord [245], possibly through an interaction with p62 observed in several ALS inclusions [246]. The RNA binding protein 45 (RBM45) modulates the antioxidant response in ALS by interacting with KEAP1. Under OS RBM45 nuclear exit occurs (along with cytoplasmic inclusion formation). Once in the cytoplasm, it binds and stabilizes KEAP1, thus interfering with NRF2 stabilization. This in turn results in less NRF2 nuclear entry in response to OS and reduces the cellular antioxidant response, thus contributing to the pathobiology of ALS. Experimental evidence revealed a significant increase in KEAP1-RBM45 binding in cytosolic fractions from ALS lumbar postmortem spinal cord compared to controls; in addition, it has been demonstrated that RBM45 can bind p62 and that RBM45-p62 cytoplasmic colocalization was increased in ALS spinal cords [247]. Nevertheless, as mentioned above, p62 also regulates cell survival via the NRF2 antioxidant response pathway: in particular, the interaction between p62 and KEAP1 is essential for p62-dependent NRF2 signalling [199, 248, 249]. Thus, since several mutations affecting the functional domains of p62 have been identified in patients with ALS and frontotemporal dementia, it has been demonstrated that p62 variants exhibit reduced KEAP1-binding, preventing NRF2 from entering the nucleus and promoting protective genes, and predisposing the cell death upon exposure to ROS [250, 251].
In an effort to clarify the role of the KEAP1-NRF2 pathway in ALS, it has been revealed that the transfection of the mouse MN-like hybrid cell line (NSC-34) with the human SOD1G93A gene causes damage in the Nrf2/ARE signalling and a reduced ability of cells to react to OS. More specifically, cell soma became round, neurites were shorter, and decreased in number, and OS resulted increased. In addition, they found that both the transcript and the protein levels of Nrf2 and the detoxifying/antioxidant enzymes HO1 and NQO1 were significantly decreased [252]. Previously, a study based on proteomic analysis had revealed marked transcriptional repression of other Nrf2-induced antioxidant proteins including CK2, ERK1/2, PKC, GPx, Mgst1, peroxiredoxin 3 (Prdx3), in both preclinical models and in human spinal MNs from ALS patients [253]. These data suggested that pharmacological stimulation of the NRF2/ARE signalling could be a valuable lever to a new therapeutic approach in ALS [253, 254]. More recently, it has been demonstrated that NSC-34 cells expressing TDP-43 mutants also exhibit shortened neurites, increased OS, and decreased HO1 level, and these effects can be reversed by the UPS inhibitor MG132, but not by the Nrf2 activator sulforaphane [255, 256], probably because MG132 induction of HO1 is Nrf2 independent; however, how mutant TDP-43 reduces HO1 level and prevents sulforaphane from activating Nrf2 signalling remains unclear. Similarly to cell lines, primary embryonic MN cultures from SOD1G93A ALS mice showed a diminished Nrf2 nuclear expression and downregulation of the enzymes involved in GSH biosynthesis, associated with increased susceptibility to Nerve Growth Factor (NGF)-induced apoptosis [257]. Surprisingly, extensive down-regulation of miRNAs such as miR-27a, miR-34a, miR-142-5p, and miR-7, involved in cytoprotection against OS and some of them also capable of targeting Nrf2 directly, has been detected in the muscle, CSF, MN progenitors, and blood as well as in post mortem specimens (brain, brain stem, and the spinal cord) of both sALS and fALS patients, unlike healthy controls [258,259,260,261,262]. Such reduction is probably due to a dysregulated miRNA biogenesis under cellular stress and represents a common molecular denominator for multiple forms of human ALS [263].
In contrast to human tissue and cultured cells, it has been demonstrated that Nrf2 activity is consistently elevated in the spinal cord of SOD1G93A rodent models of ALS, and the increase of Nrf2, thioredoxin, HSP-70, HO1, NQO1, GCLC, and GCLM protein levels follows disease progression in the lumbar spinal cord but not cortex [264, 265]. Interestingly, both Nrf2 and HO1 co-localized with reactive astrocytes, the major GSH suppliers for neighboring neurons, in the degenerating spinal cord of SOD1G93A rats, and this was interpreted as a reactive attempt to prevent cell death; moreover, crossing SOD1G93A mice with mice overexpressing Nrf2 selectively in astrocytes significantly delayed disease onset and extended survival, thus reversing the toxicity of astrocytes expressing human SOD1G93A mutation to co-cultured MNs [266, 267]. Furthermore, it has been found that modulation of the Nrf2 signalling pathway by triterpenoids compounds results in a significant increase in survival in the SOD1G93A mouse model of ALS, thereby indicating that activation of Nrf2 has a neuroprotective effect [268]. In addition, cross-breeding ALS-transgenic mice with ARE-human placental alkaline phosphatase (hPAP) reporter mice allowed to reveal that Nrf2 activity was more intense in skeletal muscle than in the spinal cord, and evident since before motor symptom onset [264]. These data were in line with the hypothesis that neuromuscular junctions, which are the synapses between MNs and muscle fiber, could represent the starting site of MN dysfunction in ALS [269]. However, contrary to expectations, the deletion of the Nfe2l2 gene had a moderate impact on the course of the disease in SOD1G93A mice and negatively affected only a few Nrf2-regulated antioxidant enzymes, thus suggesting that several Nrf2-target genes can also be regulated independently of Nrf2 in ALS mice [270, 271].
KEAP1-NRF2 system as a potential therapeutic target in ALS
The presence in ALS of increased levels of OS markers and impaired antioxidant defense in the brain and peripheral tissues [25] together with the central role of NRF2 in inducing target genes to counteract OS [272] make the KEAP1-NRF2 system a suitable therapeutic target for drugs and small molecules.
Various chemical inducers of the NRF2 activity have been identified in the last two decades, but several of these compounds still have significant drawbacks associated to their clinical potential: suboptimal pharmacokinetics (concentration vs. time) and pharmacodynamics (effect vs. time), poor target selectivity, different efficacy between animal models and human pathologies, and safety issues [66].
The main NRF2-activating drug candidates that have been explored, and many of which have reached different stages of clinical trials, are presented in Table 1.
KEAP1-dependent NRF2 activators
Electrophilic compounds
The most known NRF2 activators are electrophiles – electron-deficient species able to form covalent bonds with electron-rich nucleophiles through a variety of chemical pathways – which can covalently bind and modify cysteine residues in KEAP1 [273, 274].
Cyanoenone triterpenoids
This class of synthetic pentacyclic triterpenoids derives from the natural compound oleanolic acid [268, 275], primarily reacts with C151 in KEAP1 by interrupting KEAP1-CUL3 interaction [123], and includes the strongest NRF2 activators currently known [276]. One of them, bardoxolone methyl (BARD, CDDO-Me, RTA402) is in the late stages of clinical trials for several conditions related to advanced chronic kidney disease – including Alport syndrome and an autosomal dominant polycystic kidney disease – and to pulmonary arterial hypertension (PAH). The efficacy of a second-generation derivative, omaveloxolone (RTA408), has recently been assessed in a preclinical model for diabetic chronic non-healing wounds [277] and, interestingly, omaveloxolone is now being tested in phase-II clinical trial for the Friedreich’s ataxia (FRDA), a neurodegenerative condition in which NRF2 activation is suppressed [278, 279].
Fumaric acid esters
Fumaric acid esters are the most investigated NRF2 activators. These electrophilic modulators mainly interact with C151 in KEAP1, thereby inhibiting NRF2 ubiquitination and increasing the activity of ARE-mediated antioxidant and detoxifying enzymes [280]. Among them, the most clinically successful NRF2 activator is dimethylfumarate (DMF), authorized in Germany in 1994 for the treatment of psoriasis [281]. It was clinically approved by both the Food and Drug Administration (FDA) and the European Medicine Agency (EMA) in 2014 for the treatment of relapsing-remitting multiple sclerosis (MS), showing a significant efficacy over 2 years versus placebo and a favorable benefit-risk profile in two phase-III clinical trials, although with several adverse effects such as nausea, diarrhea, abdominal pain and leukopenia in some patients [282, 283]. Since DMF is metabolized by intestinal esterases to monomethylfumarate (MMF), novel MMF derivative compounds with greater efficacy, bioavailability, and reduced side effects are currently under investigation in preclinical studies, including VCB102 compound for psoriasis, and VCB101 compound for MS. In addition, CAT4001 compound has been shown to decrease ROS production, normalize mitochondrial length, and improve mitochondrial function in dorsal root ganglion (DRG) derived neurons from FRDA’s mice [284], thus expanding the therapeutic potential also in the field of neurodegenerative diseases. Furthermore, two other MMF derivatives are being tested in phase-II or -III clinical trials for psoriasis – XP23829 compound, and for MS – ALK8700/BIIB098 compound.
Hydroxylamine
Hydroxylamine is a synthetic NRF2 activator that protects against OS by targeting KEAP1. Several substituted derivatives of hydroxylamine are known and, it has been shown that one of them, N-tert-butyl hydroxylamine, protects cells from OS and mitochondrial damage in an in vitro model of age-related macular degeneration (AMD) [285]. In addition, most recently, another di-substituted hydroxylamine, named OT551, able to cross blood-brain barrier (BBB) and inhibit OS and inflammation, has been developed and has shown efficacy in a phase-II clinical trial on age-related macular degeneration.
Nitro fatty acids
Nitro-fatty acids (NO2-FAs) are endogenous signalling mediators with strong anti-fibrotic and anti-inflammatory activities [286], able to promote NRF2 activation through the interaction and reversible post-translational modification of C273 and C288 present in the stress sensing domain (IVR) of KEAP1 [287]. Two phase-II clinical trials have just been completed to test the efficacy of CXA10 compound as a potential treatment for PAH and primary focal segmental glomerulosclerosis. Moreover, novel nitroalkenes derived from α-tocopherol (NATOH, NATxME, and NATx0) are being characterized in both in vitro and in vivo models [288], thus opening the way to new therapeutic strategies.
Sulforaphane
Several natural electrophilic activating compounds of NRF2 have been identified, including epigallocatechin 3-gallate, quercetin, α-lipoic acid, and sulforaphane (SFN) [289]. The last one is an organo-sulfur compound first identified in broccoli [290] able to modify C151 in KEAP1, therefore inducing the antioxidant NRF2-target enzyme NQO1 [158]. A phase-II clinical study currently underway will evaluate if sulforaphane improves core symptoms in patients with autism spectrum disorders [291]. A very important aspect of this compound is that it can cross BBB and it is able to protect cells from OS in many preclinical models of neurologic diseases, such as hypoxic-ischemic injury, AD, PD [292]. In addition, in an attempt to have a dual ‘drug-prodrug’ mechanism of action, a melatonin-SFN hybrid has been designed, which demonstrated to induce neuroprotection in cortical neurons subjected to OS [293]. Furthermore, to improve the stability of SFN, a cyclodextrin formulation, SFX-01, has been developed for which two phase-II clinical trials have just been completed, evaluating the safety and efficacy in the field of ER+ metastatic breast cancer and subarachnoid hemorrhage [294, 295].
TFM735
This compound was identified by high-throughput screening analysis as an activator of NRF2 through a C151-dependent mechanism [296]. TFM735 was shown to inhibit T cell proliferation and improve experimental autoimmune encephalomyelitis (EAE) in a mouse model of MS [297].
Non-electrophilic compounds
Also known as KEAP1-NRF2 protein-protein interaction (PPI) inhibitors, they comprise several classes of compounds, including naphthalene bis-sulfonamides, tetrahydroisoquinolines, and molecules that present an oxadiazole motif [66]. Evidence has shown their capability to increase the expression of NRF2 target genes in cellular and in vivo models of chronic obstructive pulmonary disease (COPD) [298], and to interfere with the direct PPI between KEAP1 and NRF2 or the PPI between KEAP1 and CUL3 [299, 300]. It had previously been shown that the amino acid sequence DEETGE is essential for NRF2-KEAP1 interaction [124]. To increase cell penetrance, the DEETGE motif was fused with the Tat sequence of the human immunodeficiency virus together with the cleavage sequence of calpain (Cal). The intracerebroventricular (ICV) injection of the DEETGE-Cal-Tat peptide showed an increased expression of Nrf2- regulated genes and marked neuroprotective and cognitive-preserving effects in mice subjected to brain injury [301]. Similarly, ICV pretreatment or peripheral post-treatment with the DEETGE-Cal-Tat peptide was also able to decrease Keap1-Nrf2 interaction in the rat hippocampal CA1 region after global cerebral ischemia (GCI) and, as a consequence, to induce Nrf2 target genes and a powerful neuroprotection of hippocampal-dependent cognitive function after GCI [302].
Although several PPI inhibitors with improved selectivity have recently been identified, using combined approaches that brought together protein crystallography, ligand NMR spectroscopy, and computational chemistry, unfortunately, none of them has been found to be able to cross the BBB and many are quite large in size and contain carboxylic acids, features which usually prevent or decrease CNS permeability [299].
KEAP1-independent NRF2 activators
Given the complex regulation of the KEAP1-NRF2 system, it is not surprising that new pharmacological strategies are being explored, aimed at inducing NRF2 activity also in a KEAP1-independent manner.
BACH1 inhibitors
One member (HPP-4382) of the synthetic small molecules targeting BACH1 was able to induce the antioxidant enzyme HO1 through NRF2 activation in human lung fibroblasts [303]. Another compound, HPP971, elevated reduced GSH levels and protected human astrocytes from H2O2-induced cell death in vitro and induced HO1 expression in a murine EAE model of MS, attenuating loss of motor functions [304]. Furthermore, HPP971 has completed two phase-I studies for the treatment of several diseases, including blood, bone, eye, kidney, and lung diseases, where it was well tolerated [66].
HRD1 inhibitors
Pharmacological inhibition of HRD1, a negative regulator of NRF2 implicated in its ubiquitination and degradation, was tested in human liver tissues and in an animal model for the liver cirrhosis characterized by increased ER stress and ROS: LS-102 alleviated liver cirrhosis enhancing NRF2 signalling pathway [228].
GSK-3β inhibitors
GSK-3β regulates multiple critical intracellular signalling pathways and it has been implicated in the pathogenesis of numerous diseases, including cancer [305], myocardial diseases [306], and neurodegenerative diseases [307]. In the last years, several preclinical studies and clinical trials have been effectuated using different GSK-3β inhibitors, actually with few encouraging results. Among these compounds, nordihydroguaiaretic acid showed no significant effects in two phase-II clinical trials for the treatment of prostate cancer and CNS tumors. Most recently, terameprocol reached phase I clinical trial for the treatment of high-grade glioma, still ongoing. In addition, a new phase-III clinical trial was started two years ago to evaluate the effects of enzastaurin in patients with diffuse large B-cell lymphoma.
p62 activators
Two compounds able to increase p62 levels, trehalose and rapamycin, have been demonstrated to increase nuclear translocation of NRF2 in a p62-dependent manner and to enhance expression of its downstream antioxidant factors, HO1 and NQO1, in a hepatoma cellular system [308] and in a preclinical model of FRDA [309]. These compounds have been also tested in two now completed phase-II and -III trials for the treatment of systemic lupus erythematosus [310] and diabetes mellitus type 1 [311] respectively, showing progressive improvement in disease activity thereby opening the way for new treatment perspectives. Unfortunately, another phase-II/III clinical study for the treatment of autosomal dominant polycystic kidney disease was stopped because it failed to show clinical benefits to patients. A multi-center phase-II clinical trial, started a few years ago to investigate the biological and clinical effects of rapamycin in people with ALS, is currently underway and will provide important information for further potential trials [312], thus opening new therapeutic perspectives for ALS.
Conclusions
High levels of OS can be considered a result of the imbalance between oxidative species and antioxidant defense systems, and indeed represent a hallmark of many acute and chronic diseases, including ALS. In this context, it is now widely recognized that the KEAP1-NRF2 system, which is essential for the maintenance of redox homeostasis, is impaired in ALS. The structural organization and functionality of these two players, under physiological conditions and in response to OS, present a tight regulation at multiple levels and involve different partners. Evidence has determined that elevated oxidative damage to proteins, lipids, and nucleic acids is a distinctive characteristic of the motor cortex and spinal cord of ALS patients, together with high levels of KEAP1 and a decrease in the NRF2-target genes and related enzymes.
Dysregulation of KEAP1-NRF2 activity has been extensively demonstrated in cellular and animal models of ALS and in postmortem ALS motor cortex and spinal cord samples, so dysregulation of the NRF2/ARE antioxidant and cytoprotective pathway could be a possible mechanism underlying the progressive neurodegeneration in ALS and severity of the disease. Since NRF2 activation induces the antioxidant and detoxifying response to restore redox homeostasis state, in recent years there has been a growing interest in identifying and evaluating new compounds capable of targeting the KEAP1-NRF2 system, to induce cytoprotective response against OS. A variety of NRF2 activators, both KEAP1-dependent and -independent compounds, have been identified and several of them are currently in clinical development. Some of these compounds have promising therapeutic effects in animal models and in several clinical trials for various pathologies including chronic neurological diseases. Nonetheless, to date the only FDA- and EMA-approved NRF2 activator for the treatment of relapsing-remitting MS and psoriasis is DMF, and its therapeutic potential in ALS patients in terms of efficacy, safety and tolerability, is now being evaluated in a phase-II clinical trial [313].
Despite many advances in the attempt to activate NRF2 through different pharmacological approaches, much remains to be done to fully benefit from the various drug candidates able to modulate the cellular antioxidant response mediated by NRF2. In particular, the suboptimal pharmacokinetics and pharmacodynamics of these compounds and their poor ability to cross the BBB could represent considerable obstacles for a significant therapeutic efficacy. In the last decade, studies on the condition of BBB in neurodegenerative diseases, including ALS, have demonstrated the presence of vascular disruption, a condition that could theoretically favour the entry of drugs into brain regions affected by neurodegeneration. However, it has been found that the pathological degradation of BBB hinders the proper delivery and action of therapeutic agents, due to a series of functional, structural, and biochemical changes at both the endothelial level and, consequently, in the perivascular and interstitial space [314]. This background suggests that the optimal condition for a drug to be more efficient in carrying out its therapeutic action would be to cross a BBB with good vascular integrity and without further damage in the neuronal functional state. This would be especially true for drugs relying on a fine functional remodulation of a well preserved cellular system, like the NRF2 activators on the KEAP1-NRF2 system. Thus, studies on the combination of strategies that can penetrate a normal BBB or on the dynamics of BBB disruption, as well as on the identification of biomarkers of brain vascular injury, might pave the way to new drug delivery approaches targeting BBB in the near future.
Furthermore, as specified above, MNs are not isolated units and also rely on glial cells for antioxidant protection through the NRF2/ARE signalling; in fact, recent evidence has shown that boosting astrocytic Nrf2 can have protective effects both in ALS mouse models and in vitro cultures. The fact that NRF2 is readily inducible in cultured astrocytes suggests that most pharmacological NRF2 activators may specifically target astrocytes in vivo, although there is still limited direct evidence for this. In addition, given that microglia clearly exhibit functional NRF2 signalling [315], it could be an important target for NRF2 induction in neurodegeneration. However, there are few data on NRF2 activation in microglia in human tissues or animal models of ALS, and NRF2 signaling has not yet been evaluated in oligodendrocytes, although this may be relevant in the case of ALS which also involves demyelination [316]. Therefore, it would be crucial to understand and outline the specific contribution of each cell type – MN, glia, or others – to ALS, in order to strategically target them with the NRF2 activators, thereby restoring neuronal homeostasis and survival, and functional MN-glia interactions (Fig. 6).

Activation of the NRF2 signalling as a potential treatment for ALS
As NRF2 enhances cell survival under stress, we can expect that increased NRF2 activity may promote tumorigenesis through stress protection [317, 318]. Dodson and colleagues have clearly reviewed how complex it is to target NRF2 in diseases [319]. In particular, they pointed out that although NRF2 has been traditionally considered as a tumor suppressor because of its cytoprotective functions [320], a growing number of studies have shown a strong NRF2 activation at certain stages of cancer [318, 321]. Another study suggested the existence of a correlation between NRF2 accumulation and the development of a multisystemic disorder, characterized by immunodeficiency and neurological symptoms [322]. In this light, we want to emphasize how a timely and appropriately regulated manipulation of the KEAP1-NRF2 system is crucial to develop effective and safe therapeutic treatment for neurodegenerative diseases, such as ALS, which involve an imbalance in the OS response. Therefore, while aware that the pharmacological modulation of the NRF2/ARE pathway could undeniably offer new therapeutic opportunities for diseases related to OS, we should at the same time consider that most of the currently known activators of NRF2 are not particularly specific, leading to an increased risk of “off target” toxic effects. It follows that an efficient modulator of the KEAP1-NRF2 system should be set, not only with significant efficacy and bioavailability, but also with high specificity [323].
Recently, population-based studies have revealed that ALS is an age-related disease similar to other age-associated neurodegenerative diseases like PD and AD [324]. Both aging and neurodegenerative disorders, such as ALS, share a multifactorial process underlying their progressive evolution, which is genetically determined and epigenetically influenced by the environment. Nonetheless, the existence of a biological interplay among OS, aging, and ALS has been presumed for more than two decades [325] and it is now becoming more evident [326]. Thus, in the course of aging various potentially pathogenic mechanisms, including the increase in oxidative damage, could on the one hand influence the quality of aging and on the other favour the onset and evolution of a definite neurodegenerative disease. Although with some controversy, the accumulated evidence directly supports an age-related decline in the ability to respond to OS with the activation of the NRF2/ARE signalling pathway, thereby reducing the expression of its target antioxidant genes [327]. However, the phenomena that induce OS attributable to the patient’s aging, in addition to being variable in terms of intensity among individuals, are difficult to distinguish from those produced by the compromised antioxidant defense systems due to a neurodegenerative disease, such as ALS. It follows that decoding the precise relationship between aging and ALS could allow to understand whether the effects of aging, including OS, might be a prerequisite to developing the MN disease [326].
In this regard, it would be interesting to evaluate if and which OS markers may directly or indirectly reflect the inadequate functioning of the KEAP1-NRF2 system in counteracting OS, and where they are already present at high levels in young adults. Indeed, conceptually, it would be critical to monitor the progressive pathological changes of these markers throughout adulthood, so to identify subjects possibly at risk of developing a neurodegenerative process favoured by the presence of OS induced, at least in part, by an imbalance of the KEAP1-NRF2 system. However, no single OS marker is representative in determining how healthy an individual is, or in predicting his life expectancy, while a set of clinical biomarkers, including lipid peroxidation and protein oxidation products, and antioxidative acting enzymes, could be used to delineate the oxidation status of a person [328].
As a matter of fact, taking into full account the intricacy of the numerous regulatory mechanisms involved in OS control as well as the complex interplay between the many events that contribute to the progressive degeneration and death of MNs, future neuroprotective pharmacological therapies will likely need to aim for an integrated treatment, acting on several levels to limit the progression of the disease. An adequate and hopefully incisive therapeutic action will therefore depend on a finely orchestrated balance between the efficacy of the single treatment for the specific pathogenetic mechanism and the control of potentially noxious interactions among the different therapies. In that sense, while still representing a challenging goal to be achieved, the identification of specific prognostic biomarkers and also those for the evaluation of therapeutic efficacy in ALS will have a fundamental role in designing and monitoring new treatment strategies in the complex scenario of the neurodegenerative process.
Availability of data and materials
Not applicable.
Abbreviations
- 3-NT:
-
3-nitrotyrosine
- 4-HNE:
-
4-hydroxy-nonenal
- 8-OHdG:
-
8- hydroxy-2′-deoxyguanosine
- AhR:
-
aryl hydrocarbon receptor
- AKT:
-
protein kinase B
- AMP:
-
adenosine monophosphate
- AMPK:
-
AMP-activated protein kinase
- ARE:
-
antioxidant responsive element
- BACH1:
-
broad complex-tramtrack-bric à brac and cap ‘n’ collar homolog 1
- BACH2:
-
broad complex-tramtrack-bric à brac and cap ‘n’ collar homolog 2
- BBB:
-
blood-brain barrier
- BRCA1:
-
breast cancer susceptibility gene 1
- BTB:
-
Broad complex/Tramtrack/Bric à brac
- bZIP:
-
basic region leucine zipper
- C9orf72:
-
chromosome 9 open reading frame 72
- Cal:
-
calpain
- CAT:
-
catalase
- CAV1:
-
caveolin-1
- CBP:
-
CREB-binding protein
- CHD6:
-
chromo-ATPase/helicase DNA-binding protein 6
- CK2:
-
casein kinase 2
- CNC:
-
cap ‘n’ collar
- CNS:
-
central nervous system
- CREB:
-
cAMP response element-binding protein
- CRIF1:
-
CR6-interacting Factor 1
- CRM1:
-
chromosomal region maintenance 1
- CSF:
-
cerebrospinal fluid
- CUL3:
-
cullin 3
- DEM:
-
diethyl maleate
- DGR:
-
Double glycine repeat
- DLG:
-
Aspartic acid-Leucine-Glycine
- DMF:
-
dimethylfumarate
- DPP3:
-
dipeptidyl-peptidase 3
- DSAPGS:
-
Aspartic acid-Serine-Alanine-Proline-Glycine-Serine
- DSGIS:
-
Aspartic acid-Serine-Glycine-Isoleucine-Serine
- ERK1/2:
-
extracellular signal-regulated kinase 1/2
- ETGE:
-
Glutamic acid-Threonine-Glycine-Glutamic acid
- FUS:
-
fused in Sarcoma
- GBE:
-
glycogen branching enzyme
- GCL:
-
glutamate-cysteine ligase
- GCLM:
-
glutamate-cysteine ligase modifier
- GPx:
-
glutathione peroxidase
- GSH:
-
glutathione
- GSK-3β:
-
glycogen synthase kinase-3 beta
- H2O2 :
-
hydrogen peroxide
- HIF-1α:
-
hypoxia-inducible factor 1 alpha
- HO1:
-
heme oxygenase 1
- •HO2 :
-
hydroperoxyl radical
- H2O2 :
-
hydrogen peroxide
- hPAP:
-
human placental alkaline phosphatase
- HRD1:
-
synoviolin
- IVR:
-
central intervening region
- JNK1/2:
-
c-jun N-terminal kinase 1/2
- KEAP1:
-
Kelch-like ECH (erythroid cell-derived protein with CNC homology)-associated protein 1
- MAPKs:
-
mitogen-activated protein kinases
- MDA:
-
malondialdehyde
- MEFs:
-
mouse embryonic fibroblasts
- Mgst1:
-
microsomal glutathione S-transferase 1
- miRNA:
-
microRNA
- MMF:
-
monomethylfumarate
- NAPDH:
-
nicotinamide adenine dinucleotide phosphate
- NEDD8:
-
neural precursor cell expressed, developmentally down-regulated 8
- Neh:
-
Nrf2-ECH homology
- NES:
-
Nuclear Export Signal
- NLS:
-
Nuclear Localization Signal
- NFE2L2:
-
Nuclear factor erythroid 2 like 2 gene
- NF-κB:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- NGF:
-
Nerve Growth Factor
- NO• :
-
nitric oxide radical
- •NO2 :
-
nitrogen dioxide radical
- NOTCH1:
-
neurogenic locus notch homolog protein 1
- NPC:
-
nuclear pore complex
- NQO1:
-
NADPH quinone dehydrogenase
- NRF2:
-
nuclear factor erythroid 2-related factor 2
- O2 :
-
molecular oxygen
- O2 •- :
-
superoxide anion
- •OH:
-
hydroxyl radical
- ONOO− :
-
peroxynitrite
- OS:
-
oxidative stress
- OXPHOS:
-
oxidative phosphorylation
- PALB2:
-
partner and localizer of BRCA2
- PGAM5:
-
phosphoglycerate mutase 5
- PHKA1:
-
phosphorylase b kinase subunit A1
- PI3K:
-
phosphoinositide 3-kinase
- PKC:
-
protein kinase C
- PNS:
-
peripheral nervous system
- ProTα:
-
prothymosin alpha
- RAC3:
-
receptor-associated coactivator 3
- RBM45:
-
RNA binding protein 45
- RBX1:
-
RING box protein 1
- RNS:
-
reactive nitrogen species
- •ROO:
-
peroxyl radical
- ROS:
-
reactive oxygen species
- RS• :
-
thiyl radicals
- RSOH:
-
sulfenic acid
- RS(O) SR:
-
thiosulfinate
- RS(O2) SR:
-
thiosulfonate
- RSS:
-
reactive sulfur species
- RSSR:
-
disulfides
- RXRα :
-
retinoid X receptor alpha
- SFN:
-
sulforaphane
- SIAH2:
-
seven in absentia homolog 2
- Sirt1:
-
Sirtuin 1
- sMaf:
-
small musculoaponeurotic fibrosarcoma
- SOD1:
-
superoxide dismutase 1
- SQSTM1:
-
sequestosome-1 gene
- TARDBP:
-
TAR DNA-binding protein 43 gene
- TCTP:
-
translationally controlled tumor protein
- TDP-43:
-
TAR DNA-binding protein 43
- β-TrCP:
-
β-transducin repeat-containing protein
- Ub:
-
ubiquitin
- UCH-L1:
-
ubiquitin carboxyl-terminal hydrolase isoenzyme L1
- UPS:
-
ubiquitin-proteasome system
- XRE:
-
xenobiotic response element
References
Chiurchiù V, Orlacchio A, Maccarrone M. Is modulation of oxidative stress an answer? The state of the art of redox therapeutic actions in neurodegenerative diseases. Oxid Med Cell Longev. 2016;2016:1–11.
Sbodio JI, Snyder SH, Paul BD. Redox mechanisms in neurodegeneration: from disease outcomes to therapeutic opportunities. Antioxid Redox Signal. 2019;30:1450–99.
Bjorklund G, Stejskal V, Urbina MA, Dadar M, Chirumbolo S, Mutter J. Metals and Parkinson’s disease: mechanisms and biochemical processes. Curr Med Chem. 2018;25:2198–214.
Calderón-Garcidueñas L. Smoking and cerebral oxidative stress and air pollution: a dreadful equation with particulate matter involved and one more powerful reason not to smoke anything! J Alzheimers Dis. 2016;54:109–12.
Couillard C, Pomerleau S, Ruel G, Archer WR, Bergeron J, Couture P, et al. Associations between hypertriglyceridemia, dietary fat intake, oxidative stress, and endothelial activation in men. Nutrition. 2006;22:600–8.
Grant SS, Hung DT. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence. 2013;4:273–83.
Kim J-J, Kim Y-S, Kumar V. Heavy metal toxicity: An update of chelating therapeutic strategies. J Trace Elem Med Biol. 2019;54:226–31.
Larsen EL, Cejvanovic V, Kjaer LK, Pedersen MT, Popik SD, Hansen LK, et al. Clarithromycin, trimethoprim, and penicillin and oxidative nucleic acid modifications in humans: randomised, controlled trials: Antibiotics and oxidative stress. Br J Clin Pharmacol. 2017;83:1643–53.
Marrakchi M, Liu X, Andreescu S. Oxidative stress and antibiotic resistance in bacterial pathogens: state of the art, methodologies, and future trends. Adv Exp Med Biol. 2014;806:483–98. https://doi.org/10.1007/978-3-319-06068-2_23.
Massicot F, Martin C, Dutertre-Catella H, Ellouk-Achard S, Pham-Huy C, Thevenin M, et al. Modulation of energy status and cytotoxicity induced by FK506 and cyclosporin A in a renal epithelial cell line. Arch Toxicol. 1997;71:529–31.
Talarico C, Dattilo V, D’Antona L, Barone A, Amodio N, Belviso S, et al. SI113, a SGK1 inhibitor, potentiates the effects of radiotherapy, modulates the response to oxidative stress and induces cytotoxic autophagy in human glioblastoma multiforme cells. Oncotarget. 2016;7:15868–84.
Baardman J, Verberk SGS, Prange KHM, van Weeghel M, van der Velden S, Ryan DG, et al. A Defective Pentose Phosphate Pathway Reduces Inflammatory Macrophage Responses during Hypercholesterolemia. Cell Rep. 2018;25:2044–2052.e5.
Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E. From discoveries in ageing research to therapeutics for healthy ageing. Nature. 2019;571(7764):183–92. https://doi.org/10.1038/s41586-019-1365-2. Epub 2019 Jul 10.
Ceriello A. Possible role of oxidative stress in the pathogenesis of hypertension. Diabetes Care. 2008;31:S181–4.
Dikalov S, Itani H, Richmond B, Vergeade A, Rahman SMJ, Boutaud O, Blackwell T, Massion PP, Harrison DG, Dikalova A. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am J Physiol Heart Circ Physiol. 2019;316(3):H639–H646. https://doi.org/10.1152/ajpheart.00595.2018. Epub 2019 Jan 4. Erratum in: Am J Physiol Heart Circ Physiol. 2019;316(4):H939.
Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95.
Kudryavtseva AV, Krasnov GS, Dmitriev AA, Alekseev BY, Kardymon OL, Sadritdinova AF, et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget. 2016;7:44879–905.
Liao CY, Kennedy BK. SIRT6, oxidative stress, and aging. Cell Res. 2016;26(2):143–4. https://doi.org/10.1038/cr.2016.8. Epub 2016 Jan 19.
Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166:555–66.
Commoner B, Townsend J, Pake GE. Free radicals in biological materials. Nature. 1954;174(4432):689–91. https://doi.org/10.1038/174689a0.
McCord JM. The evolution of free radicals and oxidative stress. Am J Med. 2000;108:652–9.
Pham-Huy LA, He H, Pham-Huy C. Free Radicals, Antioxidants in Disease and Health. FREE Radic Antioxid. 2008;4:8.
Singh A, Kukreti R, Saso L, Kukreti S. Oxidative stress: a key modulator in neurodegenerative diseases. Molecules. 2019;24:1583.
Bartz RR, Piantadosi CA. Clinical review: oxygen as a signaling molecule. Crit Care. 2010;14:234.
Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, et al. Oxidative stress in neurodegenerative diseases. Mol Neurobiol. 2016;53:4094–125.
Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc). 2005;70(2):200–14. https://doi.org/10.1007/s10541-005-0102-7.
Ježek P, Hlavatá L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol. 2005;37:2478–503.
Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–72.
Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–67.
Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol. 2009;554:165–81. https://doi.org/10.1007/978-1-59745-521-3_11.
Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44.
Mailloux RJ, McBride SL, Harper M-E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem Sci. 2013;38:592–602.
Venditti P, Di Stefano L, Di Meo S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion. 2013;13:71–82.
Vicente-Gutierrez C, Bonora N, Bobo-Jimenez V, Jimenez-Blasco D, Lopez-Fabuel I, Fernandez E, et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat Metab. 2019;1:201–11.
Giles G, Nasim M, Ali W, Jacob C. The reactive sulfur species concept: 15 years on. Antioxidants. 2017;6:38.
Bian K. Nitric oxide NO - biogeneration regulation and relevence to human diseases. Front Biosci. 2003;8:d264–78.
Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424.
Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: redox pathways in molecular medicine. Proc Natl Acad Sci. 2018;115:5839–48.
Persinger RL, Poynter ME, Ckless K, Janssen-Heininger YM. Molecular mechanisms of nitrogen dioxide induced epithelial injury in the lung. Mol Cell Biochem. Springer. 2002;234:71–80.
Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, et al. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–7.
Gaut JP, Byun J, Tran HD, Lauber WM, Carroll JA, Hotchkiss RS, et al. Myeloperoxidase produces nitrating oxidants in vivo. J Clin Invest. 2002;109:1311–9.
Mishanina TV, Libiad M, Banerjee R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat Chem Biol. 2015;11:457–64.
Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JCB, Kowaltowski AJ, et al. mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal. 2013;18:2029–74.
Lacza Z, Kozlov AV, Pankotai E, Csordás A, Wolf G, Redl H, et al. Mitochondria produce reactive nitrogen species via an arginine-independent pathway. Free Radic Res. 2006;40:369–78.
Libiad M, Yadav PK, Vitvitsky V, Martinov M, Banerjee R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J Biol Chem. 2014;289:30901–10.
Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol. 2016;17:308–21.
Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20:148–60.
Di Domenico F, Pupo G, Giraldo E, Badìa M-C, Monllor P, Lloret A, et al. Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic Biol Med. 2016;91:1–9.
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–67.
Kumar A, Ratan RR. Oxidative stress and huntington’s disease: the good, the bad, and the ugly. J Huntingt Dis. 2016;5:217–37.
Perluigi M, Poon HF, Maragos W, Pierce WM, Klein JB, Calabrese V, et al. Proteomic analysis of protein expression and oxidative modification in R6/2 transgenic mice: a model of Huntington Disease. Mol Cell Proteomics. 2005;4:1849–61.
Sorolla MA, Reverter-Branchat G, Tamarit J, Ferrer I, Ros J, Cabiscol E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med. 2008;45:667–78.
Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P, et al. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate α-synuclein fibrilization. Nat Chem Biol. 2006;2:249–53.
Chung SY, Kishinevsky S, Mazzulli JR, Graziotto J, Mrejeru A, Mosharov EV, et al. Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and α-Synuclein accumulation. Stem Cell Rep. 2016;7:664–77.
Puspita L, Chung SY, Shim J. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain. 2017;10:53.
Bacman SR, Bradley WG, Moraes CT. Mitochondrial involvement in amyotrophic lateral sclerosis. Mol Neurobiol. 2006;33:19.
Barber SC, Mead RJ, Shaw PJ. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta. 2006;1762(11-12):1051–67. https://doi.org/10.1016/j.bbadis.2006.03.008. Epub 2006 Apr 4.
Ilieva EV, Ayala V, Jove M, Dalfo E, Cacabelos D, Povedano M, et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111–23.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Olin-Sandoval V, Yu JSL, Miller-Fleming L, Alam MT, Kamrad S, Correia-Melo C, et al. Lysine harvesting is an antioxidant strategy and triggers underground polyamine metabolism. Nature. 2019;572:249–53.
Reddi AR, Culotta VC. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell. 2013;152:224–35.
Veskovic M, Mladenovic D, Jorgacevic B, Stevanovic I, de Luka S, Radosavljevic T. Alpha-lipoic acid affects the oxidative stress in various brain structures in mice with methionine and choline deficiency. Exp Biol Med. 2015;240:418–25.
Jayedi A, Rashidy-Pour A, Parohan M, Zargar MS, Shab-Bidar S. Dietary antioxidants, circulating antioxidant concentrations, total antioxidant capacity, and risk of all-cause mortality: a systematic review and dose-response meta-analysis of prospective observational studies. Adv Nutr. Oxford University Press. 2018;9:701–16.
Lobo V, Patil A, Phatak A, Chandra N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn Rev. 2010;4(8):118–26. https://doi.org/10.4103/0973-7847.70902.
Fang Y-Z, Yang S, Wu G. Free radicals, antioxidants, and nutrition. Nutrition. 2002;18:872–9.
Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov. 2019;18:295–317.
Johnson DA, Johnson JA. Nrf2—a therapeutic target for the treatment of neurodegenerative diseases. Free Radic Biol Med. 2015;88:253–67.
Paladino S, Conte A, Caggiano R, Pierantoni GM, Faraonio R. Nrf2 Pathway in Age-Related Neurological Disorders: Insights into MicroRNAs. Cell Physiol Biochem. 2018;47:1951–76.
Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344(22):1688–700. https://doi.org/10.1056/NEJM200105313442207.
Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2017;13:96–104.
Chia R, Chiò A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018;17:94–102.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. https://doi.org/10.1016/j.neuron.2011.09.011. Epub 2011 Sep 21.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59-62. https://doi.org/10.1038/362059a0. Erratum in: Nature. 1993;364(6435):362.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72.
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11.
Blokhuis AM, Groen EJN, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol (Berl). 2013;125:777–94.
Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol (Berl). 2012;124:339–52.
Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585–91. https://doi.org/10.1056/NEJM199403033300901.
Dash RP, Babu RJ, Srinivas NR. Two decades-long journey from Riluzole to Edaravone: revisiting the clinical pharmacokinetics of the only two amyotrophic lateral sclerosis therapeutics. Clin Pharmacokinet. 2018;57:1385–98.
Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347(9013):1425–31. https://doi.org/10.1016/s0140-6736(96)91680-3.
Mullard A. FDA approves first deuterated drug: Nature Publishing Group; 2017.
Petrov D, Mansfield C, Moussy A, Hermine O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front Aging Neurosci. 2017;9:68. https://doi.org/10.3389/fnagi.2017.00068.
Yoshino H. Edaravone for the treatment of amyotrophic lateral sclerosis. Expert Rev Neurother. 2019;19:185–93.
Bonafede R, Mariotti R. ALS Pathogenesis and Therapeutic Approaches: The Role of Mesenchymal Stem Cells and Extracellular Vesicles. Front Cell Neurosci. 2017;11:80. https://doi.org/10.3389/fncel.2017.00080.
Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206.
Wood LK, Langford SJ. Motor neuron disease: a chemical perspective. J Med Chem. 2014;57:6316–31.
Chi L, Ke Y, Luo C, Gozal D, Liu R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience. 2007;144:991–1003.
Bowling AC, Schulz JB, Brown RH Jr, Beal MF. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem. 1993;61(6):2322–5. https://doi.org/10.1111/j.1471-4159.1993.tb07478.x.
Shaw PJ, Ince PG, Falkous G, Mantle D. Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann Neurol. 1995;38:691–5.
Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–74.
Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:644–54.
Pedersen WA, Fu W, Keller JN, Markesbery WR, Appel S, Smith RG, et al. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann Neurol. 1998;44:819–24.
Shibata N, Nagai R, Uchida K, Horiuchi S, Yamada S, Hirano A, et al. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001;917:97–104.
Ferrante RJ, Shinobu LA, Schulz JB, Matthews RT, Thomas CE, Kowall NW, et al. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann Neurol. 1997;42:326–34.
Bogdanov M, Brown RH, Matson W, Smart R, Hayden D, O’Donnell H, et al. Increased oxidative damage to DNA in ALS patients. Free Radic Biol Med. 2000;29:652–8.
Ihara Y, Nobukuni K, Takata H, Hayabara T. Oxidative stress and metal content in blood and cerebrospinal fluid of amyotrophic lateral sclerosis patients with and without a Cu, Zn-superoxide dismutase mutation. Neurol Res. 2005;27:105–8.
Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology. 2004;62:1758–65.
Smith RG, Henry YK, Mattson MP, Appel SH. Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol. 1998;44(4):696–9. https://doi.org/10.1002/ana.410440419.
Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neurosci Lett. 1999;260:204–6.
Shaw IC, Fitzmaurice PS, Mitchell JD, Lynch PG. Studies on cellular free radical protection mechanisms in the anterior horn from patients with amyotrophic lateral sclerosis. Neurodegeneration. 1995;4(4):391–6. https://doi.org/10.1006/neur.1995.0047.
Fujita K, Yamauchi M, Shibayama K, Ando M, Honda M, Nagata Y. Decreased cytochrome c oxidase activity but unchanged superoxide dismutase and glutathione peroxidase activities in the spinal cords of patients with amyotrophic lateral sclerosis. J Neurosci Res. 1996;45:276–81.
Przedborski S, Donaldson D, Jakowec M, Kish SJ, Guttman M, Rosoklija G, et al. Brain superoxide dismutase, catalase, and glutathione peroxidase activities in amyotrophic lateral sclerosis. Ann Neurol. 1996;39:158–65.
Przedborski S, Donaldson DM, Murphy PL, Hirsch O, Lange D, Naini AB, et al. Blood superoxide dismutase, catalase and glutathione peroxidase activities in familial and sporadic amyotrophic lateral sclerosis. Neurodegener J Neurodegener Disord Neuroprotection Neuroregeneration. 1996;5:57–64.
Babu GN, Kumar A, Chandra R, Puri SK, Singh RL, Kalita J, Misra UK. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem Int. 2008;52(6):1284–9. https://doi.org/10.1016/j.neuint.2008.01.009. Epub 2008 Jan 20.
Cova E, Bongioanni P, Cereda C, Metelli MR, Salvaneschi L, Bernuzzi S, et al. Time course of oxidant markers and antioxidant defenses in subgroups of amyotrophic lateral sclerosis patients. Neurochem Int. 2010;56:687–93.
Weiduschat N, Mao X, Hupf J, Armstrong N, Kang G, Lange DJ, et al. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci Lett. 2014;570:102–7.
Ikawa M, Okazawa H, Tsujikawa T, Matsunaga A, Yamamura O, Mori T, et al. Increased oxidative stress is related to disease severity in the ALS motor cortex: A PET study. Neurology. 2015;84:2033–9.
Wang Z, Bai Z, Qin X, Cheng Y. Aberrations in oxidative stress markers in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Oxid Med Cell Longev. 2019;2019:1–9.
Andrus PK, Fleck TJ, Gurney ME, Hall ED. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 2002;71:2041–8.
Casoni F, Basso M, Massignan T, Gianazza E, Cheroni C, Salmona M, et al. Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: POSSIBLE MULTIFUNCTIONAL ROLE IN THE PATHOGENESIS. J Biol Chem. 2005;280:16295–304.
Liu D, Wen J, Liu J, Li L. The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 1999;13:2318–28.
Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol. 2002;52:448–57.
Poon HF, Hensley K, Thongboonkerd V, Merchant ML, Lynn BC, Pierce WM, et al. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice—a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;39:453–62.
Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–9.
Deshmukh P, Unni S, Krishnappa G, Padmanabhan B. The Keap1–Nrf2 pathway: promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys Rev. 2017;9:41–56.
Canning P, Cooper CDO, Krojer T, Murray JW, Pike ACW, Chaikuad A, et al. Structural basis for Cul3 protein assembly with the BTB-Kelch Family of E3 ubiquitin ligases. J Biol Chem. 2013;288:7803–14.
Dhanoa BS, Cogliati T, Satish AG, Bruford EA, Friedman JS. Update on the Kelch-like (KLHL) gene family. Hum Genomics. 2013;7:13.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86.
Zhang DD, Lo S-C, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–53.
Kobayashi A, Kang M-I, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-Based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9.
Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015;88:101–7.
Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–91.
Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, et al. Structure of the BTB Domain of Keap1 and Its Interaction with the Triterpenoid Antagonist CDDO. Xu E, editor. PLoS ONE. 2014;9:e98896.
Lo S-C, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–17.
Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–52.
Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C, et al. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci. 2010;107:2842–7.
Fukutomi T, Takagi K, Mizushima T, Ohuchi N, Yamamoto M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex Degron and Keap1. Mol Cell Biol. 2014;34:832–46.
Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, et al. Different electrostatic potentials define ETGE and DLG Motifs as hinge and latch in oxidative stress response. Mol Cell Biol. 2007;27:7511–21.
Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol Cell Biol. 2006;26:2887–900.
Chen W, Jiang T, Wang H, Tao S, Lau A, Fang D, et al. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid Redox Signal. 2012;17:1670–5.
Fan Z, Wirth A-K, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6:e371.
Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624.
Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79.
Reddy NM, Kleeberger SR, Bream JH, Fallon PG, Kensler TW, Yamamoto M, et al. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene. 2008;27:5821–32.
Thimmulappa RK. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–95.
Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26.
Lee J-M, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–38.
Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu R-M, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci. 2004;101:3381–6.
Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci. 1996;93:13943–8.
Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci. National Acad Sciences; 1994;91:9926–9930.
Dinkova-Kostova AT, Kostov RV, Kazantsev AG. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018;285:3576–90.
Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29:1727–45.
Katoh Y, Iida K, Kang M-I, Kobayashi A, Mizukami M, Tong KI, et al. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys. 2005;433:342–50.
McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “Tethering” mechanism: a TWO-SITE INTERACTION MODEL FOR THE Nrf2-Keap1 COMPLEXA TWO-SITE INTERACTION MODEL FOR THE Nrf2-Keap1 COMPLEX. J Biol Chem. 2006;281:24756–68. J Biol Chem. 2006;281:24756–68.
McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 Degron and the Redox-insensitive Neh6 Degron. J Biol Chem. 2004;279:31556–67.
Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription: Synergistic activation of Nrf2 by CBP. Genes Cells. 2001;6:857–68.
Zhu M, Fahl WE. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem Biophys Res Commun. 2001;289:212–9.
Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, et al. RXR inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 Domain of NRF2. Cancer Res. 2013;73:3097–108.
Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32:3765–81.
Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/ -TrCP axis. Mol Cell Biol. 2012;32:3486–99.
Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/ -TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–33.
Motohashi H, Katsuoka F, Engel JD, Yamamoto M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci. 2004;101:6379–84.
Nioi P, Nguyen T, Sherratt PJ, Pickett CB. The carboxy-terminal Neh3 Domain of Nrf2 Is required for transcriptional activation. Mol Cell Biol. 2005;25:10895–906.
Tao S, Liu P, Luo G, Rojo dela Vega M, Chen H, Wu T, et al. p97 Negatively Regulates NRF2 by Extracting Ubiquitylated NRF2 from the KEAP1-CUL3 E3 Complex. Mol Cell Biol. 2017;37:e00660-16.
Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-Based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–86.
Silva-Islas CA, Maldonado PD. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol Res. 2018;134:92–9.
Baird L, Lleres D, Swift S, Dinkova-Kostova AT. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc Natl Acad Sci. 2013;110:15259–64.
Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–51.
Harder B, Jiang T, Wu T, Tao S, de la Vega MR, Tian W, et al. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem Soc Trans. 2015;43:680–6.
Theodore M, Kawai Y, Yang J, Kleshchenko Y, Reddy SP, Villalta F, et al. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008;283:8984–94.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. Wiley Online Library. 2003;8:379–91.
Hirotsu Y, Katsuoka F, Funayama R, Nagashima T, Nishida Y, Nakayama K, et al. Nrf2–MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012;40:10228–39.
Gan L, Johnson JA. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim Biophys Acta BBA - Mol Basis Dis. 1842;2014:1208–18.
Pajares M, Jiménez-Moreno N, García-Yagüe ÁJ, Escoll M, de Ceballos ML, Van Leuven F, et al. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy. 2016;12:1902–16.
Uruno A, Yagishita Y, Katsuoka F, Kitajima Y, Nunomiya A, Nagatomi R, et al. Nrf2-mediated regulation of skeletal muscle glycogen metabolism. Mol Cell Biol. 2016;36:1655–72.
Li W, Yu S-W, Kong A-NT. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem. 2006;281:27251–63.
Sun Z, Wu T, Zhao F, Lau A, Birch CM, Zhang DD. KPNA6 (Importin 7)-mediated nuclear import of Keap1 represses the Nrf2-dependent antioxidant response. Mol Cell Biol. 2011;31:1800–11.
Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–49.
Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108–46.
Petri S, Körner S, Kiaei M. Nrf2/ARE signaling pathway: key mediator in oxidative stress and potential therapeutic target in ALS. Neurol Res Int. 2012;2012:1–7.
Kang HJ, Hong YB, Kim HJ, Rodriguez OC, Nath RG, Tilli EM, et al. Detoxification: a novel function of BRCA1 in tumor suppression? Toxicol Sci. 2011;122:26–37.
Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of nf-e2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: DIRECT CROSS-TALK BETWEEN PHASE I AND II DRUG-METABOLIZING ENZYMES. J Biol Chem. 2005;280:20340–8.
Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM, MacEwan DJ. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood. 2012;120:5188–98.
Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-induced expression of NAD(P)H:Quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol. 2008;181:6730–7.
Wang Q, Li J, Yang X, Sun H, Gao S, Zhu H, et al. Nrf2 is associated with the regulation of basal transcription activity of the BRCA1 gene. Acta Biochim Biophys Sin. 2013;45:179–87.
Koundouros N, Poulogiannis G. Phosphoinositide 3-Kinase/Akt signaling and redox metabolism in cancer. Front Oncol. 2018;8:160.
Wakabayashi N, Skoko JJ, Chartoumpekis DV, Kimura S, Slocum SL, Noda K, et al. Notch-Nrf2 Axis: regulation of Nrf2 gene expression and cytoprotection by Notch signaling. Mol Cell Biol. 2014;34:653–63.
Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011;286:40725–33.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. https://doi.org/10.1016/s0092-8674(04)00045-5.
Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2008;19:92–105.
Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–34.
Kabaria S, Choi DC, Chaudhuri AD, Jain MR, Li H, Junn E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic Biol Med. 2015;89:548–56.
Goldstein LD, Lee J, Gnad F, Klijn C, Schaub A, Reeder J, et al. Recurrent loss of NFE2L2 Exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Rep. 2016;16:2605–17.
Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol. 2013;85:705–17.
Chen W, Sun Z, Wang X-J, Jiang T, Huang Z, Fang D, et al. Direct interaction between Nrf2 and p21Cip1/WAF1 upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34:663–73.
Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med. 2013;210:1529–44.
Lo S-C, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res. 2008;314:1789–803.
Lo S-C, Hannink M. PGAM5, a Bcl-XL-interacting Protein, Is a Novel Substrate for the Redox-regulated Keap1-dependent Ubiquitin Ligase Complex. J Biol Chem. American Society for Biochemistry and Molecular Biology. 2006;281:37893–903.
Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol. 2012;32:1506–17.
Hast BE, Goldfarb D, Mulvaney KM, Hast MA, Siesser PF, Yan F, et al. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res. 2013;73:2199–210.
Prajapati SC, Chauhan SS. Dipeptidyl peptidase III: a multifaceted oligopeptide N-end cutter: Dipeptidyl peptidase III. FEBS J. 2011;278:3256–76.
Karapetian RN, Evstafieva AG, Abaeva IS, Chichkova NV, Filonov GS, Rubtsov YP, et al. Nuclear oncoprotein prothymosin ␣ is a partner of Keap1: implications for expression of oxidative stress-protecting genes. MOL CELL BIOL. 2005;25:11.
Hiemstra S, Niemeijer M, Koedoot E, Wink S, Klip JE, Vlasveld M, et al. Comprehensive landscape of Nrf2 and p53 pathway activation dynamics by oxidative stress and DNA damage. Chem Res Toxicol. 2017;30:923–33.
Padmanabhan B, Nakamura Y, Yokoyama S. Structural analysis of the complex of Keap1 with a prothymosin α peptide. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2008;64:233–8.
McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci. 2010;107:18838–43.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23.
Katsuragi Y, Ichimura Y, Komatsu M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr Opin Toxicol. 2016;1:54–61.
Ichimura Y, Waguri S, Sou Y, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–31.
Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. ASBMB. 2010;285:22576–91.
Blank V, Andrews NC. The Maf transcription factors: regulators of differentiation. Trends Biochem Sci. 1997;22(11):437–41. https://doi.org/10.1016/s0968-0004(97)01105-5.
Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294:1–12.
Li W, Yu S, Liu T, Kim J-H, Blank V, Li H, et al. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim Biophys Acta BBA - Mol Cell Res. 1783;2008:1847–56.
Huang H-C, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–74.
Steinberg SF. Mechanisms for redox-regulation of protein kinase C. Front Pharmacol. 2015;6:128. https://doi.org/10.3389/fphar.2015.00128.
Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15.
Pi J, Bai Y, Reece JM, Williams J, Liu D, Freeman ML, et al. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radic Biol Med. 2007;42:1797–806.
Apopa PL, He X, Ma Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J Biochem Mol Toxicol. 2008;22:63–76.
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–209.
Yuan X, Xu C, Pan Z, Keum Y-S, Kim J-H, Shen G, et al. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells: ACTIVATION OF Nrf2-ARE SIGNALING BY BHA AND MAPK. Mol Carcinog. 2006;45:841–50.
Sun Z, Huang Z, Zhang DD. Phosphorylation of Nrf2 at Multiple Sites by MAP Kinases Has a Limited Contribution in Modulating the Nrf2-Dependent Antioxidant Response. Morty RE, editor. PLoS ONE. 2009;4:e6588.
Lage R, Diéguez C, Vidal-Puig A, López M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14:539–49.
Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol. 2016;36:1931–42.
Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3β Inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–51.
Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP AUGMENTS PROMOTER-SPECIFIC DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009;29:2658–72.
Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114(Pt13):2363–73.
Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. https://doi.org/10.1146/annurev.biochem.70.1.81.
Ganner A, Pfeiffer Z-C, Wingendorf L, Kreis S, Klein M, Walz G, et al. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem Biophys Res Commun. 2020;524:895–902.
Marzec JM, Christie JD, Reddy SP, Jedlicka AE, Vuong H, Lanken PN, et al. Functional polymorphisms in the transcription factor NRF2NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007;21:2237–46. in humans increase the risk of acute lung injury. FASEB J. 2007;21:2237–46.
Suzuki T, Shibata T, Takaya K, Shiraishi K, Kohno T, Kunitoh H, et al. Regulatory nexus of synthesis and degradation deciphers cellular Nrf2 expression levels. Mol Cell Biol. 2013;33:2402–12.
Yu S, Khor TO, Cheung K-L, Li W, Wu T-Y, Huang Y, et al. Nrf2 Expression Is Regulated by Epigenetic Mechanisms in Prostate Cancer of TRAMP Mice. Lau ATY, editor. PLoS ONE. 2010;5:e8579.
Sangokoya C, Telen MJ, Chi J-T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116:4338–48.
Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res Treat. 2011;129:983–91.
Singh B, Ronghe AM, Chatterjee A, Bhat NK, Bhat HK. MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis. 2013;34:1165–72.
Narasimhan M, Patel D, Vedpathak D, Rathinam M, Henderson G, Mahimainathan L. Identification of novel microRNAs in Post-Transcriptional Control of Nrf2 Expression and Redox Homeostasis in Neuronal, SH-SY5Y Cells. Tsuji Y, editor. PLoS ONE. 2012;7:e51111.
Wang N, Zhang L, Lu Y, Zhang M, Zhang Z, Wang K, et al. Down-regulation of microRNA-142-5p attenuates oxygen-glucose deprivation and reoxygenation-induced neuron injury through up-regulating Nrf2/ARE signaling pathway. Biomed Pharmacother. 2017;89:1187–95.
Li N, Muthusamy S, Liang R, Sarojini H, Wang E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech Ageing Dev. 2011;132:75–85.
Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–49.
Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–22.
Kang HJ, Hong YB, Kim HJ, Bae I. CR6-interacting factor 1 (CRIF1) regulates NF-E2-related factor 2 (NRF2) protein stability by proteasome-mediated degradation. J Biol Chem. 2010;285:21258–68.
Baba K, Morimoto H, Imaoka S. Seven in Absentia Homolog 2 (Siah2) protein is a regulator of NF-E2-related factor 2 (Nrf2). J Biol Chem. 2013;288:18393–405.
Li W, Liu H, Zhou J-S, Cao J-F, Zhou X-B, Choi AMK, et al. Caveolin-1 inhibits expression of antioxidant enzymes through direct interaction with nuclear erythroid 2 p45-related factor-2 (Nrf2). J Biol Chem. 2012;287:20922–30.
Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H: quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–900.
Hoshino H, Kobayashi A, Yoshida M, Kudo N, Oyake T, Motohashi H, et al. Oxidative stress abolishes leptomycin B-sensitive nuclear export of transcription repressor Bach2 that counteracts activation of Maf recognition element. J Biol Chem. 2000;275:15370–6.
Kawai Y, Garduño L, Theodore M, Yang J, Arinze IJ. Acetylation-deacetylation of the transcription factor Nrf2 (Nuclear Factor Erythroid 2-related Factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J Biol Chem. 2011;286:7629–40.
Keum Y-S, Yu S, Chang PP-J, Yuan X, Kim J-H, Xu C, et al. Mechanism of action of sulforaphane: inhibition of p38 Mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element–mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 2006;66:8804–13.
Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2—an update. Free Radic Biol Med. 2014;66:36–44.
Jain AK, Jaiswal AK. GSK-3β acts upstream of fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–10.
Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–38.
McBean G. Cysteine, glutathione, and thiol redox balance in astrocytes. Antioxidants. 2017;6:62.
Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell. 2014;158:15–24.
Nave K-A, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol. 2014;30:503–33.
Baxter PS, Hardingham GE. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic Biol Med. 2016;100:147–52.
Mead RJ, Higginbottom A, Allen SP, Kirby J, Bennett E, Barber SC, et al. S[+] Apomorphine is a CNS penetrating activator of the Nrf2-ARE pathway with activity in mouse and patient fibroblast models of amyotrophic lateral sclerosis. Free Radic Biol Med. 2013;61:438–52.
Sarlette A, Krampfl K, Grothe C, Dengler R, Petri S. Nuclear erythroid 2YRelated Factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2008;67:8.
Tanji K, Maruyama A, Odagiri S, Mori F, Itoh K, Kakita A, et al. Keap1 is localized in neuronal and glial cytoplasmic inclusions in various neurodegenerative diseases. J Neuropathol Exp Neurol. 2013;72:18–28.
Goode A, Rea S, Sultana M, Shaw B, Searle MS, Layfield R. ALS-FTLD associated mutations of SQSTM1 impact on Keap1-Nrf2 signalling. Mol Cell Neurosci. 2016;76:52–8.
Bakkar N, Kousari A, Kovalik T, Li Y, Bowser R. RBM45 modulates the antioxidant response in amyotrophic lateral sclerosis through interactions with KEAP1. Mol Cell Biol. 2015;35:2385–99.
Copple IM, Lister A, Obeng AD, Kitteringham NR, Jenkins RE, Layfield R, et al. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem. 2010;285:16782–8.
Lau A, Wang X-J, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–85.
Deng Z, Lim J, Wang Q, Purtell K, Wu S, Palomo GM, et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2020;16:917–31.
Foster A, Rea S. The role of sequestosome 1/p62 protein in amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis. Neural Regen Res. 2020;15:2186.
Wang F, Lu Y, Qi F, Su Q, Wang L, You C, et al. Effect of the human SOD1-G93A gene on the Nrf2/ARE signaling pathway in NSC-34 cells. Mol Med Rep. 2014;9:2453–8.
Wood-Allum CA. Impairment of mitochondrial anti-oxidant defence in SOD1-related motor neuron injury and amelioration by ebselen. Brain. 2006;129:1693–709.
Kirby J, Halligan E, Baptista MJ, Allen S, Heath PR, Holden H, et al. Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain. 2005;128:1686–706.
Duan W, Guo Y, Jiang H, Yu X, Li C. MG132 enhances neurite outgrowth in neurons overexpressing mutant TAR DNA-binding protein-43 via increase of HO-1. Brain Res. 2011;1397:1–9.
Duan W, Li X, Shi J, Guo Y, Li Z, Li C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience. 2010;169:1621–9.
Pehar M, Vargas MR, Robinson KM, Cassina P, Diaz-Amarilla PJ, Hagen TM, et al. Mitochondrial superoxide production and nuclear factor erythroid 2-Related Factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J Neurosci. 2007;27:7777–85.
Joilin G, Leigh PN, Newbury SF, Hafezparast M. An overview of MicroRNAs as biomarkers of ALS. Front Neurol. 2019;10:186.
Kovanda A, Leonardis L, Zidar J, Koritnik B, Dolenc-Groselj L, Ristic Kovacic S, et al. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci Rep. 2018;8:5609.
Ricci C, Marzocchi C, Battistini S. MicroRNAs as biomarkers in amyotrophic lateral sclerosis. Cells. 2018;7:219.
Rizzuti M, Filosa G, Melzi V, Calandriello L, Dioni L, Bollati V, et al. MicroRNA expression analysis identifies a subset of downregulated miRNAs in ALS motor neuron progenitors. Sci Rep. 2018;8:10105.
Waller R, Wyles M, Heath PR, Kazoka M, Wollff H, Shaw PJ, et al. Small RNA sequencing of sporadic amyotrophic lateral sclerosis cerebrospinal fluid reveals differentially expressed miRNAs related to neural and glial activity. Front Neurosci. 2018;11:731.
Emde A, Eitan C, Liou L, Libby RT, Rivkin N, Magen I, et al. Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: a new mechanism for ALS. EMBO J. 2015;34:2633–51.
Kraft A, Resch J, Johnson D, Johnson J. Activation of the Nrf2–ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp Neurol. 2007;207:107–17.
Mimoto T, Miyazaki K, Morimoto N, Kurata T, Satoh K, Ikeda Y, et al. Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice. Brain Res. 2012;1446:109–18.
Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574–81.
Vargas MR, Pehar M, Cassina P, Martínez-Palma L, Thompson JA, Beckman JS, et al. Fibroblast Growth Factor-1 Induces Heme Oxygenase-1 via Nuclear Factor Erythroid 2-related Factor 2 (Nrf2) in Spinal Cord Astrocytes: CONSEQUENCES FOR MOTOR NEURON SURVIVAL. J Biol Chem. 2005;280:25571–9.
Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol Med. 2011;51:88–96.
Pollari E, Goldsteins G, Bart G, Koistinaho J, Giniatullin R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:131. https://doi.org/10.3389/fncel.2014.00131.
Guo Y, Zhang Y, Wen D, Duan W, An T, Shi P, et al. The modest impact of transcription factor Nrf2 on the course of disease in an ALS animal model. Lab Invest. 2013;93:825–33.
Vargas MR, Burton NC, Gan L, Johnson DA, Schäfer M, Werner S, et al. Absence of Nrf2 or Its Selective Overexpression in Neurons and Muscle Does Not Affect Survival in ALS-Linked Mutant hSOD1 Mouse Models. Borchelt DR, editor. PLoS ONE. 2013;8:e56625.
Vomund S, Schäfer A, Parnham M, Brüne B, von Knethen A. Nrf2, the master regulator of anti-oxidative responses. Int J Mol Sci. 2017;18:2772.
Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15:162–73.
Satoh T, McKercher SR, Lipton SA. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med. 2013;65:645–57.
Sporn MB, Liby KT, Yore MM, Fu L, Lopchuk JM, Gribble GW. New synthetic triterpenoids: potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J Nat Prod. 2011;74:537–45.
Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci. 2005;102:4584–9.
Rabbani PS, Ellison T, Waqas B, Sultan D, Abdou S, David JA, et al. Targeted Nrf2 activation therapy with RTA 408 enhances regenerative capacity of diabetic wounds. Diabetes Res Clin Pract. 2018;139:11–23.
Lynch DR, Farmer J, Hauser L, Blair IA, Wang QQ, Mesaros C, et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann Clin Transl Neurol. 2019;6:15–26.
Paupe V, Dassa EP, Goncalves S, Auchère F, Lönn M, Holmgren A, et al. Impaired Nuclear Nrf2 Translocation Undermines the Oxidative Stress Response in Friedreich Ataxia. Andreu AL, editor. PLoS ONE. 2009;4:e4253.
Linker RA, Lee D-H, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–92.
Mrowietz C. Altmeyer, THE PARTICIPANTS IN THE GERMAN MULTICENTRE STUDY. Treatment of psoriasis with fumaric acid esters: results of a prospective multicentre study. Br J Dermatol. 1998;138:456–60.
Fernández Ó, Giovannoni G, Fox RJ, Gold R, Phillips JT, Potts J, et al. Efficacy and safety of delayed-release dimethyl fumarate for relapsing-remitting multiple sclerosis in prior interferon users: an integrated analysis of DEFINE and CONFIRM. Clin Ther. 2017;39:1671–9.
Fox RJ, Gold R, Phillips JT, Okwuokenye M, Zhang A, Marantz JL. Efficacy and tolerability of delayed-release dimethyl fumarate in Black, Hispanic, and Asian patients with relapsing-remitting multiple sclerosis: Post Hoc integrated analysis of DEFINE and CONFIRM. Neurol Ther. 2017;6:175–87.
Reilly JF, Yañez G, Bista P, Picarella D, Lee D, Vu C, et al. CAT-4001 improves mitochondrial function in a Friedreich’s ataxia model. Italy: Proc 2nd Int Ataxia Res Conf Pisa; 2017. p. 27–30.
Voloboueva LA, Killilea DW, Atamna H, Ames BN. N-tert-butyl hydroxylamine, a mitochondrial antioxidant, protects human retinal pigment epithelial cells from iron overload: relevance to macular degeneration. FASEB J. Wiley Online Library. 2007;21:4077–86.
Freeman BA, O’Donnell VB, Schopfer FJ. The discovery of nitro-fatty acids as products of metabolic and inflammatory reactions and mediators of adaptive cell signaling. Nitric Oxide. 2018;77:106–11.
Kansanen E, Bonacci G, Schopfer FJ, Kuosmanen SM, Tong KI, Leinonen H, et al. Electrophilic nitro-fatty acids activate NRF2 by a KEAP1 cysteine 151-independent mechanism. J Biol Chem. 2011;286:14019–27.
Rodriguez-Duarte J, Dapueto R, Galliussi G, Turell L, Kamaid A, Khoo NKH, et al. Electrophilic nitroalkene-tocopherol derivatives: synthesis, physicochemical characterization and evaluation of anti-inflammatory signaling responses. Sci Rep. 2018;8:12784.
Cores Á, Piquero M, Villacampa M, León R, Menéndez JC. NRF2 regulation processes as a source of potential drug targets against neurodegenerative diseases. Biomolecules. 2020;10:904.
Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci. 1992;89:2399–403.
Singh K, Connors SL, Macklin EA, Smith KD, Fahey JW, Talalay P, et al. Sulforaphane treatment of autism spectrum disorder (ASD). Proc Natl Acad Sci. 2014;111:15550–5.
Robledinos-Antón N, Fernández-Ginés R, Manda G, Cuadrado A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid Med Cell Longev. 2019;2019:9372182. https://doi.org/10.1155/2019/9372182.
Egea J, Buendia I, Parada E, Navarro E, Rada P, Cuadrado A, et al. Melatonin-sulforaphane hybrid ITH12674 induces neuroprotection in oxidative stress conditions by a ‘drug-prodrug’ mechanism of action: A designed dual-acting melatonin-sulforaphane hybrid. Br J Pharmacol. 2015;172:1807–21.
Howell SJ, Campone M, Cortés J, Duhoux FP, Ross S, Morris T, et al. Final results of the STEM trial: SFX-01 in the treatment and evaluation of ER+ Her2-metastatic breast cancer (mBC). Ann Oncol. Elsevier. 2019;30:v122.
Zolnourian AH, Franklin S, Galea I, Bulters DO. Study protocol for SFX-01 after subarachnoid haemorrhage (SAS): a multicentre randomised double-blinded, placebo controlled trial. BMJ Open. 2020;10:e028514.
Hirotsu Y, Katsuoka F, Itoh K, Yamamoto M. Nrf2 degron-fused reporter system: a new tool for specific evaluation of Nrf2 inducers: Nrf2 degron-fused reporter system. Genes Cells. 2011;16:406–15.
Higashi C, Kawaji A, Tsuda N, Hayashi M, Saito R, Yagishita Y, et al. The novel Nrf2 inducer TFM-735 ameliorates experimental autoimmune encephalomyelitis in mice. Eur J Pharmacol. 2017;802:76–84.
Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, et al. Monoacidic inhibitors of the Kelch-like ECH-Associated Protein 1: nuclear factor Erythroid 2-related factor 2 (KEAP1:NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery. J Med Chem. 2016;59:3991–4006.
Pallesen JS, Tran KT, Bach A. Non-covalent small-molecule Kelch-like ECH-associated protein 1–nuclear factor Erythroid 2-related Factor 2 (Keap1–Nrf2) inhibitors and their potential for targeting central nervous system diseases. J Med Chem. 2018;61:8088–103.
Schmoll D, Engel CK, Glombik H. The Keap1–Nrf2 protein–protein interaction: a suitable target for small molecules. Drug Discov Today Technol. 2017;24:11–7.
Zhao J, Redell JB, Moore AN, Dash PK. A novel strategy to activate cytoprotective genes in the injured brain. Biochem Biophys Res Commun. 2011;407:501–6.
Tu J, Zhang X, Zhu Y, Dai Y, Li N, Yang F, et al. Cell-permeable peptide targeting the Nrf2-Keap1 interaction: a potential novel therapy for global cerebral ischemia. J Neurosci Off J Soc Neurosci. 2015;35:14727–39.
Attucks OC, Jasmer KJ, Hannink M, Kassis J, Zhong Z, Gupta S, et al. Induction of Heme Oxygenase I (HMOX1) by HPP-4382: A Novel Modulator of Bach1 Activity. Yang C-M, editor. PLoS ONE. 2014;9:e101044.
Kim S-K, Kassis J, Attucks O, Zhong Z, Freeman J, Gupta S, et al. The novel Bach1 inhibitor HPP971 uniquely activates Nrf2 and reduces disease severity in a mouse model of experimental autoimmune encephalomyelitis. ACTRIMS-ECTRIMS Meet. 2014.
Luo J. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009;273:194–200.
Lal H, Ahmad F, Woodgett J, Force T. The GSK-3 family as therapeutic target for myocardial diseases. Circ Res. 2015;116:138–49.
Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease: GSK3 and Alzheimer’s disease. J Neurochem. 2008;104:1433–9.
Mizunoe Y, Kobayashi M, Sudo Y, Watanabe S, Yasukawa H, Natori D, et al. Trehalose protects against oxidative stress by regulating the Keap1–Nrf2 and autophagy pathways. Redox Biol. 2018;15:115–24.
Calap-Quintana P, Soriano S, Llorens JV, Al-Ramahi I, Botas J, Moltó MD, et al. TORC1 Inhibition by Rapamycin Promotes Antioxidant Defences in a Drosophila Model of Friedreich’s Ataxia. Palau F, editor. PLOS ONE. 2015;10:e0132376.
Lai Z-W, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. The Lancet. 2018;391:1186–96.
Piemonti L, Maffi P, Monti L, Lampasona V, Perseghin G, Magistretti P, et al. Beta cell function during rapamycin monotherapy in long-term type 1 diabetes. Diabetologia. 2011;54:433–9.
Mandrioli J, D’Amico R, Zucchi E, Gessani A, Fini N, Fasano A, et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine (Baltimore). 2018;97:e11119.
Vucic S, Ryder J, Mekhael L, Rd H, Mathers S, Needham M, et al. Phase 2 randomized placebo controlled double blind study to assess the efficacy and safety of tecfidera in patients with amyotrophic lateral sclerosis (TEALS Study): Study protocol clinical trial (SPIRIT Compliant). Medicine (Baltimore). 2020;99:e18904.
Sweeney MD, Sagare AP, Zlokovic BV. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–50.
Vilhardt F, Haslund-Vinding J, Jaquet V, McBean G. Microglia antioxidant systems and redox signalling. Br J Pharmacol. 2017;174:1719–32.
Zhou T, Ahmad TK, Gozda K, Truong J, Kong J, Namaka M. Implications of white matter damage in amyotrophic lateral sclerosis (Review). Mol Med Rep. 2017;16:4379–92.
Martín-Montalvo A, Villalba JM, Navas P, de Cabo R. NRF2, cancer and calorie restriction. Oncogene. 2011;30:505–20.
Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–71.
Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E, Zhang DD. Modulating NRF2 in disease: timing is everything. Annu Rev Pharmacol Toxicol. 2019;59:555–75.
Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med. 2016;22:578–93.
Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420.
Huppke P, Weissbach S, Church JA, Schnur R, Krusen M, Dreha-Kulaczewski S, et al. Activating de novo mutations in NFE2L2 encoding NRF2 cause a multisystem disorder. Nat Commun. 2017;8:818.
Wu S, Lu H, Bai Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019;8:2252–67.
Logroscino G, Tortelli R, Rizzo G, Marin B, Preux PM, Malaspina A. Amyotrophic Lateral Sclerosis: An Aging-Related Disease. Curr Geriatr Rep. 2015;4:142–53.
Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47.
Pandya VA, Patani R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: a cellular perspective. Brain. 2020;143:1057–72.
Zhang H, Davies KJA, Forman HJ. Oxidative stress response and Nrf2 signaling in aging. Free Radic Biol Med. 2015;88:314–36.
Voss P, Siems W. Clinical oxidation parameters of aging. Free Radic Res. 2006;40:1339–49.
Acknowledgements
We are grateful to Mrs. Michelle van der Schoot for language editing and reviewing.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SB led the writing of the manuscript, devised all the figures and table and edited the manuscript. MF contributed to the editing of the manuscript. MC supervised the writing and co-edited the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bono, S., Feligioni, M. & Corbo, M. Impaired antioxidant KEAP1-NRF2 system in amyotrophic lateral sclerosis: NRF2 activation as a potential therapeutic strategy. Mol Neurodegeneration 16, 71 (2021). https://doi.org/10.1186/s13024-021-00479-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-021-00479-8