
Abstract
Alzheimer’s Disease (AD) is the most prevalent progressive neurodegenerative disease; to date, no AD therapy has proven effective in delaying or preventing the disease course. In the search for novel therapeutic targets in AD, it has been shown that increased chronic psychologic stress is associated with AD risk. Subsequently, biologic pathways underlying psychologic stress have been identified and shown to be able to exacerbate AD relevant pathologies. In this review, we summarize the literature relevant to the association between psychologic stress and AD, focusing on studies investigating the effects of stress paradigms on transgenic mouse models of Amyloid-β (Aβ) and tau pathologies. In recent years, a substantial amount of research has been done investigating a key stress-response mediator, corticotropin-releasing hormone (CRH), and its interactions with AD relevant processes. We highlight attempts to target the CRH signaling pathway as a therapeutic intervention in these transgenic mouse models and discuss how targeting this pathway is a promising avenue for further investigation.
Similar content being viewed by others


Background
Alzheimer’s Disease (AD) is a progressive neurodegenerative process that is currently the sixth leading cause of death in the United States [1]. The AD brain is characterized by the aggregation of misfolded proteins and marked neuronal loss [2]. The hallmark pathologic features of AD, intracellular neurofibrillary tau tangles (NFT’s), extracellular Amyloid-β (Aβ) plaques, synaptic and neuronal loss and reactive gliosis, have all been extensively studied. Despite this enormous scientific effort, many aspects of the disease remain poorly understood [3]. Although genetic factors clearly play a role in AD, studies of monozygotic twins have suggested that non-genetic risk factors are also involved [4, 5]. As non-genetic risk factors may be more easily modifiable than genetic risk factors, it is worthwhile to explore these pathways in search of therapeutic strategies for the treatment or prevention of AD. One non-genetic risk factor that has been associated with increased risk for AD is increased chronic psychologic stress. Epidemiologic studies have shown that individuals predisposed to experiencing psychologic stress, and those affected with diseases associated with chronic stress such as Major Depression and Post-traumatic Stress Disorder (PTSD), have an increased risk of developing AD (Table 1) [6,7,8,9,10,11,12].
Although these studies associating psychologic stress and AD are intriguing, like most epidemiologic studies of AD these studies are subject to many confounds that could dampen enthusiasm for further research let alone for guiding a therapeutic development program. Indeed, unless based on autopsy proven AD or other modalities such as amyloid imaging that are now available to enable more accurate diagnosis of AD in living patients, most epidemiologic studies likely included a relatively high percentage of individuals (20% or more) with other forms of dementia and even the control group (>20%), if over age 65, is likely to have amyloid pathology [13, 14]. Thus, these studies are conservatively viewed as epidemiologic associations with dementia and not necessarily AD. Further, epidemiologic studies of psychologic stress are particularly challenging, as it is difficult to quantify stress in a way that is potentially reflective of the biological processes associated with this relatively subjective phenotype. Finally, in a disease like AD with a long prodromal state, it can be challenging to link early or mid-life risk factors with development of the disease late in life.
Given these and other potential confounds, epidemiologic data likely needs to be combined with biological or genetic data to serve as a basis for developing novel AD therapies. In this review we will evaluate the biological underpinnings that suggest the association between AD and stress may be strong enough to warrant investigation as a possible avenue for AD therapy. The course of molecular research into stress and AD began largely with investigation into stress-steroids, namely cortisol, but more recently has shifted towards Corticotropin-Releasing Hormone (CRH), a neuropeptide which exerts its effects primarily through activation of the CRHR1 receptor [15]. These data have shown that stress mediators are able to regulate AD relevant processes and provide converging evidence that activation of the stress response pathway may be able to exacerbate AD pathologies.
Main text
Stress signaling
When approaching the biology underlying the stress response, it is important to consider all of the potential contributors if we are to determine what pathways mediate the effects of stress on AD relevant processes. CRH and the CRH family peptides urocortin (UCN) 1, 2, and 3 are the main coordinators of the response to psychologic stress. These peptides exert their effects through binding and activating the CRHR1 and CRHR2 receptors in the central nervous system (CNS) and the periphery [15,16,17]. The classical stress response pathway begins with the perception of stress leading to the release of CRH from the paraventricular nuclei (PVN) of the hypothalamus, binding to CRHR1 Receptors in the anterior pituitary and causing the release of Adrenocorticotropic Hormone (ACTH) into the systemic circulation. ACTH then binds to MC2 receptors in the adrenal cortices, inducing glucocorticoid release [18]. This cascade is known as the Hypothalamic-Pituitary-Adrenal (HPA) axis [18, 19].
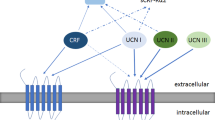
In addition to the CRHR1 present in the anterior pituitary, the CRHR1 is also widely expressed throughout the brain (Fig. 1). Its signaling in these locations is thought to cause many of the psychologic effects of stress and is also implicated in learning and memory [20,21,22]. Indeed, CRHR1 is expressed on both glutamatergic and dopaminergic neurons and can have either anxiogenic or anxiolytic effects depending on the brain region [23]. It appears that stress, up to a certain threshold, is stimulating and induces rapid memory formation; above that threshold, stress becomes overwhelming and deleterious [23, 24]. CRH expressing neurons are distributed throughout the CNS. Of special relevance in AD is the expression of CRH in the cortex and hippocampal-amygdalar complex, areas which also express the CRHR1 receptor [15]. The role of the CRHR2 receptor is not yet fully understood; it has been implicated in the anorexic effects of stress, and theories as to its role vary from functioning as a counterbalance to CRHR1 stress signaling, to being a part of a different stress pathway that responds to inescapable and unpredictable stressors [16]. The CRHR2 is expressed in discrete areas of the brain, but is widely expressed in the periphery (Fig. 1) [25]. The urocortins, similarly to the CRHR2, are expressed discretely within the brain. Both CRHR1 and CRHR2 are G-Protein Coupled Receptors that signal primarily through the Gs, Gq, and MAPK/ERK pathways [15]. Activation of these pathways can lead to increased intracellular calcium levels [26], an effect that may be worthwhile to explore considering the hypothesized role of calcium dyshomeostasis in AD [27]. In terms of specificity towards CRHR1 and 2, i) CRH has specificity for the CRHR1, ii) urocortin 1 can activate both CRHR1 and CRHR2, and iii) urocortin 2 and 3 are unique in that they show specificity for CRHR2 and do not appear to have affinity for CRHR1 at physiologic concentrations [15, 28] (Fig. 2). Overall, CRH and the urocortins are proposed to act as neuromodulators within the CNS to orchestrate various components of the psychologic stress response [17].


CRH-family peptides and receptor affinities [17]
Cortisol and AD
In the 1990’s and early 2000’s, an appreciable amount of study focused on the role of cortisol and corticosteroids in AD; this was largely driven by several observations. One of the direct effects of psychologic stress is an increased level of circulating glucocorticoids, mainly cortisol in humans and corticosterone in mice [18]. Cushing’s syndrome, a state of excess glucocorticoids seen in patients with pituitary tumors or those who have received large amounts of exogenous steroids, is associated with atrophy of the hippocampus, a feature shared with both conditions of chronic stress and AD [29,30,31]. The hypothesized role that glucocorticoids play in AD has been reviewed elsewhere [31,32,33], but we will make note of the most critical data. AD patients have been shown to have mildly increased levels of cortisol when compared to controls (Table 2) [34, 35] and corticosteroids have been shown to be able to induce Aβ plaque load and hyperphosphorylated tau levels in APP overexpressing mouse models [36,37,38,39], effects that were able to be reduced with a glucocorticoid receptor (GR) antagonist [40].
The hippocampus is a key negative regulator of the HPA axis, with increased glucocorticoid levels causing increased GR signaling in the hippocampus leading to inhibition of CRH release from the hypothalamus [18]. One hypothesis for the increased levels of glucocorticoids found in AD is that hippocampal damage may cause a loss of negative feedback on hypothalamic CRH release. AD patients have also been found to have altered responses to a CRH stimulation test, with higher levels of cortisol than controls [41, 42], suggesting that AD patients may have a more complex alteration in HPA axis activity.
CRH and AD
In the early 2000’s, investigation into the stress response and AD began to branch into the CRH signaling pathway. This occurred simultaneously with a large effort to target CRH signaling in various stress related disorders (Major Depression, PTSD, addiction). Since this time, a substantial amount of data implicating CRH signaling in both Aβ production and tau phosphorylation has been produced.
Aβ
An increasing amount of evidence shows that stress and specifically CRH-CRHR1 signaling can increase Aβ peptide production. In primary neuronal cultures (PNC’s), CRH treatment was shown to increase levels of total secreted Aβ peptide, an effect that was partially blocked by the small molecule CRHR1 antagonists Antalarmin and R121919 [43]. Our group was able to replicate the CRH-induced increases in Aβ in both PNC’s and a CRHR1 overexpressing SH-SY5Y cell line, but we saw similar increases upon treatment with the peptide CRHR1 antagonist Astressin as were seen with CRH treatment [44]. Recent work showing that adrenergic GPCR’s are able to affect APP processing led to our hypothesis that the CRHR1 may also be able to directly regulate these processes [45]. An in vitro γ-secretase assay showed that CRH treatment can modulate γ-secretase activity towards increased Aβ42 production [44] and that upon CRH treatment, CRHR1 is internalized by a β-arrestin 2 mediated interaction, where it then colocalized with γ-secretase intracellularly. This fit well with previous data that has shown that β-arrestin regulates Aβ production via y-secretase [46]. Furthermore, we showed that the peptide CRHR1 antagonists Astressin and α-helical CRH were able to induce rapid internalization of cells overexpressing the CRHR1. This data led to the hypothesis that CRHR1 antagonists may modulate γ-secretase activity towards increased Aβ production through inducing internalization of the receptor. Evaluation of the Gs coupled pathway of CRHR1 has shown that pretreatment with the Protein Kinase A (PKA) inhibitor H-89 was able to significantly reduce Aβ40 and Aβ42 production in a CRH treated PNC model [43]. Through these investigations, a picture begins to form that includes three possible mechanisms for CRH induced Aβ increases.
-
1.
CRHR1 interactions with β-arrestin 2 and γ-secretase: demonstrated by the data that CRHR1 antagonists do not stimulate the receptor, but do induce its internalization and increase Aβ40 and Aβ42 production in vitro [44].
-
2.
CRH directly modulating γ -secretase activity: demonstrated by data that CRH treatment increases Aβ42 production in an in vitro γ–secretase assay [44].
-
3.
Classical CRHR1 G s /G q pathway signaling: demonstrated by data that treatment with PKA inhibitor H-89 is able to reduce CRH induced increases in Aβ40 and Aβ42 in vitro [43].
Multiple stress paradigms have been able to induce increases in Aβ40 and Aβ42 levels and plaque load in the brains of wild type (WT) and mutant APP overexpressing transgenic mouse models (Table 3).
Studies investigating the mechanisms through which stress mediates these effects on Aβ have focused mainly on CRHR1 and GR signaling. While acute restraint stress was able to induce increases in total soluble Aβ levels in the brain interstitial fluid (ISF) of Tg2576 mice, these increases were not reproduced with administration of corticosterone to the mice. These increases were able to be induced by the administration of CRH and blocked by administration of the CRHR1 antagonist α-helical CRH [47]. These findings suggest that acute stress-induced increases in Aβ40 and Aβ42 in the brain may be due to a CRH-mediated effect, which agrees with previous data showing that corticosteroid-induced increases in brain Aβ40 and Aβ42 levels take place 48–72 h after administration [36]. In addition, it was found that chronic isolation stress caused increases in both GR and CRHR1 expression in the cortex and hippocampus, along with increased corticosterone levels and plaque burden in the Tg2576 mouse model [48]. In a double transgenic Tg2576 and CRH overexpressing mouse model, it was shown that overexpression of CRH was related with large increases in brain Aβ40 and Aβ42 levels along with increased plaque burden in the cortex [49].
While the role of the urocortins in stress-induced Aβ production has been largely unexplored, one study found that injection of urocortin 1 (an agonist for both CRHR1 and CRHR2) into the amygdala of male Wistar rats induced increases in amygdalar Aβ40 levels, while 5 h of acute restraint stress in the rats showed increases in both Aβ40 and Aβ42 [50]. A double transgenic Tg2576 and homozygous CRHR1 knockout mouse line (supplemented with corticosterone for health maintenance) showed highly significant decreases (>60%) in basal levels of Aβ40, Aβ42, and APP C-Terminal Fragment (α and β) levels in the brain, suggesting that a CRHR1-mediated mechanism contributes to an appreciable amount of Aβ processing [51].
Tau
While there is a substantial amount of evidence that CRH signaling can affect Aβ production and Aβ plaque deposition, most data connecting the CRH signaling pathway with tau-related processes is shown through studies on tau phosphorylation and not aggregate deposition. The neurofibrillary tangles found in the AD brain are composed of hyperphosphorylated, cleaved, and conformationally altered tau protein [52]. Phosphorylation of tau at certain residues causes its dissociation from microtubules and enables aggregates to form [52].
Multiple stress paradigms have been able to increase tau phosphorylation in mice [53,54,55,56,57,58,59,60] (Table 4) and it was shown that a CRH KO mouse model did not show the stress induced increases in hyperphosphorylated tau that were seen in WT mice [57].
Furthermore, it was shown that stress induced tau phosphorylation could not be replicated by administration of corticosterone and that CRHR1 antagonist NBI27914 was able to decrease the effect of stress on tau phosphorylation [54] at certain phospho-epitopes. Mice that had undergone adrenalectomy also replicated the stress-induced increases in tau phosphorylation. These data provide several pieces of evidence pointing away from a glucocorticoid mediated mechanism and towards one mediated by CRH [55]. It was also shown that a CRHR1 knockout mouse line did not replicate these increases in stress induced tau phosphorylation, while their CRHR2 knockout counterparts actually had a hyperactive response to stress when compared to WT mice, lending evidence to the hypothesis that the CRHR2 may have a regulatory role over CRHR1 signaling [55]. A CRHR1 and CRHR2 double knockout mouse maintained the lack of response seen with the CRHR1 single knockout mouse [59]. Lastly, a CRH overexpressing mouse model showed increased tau phosphorylation at multiple sites when compared with controls [56]. Unfortunately, the vast majority of these studies on stress and tau pathology have used WT mice and only measure tau phosphorylation and not aggregate formation. While increased phosphorylation of tau frequently correlates with increased aggregation and neurodegeneration in models of tau pathology, further evaluation of the CRH signaling pathway in deposit-forming models of tau pathology is necessary. As transgenic models of tau aggregation are actually models of Frontotemporal Dementia (FTD), these studies could additionally link stress to other dementias or neurodegenerative processes.
Interventions targeting the CRH signaling pathway
With evidence showing that both CRH and glucocorticoids can exacerbate AD pathology in the brains of transgenic mouse models, it follows that abnormalities in the HPA axis may play a role in the development and progression of AD. Targeting CRH could be the most efficacious single intervention within the HPA axis, as an intervention blocking the effects of CRH would decrease downstream glucocorticoid release, potentially blocking both CRH-mediated and glucocorticoid-mediated effects on Aβ production and tau phosphorylation.
Overactive CRH signaling has been implicated in anxiety, major depression, and inflammatory disorders and an effective intervention targeting the CRH signaling pathway is highly sought in these fields [16, 17, 61, 62]. Despite preclinical studies demonstrating their potential efficacy, for unknown reasons, the CRHR1 antagonists GSK511679 and Pexacerfont were not shown to be effective in clinical trials investigating their effects in alcoholism and generalized anxiety disorder [63]. While there has yet to be a clinical trial investigating an anti-CRH therapy in any neurodegenerative disease, there has been substantial effort to implement CRHR1 antagonists preclinically in mouse models of Aβ and tau pathologies (Table 5). CRHR1 antagonists have shown some efficacy in decreasing stress induced Aβ pathologies, with no effect in an acute stress paradigm [44], but alleviating effects in chronic stress paradigms [43] as well as in unstressed APP overexpressing mice [64]. Studies have utilized both peptide and small molecule CRHR1 antagonists (Astressin, Antalarmin, α-helical CRH, R121919, NBI27914) in the various aforementioned stress paradigms. One study found that even in unstressed mice, the CRHR1 antagonist R121919 was able to significantly decrease amyloid deposits in the brain. The effects of the CRHR1 inhibitors on tau showed either decreased tau inclusions in the cortex of PS19 tau mice [54] or decreased tau phosphorylation [43, 59], but, in contrast to the work done in various APP overexpressing mouse models, the investigation into stress and tau aggregation is limited to a single study.
Although there has been some proof of efficacy for CRHR1 antagonists in decreasing AD pathology in mouse models, one should question the probability of their success in human trials, as these antagonists have a history of having promising effects in rodent models and then failing in the clinic [61, 62, 65, 66]. In addition, CRHR1 antagonists can induce the internalization of CRHR1 and cause increases in Aβ40 and Aβ42 in in vitro models of the human CRHR1, drawing some concern to their use as an AD prevention. We believe that the strength of the literature shows that the CRH signaling pathway is an interesting target that merits further investigation into possible therapeutic interventions, but we posit that direct targeting of CRH will provide stronger and more worthwhile effects than receptor-based interventions. Direct targeting of CRH could be accomplished through several methods including, but not limited to, CRISPR-CAS9, antisense oligonucleotides, or direct immunotargeting of the CRH peptide. Any investigator attempting to develop one of these methods can do so with the knowledge that the creation of a therapy able to effectively downregulate CRH signaling would have broad impact in many conditions of chronic stress and possibly AD.
Impact of psychologic stress on other neurodegenerative diseases
In addition to AD, psychologic stress has been explored as a risk factor and exacerbating component in Parkinson’s Disease, Huntington’s Disease and vascular dementia. There are several comprehensive reviews available on the impact of psychologic stress and Major Depression on neurodegenerative disease [32, 67, 68], and it is likely that stress acts through unique mechanisms in each disease. Similarly to AD, increased psychologic stress has been associated epidemiologically with an increased risk of Parkinson’s Disease [69, 70]. Stress paradigms have been shown to exacerbate the phenotype of several mouse and rat models of Parkinson’s [71,72,73,74,75,76], yet there do not appear to be studies attempting to target stress pathways therapeutically within these models. Furthermore, early reports have shown that stress paradigms have a detrimental effect on the phenotype of the R6/1 mouse model of Huntington’s [77, 78]. Further investigation into the mechanisms of how stress signaling affects these proteinopathies is needed, and should also grow to include other neurodegenerative proteinopathies such as Amyotrophic Lateral Sclerosis (ALS) and FTD. Indeed, as mentioned earlier some of the work already done on tau mouse models may be applicable to FTD. Lastly, it is pertinent to mention that the risk of both stroke and vascular dementia have also been associated with increased psychologic stress levels [11, 79, 80]. As patients with all of the neurodegenerative conditions mentioned suffer from stressful neuropsychiatric comorbidities [67, 81], interventions targeting stress signaling pathways could have both preventative and therapeutic effects on these diseases.
Conclusions
Considerable epidemiologic data suggests that chronic psychologic stress can be a risk factor in AD, or at least dementia. Studies in animal and cell models provide biological underpinnings to support this association and identify CRH as the logical therapeutic target. Based on current data, successful antagonism of CRH may result in beneficial effects on both Aβ amyloid and tau pathology, an effect that would be predicted to have a major impact on AD. More speculatively, such therapies could improve the harmful negative behavioral symptoms that accompany chronic stress. Indeed, normal stress-signaling is not always deleterious and plays a role in learning and memory. Empirical studies will need to be carried out in order to determine if one can target CRH directly in a manner that has beneficial effects and avoids side effects due to under-stimulation of the pathway. Given the need for novel targets in AD, we believe that additional preclinical studies that explore the potential impact of targeting CRH could pave the way for increased interest in this pathway as a target in AD.
Abbreviations
- ACTH:
-
Adrenocorticotropic Hormone
- AD:
-
Alzheimer’s Disease
- ALS:
-
Amyotrophic Lateral Sclerosis
- Aβ:
-
Amyloid β
- CRH:
-
Corticotropin-releasing hormone
- CRHR1:
-
Corticotropin-Releasing Hormone Receptor 1
- CRHR2:
-
Corticotropin-Releasing Hormone Receptor 2
- FTD:
-
Frontotemporal Dementia
- GR:
-
Glucocorticoid Receptor
- HPA:
-
Hypothalamic-Pituitary-Adrenal
- NFT:
-
Neurofibrillary tangles
- PKA:
-
Protein Kinase A
- PTSD:
-
Post-traumatic Stress Disorder
- PVN:
-
Paraventricular nuclei
- UCN:
-
Urocortin
- WT:
-
Wild type
References
2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12:459–509.
Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–48.
Golde TE, Borchelt DR, Giasson BI, Lewis J. Thinking laterally about neurodegenerative proteinopathies. J Clin Invest. 2013;123:1847–55.
Gatz M, Reynolds CA, Finkel D, Pedersen NL, Walters E. Dementia in Swedish twins: predicting incident cases. Behav Genet. 2010;40:768–75.
Iacono D, Volkman I, Nennesmo I, Pedersen NL, Fratiglioni L, Johansson B, et al. Neuropathologic assessment of dementia markers in identical and fraternal twins: AD and non-AD pathologies in older twins. Brain Pathol. 2014;24:317–33.
Byers AL, Yaffe K. Depression and risk of developing dementia. Nat Rev Neurol. 2011;7:323–31.
Kaup AR, Byers AL, Falvey C, Simonsick EM, Satterfield S, Ayonayon HN, et al. Trajectories of depressive symptoms in older adults and risk of dementia. JAMA Psychiatry. 2016;73:525.
Wilson RS, Evans DA, Bienias JL, De Leon CM, Schneider JA, Bennett DA. Proneness to psychological distress is associated with risk of Alzheimer’s disease. Neurology. 2003;61:1479–85.
Johansson L, Guo X, Duberstein PR, Hällström T, Waern M, Östling S, et al. Midlife personality and risk of Alzheimer disease and distress a 38-year follow-up. Neurology. 2014;83:1538–44.
Xu W, Tan L, Wang H-F, Jiang T, Tan M-S, Tan L, et al. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 2015;jnnp-2015-310548.
Andel R, Crowe M, Hahn EA, Mortimer JA, Pedersen NL, Fratiglioni L, et al. Work-related stress may increase the risk of vascular dementia. J Am Geriatr Soc. 2012;60:60–7.
Tsolaki M, Papaliagkas V, Kounti F, Messini C, Boziki M, Anogianakis G, et al. Severely stressful events and dementia: a study of an elderly Greek demented population. Psychiatry Res. 2010;176:51–4.
Bangen KJ, Clark AL, Werhane M, Edmonds EC, Nation DA, Evangelista N, et al. Cortical amyloid burden differences across empirically-derived mild cognitive impairment subtypes and interaction with APOE ɛ4 genotype. Brandt J, editor. J. Alzheimers Dis. 2016;52:849–61.
Loring DW, Goldstein FC, Chen C, Drane DL, Lah JJ, Zhao L, et al. False-positive error rates for reliable digit span and auditory verbal learning test performance validity measures in amnestic mild cognitive impairment and early Alzheimer disease. Arch Clin Neuropsychol. 2016;31:313–31.
Dautzenberg FM, Hauger RL. The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci. 2002;23:71–7.
Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM. Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann N Y Acad Sci. 2009;1179:120–43.
Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol. Disord.-Drug targets former. Curr. Drug targets-CNS Neurol. Disord. 2006;5:453–79.
Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, et al. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. In: Terjung R, editor. Compr. Physiol. [Internet]. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2016 [cited 2016 Sep 30]. p. 603–21. Available from: http://doi.wiley.com/10.1002/cphy.c150015.
Rivier C, Vale W. Modulation of stress-induced ACTH release by corticotropin-releasing factor, catecholamines and vasopressin. Nature. 1983;305:325–7.
Potter E, Sutton S, Donaldson C, Chen R, Perrin M, Lewis K, et al. Distribution of corticotropin-releasing factor receptor mRNA expression in the rat brain and pituitary. Proc Natl Acad Sci U S A. 1994;91:8777–81.
Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;15:6340–50.
Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, et al. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212.
Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM, et al. Glutamatergic and dopaminergic neurons mediate Anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333:1903–7.
Sapolsky RM. Stress and the brain: individual variability and the inverted-U. Nat Neurosci. 2015;18:1344–6.
Bamberger CM, Bamberger A. The peripheral CRH/urocortin system. Ann N Y Acad Sci. 2000;917:290–6.
Inda C, dos Santos Claro PA, Bonfiglio JJ, Senin SA, Maccarrone G, Turck CW, et al. Different cAMP sources are critically involved in G protein–coupled receptor CRHR1 signaling. J Cell Biol. 2016;214:181–95.
Agostini M, Fasolato C. When, where and how? Focus on neuronal calcium dysfunctions in Alzheimer’s Disease. Cell Calcium [Internet]. 2016 [cited 2016 Oct 11]; Available from: http://linkinghub.elsevier.com/retrieve/pii/S0143416016300999.
Hoare SRJ, Sullivan SK, Schwarz DA, Ling N, Vale WW, Crowe PD, et al. Ligand affinity for amino-terminal and Juxtamembrane domains of the Corticotropin releasing factor type I receptor: Regulation by G-protein and Nonpeptide antagonists. Biochemistry (Mosc). 2004;43:3996–4011.
Höschl C, Hajek T. Hippocampal damage mediated by corticosteroids—a neuropsychiatric research challenge. Eur Arch Psychiatry Clin Neurosci. 2001;251:81–8.
Sousa N, Lukoyanov NV, Madeira MD, Almeida OFX, Paula-Barbosa MM. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience. 2000;97:253–66.
Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–45.
Pardon M-C, Rattray I. What do we know about the long-term consequences of stress on ageing and the progression of age-related neurodegenerative disorders? Neurosci Biobehav Rev. 2008;32:1103–20.
Landfield PW, Blalock EM, Chen K-C, Porter NM. A new glucocorticoid hypothesis of brain aging: implications for Alzheimer’s disease. Curr Alzheimer Res. 2007;4:205–12.
Popp J, Wolfsgruber S, Heuser I, Peters O, Hüll M, Schröder J, et al. Cerebrospinal fluid cortisol and clinical disease progression in MCI and dementia of Alzheimer’s type. Neurobiol Aging. 2015;36:601–7.
Ennis GE, An Y, Resnick SM, Ferrucci L, O’Brien RJ, Moffat SD. Long-term cortisol measures predict Alzheimer disease risk. Neurology. 2016:10–1212.
Green KN. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26:9047–56.
Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, et al. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiatry. 2009;14:95–105.
Pinheiro S, Silva J, Mota C, Vaz-Silva J, Veloso A, Pinto V, et al. Tau Mislocation in glucocorticoid-triggered hippocampal pathology. Mol Neurobiol. 2016;53:4745–53.
Sotiropoulos I, Catania C, Pinto LG, Silva R, Pollerberg GE, Takashima A, et al. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J Neurosci. 2011;31:7840–7.
Baglietto-Vargas D, Medeiros R, Martinez-Coria H, LaFerla FM, Green KN. Mifepristone alters amyloid precursor protein processing to preclude amyloid Beta and Also reduces tau pathology. Biol Psychiatry. 2013;74:357–66.
Näsman B, Olsson T, Fagerlund M, Eriksson S, Viitanen M, Carlström K. Blunted adrenocorticotropin and increased adrenal steroid response to human corticotropin-releasing hormone in Alzheimer’s disease. Biol Pyschiatry. 1996;39:311–8.
Bernardi F, Lanzone A, Cento RM, Spada RS, Pezzani I, Genazzani AD, et al. Allopregnanolone and dehydroepiandrosterone response to corticotropin-releasing factor in patients suffering from Alzheimer’s disease and vascular dementia. Eur J Endocrinol. 2000;142:466–71.
Dong H, Wang S, Zeng Z, Li F, Montalvo-Ortiz J, Tucker C, et al. Effects of corticotrophin-releasing factor receptor 1 antagonists on amyloid-β and behavior in Tg2576 mice. Psychopharmacology. 2014;231:4711–22.
Park H-J, Ran Y, Jung JI, Holmes O, Price AR, Smithson L, et al. The stress response neuropeptide CRF increases amyloid- production by regulating -secretase activity. EMBO J. 2015;34:1674–86.
Chen Y, Peng Y, Che P, Gannon M, Liu Y, Li L, et al. α 2A adrenergic receptor promotes amyloidogenesis through disrupting APP-SorLA interaction. Proc. Natl. Acad. Sci. 2014;111:17296–301.
Thathiah A, Horré K, Snellinx A, Vandewyer E, Huang Y, Ciesielska M, et al. β-arrestin 2 regulates Aβ generation and γ-secretase activity in Alzheimer’s disease. Nat. Med. 2012;19:43–9.
Kang JE, Cirrito JR, Dong H, Csernsnsky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc Natl Acad Sci. 2007;104:10673–8.
Dong H, Yuede CM, Yoo H-S, Martin MV, Deal C, Mace AG, et al. Corticosterone and related receptor expression are associated with increased β-amyloid plaques in isolated Tg2576 mice. Neuroscience. 2008;155:154–63.
Dong H, Murphy KM, Meng L, Montalvo-Ortiz J, Zeng Z, Kolber BJ, et al. Corticotrophin releasing factor accelerates neuropathology and cognitive decline in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2012;28:579–92.
Ray B, Gaskins DL, Sajdyk TJ, Spence JP, Fitz SD, Shekhar A, et al. Restraint stress and repeated corticotrophin-releasing factor receptor activation in the amygdala both increase amyloid-β precursor protein and amyloid-β peptide but have divergent effects on brain-derived neurotrophic factor and pre-synaptic proteins in the prefrontal cortex of rats. Neuroscience. 2011;184:139–50.
Campbell SN, Zhang C, Roe AD, Lee N, Lao KU, Monte L, et al. Impact of CRFR1 ablation on amyloid-β production and accumulation in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2015;45:1175–84.
Iqbal K, Liu F, Gong C-X. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol. 2015;12:15–27.
Rothman SM, Herdener N, Frankola KA, Mughal MR, Mattson MP. Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Aβ and pTau in a mouse model of Alzheimer’s disease. Brain Res. 2013;1529:200–8.
Carroll JC, Iba M, Bangasser DA, Valentino RJ, James MJ, Brunden KR, et al. Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a Corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of Tauopathy. J Neurosci. 2011;31:14436–49.
Rissman RA, Lee K-F, Vale W, Sawchenko PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. 2007;27:6552–62.
Campbell SN, Zhang C, Monte L, Roe AD, Rice KC, Taché Y, et al. Increased tau phosphorylation and aggregation in the hippocampus of mice overexpressing corticotropin-releasing factor. J Alzheimers Dis. 2015;43:967–76.
Filipcik P, Novak P, Mravec B, Ondicova K, Krajciova G, Novak M, et al. Tau protein phosphorylation in diverse brain areas of normal and CRH deficient mice: up-Regulation by stress. Cell Mol Neurobiol. 2012;32:837–45.
Le MH, Weissmiller AM, Monte L, Lin PH, Hexom TC, Natera O, et al. Functional Impact of Corticotropin-Releasing Factor Exposure on Tau Phosphorylation and Axon Transport. Ryabinin AE, editor. PLOS ONE. 2016;11:e0147250.
Rissman RA, Staup MA, Lee AR, Justice NJ, Rice KC, Vale W, et al. Corticotropin-releasing factor receptor-dependent effects of repeated stress on tau phosphorylation, solubility, and aggregation. Proc Natl Acad Sci. 2012;109:6277–82.
Kvetnansky R, Novak P, Vargovic P, Lejavova K, Horvathova L, Ondicova K, et al. Exaggerated phosphorylation of brain tau protein in CRH KO mice exposed to repeated immobilization stress. Stress. 2016;19:395–405.
Kehne JH, Cain CK. Therapeutic utility of non-peptidic CRF1 receptor antagonists in anxiety, depression, and stress-related disorders: evidence from animal models. Pharmacol Ther. 2010;128:460–87.
Lowery EG, Thiele TE. Pre-clinical evidence that corticotropin-releasing factor (CRF) receptor antagonists are promising targets for pharmacological treatment of alcoholism. CNS Neurol. Disord.-Drug targets former. Curr. Drug targets-CNS Neurol. Disord. 2010;9:77–86.
Kwako LE, Spagnolo PA, Schwandt ML, Thorsell A, George DT, Momenan R, et al. The corticotropin releasing hormone-1 (CRH1) receptor antagonist pexacerfont in alcohol dependence: a randomized controlled experimental medicine study. Neuropsychopharmacology. 2015;40:1053–63.
Zhang C, Kuo C-C, Moghadam SH, Monte L, Campbell SN, Rice KC, et al. Corticotropin-releasing factor receptor-1 antagonism mitigates beta amyloid pathology and cognitive and synaptic deficits in a mouse model of Alzheimer’s disease. Alzheimers Dement. 2016;12:527–37.
Contoreggi C, Lee MR, Chrousos G. Addiction and corticotropin-releasing hormone type 1 receptor antagonist medications: Antistress drugs as treatments for addiction. Ann N Y Acad Sci. 2013;1282:107–18.
Spierling SR, Zorrilla EP. Don’t stress about CRF: assessing the translational failures of CRF1antagonists. Psychopharmacology (Berl.) [Internet]. 2017 [cited 2017 Mar 12]; Available from: http://link.springer.com/10.1007/s00213-017-4556-2.
Réus GZ, Titus SE, Abelaira HM, Freitas SM, Tuon T, Quevedo J, et al. Neurochemical correlation between major depressive disorder and neurodegenerative diseases. Life Sci. 2016;158:121–9.
Bao A-M, Meynen G, Swaab DF. The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res Rev. 2008;57:531–53.
Austin KW, Ameringer SW, Cloud LJ. An integrated review of psychological stress in Parkinson’s disease: biological mechanisms and symptom and health outcomes. Park Dis. 2016;2016:1–15.
Hou G, Tian R, Li J, Yuan T-F. Chronic stress and Parkinson’s disease. CNS Neurosci Ther. 2014;20:1–2.
Smith LK, Jadavji NM, Colwell KL, Katrina Perehudoff S, Metz GA. Stress accelerates neural degeneration and exaggerates motor symptoms in a rat model of Parkinson’s disease. Eur J Neurosci. 2008;27:2133–46.
Lauretti E, Di Meco A, Merali S, Praticò D. Chronic behavioral stress exaggerates motor deficit and neuroinflammation in the MPTP mouse model of Parkinson’s disease. Transl Psychiatry. 2016;6:e733.
Kibel A, Drenjančević-Perić I. Impact of glucocorticoids and chronic stress on progression of Parkinson’s disease. Med Hypotheses. 2008;71:952–6.
Wu Q, Yang X, Zhang Y, Zhang L, Feng L. Chronic mild stress accelerates the progression of Parkinson’s disease in A53T α-synuclein transgenic mice. Exp Neurol. 2016;285:61–71.
Sugama S, Kakinuma Y. Loss of dopaminergic neurons occurs in the ventral tegmental area and hypothalamus of rats following chronic stress: possible pathogenetic loci for depression involved in Parkinson’s disease. Neurosci Res. 2016;111:48–55.
Sugama S, Sekiyama K, Kodama T, Takamatsu Y, Takenouchi T, Hashimoto M, et al. Chronic restraint stress triggers dopaminergic and noradrenergic neurodegeneration: possible role of chronic stress in the onset of Parkinson’s disease. Brain Behav Immun. 2016;51:39–46.
Mo C, Renoir T, Pang TYC, Hannan AJ. Short-term memory acquisition in female Huntington’s disease mice is vulnerable to acute stress. Behav Brain Res. 2013;253:318–22.
Mo C, Renoir T, Hannan AJ. Effects of chronic stress on the onset and progression of Huntington’s disease in transgenic mice. Neurobiol Dis. 2014;71:81–94.
Kronenberg G, Schöner J, Nolte C, Heinz A, Endres M, Gertz K. Charting the perfect storm: emerging biological interfaces between stress and stroke. Eur. Arch. Psychiatry Clin. Neurosci. [Internet]. 2017 [cited 2017 May 13]; Available from: http://link.springer.com/10.1007/s00406-017-0794-x.
Stuller KA, Jarrett B, DeVries AC. Stress and social isolation increase vulnerability to stroke. Exp Neurol. 2012;233:33–9.
van der Linde RM, Dening T, Stephan BCM, Prina AM, Evans E, Brayne C. Longitudinal course of behavioural and psychological symptoms of dementia: systematic review. Br J Psychiatry. 2016;209:366–77.
Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102.
Lee K-W, Kim J-B, Seo J-S, Kim T-K, Im J-Y, Baek I-S, et al. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem. 2009;108:165–75.
Huang H-J, Liang K-C, Ke H-C, Chang Y-Y, Hsieh-Li HM. Long-term social isolation exacerbates the impairment of spatial working memory in APP/PS1 transgenic mice. Brain Res. 2011;1371:150–60.
Borchelt DR, Ratovitski T, Van Lare J, Lee MK, Gonzales V, Jenkins NA, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–45.
Rothman SM, Herdener N, Camandola S, Texel SJ, Mughal MR, Cong W-N, et al. 3xTgAD mice exhibit altered behavior and elevated Aβ after chronic mild social stress. Neurobiol. Aging. 2012;33:830.e1–830.e12.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–21.
Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW. Overproduction of corticotropin-releasing factor in transgenic mice: a genetic model of anxiogenic behavior. J Neurosci. 1994;14:2579–84.
Baglietto-Vargas D, Chen Y, Suh D, Ager RR, Rodriguez-Ortiz CJ, Medeiros R, et al. Short-term modern life-like stress exacerbates Aβ-pathology and synapse loss in 3xTg-AD mice. J Neurochem. 2015;134:915–26.
Justice NJ, Huang L, Tian J-B, Cole A, Pruski M, Hunt AJ, et al. Posttraumatic stress disorder-like induction elevates -amyloid levels, which directly activates Corticotropin-releasing factor neurons to exacerbate stress responses. J Neurosci. 2015;35:2612–23.
Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S Tauopathy mouse model. Neuron. 2007;53:337–51.
Acknowledgements
Hyo-Jin Park (University of Florida) provided the basis for this work within the lab.
Funding
NIH U01AG046139, R01AG018454, P50AG047266
Availability of data and materials
Not applicable.
Authors’ contributions
HF did the majority of the appropriate literature search and manuscript production. VT edited the manuscript and provided illustrations. CC edited the manuscript providing tau protein expertise. EK edited the manuscript providing HPA axis expertise. TG extensively edited the manuscript providing AD expertise. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Futch, H.S., Croft, C.L., Truong, V.Q. et al. Targeting psychologic stress signaling pathways in Alzheimer’s disease. Mol Neurodegeneration 12, 49 (2017). https://doi.org/10.1186/s13024-017-0190-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-017-0190-z