
Abstract
Neuropsychiatric symptoms (NPSs) are common in patients with Alzheimer’s disease (AD) and are associated with accelerated cognitive impairment and earlier deaths. This review aims to explore the neural pathogenesis of NPSs in AD and its association with the progression of AD. We first provide a literature overview on the onset times of NPSs. Different NPSs occur in different disease stages of AD, but most symptoms appear in the preclinical AD or mild cognitive impairment stage and develop progressively. Next, we describe symptom-general and -specific patterns of brain lesions. Generally, the anterior cingulate cortex is a commonly damaged region across all symptoms, and the prefrontal cortex, especially the orbitofrontal cortex, is also a critical region associated with most NPSs. In contrast, the anterior cingulate-subcortical circuit is specifically related to apathy in AD, the frontal-limbic circuit is related to depression, and the amygdala circuit is related to anxiety. Finally, we elucidate the associations between the NPSs and AD by combining the onset time with the neural basis of NPSs.
Similar content being viewed by others
Background
As the worldwide population ages, over 50 million people are now living with dementia, and this number is set to increase to 152 million by 2050 [1]. Dementia has reached epidemic proportions, with major social, medical, and economic burdens [2]. The leading cause of dementia is Alzheimer’s disease (AD), whose main clinical manifestation is cognitive impairment, but 80% of AD patients also show various behavioural and psychological symptoms, collectively known as neuropsychiatric symptoms (NPSs) [3]. These symptoms are associated with more rapid progression to severe dementia and an earlier death [4]. They also adversely reduce the quality of life of patients and caregivers [5].
Cross-sectional and longitudinal studies have indicated that different NPSs occur mostly at different stages of AD [6,7,8]. For example, hallucinations seem to be more common in patients with severe AD, while irritability tends to occur in the early stages of the disease [7]. Even if some NPSs seem to appear together and share some of the same pathological features (for example, both depression and apathy are expressed as loss of interest and motivation), they have different pathological mechanisms. A clear understanding of the pathological mechanisms of differential NPSs is crucial for the early detection and treatment of the disease and the NPSs.
Many empirical studies have been conducted to understand the neural pathogenesis of NPSs in AD and its role in the progression of AD primarily using neuroimaging techniques. Yet, there has only been a small, though growing, number of reviews on this body of literature. Moreover, most of these reviews were merely of qualitative with regard to the brain regions associated with NPSs [9,10,11,12]. In our current efforts, we attempted to provide a more comprehensive review focusing on the quantitative aspects of relevant reports available in the literature. In doing so, we will manage to summarize the number of significant associations between NPSs and brain regions to describe quantitatively symptom-general and -specific patterns of brain lesions, so that we can determine the core damage regions of each symptom, with more detailed pathological information of NPSs in AD.
Accurate assessments of NPSs are the basis of NPSs pathogenesis neuroimaging research. Although these assessments are well-established and available in the NPSs literature with or without the use of neuroimaging, reviews of NPSs have only briefly summarized these NPSs test [13, 14]. We believe that a more detailed description of these tests is needed, including the applicable population of the tests, their advantages and disadvantages, among others. More importantly to the study of AD, we cannot ignore the problem of assessing NPSs in AD patients, accurate assessment of whose neurological and behavioural symptoms is critical and yet compounded with NPSs. Finally, a number of researches have shown that the onset time and the association of NPSs with different cognitive domains are variable, better understanding of which helps to comprehend the association between NPSs and AD. To the best of our knowledge, such issues have not been systematically summarized in review.
Overall, the current understanding of the pathological mechanism of NPSs in patients with AD is limited, especially the relationship between NPSs and AD, so a systematic review is needed to clarify these problems. We therefore provide an extensive review to 1) summarize the clinical assessment, onset time, and association with cognitive impairment of NPSs, and 2) quantitatively describe symptom-general and -specific patterns of brain lesions and brain circuits; and 3) elucidate the associations between the NPSs and AD.
Main text
Methods
Search strategy and selection criteria
We performed a systematic literature review following the guidelines for Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA, http://www.prisma-statement.org/). Research papers published up to March 2020 were identified in the databases PubMed and PsycINFO databases, using the following terms: ‘Alzheimer disease’ or ‘mild cognitive impairment’ AND ‘neuropsychiatric symptoms’, ‘apathy’, ‘delusions’, ‘depression’, ‘agitation’, ‘hallucination’, ‘anxiety’, ‘euphoria’, ‘disinhibition’, ‘irritability’, ‘aberrant motor behavior’, ‘sleep disturbances’, ‘appetite disturbances’, or ‘eating disorder’. The selection criteria were as follows: (1) To limit the heterogeneity, we focused only on AD and excluded studies that included non-AD dementia and non-amnesic MCI; (2) To ensure the reliability of the research results, articles with a sample size less than 10 were excluded; (3) Articles with subjects younger than 50 years of age were excluded.
Study quality assessment
All included original manuscripts were assessed by two independent reviewers to avoid possible bias and reporting quality using the Joanna Briggs Institute-Qualitative Assessment and Review instrument (JBI-QARI) [15]. Six papers were considered low quality and were excluded. Twenty-five papers comparing the pathological mechanisms of subjects with and without NPSs, without matching confounding variables such as age and cognitive ability, but without methodological problems, were rated as of moderate quality. The rest of the papers were rated as high quality with low bias.
Finally, 114 studies were included, including 66 neuroimaging studies to explore the pathological mechanism of NPSs and 48 non-imaging studies to summarize clinical assessments of NPSs (N = 25), onset times of NPSs (N = 8), and the associations of NPSs with cognitive impairment (N = 15). sFigure 1 summarizes the process for study selection and inclusion. AD is defined by the standard diagnostic criteria, primarily NINCDS-ADRDA (approximately 78%), followed by the CDR, DSM-IV, and CERAD. Amnestic MCI (aMCI) is defined by Petersen criteria.
Neuropsychiatric symptoms in Alzheimer’s disease
In this section, we will first summarize the existing clinical assessment scales of NPSs and discuss the main problems these assessments have when used to measure NPSs in AD patients. Then, we will summarize the results on the onset times of NPSs. Final, we will summarize the report findings on the association between NPSs and the cognitive impairment.
Clinical assessment of neuropsychiatric symptoms
We summarized the scales for measuring neuropsychiatric and behavioural symptoms, listed the measured symptoms, applicable population, and described each scale in Table 1. Multiple instruments are available for assessing NPSs in AD, but there are several problems that need to attention when selecting a measurement scale. First, since NPSs overlap with dementia symptoms, attempts should be made to rule out the effects of cognitive impairment on the measures. For example, the Behavioral Pathology in Alzheimer’s Disease Rating scale (BEHAVE-AD) is commonly used to assess non-cognitive behavioural disorders in patients with AD [16]. Secondly, some scales are only applicable to subjects with a certain level of cognition. For example, the Hamilton depression scale (HAMD) is only used to assess patients with mild dementia [17]; while the Depressive Signs Scale (DSS) cannot assess depressive symptoms in patients with mild or moderate dementia [18]. In addition, multiple NPSs tend to occur simultaneously (e.g. apathy and depression), and the functional relationships among these different NPSs are not clear. It is recommended to adopt a scale that can measure multiple NPSs simultaneously, and all of them should be independent measurements, such as Neuropsychiatric Inventory (NPI) scale [19]. Finally, the patient and the caregiver may exaggerate or conceal the severity of the symptoms because of the pathological injury and the caregiver’s emotions, respectively, so the instrument should be graded based on information from as many sources as possible, such as the Dimensional Apathy Scale [20].
Therefore, although many scales have been developed to measure neuropsychiatric and behavioural symptoms, scales that can accurately measure different NPSs in patients with AD are still inadequate or lacking. In practice, we should carefully select appropriate scales according to the population to be assessed and their needs.
The onset time of neuropsychiatric symptoms
Table 2 summarizes the study findings that provide information on the onset time of NPSs. For example, one study suggested that the prevalence of delusion was significantly increased in mild AD compared to aMCI [21]; and another study showed that the prevalence of aberrant motor behaviours, delusion, hallucinations and sleep disturbances were significantly higher in moderate AD than in mild AD [22]. For the purpose of this study, the AD continuum was divided into four stages as aMCI (or preclinical AD), mild AD, moderate AD, and severe AD, and these findings are summarized in sTable 1 and Fig. 1. sTable 1 shows that several studies have found that the prevalence of NPSs are significantly higher at a certain stage than at its previous one. Each rise of the curve in Fig. 1 represents at least one study suggesting a significant increase in the prevalence of the NPSs compared to the previous stage.

Schematic diagram of the onset time of NPSs. Disease progression is divided into five stages: healthy stage, preclinical AD or aMCI, mild AD, moderate AD, and severe AD. Each rise of the curve represents a significant increase in the prevalence of the NPSs compared to the previous stage. Abbreviations: AD, Alzheimer’s disease; aMCI, amnestic mild cognitive impairment
Most NPSs occur in the preclinical AD or aMCI phase, including apathy, depression, anxiety, irritability, agitation, sleep disturbances, and abnormal motor behaviour (Fig. 1, sTable 1). Delusions and eating disturbances occur in the mild AD phase. Finally, disinhibition, hallucinations, and euphoria occur in the moderate AD phase. In addition to disinhibition and appetite changes, the onset of the other NPSs is progressive, meaning the prevalence of these symptoms continues increasing as the disease progresses.
Neuropsychiatric symptoms and cognitive dysfunction in AD
By collecting and sorting out the literature findings on the association between NPSs and cognitive decline, we found that NPSs were closely related to global cognitive impairment [27, 28] and activities of daily living decline [29, 30]. In general, different NPSs in patients with AD were related to specific cognitive impairment (Table 3, sTable 2). Mental symptoms and agitation seem to be associated with more cognitive domains and more rapid cognitive decline, all accompanied by impaired language and memory function [28, 29, 31,32,33,34]. In addition, delusions are associated with decreased executive function, reasoning ability, and conceptualization [31]; hallucinations are associated with decreased visuospatial function [33]; while agitation is associated with decreased executive function, visuospatial function, and conceptualization [29, 32, 35]. Apathy is closely related to executive function [29, 36,37,38] and comportment (which stands for Social Behavior) [29, 32]. Depression are accompanied by a decline in executive [38, 39] and memory function [39, 40]. Abnormal motor behaviour is associated with executive and language impairment. Disinhibition and sleep disturbances are related only to executive and memory functions, respectively [29, 35, 41]. However, we did not find that euphoria, irritability, or eating disturbances were associated with any specific cognitive domains. Hence, executive function is the most closely related to NPSs among all cognitive domains, implying the executive function deficits and some NPSs may stem from common neurobiological mechanisms.
Neuroimaging findings in neuropsychiatric symptoms
We summarized the neuroimaging findings of NPSs in AD (Table 4), of which approximately two-thirds used the NPI scale to evaluate NPSs and three-quarters used the NICDS-ADRDA criteria for the diagnosis of AD. Based on the reports we identified for this review, we defined the frequency of the lesion regions in the NPS-specific brain lesion pattern, and the high frequency represented that the region was most affected by the symptom (Figs. 2, 3, 4 and 5, sFigure 2–5). “Lesion” was defined as a pathological lesion associated with NPSs, including gray matter volume atrophy, cortical thinning, decreased white matter integrity, decreased metabolism, and increased Aβ deposition. In addition, brain circuits for apathy, depression, and anxiety in AD patients were also reviewed in this section.

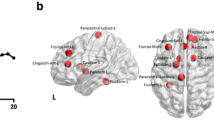
The brain lesion pattern and anterior cingulate-subcortical circuit of apathy. a The lesion brain region with the highest frequency of apathy is the anterior cingulate cortex (especially on the left), followed by the left medial frontal, medial orbitofrontal, medial thalamus, left lateral orbitofrontal, left superior and ventrolateral frontal regions, as well as the parietal, the head of the left caudate nucleus, putamen, and other regions of the frontal lobe. b The anterior cingulate-subcortical circuit begins in the anterior cingulate cortex and projects to the ventral striatum, which includes the nucleus accumbens, ventral putamen, ventromedial caudate, and olfactory tubercle. The ventral striatum has circuit linkages to the ventral pallidum and rostrodorsal substantia nigra. Then the ventral pallidum provides limited input to the mediodorsal thalamus. The anterior cingulate circuit is closed with projections from the dorsal portion of the magnocellular mediodorsal thalamus to the anterior cingulate. Abbreviations: GP: globus pallidus, SN: substantia nigra. b A visual adaptation of a figure from Nobis et al. [13], with permission

The brain lesion patterns and frontal-limbic circuit of depression in AD. a The brain region with the highest frequency of depression lesions is the superior frontal lobe, followed by the left inferior temporal and other frontal regions, as well as other temporal regions, the anterior cingulate, entorhinal, right hippocampal, caudate nucleus, lentiform nucleus, fusiform, right posterior cingulate, precuneus, supramarginal and parietal lobe. b The frontal-limbic circuit is composed of a dorsal part dominated by the dorsolateral prefrontal cortex and ventral part dominated by the subgenual cingulate and inferior temporal cortex. A direct projection from the subgeniual cingulate to the dorsolateral prefrontal cortex and a bidirectional indirect pathway through multiple marginal regions, including the posterior cingulate, hypothalamus, hippocampus, and insula are delineated. Abbreviations: rACC = rostral anterior cingulate; BG = basal ganglia; Th = thalamus. b A visual adaptation of a figure from Mayberg et al. [99], with permission

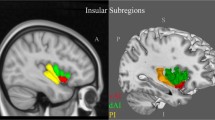
The brain lesion pattern and amygdala circuit of anxiety in AD. a The anterior and posterior cingulate cortex, entorhinal cortex, parahippocampal gyrus and insula cortex are the highest frequency of anxiety lesion regions, and the second is the amygdala, right precuneus, inferior parietal lobule, left anterior superior temporal, putamen, middle cingulate cortex and the frontal lobe. b The afferent arm of the anxiety circuit includes the exteroceptive sensory systems of the brain, which convey the sensory information contained in anxiety-inducing stimuli to the dorsal thalamus. An exception is the olfactory system, which carries information through the amygdala and entorhinal cortex, not the thalamus. Visceral afferent pathways alter the function of the locus coeruleus and amygdala. The thalamus relays sensory information to the primary sensory receptive areas of the cortex, which project to adjacent unimodal and polymodal cortical association areas. The cortical association areas send projections to the amygdala, entorhinal cortex, orbitofrontal cortex, and cingulate gyrus. The efferent pathways involving the amygdala, locus coeruleus, hypothalamus, periaqueductal gray, and striatum mediate autonomic, neuroendocrine, and skeletal-motor responses are associated with anxiety. Abbreviations: BNST = bed nucleus of the stria terminalis. b A visual adaptation of a figure from Charney et al. [100], with permission

The brain lesion patterns of other neuropsychiatric symptoms in AD. The degree of damage to different regions varies with different symptoms, while the anterior cingulate cortex (black box) is an area of common damage for all symptoms and is the most common damaged area for agitation, irritability, disinhibition, and eating disturbances. In addition, delusions are closely associated with damage to the orbitofrontal and superior temporal lobes, followed by the occipital and other areas of the frontotemporal lobes. Hallucinations are associated with damage to the left superior frontal lobe, followed by the occipital, parietal, and dorsolateral prefrontal lobes. Agitation is associated with damage to the posterior cingulate gyrus, followed by the middle cingulate gyrus and insula. Irritability is closely associated with damage to the right insula. Disinhibition is also closely associated with damage to the insula and the middle frontal lobe, cingulate other regions. Aberrant motor behaviour and eating disturbances mainly affect the orbitofrontal area, and sleep disturbances are also associated with the right middle frontal gyrus and hypothalamus
Apathy in AD
Apathy altered anterior cingulate cortex in AD
White matter studies on apathy consistently show that patients with low fractional anisotropy (FA)—a measure representing white matter integrity and information transfer speed—of the anterior cingulate cortex (ACC) are more likely to present with apathy symptoms [49,50,51]. In addition, FA in the right thalamus and bilateral parietal lobes and white matter hyperintensities in the frontal lobe were correlated with apathy [51]. Gray matter studies on apathy showed that gray matter atrophy in the bilateral ACC and left medial frontal cortex [45]. Moreover, decreased gray matter density in the bilateral ACC, frontal cortex, head of the left caudate nucleus and bilateral putamen [46], and decreased cortical thickness in the left caudal ACC, left orbitofrontal cortex (OFC), left superior, and ventrolateral frontal region [48] and inferior temporal region [47] were all correlated with the severity of apathy.
Positron emission tomography (PET) studies have demonstrated that the AD patients with apathy had glucose hypometabolism in the ACC, OFC, ventral striatum, and medial thalamus [53, 54], higher neurofibrillary tangles burden in the ACC [55, 56], and higher amyloid-β (Aβ) deposition in the bilateral frontal lobe and right ACC [52].
Anterior cingulate circuit lesions cause apathy in AD
Apathy has been conceptualized as a motivational barrier or defect in goal-directed behaviour [101]. As reported, the normal motivational behaviour is related to the anterior cingulate-subcortical circuit [102]. The anterior cingulate circuit, linking the ventral striatum to the thalamus via the rostromedial ventrolateral globus pallidus interna and ventral pallidum, originates in the ACC and medial OFC [13, 103, 104]. The disruption of this circuit may be crucially involved in effort-based decision making and executive functions [13]. In particular, lesions to the medial OFC and ventral striatum can lead to the inability to connect emotions with ongoing or upcoming behaviour [105].
Summary
Apathy is the most common NPS in AD and has been the focus of past research on NPSs. Although the regions of apathy lesions are not the same in different studies, it is consistently shown that apathy is closely related to changes in the structure and function of the medial frontal cortex and the ACC in AD. Meanwhile, subcortical alterations in the ventral striatum, medial thalamus, and ventral pallidum are also related to apathy. The imaging findings in the regions of apathy lesion supports the association of apathy with anterior cingulate circuit lesions in AD (Fig. 2, sFigure 2).
Depression in AD
Cortical and subcortical limbic brain regions abnormalities
Depression associated gray matter volume atrophy and cortical thinning mainly occur in the frontal and temporal lobes, especially in the left dorsolateral prefrontal, right medial prefrontal, OFC, ACC, and inferior temporal gyrus [40, 57, 59, 60]. The severity of depression was also associated with gray matter changes in the right hippocampal [58], entorhinal [57], left parietal [60], and striatal [61] regions. Similarly, depression causes white matter lesions in the frontal, parietal, and temporal lobes [62]. Hypometabolism in the bilateral superior frontal, left anterior cingulate, and dorsolateral prefrontal regions have been noted in patients with depression [54, 64, 65]. The presence of depression was also associated with the accumulation of Aβ in the frontal lobe in aMCI [63].
Frontal-limbic circuits abnormalities and depression in AD
Most research shows that depression is associated with frontal-striatal and subcortical limbic circuits in AD [106, 107]. Mayberg’s frontal-limbic model of depression involves the dorsal, ventral, and rostral compartments [108]. The disturbances in the dorsal compartment, which includes the dorsolateral prefrontal, dorsal ACC, and posterior cingulate cortex, cause attentional and cognitive disturbances. The ventral compartment, which consists of the paralimbic cortical, subcortical, and brainstem regions, is associated with the vegetative and somatic symptoms of depression, such as insomnia and loss of appetite. The rostral ACC connects the dorsal and ventral compartments and plays an important regulatory role in the whole network.
Other researchers believe that the hippocampus is the most common area of structural brain changes in depression [58, 109], and is associated with prefrontal cortex damage. A hippocampal–prefrontal cortex model was proposed, in which the hippocampus—a central part of memory function—regulates mood disorders and cognitive dysfunction in depressed patients [110]. The model also emphasizes the role of the limbic system, such as the cingulate gyrus and amygdala.
Summary
Depression may precede a cognitive decline in AD [8] and accelerate the rate of cognitive decline [111]. Both depression and apathy were associated with structural brain changes in the frontal, temporal, and occipital lobes, but apathy was more associated with the anterior cingulate-subcortical circuit, and depression was more associated with neuropathology in the frontal-subcortical limbic circuits in AD (Fig. 3, sFigure 3). The subcortical limbic system of depression mainly includes the hippocampus, amygdala, locus ceruleus, substantia nigra, and hypothalamus [106].
Anxiety in AD
Subcortical brain region lesions in AD anxiety
An anxiety state predicts a decreased entorhinal cortical volume [67] and may be associated with amygdala atrophy [66]. The severity of anxiety was also associated with hyperperfusion of the bilateral ACC, decreased gray matter volume in the right precuneus, inferior parietal, left parahippocampal, posterior cingulate gyrus, left insula, and bilateral putamen lobes [68, 69], and hypometabolism in the bilateral entorhinal, anterior hippocampal, left superior temporal and insula regions [72]. 18F-Florbetapir-PET studies show the patients with anxiety had higher Aβ deposition in the precuneus—posterior cingulate, frontal, parietal, anterior cingulate cortex and global cerebral [71]. The locus coeruleus in the hypothalamus is also thought to be the centre of the anxiety-related network [25], and anxiety cells are enriched in the CA1 in the ventral hippocampus [112].
Amygdala-medial prefrontal cortex mediated anxiety in AD
Anxiety can be thought of as a set of expected emotional, cognitive, and behavioural changes to the uncertainty of potential future threats, accompanied by fear [113]. The amygdala plays a pivotal role in the transmission and interpretation of fear and anxiety because it receives extensive afferents from the thalamus and extracortical sensory systems and as a subcortical visceral afferent pathway (Fig. 4) [100]. The neuronal interactions between the amygdala and cortical regions, such as the OFC, provide a framework for the initiation of coping behaviors based upon the nature of the threat and prior experiences [114]. Grupe DW and Nitschke JB [115] developed the ‘Uncertainty and Anticipation Model of Anxiety’, which emphasizes that activity in the dorsomedial prefrontal regions and OFC reflects probabilistic estimates of future events and expected costs, respectively, and mainly include the amygdala, bed nucleus of the stria terminalis (BNST), ventromedial prefrontal cortex, OFC, anterior mid-cingulate cortex and anterior insula.
An alternative network is also proposed. Under this network, the hippocampus receives convergent, integrated inputs from all sensory systems through the projections of the entorhinal cortex [116], and it works with the entorhinal cortex on situational fear conditioning. Projections from the hippocampus to the BNST and projections from the BNST to hypothalamic and brainstem sites may be involved in the expression of contextual fear conditioning. Theta oscillations within the hippocampus-amygdala-medial prefrontal cortex circuit are associated with anxious behavior [117]. Both this circuit and the ‘Uncertainty and Anticipation Model of Anxiety’ suggest that the amygdala plays a pivotal role in the assessment of, and response to, danger.
Summary
Anxiety is more common in individuals with dementia than in those without dementia [21], and it has been described as a risk factor for AD [118]. Anxiety is primarily associated with damage to the subcortical regions in AD: the amygdala plays an important role in risk assessment and response, the locus coeruleus plays an important role in the efferent response system, and the hypothalamus plays an important role in the integration of autonomic and neuroendocrine responses to threats. The anterior mid-cingulate cortex is closely linked to these brain regions and plays a central role in a series of maladaptive responses to uncertainty (Fig. 4, sFigure 4).
Delusions and hallucinations in AD
Frontotemporal region lesions in AD delusion
Delusions are characterized by asymmetrical brain structure change in the frontal and temporal regions as well as mainly atrophy in the right temporal lobe and left frontal lobe [74, 75, 119]. In addition, delusions are associated with gray matter change in the right hippocampus, left frontal lobe, right frontoparietal cortex, and left claustrum [46, 73], and white matter changes in the bilateral frontal, parieto-occipital region, left basal ganglia, the body of the corpus callosum and the superior temporal gyrus [77,78,79]. The severity of delusions is associated with hypometabolism in the frontal lobe, especially in the right lateral frontal cortex, ACC and OFC [34, 80, 82], but the association with metabolism in the temporal lobe and occipital lobe is inconsistent. Some studies showed delusions had hypometabolism in the bilateral temporal cortex and the left medial occipital region [34, 81], while other studies showed delusions had higher metabolism in the superior temporal, left inferior temporal gyrus and inferior parietal lobe [80, 81] or no connection [82]. The discrepancies among previous studies are likely due to sex differences. One study showed that women with delusions had frontotemporal atrophy in AD, but men with delusions did not have brain atrophy compared to men without delusions [76].
Anterior-posterior neural network lesions in AD hallucination
Hallucinations are associated with atrophy of the gray matter in the anterior right insula, left superior frontal gyrus, lingual gyrus and lateral occipital lobes in AD [47, 84], as well as hypometabolism in the right ventral and dorsolateral prefrontal areas [84]. Patients with hallucinations also had hypoperfusion in the parietal lobe [87]. The damaged brain areas associated with hallucinations mainly include the anterior (e.g., dorsolateral prefrontal area) -posterior (e.g., occipital lobes) neural network and the anterior insula. It is worth noting that these studies do not distinguish between the types of hallucinations. The most common form of hallucinations is visual in AD [120]. Visual hallucinations are mainly caused by atrophy and white matter lesions in the occipital lobe [85, 86]. These lesions are related to the disturbance in the lateral frontal cortex, namely, the ventral visual stream system [121].
Summary
Both delusions and hallucinations occur in the middle or late AD stage, and hallucinations may be more common in severe dementia [122]. Compared to delusions, only a limited number of studies have looked specifically at hallucinations in AD, so researchers seem to focus more on delusions (despite conflicting evidence and patients clinging to false beliefs) [123]. The brain lesions of delusions mainly occur in the frontotemporal lobe, accompanied by lateralization and sex differences, while brain lesions of hallucinations mainly occur in the anterior-posterior neural network and anterior insula region (Fig. 5, sFigure 5).
Other neuropsychiatric symptoms in AD
Hyperactivity syndrome in AD
Hyperactivity syndrome includes agitation, disinhibition, irritability, euphoria, and aberrant motor behavior [124], which is related to increased functional connectivity in the anterior cingulate cortex and right insula areas of the salience network [90]. Other neuroimaging findings support this view, especially for the agitation. Diffusion tensor imaging studies showed that irritability and agitation are related to the decreased white matter integrity in the ACC, which is a core component of the salience network [125]. Magnetic resonance imaging studies showed that agitation is related to greater atrophy in the frontal, cingulate, insular, amygdala, and hippocampal regions [46, 88] while aberrant motor behaviour is related to greater atrophy in the amygdala [66]. These are predominantly frontolimbic regions and compose many components of the significance network. PET studies showed that aberrant motor behaviour is associated with hypometabolism of the striatum and frontotemporal lobes and hypermetabolism of the OFC [92, 93]. In addition, a study found that the severity of the agitation is correlated with the atrophy score of the posterior temporal lobe [89].
Eating disturbance in AD
AD patients sometimes suffer from eating disturbances and weight loss, and nearly half of patients with AD experience appetite changes in the mild stage [126]. A longitudinal study found that patients had accelerated weight loss as many as 6 years before the diagnosis of AD [127]. A functional neuroimaging study found hypoperfusion of the ACC, OFC, and left middle mesial temporal cortices can predict appetite disturbances [98]. Other neuroimaging studies found weight loss is associated with atrophy of the mesial temporal cortex [96] and hypometabolism of the ACC [97]. In addition, the amygdala and OFC affect the internal balance between hunger and satiety and external motivational control of appetite [128]. Therefore, eating disturbances may be related to the network of the ventral (orbitobasal) frontal cortex, medial temporal cortex, and amygdala in AD.
Sleep disturbances in AD
Common sleep disturbances in AD include fragmentation of sleep at night, decreased duration of sleep at night, daytime sleepiness, and inversion of the sleep-wake cycle [129]. The relationship between sleep and AD is complex and bidirectional, and the underlying mechanism is the interaction between sleep and Aβ—sleep disturbances increase the generation of Aβ and decrease the elimination of Aβ; and once Aβ accumulates, there is increased sleep disturbance [130, 131]. Amyloid deposition appears to be associated with decreased sleep quality, but not with changes in sleep quantity in the preclinical stage of AD [132]. Worse sleep quality increases the Aβ burden in the precuneus [133], and a shorter sleep quantity at night increases the Aβ burden in the right hippocampus and thalamus in healthy older peoples [134]. In addition, tau pathology as the second hallmark of AD can also cause sleep disturbances. Sleep regulating areas mainly include the brain stem, thalamus, hypothalamus, midbrain and basal forebrain [135]. Many of these areas show tau pathology at pretangle stages or stages by Braak staging, before any cortical tau or amyloid pathology development [136]. Orexin, as an important sleep-wake regulatory marker, increases in the cerebrospinal fluid in patients with moderate to severe AD and is positively correlated with tau protein levels [137]. Hence, tau pathology may also play an important role in sleep disturbances in AD.
Summary
Dysfunction of the orbitofrontal–subcortical circuit is characterized by personality changes including disinhibition, agitation, and irritability; this circuit connects the frontal monitoring systems to the limbic system [104]. Few studies on the relationships between other NPSs and neuroimaging have been conducted in AD, but they all are associated with a lesion in the ACC (Fig. 5, sFigure 5).
Relationship between NPSs and Alzheimer’s disease
Behavioral and neuropsychiatric symptoms are associated with abnormalities in specific brain regions, such as the prefrontal and subcortical limbic regions, which disrupt the normal balance of neurotransmission. According to neuroimmunoregulation theory, this, in turn, is associated with inflammatory pathways that lead to microglial activation and aggregation and the formation of neurofibrillary tangles, ultimately triggering neuronal loss [138]. In addition, NPSs are related to the dysfunction of various neurotransmitter pathways related to AD, including the dopamine system, the serotonin system, and the cholinergic system [139]. In the current section, we will further discuss the molecular and cellular changes associated with stages of AD progression and their relationship to NPSs.
NPSs in preclinical Alzheimer’s disease
NPSs are variable and sporadic throughout the course of the disease, but an important group appears early (Fig. 1), especially emotional symptoms (e.g., depression, anxiety, and apathy), before the clinical diagnosis of cognitive impairment [140, 141].
The presence of microglial activation and inflammatory signals in patients with AD prior to “clinical diagnosis” may explain the occurrence of NPSs in the early stages of the disease. Activation of microglia has been shown to be associated with deficits in social interaction [142]. Meanwhile, apathy, anxiety, depression, and agitation were associated with increased pro-inflammatory cytokines (systemic tumor necrosis factor α) detected in the serum of AD patients [143, 144]. Similarly, another study found an association between the levels of diverse cytokines present in CSF of patients with dementia, discovering that anti-inflammatory interleukin-6 (IL-6) cytokine levels were inversely proportional to anxiety scores in AD patients [145]. Importantly, Ledo et al. found that depressive-like behavior induced by Alzheimer’s Aβ oligomers in mice is mediated by inflammation through microglial cell activation in the hippocampus, decreasing 5-HT levels in the hippocampus and prefrontal cortex [146].
Depression is also influenced by the reduction of dopamine and serotonin in the brain, while AD has been associated with loss of serotonergic neurons and a decrease in 5-hydrotryptamine (5-HT) levels in the postmortem brains with this disease [147, 148]. In a healthy brain, dopamine is constantly released into the hippocampus, which links emotional feelings with cognitive processes [149, 150]. In AD, a decrease in dopamine levels coupled with a decrease in serotonin triggers depression, which is regarded as a prodromal symptom of AD. In addition, late-life depression and AD share common genetic factors, including brain-derived neurotrophic factor, apolipoprotein E, interleukin-1, and methylenetetrahydrofolate reductase, while inflammatory pathways are activated in both disorders [151, 152]. In this context, the changes produced by late-life depression seem to have an impact on the hippocampus, inducing inflammatory events that activate microglia, which trigger the overproduction of pro-inflammatory cytokines, as described in earlier time about the conceptual scheme of the neuroimmunomodulation theory [138].
NPSs in mild to severe Alzheimer’s disease
As AD progresses, most NPSs present in the early stages of AD become more severe and common, and some psychiatric and behavioral symptoms begin to appear. One hypothesis that has been suggested is that AD progressive cholinergic loss (resulting in a loss of inhibition of the dopamine system), in the context of a relatively spared dopaminergic system, may increase the tendency of AD patients to develop psychosis because of a relative striatal hyperdopaminergia [153]. Available evidence suggested that striatal dopamine (D2/D3) receptors are increased in AD patients with delusional compared with AD patients without delusions [154], and that higher striatal D2 receptors are associated with wandering behavior [92].
In addition, several studies have shown a correlation between serotonin deficiency and NPSs. In patients with AD, lower levels of serotonin1A receptors were associated with more severe depressive symptoms [155], and lower concentrations of serotonin in the temporal cortex were associated with hyperactivity and psychosis [148]. The depressed AD patients showed larger and more extensive reductions in serotonin transporters including the midbrain, nucleus accumbens, and thalamus [156]. Further, the study showed glucose metabolism in the right dorsolateral prefrontal cortex was positively correlated with 5-HT transporter ([11C]-DASB) levels in the striatum in AD patients, suggesting that subcortical serotonergic dysfunction may affect cortical function in regions involved in affective processing such as dorsolateral prefrontal cortex. For example, prefrontal cortex interactions with the hypothalamus mediate reward aspects of eating such as food cravings [157].
In the final stage of AD, the pathology of all NPSs becomes complicated and difficult to treat. Worsening mental symptoms (delusions and hallucinations) cause confusion between reality and morbid fantasies, and patients exhibit severe abnormal motor behaviour, often characterized by scratching, which can lead to recurrent hyperfascial skin infections. These destructive NPSs accelerate the death of AD patients.
Summary
We believe that there may be two connective mechanisms between NPSs and AD: (A) NPSs arise as a consequence of AD pathology. AD affects key brain regions of underlying behavior, emotion, or mental, so NPSs may be a direct non-cognitive manifestation of AD neurodegenerative disease [158]. AD-related cognitive decline may also develop into depression, anxiety, or similar NPSs as a psychological response. Other NPSs in the AD stage are caused by AD through reverse causality or psychological responses, and the onset of NPSs will aggravate the pathology of AD. (B) NPSs and AD pathology arise as a consequence of some shared pathologic process. In this case, there is no causal relationship between NPSs and AD pathology, but a third factor, such as brain vascular disease or white matter change, leads to the occurrence of AD and NPSs [159, 160].
Conclusions
NPSs almost universal existence in the AD, combined with their disabling effects on patients and caregivers, is contrasted by the fact that few effective and safe treatments exist, which is mainly attributed to the following three reasons: (1) Lack of reliable and effective measurement of NPSs in AD; (2) Biomarkers associated with symptom-specific in patients with AD have not yet been developed; (3) The relationship between NPS and the pathological mechanism of AD remains unclear.
The current review provides a good complement to these treatment issues. Firstly, we summarized the detailed scale information, as well as some possible problems in the NPSs measurement process, which may be helpful for accurate assessment of NPSs. Next, we described symptom-general and -specific patterns of brain lesions. The anterior cingulate cortex is a commonly damaged region across all symptoms, and the prefrontal cortex, especially the orbitofrontal cortex, is also a critical region associated with most NPSs. This conclusion was supported by an intervention study, which found that greater reduction in orbitofrontal blood flow has been associated with a greater behavioural response to treatment with donepezil [161].
In contrast, the anterior cingulate-subcortical circuit is specifically related to apathy in AD, the frontal-limbic circuit to depression, and the amygdala circuit to anxiety. Understanding symptom-specific brain lesion networks may help track treatment response for targeted drug therapy. For example, it is important to understand whether observed network changes are the result of functional remodeling of defective networks or reflect the plasticity of compensatory circuitry complement in treatment development. Finally, we elucidated the two possible connective mechanisms between NPSs and AD: etiologic pathways and interactions, and summarized the onset time of NPSs. Different NPSs occur in different disease stages of AD, but most symptoms appear in the preclinical AD or mild cognitive impairment stage and develop progressively, which suggested that the critical treatment window for NPSs should be advanced to the early stage of AD.
There are still some limitations in the study of the pathological mechanism of NPSs in AD patients, and more studies are needed to solve them in the future. Firstly, we found the differences between the subtypes of each symptom in exploring the NPS-specific pathologic mechanisms. For example, in addition to the weight loss caused by the loss of appetite mentioned above, AD patients may also experience increased appetite, difficulty swallowing, and other symptoms of eating disturbances. The relationship between dementia stages and eating disorders may depend on the type of eating disorder. For example, people with mild AD are more likely to experience anorexia, while people with moderate AD have an increased appetite and changes in food preferences and eating habits, and people with severe AD have difficulty swallowing [126]. We suspect that in AD, the relationship between the dementia stage and other NPSs may also differ depending on the subtype of symptoms. Therefore, future studies should further explore the relationship between various symptom subtypes and the severity of dementia in order to better understand the pathological association.
In addition, we should pay attention to the pathological superposition of multiple NPSs. One type of NPSs is unlikely to appear alone and instead is most likely to occur with other types of symptoms in AD [162, 163]. Among patients with dementia, 55% report two or more symptoms, and 44% report three or more symptoms [3]. However, most studies, including some included in the current review, did not adjust the presence of other NPSs when exploring the mechanism of a particular NPS. This is a major limitation of the current review.
Some longitudinal studies showed that individuals with two NPSs had an additional 1.9-fold elevated risk of developing dementia compared with those with zero or one NPS, while those with three or more symptoms had an additional risk of 3 [118] and significantly higher odds of having functional limitations [164]. Another study also showed the number of comorbid NPSs, but not symptom clusters, are associated with an increased risk of dementia [165]. These findings suggest that understanding the comorbid pattern of NPSs will help us to further clarify the pathogenesis of NPSs in AD and contribute to clinical evaluation. However, there are few neuroimaging studies on comorbid NPSs and future studies should focus on this issue.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- NPSs:
-
Neuropsychiatric symptoms
- CSDD:
-
Cornell Scale for Depression in Dementia
- BEHAVE-AD:
-
The Behavioral Pathology in Alzheimer’s disease rating scale
- BRSD:
-
Behavior Rating Scale for Dementia
- HAMD:
-
Hamilton depression scale
- DSS:
-
Depressive Signs Scale
- HAMD:
-
Hamilton Depression Scale
- HAMA:
-
Hamilton Anxiety Scale
- NPI:
-
Neuropsychiatric Inventory
- FA:
-
Fractional anisotropy
- ACC:
-
Anterior cingulate cortex
- OFC:
-
Orbitofrontal cortex
- PET:
-
Positron emission tomography
- Aβ:
-
Amyloid-β
- BNST:
-
Bed nucleus of the stria terminalis
- MCI:
-
Mild cognitive impairment
- rACC:
-
Rostral anterior cingulate
- BG:
-
Basal ganglia
- Th:
-
Thalamus
- IL-6:
-
Interleukin-6
References
International AsD. World Alzheimer report 2019: attitudes to dementia. In: Book World Alzheimer Report 2019: Attitudes to dementia (Editor ed.^eds.). London: Alzheimer’s Disease Internationals; 2019.
Sindi S, Mangialasche F, Kivipelto M. Advances in the prevention of Alzheimer's disease. F1000prime Rep. 2015;7:50.
Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky SJJ. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. Jama. 2002;288:1475–83.
Peters ME, Schwartz S, Han D, Rabins PV, Steinberg M, Tschanz JT, et al. Neuropsychiatric symptoms as predictors of progression to severe Alzheimer’s dementia and death: the Cache County Dementia Progression Study. Am J Psychiatr. 2015;172:460–5.
Shin I-S, Carter M, Masterman D, Fairbanks L, Cummings JL. Neuropsychiatric symptoms and quality of life in Alzheimer disease. Am J Geriatr Psychiatry. 2005;13(6):469–74. https://doi.org/10.1097/00019442-200506000-00005.
Jost BC, Grossberg GT. The evolution of psychiatric symptoms in Alzheimer’s disease: a natural history study. J Am Geriatr Soc. 1996;44(9):1078–81. https://doi.org/10.1111/j.1532-5415.1996.tb02942.x.
Craig D, Mirakhur A, Hart DJ, McIlroy SP, Passmore AP. A cross-sectional study of neuropsychiatric symptoms in 435 patients with Alzheimer's disease. Am J Geriatr Psychiatry. 2005;13(6):460–8. https://doi.org/10.1097/00019442-200506000-00004.
Jicha GA, Carr SA. Conceptual evolution in Alzheimer's disease: implications for understanding the clinical phenotype of progressive neurodegenerative disease. J Alzheimers Dis. 2010;19(1):253–72. https://doi.org/10.3233/JAD-2010-1237.
Rosenberg PB, Nowrangi MA, Lyketsos CG. Neuropsychiatric symptoms in Alzheimer's disease: what might be associated brain circuits? Mol Asp Med. 2015;43:25–37.
Nowrangi MA, Lyketsos CG, Rosenberg PB. Principles and management of neuropsychiatric symptoms in Alzheimer’s dementia. Alzheimer's Res Ther. 2015;7:12.
Geda YE, Schneider LS, Gitlin LN, Miller DS, Smith GS, Bell J, et al. Neuropsychiatric symptoms in Alzheimer's disease: past progress and anticipation of the future. Alzheimers Dement. 2013;9(5):602–8. https://doi.org/10.1016/j.jalz.2012.12.001.
Tascone LS, Bottino CMC. Neurobiology of neuropsychiatric symptoms in Alzheimer's disease: a critical review with a focus on neuroimaging. Dementia Neuropsychologia. 2013;7:236–43.
Nobis L, Husain M. Apathy in Alzheimer's disease. Curr Opin Behav Sci. 2018;22:7–13. https://doi.org/10.1016/j.cobeha.2017.12.007.
Seignourel PJ, Kunik ME, Snow L, Wilson N, Stanley M. Anxiety in dementia: a critical review. Clin Psychol Rev. 2008;28(7):1071–82. https://doi.org/10.1016/j.cpr.2008.02.008.
Institute JB. Joanna Briggs institute reviewers’ manual: 2014 edition. Adelaide: The Joanna Briggs Institute; 2014.
Reisberg B, Auer SR, Monteiro IM. Behavioral pathology in Alzheimer's disease (BEHAVE-AD) rating scale. Int Psychogeriatr. 1997;8(S3):301–8. https://doi.org/10.1017/S1041610297003529.
Hamilton M. A rating scale for depression. J Neurol, Neurosurg Psychiatry Investig. 1960;23:56.
Katona C, Aldridge C. The dexamethasone suppression test and depressive signs in dementia. J Affect Disord. 1985;8(1):83–9. https://doi.org/10.1016/0165-0327(85)90076-X.
Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The neuropsychiatric inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44:2308.
Radakovic R, Abrahams S. Developing a new apathy measurement scale: dimensional apathy scale. Psychiatry Res. 2014;219:658–63.
Hwang TJ, Masterman DL, Ortiz F, Fairbanks LA, Cummings JL. Mild cognitive impairment is associated with characteristic neuropsychiatric symptoms. Alzheimer Dis Assoc Disord. 2004;18(1):17–21. https://doi.org/10.1097/00002093-200401000-00004.
Cheng S-T, Kwok T, Lam LC. Neuropsychiatric symptom clusters of Alzheimer's disease in Hong Kong Chinese: prevalence and confirmatory factor analysis of the neuropsychiatric inventory. Int Psychogeriatr. 2012;24:1465–73.
Burns A, Jacoby R, Levy R. Psychiatric phenomena in Alzheimer's disease. IV: disorders of behaviour. Br J Psychiatry. 1990;157(1):86–94. https://doi.org/10.1192/bjp.157.1.86.
Di Iulio F, Palmer K, Blundo C, Casini AR, Gianni W, Caltagirone C, et al. Occurrence of neuropsychiatric symptoms and psychiatric disorders in mild Alzheimer's disease and mild cognitive impairment subtypes. Int Psychogeriatr. 2010;22:629–40.
Ehrenberg AJ, Suemoto CK, Resende F, de Paula E, Petersen C, Leite REP, et al. Neuropathologic correlates of psychiatric symptoms in Alzheimer’s disease. J Alzheimers Dis. 2018;66:115–26.
van der Linde RM, Dening T, Stephan BC, Prina AM, Evans E, Brayne C. Longitudinal course of behavioural and psychological symptoms of dementia: systematic review. Br J Psychiatry. 2016;209(5):366–77. https://doi.org/10.1192/bjp.bp.114.148403.
Wadsworth LP, Lorius N, Donovan NJ, Locascio JJ, Rentz DM, Johnson KA, et al. Neuropsychiatric symptoms and global functional impairment along the Alzheimer’s continuum. Dement Geriatr Cogn Disord. 2012;34(2):96–111. https://doi.org/10.1159/000342119.
Lopez OL, Becker JT, Brenner RP, Rosen J, Bajulaiye O, Reynolds C. Alzheimer's disease with delusions and hallucinations: neuropsychological and electroencephalographic correlates. Neurology. 1991;41:906.
Senanarong V, Poungvarin N, Jamjumras P, Sriboonroung A, Danchaivijit C, Udomphanthuruk S, et al. Neuropsychiatric symptoms, functional impairment and executive ability in Thai patients with Alzheimer's disease. Int Psychogeriatr. 2005;17:81–90.
D’Onofrio G, Sancarlo D, Panza F, Copetti M, Cascavilla L, Paris F, et al. Neuropsychiatric symptoms and functional status in Alzheimer’s disease and vascular dementia patients. Curr Alzheimer Res. 2012;9:759–71.
Jeste DV, Wragg RE, Salmon DP, Harris MJ, Thal LJ. Cognitive deficits of patients with Alzheimer's disease with and without delusions. Am J Psychiatry. 1992;149(2):184–9.
Rozum W, Cooley B, Richens A, Matyi J, Vernon E, Tschanz J. Specific cognitive/behavioral domains predict neuropsychiatric symptoms in severe dementia; 2017.
Wilson RS, Gilley DW, Bennett DA, Beckett LA, Evans DA. Hallucinations, delusions, and cognitive decline in Alzheimer's disease. J Neurol, NeurosurgPsychiatry. 2000;69:172–7.
Sultzer DL, Leskin LP, Melrose RJ, Harwood DG, Narvaez TA, Ando TK, et al. Neurobiology of delusions, memory, and insight in Alzheimer disease. Am J Geriatr Psychiatry. 2014;22(11):1346–55. https://doi.org/10.1016/j.jagp.2013.06.005.
Chen ST, Sultzer DL, Hinkin CH, Mahler ME, Cummings JL. Executive dysfunction in Alzheimer's disease: association with neuropsychiatric symptoms and functional impairment. J Neuropsychiatry Clin Neurosci. 1998;10:426–32.
McPHERSON S, Fairbanks L, Tiken S, Cummings JL, Back-Madruga C. Apathy and executive function in Alzheimer's disease. J Int Neuropsychol Soc. 2002;8(3):373–81. https://doi.org/10.1017/S1355617702813182.
Grossi D, Santangelo G, Barbarulo AM, Vitale C, Castaldo G, Proto MG, et al. Apathy and related executive syndromes in dementia associated with Parkinson’s disease and in Alzheimer’s disease. Behav Neurol. 2013;27:515–22.
Nakaaki S, Murata Y, Sato J, Shinagawa Y, Hongo J, Tatsumi H, et al. Association between apathy/depression and executive function in patients with Alzheimer's disease. Int Psychogeriatr. 2008;20:964–75.
Pietrzak RH, Lim YY, Neumeister A, Ames D, Maruff P. Amyloid-β, anxiety, and cognitive decline in preclinical Alzheimer disease: a multicenter, prospective cohort study. JAMA Psychiatry. 2015;72(3):284–91. https://doi.org/10.1001/jamapsychiatry.2014.2476.
Son JH, Han DH, Min KJ, Kee BS. Correlation between gray matter volume in the temporal lobe and depressive symptoms in patients with Alzheimer's disease. Neurosci Lett. 2013;548:15–20.
Westerberg CE, Lundgren EM, Florczak SM, Mesulam M-M, Weintraub S, Zee PC, et al. Sleep influences the severity of memory disruption in amnestic mild cognitive impairment: results from sleep self-assessment and continuous activity monitoring. Alzheimer Dis Assoc Disord. 2010;24(4):325–33. https://doi.org/10.1097/WAD.0b013e3181e30846.
Scarmeas N, Brandt J, Albert M, Hadjigeorgiou G, Papadimitriou A, Dubois B, Sarazin M, Devanand D, Honig L, Marder K. Delusions and hallucinations are associated with worse outcome in Alzheimer disease. J Arch Neurol.. 2005;62:1601-8.
Boyle PA, Malloy PF, Salloway S, Cahn-Weiner DA, Cohen R, Cummings JL. Executive dysfunction and apathy predict functional impairment in Alzheimer disease. Am J Geriatr Psychiatry. 2003;11:214-21.
Nagata T, Shinagawa S, Ochiai Y, Kada H, Kasahara H, Nukariya K, Nakayama K: Relationship of frontal lobe dysfunction and aberrant motor behaviors in patients with Alzheimer's Int Psychogeriatr. 2010;22:463-9.
Apostolova LG, Akopyan GG, Partiali N, Steiner CA, Dutton RA, Hayashi KM, et al. Structural correlates of apathy in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007;24:91–7.
Bruen PD, McGeown WJ, Shanks MF, Venneri A. Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer's disease. Brain. 2008;131(9):2455–63. https://doi.org/10.1093/brain/awn151.
Donovan NJ, Wadsworth LP, Lorius N, Locascio JJ, Rentz DM, Johnson KA, et al. Regional cortical thinning predicts worsening apathy and hallucinations across the Alzheimer disease spectrum. Am J Geriatr Psychiatry. 2014;22(11):1168–79. https://doi.org/10.1016/j.jagp.2013.03.006.
Tunnard C, Whitehead D, Hurt C, Wahlund L, Mecocci P, Tsolaki M, et al. Apathy and cortical atrophy in Alzheimer's disease. Int J Geriatr Psychiatry. 2011;26(7):741–8. https://doi.org/10.1002/gps.2603.
Tighe SK, Oishi K, Mori S, Smith GS, Albert M, Lyketsos CG, et al. Diffusion tensor imaging of neuropsychiatric symptoms in mild cognitive impairment and Alzheimer’s dementia. J Neuropsychiatry Clin Neurosci. 2012;24(4):484–8. https://doi.org/10.1176/appi.neuropsych.11120375.
Kim JW, Lee DY, Choo IH, Seo EH, Kim SG, Park SY, et al. Microstructural alteration of the anterior cingulum is associated with apathy in Alzheimer disease. Am J Geriatr Psychiatry. 2011;19:644–53.
Ota M, Sato N, Nakata Y, Arima K, Uno M. Relationship between apathy and diffusion tensor imaging metrics of the brain in Alzheimer's disease. Int J Geriatr Psychiatry. 2012;27(7):722–6. https://doi.org/10.1002/gps.2779.
Mori T, Shimada H, Shinotoh H, Hirano S, Eguchi Y, Yamada M, et al. Apathy correlates with prefrontal amyloid β deposition in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2014;85:449–55.
Marshall GA, Monserratt L, Harwood D, Mandelkern M, Cummings JL, Sultzer DL. Positron emission tomography metabolic correlates of apathy in Alzheimer disease. Arch Neurol. 2007;64:1015–20.
Holthoff VA, Beuthien-Baumann B, Kalbe E, Lüdecke S, Lenz O, Zündorf G, et al. Regional cerebral metabolism in early Alzheimer’s disease with clinically significant apathy or depression. Biol Psychiatry. 2005;57(4):412–21. https://doi.org/10.1016/j.biopsych.2004.11.035.
Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL. Neuropathologic correlates of apathy in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;21:144–7.
Tekin S, Mega MS, Masterman DM, Chow T, Garakian J, Vinters HV, et al. Orbitofrontal and anterior cingulate cortex neurofibrillary tangle burden is associated with agitation in Alzheimer disease. Ann Neurol. 2001;49(3):355–61. https://doi.org/10.1002/ana.72.
Zahodne LB, Gongvatana A, Cohen RA, Ott BR, Tremont G, Initiative AsDN. Are apathy and depression independently associated with longitudinal trajectories of cortical atrophy in mild cognitive impairment? Am J Geriatr Psychiatry. 2013;21(11):1098–106. https://doi.org/10.1016/j.jagp.2013.01.043.
Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, et al. Automated 3D mapping of hippocampal atrophy and its clinical correlates in 400 subjects with Alzheimer's disease, mild cognitive impairment, and elderly controls. Hum Brain Mapp. 2009;30:2766–88.
Lebedev AV, Beyer MK, Fritze F, Westman E, Ballard C, Aarsland D. Cortical changes associated with depression and antidepressant use in Alzheimer and Lewy body dementia: an MRI surface-based morphometric study. Am J Geriatr Psychiatry. 2014;22:4–13. e11.
Lebedeva A, Westman E, Lebedev AV, Li X, Winblad B, Simmons A, et al. Structural brain changes associated with depressive symptoms in the elderly with Alzheimer’s disease. Neurol Neurosurg Psychiatry. 2014;85(8):930–5. https://doi.org/10.1136/jnnp-2013-307110.
Brommelhoff JA, Spann BM, Go JL, Mack WJ, Gatz M. Striatal hypodensities, not white matter hypodensities on CT, are Associated with Late-Onset Depression in Alzheimer's Disease. J Aging Res. 2011;2011(3):187219.
Lee GJ, Lu PH, Hua X, Lee S, Wu S, Nguyen K, et al. Depressive symptoms in mild cognitive impairment predict greater atrophy in Alzheimer's disease-related regions. Biol Psychiatry. 2012;71(9):814–21. https://doi.org/10.1016/j.biopsych.2011.12.024.
Chung JK, Plitman E, Nakajima S, Chow TW, Chakravarty MM, Caravaggio F, et al. Lifetime history of depression predicts increased amyloid-β accumulation in patients with mild cognitive impairment. J Alzheimers Dis. 2015;45(3):907–19. https://doi.org/10.3233/JAD-142931.
Lee HS, Choo IH, Lee DY, Kim JW, Seo EH, Kim SG, et al. Frontal dysfunction underlies depression in mild cognitive impairment: a FDG-PET study. Psychiatry Investig. 2010;7(3):208–14. https://doi.org/10.4306/pi.2010.7.3.208.
Hirono N, Mori E, Ishii K, Ikejiri Y, Imamura T, Shimomura T, et al. Frontal lobe hypometabolism and depression in Alzheimer's disease. Neurology. 1998;50:380–3.
Poulin SP, Dautoff R, Morris JC, Barrett LF, Dickerson BC, Initiative AsDN. Amygdala atrophy is prominent in early Alzheimer's disease and relates to symptom severity. Psychiatry Res Neuroimaging. 2011;194:7–13.
Mah L, Binns MA, Steffens DC, Initiative AsDN. Anxiety symptoms in amnestic mild cognitive impairment are associated with medial temporal atrophy and predict conversion to Alzheimer disease. Am J Geriatr Psychiatry. 2015;23:466–76.
Tagai K, Nagata T, Shinagawa S, Nemoto K, Inamura K, Tsuno N, et al. Correlation between both morphologic and functional changes and anxiety in Alzheimer's disease. Dement Geriatr Cogn Disord. 2014;38(3-4):153–60. https://doi.org/10.1159/000358822.
Nour AEAM, Jiao Y, Teng GJ. Neuroanatomical associations of depression, anxiety and apathy neuropsychiatric symptoms in patients with Alzheimer's disease. Acta Neurol Belgica. 2020(5). https://doi.org/10.1007/s13760-020-01349-8.
Berlow YA, Wells WM, Ellison JM, Sung YH, Renshaw PF, Harper DG. Neuropsychiatric correlates of white matter hyperintensities in Alzheimer's disease. Int J Geriatr Psychiatry. 2010;25(8):780–8. https://doi.org/10.1002/gps.2418.
Bensamoun D, Guignard R, Furst AJ, Derreumaux A, Manera V, Darcourt J, et al. Associations between neuropsychiatric symptoms and cerebral amyloid deposition in cognitively impaired elderly people. J Alzheimers Dis. 2016;49(2):387–98. https://doi.org/10.3233/JAD-150181.
Hashimoto H, Monserratt L, Nguyen P, Feil D, Harwood D, Mandelkern MA, et al. Anxiety and regional cortical glucose metabolism in patients with Alzheimer’s disease. J Neuropsychiatry Clin Neurosci. 2006;18(4):521–8. https://doi.org/10.1176/jnp.2006.18.4.521.
Serra L, Perri R, Cercignani M, Spanò B, Fadda L, Marra C, et al. Are the behavioral symptoms of Alzheimer's disease directly associated with neurodegeneration? J Alzheimers Dis. 2010;21:627–39.
Geroldi C, Bresciani L, Zanetti O, Frisoni GB. Regional brain atrophy in patients with mild Alzheimer's disease and delusions. Int Psychogeriatr. 2002;14(4):365–78. https://doi.org/10.1017/S1041610202008566.
Geroldi C, Akkawi NM, Galluzzi S, Ubezio M, Binetti G, Zanetti O, et al. Temporal lobe asymmetry in patients with Alzheimer's disease with delusions. J Neurol Neurosurg Psychiatry. 2000;69:187–91.
Whitehead D, Tunnard C, Hurt C, Wahlund L, Mecocci P, Tsolaki M, et al. Frontotemporal atrophy associated with paranoid delusions in women with Alzheimer's disease. Int Psychogeriatr. 2012;24(1):99–107. https://doi.org/10.1017/S1041610211000974.
Nakaaki S, Sato J, Torii K, Oka M, Negi A, Nakamae T, et al. Decreased white matter integrity before the onset of delusions in patients with Alzheimer’s disease: diffusion tensor imaging. Neuropsychiatr Dis Treat. 2013;9:25.
Anor CJ, O'Connor S, Saund A, Tang-Wai DF, Keren R, Tartaglia MC. Neuropsychiatric symptoms in Alzheimer disease, vascular dementia, and mixed dementia. Neurodegener Dis. 2017;17:127–34.
Lee DY, Choo IH, Kim KW, Jhoo JH, Youn JC, Lee UY, et al. White matter changes associated with psychotic symptoms in Alzheimer’s disease patients. J Neuropsychiatry Clin Neurosci. 2006;18(2):191–8. https://doi.org/10.1176/jnp.2006.18.2.191.
Mentis MJ, Weinstein EA, Horwitz B, McIntosh AR, Pietrini P, Alexander GE, et al. Abnormal brain glucose metabolism in the delusional misidentification syndromes: a positron emission tomography study in Alzheimer disease. Biol Psychiatry. 1995;38(7):438–49. https://doi.org/10.1016/0006-3223(94)00326-X.
Hirono N, Mori E, Ishii K, Kitagaki H, Sasaki M, Ikejiri Y, et al. Alteration of regional cerebral glucose utilization with delusions in Alzheimer's disease. J Neuropsychiatry Clin Neurosci. 1998;10:433–9.
Sultzer DL, Brown CV, Mandelkern MA, Mahler ME, Mendez MF, Chen ST, et al. Delusional thoughts and regional frontal/temporal cortex metabolism in Alzheimer’s disease. Am J Psychiatr. 2003;160(2):341–9. https://doi.org/10.1176/appi.ajp.160.2.341.
Mega MS, Lee L, Dinov ID, Mishkin F, Toga AW, Cummings JL. Cerebral correlates of psychotic symptoms in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2000;69(2):167–71. https://doi.org/10.1136/jnnp.69.2.167.
Blanc F, Noblet V, Philippi N, Cretin B, Foucher J, Armspach J-P, et al. Right anterior insula: core region of hallucinations in cognitive neurodegenerative diseases. PLoS One. 2014;9(12):e114774. https://doi.org/10.1371/journal.pone.0114774.
Holroyd S, Shepherd ML, Downs JH III. Occipital atrophy is associated with visual hallucinations in Alzheimer's disease. J Neuropsychiatry Clin Neurosci. 2000;12:25–8.
Lin S-H, Yu C-Y, Pai M-C. The occipital white matter lesions in Alzheimer's disease patients with visual hallucinations. Clin Imaging. 2006;30(6):388–93. https://doi.org/10.1016/j.clinimag.2006.09.025.
Kotrla KJ, Chacko RC, Harper RG, Jhingran S, Doody R. SPECT findings on psychosis in Alzheimer's disease. Am J Psychiatry. 1995;152:1470–5.
Trzepacz PT, Yu P, Bhamidipati PK, Willis B, Forrester T, Tabas L, et al. Frontolimbic atrophy is associated with agitation and aggression in mild cognitive impairment and Alzheimer's disease. Alzheimer's Dement. 2013;9:S95–S104. e101.
Hsu J-L, Lee W-J, Liao Y-C, Lirng J-F, Wang S-J, Fuh J-L. Posterior atrophy and medial temporal atrophy scores are associated with different symptoms in patients with Alzheimer’s disease and mild cognitive impairment. PLoS One. 2015;10(9):e0137121.
Balthazar ML, Pereira FR, Lopes TM, da Silva EL, Coan AC, Campos BM, et al. Neuropsychiatric symptoms in Alzheimer's disease are related to functional connectivity alterations in the salience network. Hum Brain Mapp. 2014;35(4):1237–46. https://doi.org/10.1002/hbm.22248.
Weissberger GH, Melrose RJ, Narvaez TA, Harwood D, Mandelkern MA, Sultzer DL. 18F-Fluorodeoxyglucose positron emission tomography cortical metabolic activity associated with distinct agitation behaviors in Alzheimer disease. Am J Geriatr Psychiatry. 2017;25(6):569–79. https://doi.org/10.1016/j.jagp.2017.01.017.
Meguro K, Yamaguchi S, Itoh M, Fujiwara T, Yamadori A. Striatal dopamine metabolism correlated with frontotemporal glucose utilization in Alzheimer's disease: a double-tracer PET study. Neurology. 1997;49:941–5.
Reilly TJ, Staff RT, Ahearn TS, Bentham P, Wischik CM, Murray AD. Regional cerebral blood flow and aberrant motor behaviour in Alzheimer's disease. Behav Brain Res. 2011;222(2):375–9. https://doi.org/10.1016/j.bbr.2011.04.003.
Ismail Z, Herrmann N, Francis P, Rothenburg L, Lobaugh N, Leibovitch F, et al. A SPECT study of sleep disturbance and Alzheimer’s disease. Dement Geriatr Cogn Disord. 2009;27:254–9.
Liguori C, Chiaravalloti A, Nuccetelli M, Izzi F, Sancesario G, Cimini A, et al. Hypothalamic dysfunction is related to sleep impairment and CSF biomarkers in Alzheimer’s disease. J Neurol. 2017;264(11):2215–23. https://doi.org/10.1007/s00415-017-8613-x.
Grundman M, Corey-Bloom J, Jernigan T, Archibald S, Thal L. Low body weight in Alzheimer's disease is associated with mesial temporal cortex atrophy. Neurology. 1996;46:1585–91.
Hu X, Okamura N, Arai H, Higuchi M, Maruyama M, Itoh M, et al. Neuroanatomical correlates of low body weight in Alzheimer's disease: a PET study. Prog Neuro-Psychopharmacol Biol Psychiatry. 2002;26:1285–9.
Ismail Z, Herrmann N, Rothenburg LS, Cotter A, Leibovitch FS, Rafi-Tari S, et al. A functional neuroimaging study of appetite loss in Alzheimer’s disease. J Neurol Sci. 2008;271:97–103.
Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatr. 1999;156:675–82.
Charney DS, Grillon C, Bremner JD. The neurobiological basis of anxiety and fear: circuits, mechanisms, and neurochemical interactions (part I). Neuroscientist. 1998;4(1):35–44. https://doi.org/10.1177/107385849800400111.
Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex–basal ganglia circuits. Cereb Cortex. 2006;16:916–28.
Bonelli RM, Cummings JL. Frontal-subcortical circuitry and behavior. Dialogues Clin Neurosci. 2007;9:141.
Le Heron C, Apps MAJ, Husain M. The anatomy of apathy: a neurocognitive framework for amotivated behaviour. Neuropsychologia. 2017;118:54-67.
Tekin S, Cummings JL. Frontal–subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002;53(2):647–54. https://doi.org/10.1016/S0022-3999(02)00428-2.
Levy R. Dubois BJCc: apathy and the functional anatomy of the prefrontal cortex–basal ganglia circuits. Cereb Cortex. 2006;16:916–28.
Tagariello P, Girardi P, Amore M. Depression and apathy in dementia: same syndrome or different constructs? A critical review. Arch Gerontol Geriatr. 2009;49:246–9.
Mortby ME, Maercker A, Forstmeier S. Apathy: a separate syndrome from depression in dementia? A critical review. Aging Clin Exp Res. 2012;24:305–16.
Deckersbach T, Dougherty DD, Rauch SL. Functional imaging of mood and anxiety disorders. J Neuroimaging. 2006;16(1):1–10. https://doi.org/10.1177/1051228405001474.
Campbell S, MacQueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci. 2004;29(6):417–26.
Sampath D, Sathyanesan M, Newton SS. Cognitive dysfunction in major depression and Alzheimer’s disease is associated with hippocampal–prefrontal cortex dysconnectivity. Neuropsychiatr Dis Treat. 2017;13:1509.
Rapp MA, Schnaider-Beeri M, Wysocki M, Guerrero-Berroa E, Grossman HT, Heinz A, et al. Cognitive decline in patients with dementia as a function of depression. Am J Geriatr Psychiatry. 2011;19(4):357–63. https://doi.org/10.1097/JGP.0b013e3181e898d0.
Jimenez JC, Su K, Goldberg AR, Luna VM, Biane JS, Ordek G, et al. Anxiety cells in a hippocampal-hypothalamic circuit. Neuron. 2018;97:670–683. e676.
Tuma A H , Maser J . Anxiety and the anxiety disorders. Int Clin Psychopharmacol. 1990;5(1):75.
Rempel-Clower NL. Role of orbitofrontal cortex connections in emotion. Ann N Y Acad Sci. 2007;1121:72–86.
Grupe DW, Nitschke JB. Uncertainty and anticipation in anxiety: an integrated neurobiological and psychological perspective. Nat Rev Neurosci. 2013;14:488–501.
ten Donkelaar HJ. The Limbic System. In Clinical Neuroanatomy: Brain Circuitry and Its Disorders. Berlin: Springer Berlin Heidelberg; 2011:633-710. https://doi.org/10.1007/978-3-642-19134-3_14.
Jacinto LR, Cerqueira JJ, Sousa N. Patterns of theta activity in limbic anxiety circuit preceding exploratory behavior in approach-avoidance conflict. Front Behav Neurosci. 2016;10:171.
Acosta I, Borges G, Aguirre-Hernandez R, Sosa AL, Prince M, Group DR. Neuropsychiatric symptoms as risk factors of dementia in a Mexican population: a 10/66 Dementia Research Group Study. Alzheimers Dement. 2018;14:271–9.
Rossi R, Geroldi C, Bresciani L, Zanetti O, Frisoni G. Delusions in ad are associated with less severe brain structural changes. Res Pract Alzheimer's Dis. 2003;7:168–73.
Jeste DV, Finkel SI. Psychosis of Alzheimer's disease and related dementias: diagnostic criteria for a distinct syndrome. Am J Geriatr Psychiatry. 2000;8:29–34.
Collerton D, Perry E, McKeith I. Why people see things that are not there: a novel perception and attention deficit model for recurrent complex visual hallucinations. Behav Brain Sci. 2005;28:737–57.
Bassiony MM, Lyketsos CG. Delusions and hallucinations in Alzheimer’s disease: review of the brain decade. Psychosomatics. 2003;44(5):388–401. https://doi.org/10.1176/appi.psy.44.5.388.
Reeves SJ, Gould RL, Powell JF, Howard RJ. Origins of delusions in Alzheimer's disease. Neurosci Biobehav Rev. 2012;36:2274–87.
Aalten P, Verhey FR, Boziki M, Bullock R, Byrne EJ, Camus V, et al. Neuropsychiatric syndromes in dementia. Dement Geriatr Cogn Disord. 2007;24(6):457–63. https://doi.org/10.1159/000110738.
Tighe SK, Oishi K, Mori S, Smith GS, Albert M, Lyketsos CG, et al. Diffusion tensor imaging of neuropsychiatric symptoms in mild cognitive impairment and Alzheimer’s dementia. J Neuropsychiatry Clin Neurosci. 2012;24:484–8.
Kai K, Hashimoto M, Amano K, Tanaka H, Fukuhara R, Ikeda M. Relationship between eating disturbance and dementia severity in patients with Alzheimer’s disease. PLoS One. 2015;10(8):e0133666.
Gao S, Nguyen JT, Hendrie HC, Unverzagt FW, Hake A, Smith-Gamble V, et al. Accelerated weight loss and incident dementia in an elderly African-American cohort. J Am Geriatr Soc. 2011;59(1):18–25. https://doi.org/10.1111/j.1532-5415.2010.03169.x.
Hinton EC, Parkinson JA, Holland AJ, Arana FS, C. Roberts A, Owen AM. Neural contributions to the motivational control of appetite in humans. Eur J Neurosci. 2004;20:1411–8.
Peter-Derex L, Yammine P, Bastuji H, Croisile B. Sleep and Alzheimer's disease. Sleep Med Rev. 2015;19:29–38.
Ju Y-ES, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology—a bidirectional relationship. Nat Rev Neurol. 2014;10:115.
Holth JK, Patel TK, Holtzman DM. Sleep in Alzheimer's disease–beyond amyloid. Neurobiol Sleep Circadian Rhythms. 2017;2:4–14.
Ju Y-ES, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70(5):587–93. https://doi.org/10.1001/jamaneurol.2013.2334.
Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, et al. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 2013;70:1537–43.
Shokri-Kojori E, Wang G-J, Wiers CE, Demiral SB, Guo M, Kim SW, et al. β-amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci. 2018;115(17):4483–8. https://doi.org/10.1073/pnas.1721694115.
Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63.
Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–9.
Liguori C, Romigi A, Nuccetelli M, Zannino S, Sancesario G, Martorana A, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71(12):1498–505. https://doi.org/10.1001/jamaneurol.2014.2510.
Maccioni RB, Rojo LE, Fernandez JA, Kuljis RO. The role of neuroimmunomodulation in Alzheimer's disease. Ann N Y Acad Sci. 2009;1153(1):240–6. https://doi.org/10.1111/j.1749-6632.2008.03972.x.
Tonkonogy J. The American Psychiatric Publishing textbook of Alzheimer disease and other dementias. Psychiatr Serv. 2010;61(2):206–7. https://doi.org/10.1176/ps.2010.61.2.206a.
Masters MC, Morris JC, Roe CM. “Noncognitive” symptoms of early Alzheimer disease: a longitudinal analysis. Neurology. 2015;84:617–22.
Ballard CG, Gauthier S, Cummings JL, Brodaty H, Grossberg GT, Robert P, et al. Management of agitation and aggression associated with Alzheimer disease. Nat Rev Neurol. 2009;5(5):245–55. https://doi.org/10.1038/nrneurol.2009.39.
Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17(3):400–6. https://doi.org/10.1038/nn.3641.
Lasselin J, Elsenbruch S, Lekander M, Axelsson J, Benson S. Mood disturbance during experimental endotoxemia: predictors of state anxiety as a psychological component of sickness behavior. Brain Behav Immun. 2016;57:30–7.
Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 2011;77(3):212–8. https://doi.org/10.1212/WNL.0b013e318225ae07.
Larksater M, Holmgren S, Freund-Levi Y, Aarsl D, Hjorth E. Neuropsychiatric symptoms in dementia-a role for neuroinflammation? Brain Res Bull. 2014;108:88-93.
Ledo JH, Azevedo EP, Beckman D, Ribeiro FC, Ferreira ST. Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer's amyloid-oligomers in mice. J Neurosci. 2016;36(48):12106–16. https://doi.org/10.1523/JNEUROSCI.1269-16.2016.
Kovacs GG, Klöppel S, Fischer I, Dorner S, Lindeck-Pozza E, Birner P, et al. Nucleus-specific alteration of raphe neurons in human neurodegenerative disorders. Neuroreport. 2003;14(1):73–6. https://doi.org/10.1097/00001756-200301200-00014.
Garcia-Alloza M, Gil-Bea FJ, Diez-Ariza M, Chen C-H, Francis PT, Lasheras B, et al. Cholinergic–serotonergic imbalance contributes to cognitive and behavioral symptoms in Alzheimer’s disease. Neuropsychologia. 2005;43:442–9.
Russo SJ, Nestler E. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14(9):609–25. https://doi.org/10.1038/nrn3381.
Rocchetti J, Isingrini E, Dal Bo G, Sagheby S, Menegaux A, Tronche F, et al. Presynaptic D2 dopamine receptors control long-term depression expression and memory processes in the temporal hippocampus. Biol Psychiatry. 2015;77(6):513–25. https://doi.org/10.1016/j.biopsych.2014.03.013.
Ye Q, Bai F, Zhang Z. Shared genetic risk factors for late-life depression and Alzheimer’s disease. J Alzheimers Dis. 2016;52(1):1–15. https://doi.org/10.3233/JAD-151129.
Stefano P, Concetta C, Luigi D, Marco C, Antonis P, Ioannis L, et al. Role of neurodevelopment involved genes in psychiatric comorbidities and modulation of inflammatory processes in Alzheimer's disease. J Neurol Sci. 2016;370:162–6. https://doi.org/10.1016/j.jns.2016.09.053.
Hirao K, Pontone GM, Smith GS. Molecular imaging of neuropsychiatric symptoms in Alzheimer's and Parkinson's disease. Neurosci Biobehav Rev. 2015;49:157–70. https://doi.org/10.1016/j.neubiorev.2014.11.010.
Reeves S, Brown R, Howard R, Grasby P. Increased striatal dopamine (D2/D3) receptor availability and delusions in Alzheimer disease. Neurology. 2009;72(6):528–34. https://doi.org/10.1212/01.wnl.0000341932.21961.f3.
Lai MKP, Tsang SW, Esiri MM, Francis PT, Wong TH, Chen CP. Differential involvement of hippocampal serotonin1A receptors and re-uptake sites in non-cognitive behaviors of Alzheimer's disease. Psychopharmacology. 2011;213:431–9.
Ouchi Y, Yoshikawa E, Futatsubashi M, Yagi S, Ueki T, Nakamura K. Altered brain serotonin transporter and associated glucose metabolism in Alzheimer disease. J Nucl Med. 2009;50(8):1260–6. https://doi.org/10.2967/jnumed.109.063008.
Öngür D, Price J. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex. 2000;10(3):206–19. https://doi.org/10.1093/cercor/10.3.206.
Jicha GA, Carr SA, Lovell MA. Conceptual evolution in Alzheimer's disease: implications for understanding the clinical phenotype of progressive neurodegenerative disease. J Alzheimers Dis. 2010;19:253–72.
Alexopoulos GS. The vascular depression hypothesis: 10 years later. Biol Psychiatry. 2006;60:1304–5.
Taylor WD, Aizenstein HJ, Alexopoulos GS. The vascular depression hypothesis: mechanisms linking vascular disease with depression. Mol Psychiatry. 2013;18(9):963–74. https://doi.org/10.1038/mp.2013.20.
Mega MS, Dinov ID, Lee L, O’Connor SM, Masterman DM, Wilen B, et al. Orbital and dorsolateral frontal perfusion defect associated with behavioral response to cholinesterase inhibitor therapy in Alzheimer's disease. J Neuropsychiatry Clin Neurosci. 2000;12:209.
Dechamps A, Jutand MA, Onifade C, Richard-Harston S, Bourdel-Marchasson I. Co-occurrence of neuropsychiatric syndromes in demented and psychotic institutionalized elderly. Int J Geriatr Psychiatry. 2008;23(11):1182–90. https://doi.org/10.1002/gps.2052.
Wetzels RB, Zuidema SU, de Jonghe JF, Verhey FR, Koopmans RT. Course of neuropsychiatric symptoms in residents with dementia in nursing homes over 2-year period. Am J Geriatr Psychiatry. 2010;18(12):1054–65. https://doi.org/10.1097/JGP.0b013e3181f60fa1.
Okura T, Plassman BL, Steffens DC, Llewellyn DJ, Potter GG, Langa KM. Prevalence of neuropsychiatric symptoms and their association with functional limitations in older adults in the United States: the aging, demographics, and memory study. J Am Geriatr Soc. 2010;58(2):330–7. https://doi.org/10.1111/j.1532-5415.2009.02680.x.
Mortby ME, Burns R, Eramudugolla R, Ismail Z, Anstey KJ. Neuropsychiatric symptoms and cognitive impairment: understanding the importance of co-morbid symptoms. J Alzheimers Dis. 2017;59(1):141–53. https://doi.org/10.3233/JAD-170050.
Acknowledgements
We are grateful to Kewei Chen (Banner Alzheimer’s Institute) and Chuansheng Chen (University of California, Irvine) for helpful comments on earlier version of this review.
Funding
This work was supported by National Key Research and Development Project of China (grant number 2018YFC1315200), National Science Fund for Distinguished Young Scholars (grant number 81625025), Funds for International Cooperation and Exchange of the National Natural Science Foundation of China (grant number 81820108034), National Natural Science Foundation of China (grant number 31700997, and 82071205).
Author information
Authors and Affiliations
Contributions
YJC and MXD led the writing of the manuscript, devised all the figures and edited the manuscript. ZJZ supervised the writing and co-edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declared no competing of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Dang, M. & Zhang, Z. Brain mechanisms underlying neuropsychiatric symptoms in Alzheimer’s disease: a systematic review of symptom-general and –specific lesion patterns. Mol Neurodegeneration 16, 38 (2021). https://doi.org/10.1186/s13024-021-00456-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-021-00456-1