
Abstract
Pulmonary alveolar microlithiasis (PAM) is a rare autosomal recessive lung disease caused by variants in the SLC34A2 gene encoding the sodium-dependent phosphate transport protein 2B, NaPi-2b. PAM is characterized by deposition of calcium phosphate crystals in the alveoli. Onset and clinical course vary considerably; some patients remain asymptomatic while others develop severe respiratory failure with a significant symptom burden and compromised survival. It is likely that PAM is under-reported due to lack of recognition, misdiagnosis, and mild clinical presentation. Most patients are genetically uncharacterized as the diagnostic confirmation of PAM has traditionally not included a genetic analysis. Genetic testing may in the future be the preferred tool for diagnostics instead of invasive methods. This systematic review aims to provide an overview of the growing knowledge of PAM genetics. Rare variants in SLC34A2 are found in almost all genetically tested patients. So far, 34 allelic variants have been identified in at least 68 patients. A majority of these are present in the homozygous state; however, a few are found in the compound heterozygous form. Most of the allelic variants involve only a single nucleotide. Half of the variants are either nonsense or frameshifts, resulting in premature termination of the protein or decay of the mRNA. There is currently no cure for PAM, and the only effective treatment is lung transplantation. Management is mainly symptomatic, but an improved understanding of the underlying pathophysiology will hopefully result in development of targeted treatment options. More standardized data on PAM patients, including a genetic diagnosis covering larger international populations, would support the design and implementation of clinical studies to the benefit of patients. Further genetic characterization and understanding of how the molecular changes influence disease phenotype will hopefully allow earlier diagnosis and treatment of the disease in the future.
Similar content being viewed by others

Background
Pulmonary alveolar microlithiasis (PAM) (OMIM #265100) is an autosomal recessive lung disease where calcium-phosphate concretions (microliths) are formed in the alveoli [1,2,3]. PAM was first named by the Hungarian physician Ludwig Puhr in 1933 [4]. It is caused by variants in the SLC34A2 gene (Entrez Gene ID 10568) encoding the sodium-dependent phosphate transport protein 2B, NaPi-2b [5,6,7]. The protein belongs to the sodium-transporter family SLC34, which is involved in the inorganic phosphate (Pi) homeostasis [8]. The incidence of PAM is unknown. Less than 1200 patients are described in the literature, and most descriptions are from Asia and Europe. Both familial and sporadic cases are reported. In almost all families with PAM, transmission is reported to be horizontal. In the rare case of vertical transmission, this has always been a result of consanguinity [9]. Although almost all patients in the literature who have been genetically evaluated have pathogenic variants in SLC34A2, genetic testing is not part of the routine diagnostic evaluation. However, genetic investigation is increasingly recommended [1]. This review will provide an overview of PAM with a specific focus on underlying genetic aspects.
Search strategy
A structured literature search for the genetic part of the review was performed according to preferred reporting items for systematic reviews and meta-analyses (PRISMA) 2009 guidelines [10]. The following online reference databases were used: Embase, PubMed, SCOPUS, Cochrane, and Web of Science. Searches were carried out in August 2022. The search terms used were 'pulmonary alveolar microlithiasis' AND 'SLC34A2'. Additionally, a search in The Human Gene Mutation Database (HGMD) Professional was performed in August 2022 (HGMD Professional 2022.2) [11]. Furthermore, additional articles were identified from reference lists of studies included in this review and from existing reviews.
Study selection process
The literature search yielded a total of 287 citations. Removal of duplicates, resulted in a total of 115 citations for possible inclusion. Titles and abstracts of these citations were screened by one reviewer to remove obviously irrelevant studies. One Japanese report with no abstract in English, French, or German was excluded. A conference abstract with a subsequent publication from the same authors regarding the same patients was also excluded. A total of 34 studies, including 29 original reports and five abstracts (four conference abstracts and one English abstract of a Chinese-language study), were included in the genetic part of the review (Fig. 1).

Flow diagram of inclusion of literature. A structured literature search for the part of the review concerning the spectrum of allelic variants in SLC34A2 was performed according to preferred reporting items for systematic reviews and meta-analyses (PRISMA) 2009 guidelines [10]
Diagnosis, clinical characteristics and treatment
Currently, PAM is diagnosed based on the typical radiographic appearance and detection of characteristic microliths in the bronchoalveolar lavage (BAL) fluid or a lung biopsy [1, 2, 9]. The microliths are comprised of calcareous concentric, laminated bodies typically less than 1 mm in diameter and predominantly formed of calcium and phosphorus [2]. Additional accompanying features are inflammation, fibrosis, and calcification of the lung interstitium [9, 12]. The pathophysiology of PAM is not yet fully understood. It has been proposed that the deposition of the microliths in the alveolus is caused by accumulation of phosphate from degraded surfactant phospholipids [6, 13]. Normally, phosphate will be cleared from the alveolar space by transport via NaPi-2b located in the apical membrane of the alveolar type II cell. When the transporter does not work properly, this leads to an excess of phosphate in the alveolar lumen with subsequent precipitation of extracellular calcium (Fig. 2) [5, 6, 13].

Presumed pathophysiology of PAM. Alveolar type II cell in the alveolus of the lung. Dysfunctional sodium-phosphate co-transporter (NaPi-2b) in the apical membrane leading to a decreased cell uptake of phosphate from the alveolar space and deposition of calcium-phosphate stones (microliths) due to chelation
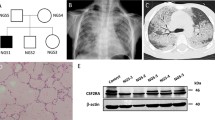
Although PAM is diagnosed at all ages, most patients are diagnosed between 10 and 30 years of age [9]. Many patients are diagnosed incidentally or in connection with familial investigations. Dyspnoea, dry cough, fatigue, and chest pain are frequent complains in symptomatic patients. Pneumothorax, clubbing, haemoptysis, hypoxia, and cyanosis have been reported [2, 3, 9, 14,15,16]. Lung function is usually normal or has a restrictive pattern [2]. PAM is generally slowly progressing, but a milder or more aggressive course might be observed [9]. The radiographic appearance is often pronounced and disproportionate to the clinical severity [15, 17]. A chest X-ray typically shows a sand-like pattern corresponding to calcifications with bilateral basal and middle zone predilection. Numerous miliary calcified nodules distributed throughout the lungs are seen on high-resolution computed tomography (HRCT) (Fig. 3) [1, 2]. The radiographic appearance is very characteristic, and in cases with typical HRCT findings, a lung biopsy is not needed to establish the diagnosis [18].

High-resolution computed tomography (HRCT) showing classical findings suggestive of pulmonary alveolar microlithiasis. A. Multiple microcalcifications, axial plane. B. Multiple microcalcifications and septal thickening (arrow), coronal plane
Extrapulmonary calcifications have been reported in PAM and may reflect a syndrome rather than a restricted lung disease [2, 3, 19,20,21,22,23,24,25,26,27,28]. Although the frequency of extrapulmonary manifestations is unknown, it is reasonable to hypothesize that this is not an uncommon finding as SLC34A2 is expressed in tissues other than lung tissue [23, 29,30,31].
To date, no effective treatment exists except lung transplantation [9]. A few case studies report beneficial effects of the bisphosphonate, etidronate, while others report no benefit of the treatment [2, 32,33,34,35]. Use of systemic corticosteroids is generally not considered to be effective, although symptomatic improvement has been reported in a few cases [24, 36,37,38]. Besides, therapeutic BAL has proven ineffective, although symptomatic improvement has been described in one case [36, 39,40,41]. Supplemental oxygen therapy should be considered in hypoxic patients. Long-term follow-up data in PAM are sparse and the prognosis is thus unknown. However, current data indicate a poor long-term prognosis [42]. Several environmental factors such as smoking, inhalation of snuff, repetitive lung infections, and cold weather have been proposed to negatively influence course of the disease [3, 5, 43, 44].
Etiology
SLC34A2: genetic aspects
SLC34A2 is located on the short arm of chromosome 4 (4p15.2). It contains 13 exons, of which the first seems to exist in several alternative versions, all non-coding. SLC34A2 encodes a protein (NaPi-2b) of 690 amino acids. The gene is highly conserved in bony vertebrates and variants are therefore likely to affect the protein functionally [7, 45,46,47]. The expression at the protein level has mainly been investigated in animals. In addition to lung tissue, the expression at the protein level has also been found in tissues such as the small intestine, the mammary glands, the liver, the bile duct, and in the epididymis [48,49,50,51,52]. In addition, SLC34A2 is expressed on the surface of different cancer types, and is a known ROS1 (ROS proto-oncogene 1, receptor tyrosine kinase) fusion partner in non-small cell lung cancer [53,54,55].
The sodium-phosphate transporter NaPi-2b
NaPi-2b (NP_006415) is a member of the transporter family SLC34, which includes the protein isoforms NaPi-2a (encoded by SLC34A1) and NaPi-2c (encoded by SLC34A3). This protein family is essential for maintaining Pi homeostasis in the human body where regulation is mediated by the intestine (NaPi-2b) and kidney (NaPi-2a, NaPi-2c) [8, 56]. NaPi-2a and NaPi-2b are both electrogenic co-transporters with a 3:1 (Na+: Pi) stoichiometry, whereas NaPi-2c is electroneutral with a 2:1 (Na+: Pi) stoichiometry [57]. The crystallographic structure has not been determined for any of the family members, not even the bacterial homologs. Thus, the present knowledge of structure and function is mainly based on indirect studies on wild-type and designed variants with different biophysical and biochemical methods [8]. The SLC34 group of eukaryotic transporters is presumed to have identical transmembrane (TM) topology [58,59,60]. The predicted topological model of the isoforms consists of 12 TM domains including two inverted repeated regions, a large extracellular loop with two N-glycosylation sites and a disulfide bridge linking the two halves of the protein, and with both C- and N-terminal regions located intracellularly. The TM domains 3–4 and 8–9 are presumed to form a substrate coordination site. Important areas for regulation and targeting are located at the C-terminal region and in the area between TM domain 10 and 11. A critical region for electrogenicity is located between TM domain 4 and 5 [8]. Recently, a three-dimensional structural model has been developed of the human NaPi-2 with the topology of the bacterial dicarboxylate co-transporter VcINDY as a template [61, 62].
Regulation of NaPi-2b expression
NaPi-2b expression is regulated by several factors (reviewed in Hernando et al. 2018 [63]). The expression in the intestine depends on dietary Pi levels with an increased level of expression in the intestinal epithelia when the dietary levels decrease [64, 65]. Interestingly, the expression of NaPi-2b in the alveolar type II cells is seemingly not influenced by dietary intake of phosphate [48]. In the intestine, NaPi-2b expression is up-regulated by estrogen, vitamin D3, and during metabolic acidosis, and the expression is suppressed by glucocorticoids, epidermal growth factor (EGF), and when the vitamin D receptor (VDR) is lacking [64, 66,67,68,69,70]. In addition, dexamethasone has been shown to down-regulate mRNA expression of NaPi-2b and decrease the uptake of phosphate in cultured alveolar type II cells from rats [71]. Contrary to this, NaPi-2b in rat lung was found not to be regulated at the mRNA level by the vitamin D analog ED-71 (1α, 25-dihydroxy-2ß-(3-hydroxypropoxy) vitamin D3) [72].
The spectrum of allelic variants in PAM
In 2006, variants in SLC34A2 were initially identified as causative for PAM [5, 6]. Since then, 34 allelic variants have been documented in the literature in at least 68 patients (49 families) [2, 3, 5, 6, 35, 43, 73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94] (Fig. 4, Table 1). Only around 5% of the patients reported have been genetically investigated. However, pathogenic allelic variants in SLC34A2 were found in more than 95% of these patients or families. In three siblings with PAM, a variant was reported in exon 2 within a sequence that, to the best of our knowledge, is not located in the coding regions of SLC34A2 [102]. Thus, this variant is not further included in this review. Genetically unresolved cases have been reported and reports have been published on a few patients without variants in SLC34A2 [103,104,105]. In one of these patients, only one pathogenic variant on a single allele was reported [105]. In addition, a cytogenetic study in a patient with myelofibrosis revealed a rearrangement of the long arms of chromosomes 4 and 5; this patient was subsequently diagnosed with PAM [106]. More efforts must be made to clarify which genetic alterations contribute to disease in these patients as the method chosen to analyse SLC34A2 may not have been sufficient. If the genetic region sequenced is restricted only to the coding part and intron–exon boundaries, variants in introns or in the promoter region may be overlooked. In addition, the detection of larger deletions requires another analytic approach.

Allelic variants in SLC34A2 in PAM patients reported in the literature [2, 3, 5, 6, 35, 43, 73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94]. Red small squares represent the number of families in which the individual variants are found. Narrow box for non-coding exon and wider box for coding exon. Exons, introns, and deletions are not drawn to scale. Variants are present in homozygous form unless otherwise stated
We performed an evaluation of the allelic variants previously reported in several standard computational prediction tools [97,98,99,100,101]. All analyzable variants were predicted to be deleterious by at least one prediction tool, which further supports the pathogenicity of the variants (Table 1). Five larger deletions are reported including whole gene deletion, a deletion spanning exon 2–6, a deletion including exon 5, a deletion spanning the last part of intron 5 and the first third of exon 6, and a 186-nucleotide deletion involving the promoter and exon 1. In addition, three splice site variants have been found in intron 5, 9 and in intron 11, respectively. Splice site variants and larger deletions most likely lead to loss of function or truncation of the protein with a decreased protein activity. Most variants (15/34) are either nonsense or frameshifts (Fig. 5), resulting in premature termination of the protein or probable decay of the mRNA, subsequently without any protein formation. The variants are distributed throughout the entire gene. Half of the missense variants and an in-frame deletion are located in presumably functionally important areas of the protein, likely leading to protein damage (Fig. 6).

Types of allelic variants reported in PAM. Splice site 9% (3 variants), nonsense 24% (8 variants), missense 29% (10 variants), large deletion 15% (5 variants), in-frame deletion 3% (1 variant), and frameshift 21% (7 variants)

Allelic variants in SLC34A2 in the literature marked on a model of NaPi-2b. Splice site variants and larger deletions (c.-6773_-6588del, the 5.5 kb deletion involving exons 2–6, c.380-345_ c.523+659del, c.524-18_559del, and the whole gene deletion) are not included in the figure. All variants are shown in the figure as dots. Light blue: missense variant, red: nonsense variant, dark blue: frameshift variant, yellow: in-frame deletion. The transmembrane (TM) domains with red color (TM domains 3–4 and 8–9) form the substrate coordination site. Areas for electrogenicity, regulation and targeting are found in the area between TM domains 4–5, 10–11, and at the C-terminal region [8, 107]. The model is made by superimposing human NaPi-2b on rat NaPi-2a predicted topology and is modified from Forster et al. 2013 [8] and Virkki et al. 2007 [96]. The protein sequences used for alignment in Clustal Omega version 1.2.4 [108]: Ensembl Transcript ID ENST00000382051.8 (Human (GRCh38.p13) assembly) and Ensembl Transcript ID ENSRNOT00000033749.6 (Rat (Rnor_6.0) assembly)
Exon 12 is most frequently involved, and almost one-third (10/34) of the allelic variants are found in the same genomic area within 129 nucleotides in exons 11–12 and intron 11. Two variants (c.1402_1404delACC and c.1390G>C) are located in or nearby four 3-nucleotide (ACC) tandem repeats, which may predispose replication errors. Furthermore, two allelic variants (c.316G>C and c.316G>A) result in the same change at the protein level (p.Gly106Arg), and two variants (c.1327delC and c.1328delT) affect another amino acid (Leu443). The amino acid positions 106 and 443 may therefore represent other hot spots for pathogenic variants in NaPi-2b.
In almost all patients, the identified variants were in the homozygous state. Only four cases are described with variants in the compound heterozygous state (combining c.316G>A and c.1238G>A, c.448G>A and c.524-1G>C, c.910A>T and c.1363T>C, and c.1048 + 1G>A and c.1390G>C) [3, 79, 81, 94]. Strikingly, these combinations of variants consist of a missense variant with either nonsense or a splice site variant on the other allele. Some of these variants have previously been described in a homozygous state in several patients [3, 6, 80, 83, 109]. A patient was reported with only one pathogenic allelic variant [105], which alone is unlikely to explain the genetic cause of the disease since PAM is considered to follow a recessive inheritance pattern and disease has never been reported in carriers.
So far, the majority (22/34) of the allelic variants have only been reported in a single patient or in one family. Four variants (c.226C>T, c.910A>T, c.1048 + 1G>A, and c.1402_1404delACC) have been described in three to five unrelated patients/families (Fig. 4, Table 2). So far, c.226C>T is only found in patients of Middle Eastern origin, c.910A>T in Chinese patients, c.1048 + 1G>A in Japanese patients, and c.1402_1404delACC in patients of European origin.
Demographics and clinical data in patients with SLC34A2 variants
Generally, patients reported with SLC34A2 variants present with typical features of PAM, including e.g., variability in age, symptoms, and clinical findings, although detailed clinical data are missing in many reports. Table 2 summarizes patient demographics, symptoms, and smoking status. Patients reported with variants come from countries all over the world, and most were adults (Fig. 7). The age span was 9 months to 69 years with a slight female predominance. The presence of variants was a consequence of consanguineous marriages in 63% of the families. This may be an underestimation as there was no information on consanguinity in approximately 40% of the families.

Documented PAM cases with known SLC34A2 variants [2, 3, 5, 6, 43, 73, 74, 77,78,79,80,81,82, 84,85,86, 88,89,90,91,92,93,94]. *In one American report, no information was available regarding country of origin and number of patients [85]. The thickness of the arrows is proportional with the number of patients
Twenty-two patients were children at time of diagnosis, and they were most often diagnosed in a familial setting or incidentally, and the diagnosis was almost exclusively based on BAL or biopsy. Approximately half of the children (9/19) were asymptomatic with normal lung function (60% (9/15)). Radiological abnormalities were reported in all children, but only around half of the reports described calcifications. Among the reports including information on age, 68% (26/38) of the adults were symptomatic. Almost 90% (22/25) of the adults had abnormal lung function. All reported radiological findings were typical for PAM and in around 20% (7/32), the diagnosis was based on radiographic findings only.
Genotype–phenotype correlation
It remains to be explored whether there is a genotype–phenotype correlation in PAM. Functional studies exploring the effect of human SLC34A2 variants are sparse, and there is no standardized criterion for clinical classification. In our recent report including 14 PAM patients, an association between disease and variant severity was found. Although an association was found, we highlight the challenge of proper classification of disease and variants and the need for confirmation in a larger number of patients [3]. Only a few case reports have been published describing patients and families with recurrent variants (Table 2). Generally, it is difficult to compare the clinical data in the case reports as the descriptions are not standardized. Furthermore, the age of the patients varied considerably and asymptomatic children and young adults may develop symptoms later in life, which complicates the phenotype evaluation in these patients. Even though data are scarce, smoking might be associated with more severe disease [3, 5, 78, 81].
Functional studies of SLC34A2 variants
Two SLC34A2 variants identified in six Japanese patients (c.1048 + 1G>A (p.?) and c.857_871delins19 (p.Ile286LysfsTer29) were found to impair phosphate transport in the presence of sodium when expressed in Xenopus laevis oocytes [6]. Recently, our group investigated four SLC34A2 variants previously reported in PAM patients (c.910A>T (p.Lys304Ter), c.1328delT (p.Leu443ArgfsTer6), c.1402_1404delACC (p.Thr468del), and c.1456C>T (p.Gln486Ter)). NaPi-2b mutant constructs were expressed in Xenopus laevis oocytes, and transport function was investigated with a 32Pi uptake assay. All mutants were found non-functional [114]. Interestingly, two previous studies of the rat and the human NaPi-2a expressed in Xenopus laevis oocytes included mutants at the same amino acid positions as c.1390G>C (p.Gly464Arg) and c.1402_1404delACC (p.Thr468del), which was later described in PAM patients [62, 115]. Amino acid substitutions with cysteine, using the substituted cysteine accessibility method (SCAM) [115] or alanine substitution [62] revealed non-functional mutants, except when the threonine corresponding to Thr467 in human NaPi-2b was substituted with cysteine. In addition, the variant c.575C>A (p.Thr192Lys) found in a Chinese family was investigated in human alveolar epithelial cells (A549 cells) and revealed signs of reduced phosphate transport function compared to normal controls [116]. Generally, data from these reports support the underlying dysfunction of NaPi-2b in PAM.
Animal models in PAM
Several conditional Slc34a2 knock-out (KO) models have been developed and have provided important knowledge of possible compensatory mechanisms of lost active Na+-dependent phosphate transport [51, 52, 117, 118]. In a study with a conditional Slc34a2 KO mouse model in the lung epithelium, a PAM phenotype with progressive radiographic lung manifestations including microlith accumulation, inflammation, and fibrosis was reported. The Slc34a2 KO mice showed no clear compensatory up-regulation of other sodium-phosphate co-transporters. However, expression of the sodium-dependent phosphate transporter Pit-1 (Slc20a1) was found slightly increased in alveolar type II cells of Slc34a2 KO mice on a low-phosphate diet. There was also evidence of microlith burden reduction in the mice during phosphate-restrictive diet. When measuring levels of calcium, phosphate, total protein, SP-D, and saturated phosphatidylcholine, which is a major component of pulmonary surfactant, all the parameters were increased in the BAL fluid of Slc34a2 KO mice compared to normal mice. Furthermore, serum SP-D and inflammatory mediating cytokine MCP-1 (monocyte chemotactic protein 1) were higher in NaPi-2b deficient mice compared to control mice, and it increased with the progression of microlith deposition. A month after microliths from Slc34a2 KO mice were instilled into the lungs of normal mice, the microcalcifications cleared completely, without any evidence of inflammation or fibrosis. The serum level of MCP-1 in these mice reached baseline at the end of the time-period suggesting MCP-1 as a potential biomarker of disease burden. Based on data from this study, the authors concluded that gene editing of NaPi-IIb expression in the lung may be a promising future therapeutic strategy in PAM [118].
Current gaps in understanding of PAM
The discovery of SLC34A2 as the causative gene in PAM has brought us a step closer to understand this heterogeneous disease, although the pathophysiology is not yet clear.
Further studies, including investigations of pathogenic variants in SLC34A2 in cells and animal models, are needed to explore the basic mechanisms of the disease. Investigation of underlying factors, including possible compensatory mechanisms such as mediation of phosphate by other transporters in the alveolar type II cell, is necessary and possible involvement of environmental factors should be explored. Furthermore, the few patients without SLC34A2 variants should be further evaluated to identify alternative genetic causes.
Except for lung transplantation, no cure or effective treatment is currently available in PAM. Neither variant-specific therapy involving e.g., systemically, or locally administered agents that could increase the quantity and the function of NaPi-2b in the lungs nor gene therapy, either via gene addition or clustered regularly interspaced short palindromic repeats (CRISPR)-based gene editing, has been tested in patients with PAM. In patients without advanced disease, gene therapy could possibly cure the patients as it is expected to be persistent in the whole lifespan of the recipient cells. You could speculate that patients with a high disease burden may not benefit from gene therapy to the same extent. In any case, detailed knowledge about the molecular consequences of the different variants identified in patients with PAM is required to be able to treat successfully based on these techniques.
To be able to develop a molecular classification, genetic testing should be performed in more patients, and the spectrum of variants should be evaluated for distinct function and distribution patterns. It is essential to explore and characterize variants in patients and compare these findings to careful clinical characterization of patients. A systematic detailed description of patient data in case reports is recommended and should, in addition to symptoms and clinical findings, include disease course, medical history, and presence of extrapulmonary calcifications, family history, consanguinity, smoking status and other possible triggers. It would indeed be desirable to have a validated disease severity classification, which would be helpful to assess disease burden, stratify patients, and to perform research. Clinical research would also benefit from an international PAM database including de-identified clinical, genetic, and demographic data.
Genetic counseling
Genetic counseling of patients with PAM is recommended. This will provide useful information for patients and their families including the possibility of genetic testing of other family members and if relevant, the possibility of prenatal/preimplantation genetic diagnostics. In extended consanguineous families with a genetically proven case of PAM, other related couples and family members could benefit from genetic counseling. Although no cure or effective treatment is currently available except for lung transplantation, diagnosis in childhood or adolescence permits early family education and genetic counseling. In addition, it will be possible to initiate more intensive supportive care earlier, including e.g., pneumococcal and influenza vaccinations, and to plan for future transplantation.
Conclusions
PAM is a rare genetic lung disease with a varying clinical course. The genetics of PAM, including the presence of a possible genotype–phenotype association, remains to be explored. Variants in SLC34A2 are found in almost all patients undergoing genetic evaluation. So far, 34 allelic variants are reported in at least 68 patients, with most variants described in only a single patient. The occurrence of consanguinity is significant. We recommend a thorough systematic clinical description together with a genetic investigation in all new cases. A clinical grade system would be useful, and clinical studies and functional and experimental studies of the variants are needed to explore future treatment strategies. Finally, since the proportion of patients with SLC34A2 variants seems to be very high, the genetic characterization may in some cases be the preferred diagnostic tool to invasive investigations, especially in the diagnostics of children.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed for the writing of this review.
Abbreviations
- PAM:
-
Pulmonary alveolar microlithiasis
- NaPi-2b:
-
The sodium-dependent phosphate transport protein 2b
- Pi:
-
Inorganic phosphate
- BAL:
-
Bronchoalveolar lavage
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- HRCT:
-
High-resolution computed tomography
- SP:
-
Surfactant Protein
- ROS1:
-
ROS proto-oncogene 1; receptor tyrosine kinase
- NaPi-2a:
-
The sodium-dependent phosphate transport protein 2a
- NaPi-2c:
-
The sodium-dependent phosphate transport protein 2c
- TM:
-
Transmembrane
- EGF:
-
Epidermal growth factor
- VDR:
-
Vitamin D receptor
- ED-71:
-
1α, 25-Dihydroxy-2ß-(3-hydroxypropoxy) vitamin D3
- A549 cells:
-
Human alveolar epithelial cells
- SCAM:
-
The substituted cysteine accessibility method
- KO:
-
Knock-out
- Slc20a1 :
-
The sodium-dependent phosphate transporter Pit-1
- MCP-1:
-
Monocyte chemotactic protein 1
References
Bendstrup E, Jonsson ALM. Pulmonary alveolar microlithiasis: no longer in the stone age. ERJ Open Res. 2020;6(3).
Jonsson AL, Simonsen U, Hilberg O, Bendstrup E. Pulmonary alveolar microlithiasis: two case reports and review of the literature. Eur Respir Rev. 2012;21(125):249–56.
Jonsson ALM, Bendstrup E, Mogensen S, Kopras EJ, McCormack FX, Campo I, et al. Eight novel variants in the SLC34A2 gene in pulmonary alveolar microlithiasis. Eur Respir J. 2020;55(2).
Puhr L. Mikrolithiasis alveolaris pulmonum [Pulmonary alveolar microlithiasis.] Virchows Arch (Pathol Anat). 1933;290:156.
Corut A, Senyigit A, Ugur SA, Altin S, Ozcelik U, Calisir H, et al. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am J Hum Genet. 2006;79(4):650–6.
Huqun, Izumi S, Miyazawa H, Ishii K, Uchiyama B, Ishida T, et al. Mutations in the SLC34A2 gene are associated with pulmonary alveolar microlithiasis. Am J Respir Crit Care Med. 2007;175(3):263–8.
SLC34A2, Gene ID: 10568 [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004—[cited 2022 09 25]. Available from: https://www.ncbi.nlm.nih.gov/gene/.
Forster IC, Hernando N, Biber J, Murer H. Phosphate transporters of the SLC20 and SLC34 families. Mol Aspects Med. 2013;34(2–3):386–95.
Castellana G, Castellana G, Gentile M, Castellana R, Resta O. Pulmonary alveolar microlithiasis: review of the 1022 cases reported worldwide. Eur Respir Rev. 2015;24(138):607–20.
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7): e1000097.
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–77.
Moran CA, Hochholzer L, Hasleton PS, Johnson FB, Koss MN. Pulmonary alveolar microlithiasis. A clinicopathologic and chemical analysis of seven cases. Arch Pathol Lab Med. 1997;121(6):607–11.
Poelma DL, Ju MR, Bakker SC, Zimmermann LJ, Lachmann BF, van Iwaarden JF. A common pathway for the uptake of surfactant lipids by alveolar cells. Am J Respir Cell Mol Biol. 2004;30(5):751–8.
Saito A, McCormack FX. Pulmonary alveolar microlithiasis. Clin Chest Med. 2016;37(3):441–8.
Castellana G, Gentile M, Castellana R, Fiorente P, Lamorgese V. Pulmonary alveolar microlithiasis: clinical features, evolution of the phenotype, and review of the literature. Am J Med Genet. 2002;111(2):220–4.
Devine OP, Harborne AC. Pneumothorax secondary to pulmonary alveolar microlithiasis. Clin Case Rep. 2018;6(4):764–5.
Ferreira Francisco FA, Pereira e Silva JL, Hochhegger B, Zanetti G, Marchiori E. Pulmonary alveolar microlithiasis. State-of-the-art review. Respir Med. 2013;107(1):1–9.
Francisco FA, Rodrigues RS, Barreto MM, Escuissato DL, Araujo Neto CA, Silva JL, et al. Can chest high-resolution computed tomography findings diagnose pulmonary alveolar microlithiasis? Radiol Bras. 2015;48(4):205–10.
Arslan A, Yalin T, Akan H, Belet U. Pulmonary alveolar microlithiasis associated with calcifications in the seminal vesicles. J Belge Radiol. 1996;79(3):118–9.
Kanat F, Teke T, Imecik O. Pulmonary alveolar microlithiasis with epididymal and periurethral calcifications causing obstructive azospermia. Int J Tuberc Lung Dis. 2004;8(10):1275.
Qublan HSAI, Al-Kaisi NS. Azoospermia associated with testicular and pulmonary microlithiasis. J Diagn Med Sonogr. 2003;19:192–4.
Castellana G, Carone D, Castellana M. Microlithiasis of seminal vesicles and severe oligoasthenospermia in pulmonary alveolar microlithiasis (PAM): report of an unusual sporadic case. Int J Fertil Steril. 2015;9(1):137–40.
Jonsson AL, Hilberg O, Bendstrup EM, Mogensen S, Simonsen U. SLC34A2 gene mutation may explain comorbidity of pulmonary alveolar microlithiasis and aortic valve sclerosis. Am J Respir Crit Care Med. 2012;185(4):464.
Badger TL, Gottlieb L, Gaensler EA. Pulmonary alveolar microlithiasis, or calcinosis of the lungs. N Engl J Med. 1955;253(17):709–15.
Coetzee T. Pulmonary alveolar microlithiasis with involvement of the sympathetic nervous system and gonads. Thorax. 1970;25(5):637–42.
Sharp ME, Danino EA. An unusual form of pulmonary calcification: microlithiasis alveolaris pulmonum. J Pathol Bacteriol. 1953;65(2):389–99.
Kiatboonsri S, Charoenpan P, Vathesatogkit P, Boonpucknavig V. Pulmonary alveolar microlithiasis: report of five cases and literature review. J Med Assoc Thai. 1985;68(12):672–7.
Samrah S, Shraideh H, Rawashdeh S, Khassawneh B. Tricuspid valve calcification in familial pulmonary alveolar microlithiasis: a case report. Ann Med Surg (Lond). 2020;55:256–9.
Feild JA, Zhang L, Brun KA, Brooks DP, Edwards RM. Cloning and functional characterization of a sodium-dependent phosphate transporter expressed in human lung and small intestine. Biochem Biophys Res Commun. 1999;258(3):578–82.
Xu H, Bai L, Collins JF, Ghishan FK. Molecular cloning, functional characterization, tissue distribution, and chromosomal localization of a human, small intestinal sodium-phosphate (Na+-Pi) transporter (SLC34A2). Genomics. 1999;62(2):281–4.
Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2008;23(1):22–44.
Ozcelik U, Yalcin E, Ariyurek M, Ersoz DD, Cinel G, Gulhan B, et al. Long-term results of disodium etidronate treatment in pulmonary alveolar microlithiasis. Pediatr Pulmonol. 2010;45(5):514–7.
Jankovic S, Pavlov N, Ivkosic A, Erceg I, Glavina-Durdov M, Tocilj J, et al. Pulmonary alveolar microlithiasis in childhood: clinical and radiological follow-up. Pediatr Pulmonol. 2002;34(5):384–7.
Mariotta S, Guidi L, Mattia P, Torrelli L, Pallone G, Pedicelli G, et al. Pulmonary microlithiasis. Report of two cases. Respiration. 1997;64(2):165–9.
Cakir E, Gedik AH, Ozdemir A, Buyukpinarbasili N, Bilgin M, Ozgen IT. Response to disodium etidronate treatment in three siblings with pulmonary alveolar microlithiasis. Respiration. 2015;89(6):583–6.
Pracyk JB, Simonson SG, Young SL, Ghio AJ, Roggli VL, Piantadosi CA. Composition of lung lavage in pulmonary alveolar microlithiasis. Respiration. 1996;63(4):254–60.
Ganesan N, Ambroise MM, Ramdas A, Kisku KH, Singh K, Varghese RG. Pulmonary alveolar microlithiasis: an interesting case report with systematic review of Indian literature. Front Med. 2015;9(2):229–38.
Flynn AAA. Pulmonary alveolar microlithiasis. Int J Case Rep Images. 2013;4(2):108–10.
Mascie-Taylor BH, Wardman AG, Madden CA, Page RL. A case of alveolar microlithiasis: observation over 22 years and recovery of material by lavage. Thorax. 1985;40(12):952–3.
Palombini BC, da Silva PN, Wallau CU, Camargo JJ. Bronchopulmonary lavage in alveolar microlithiasis. Chest. 1981;80(2):242–3.
Chalmers AG, Wyatt J, Robinson PJ. Computed tomographic and pathological findings in pulmonary alveolar microlithiasis. Br J Radiol. 1986;59(700):408–11.
Tachibana T, Hagiwara K, Johkoh T. Pulmonary alveolar microlithiasis: review and management. Curr Opin Pulm Med. 2009;15(5):486–90.
Ma T, Ren J, Yin J, Ma Z. A pedigree with pulmonary alveolar microlithiasis: a clinical case report and literature review. Cell Biochem Biophys. 2014;70(1):565–72.
Chinachoti N, Tangchai P. Pulmonary alveolar microlithiasis associated with the inhalation of snuff in Thailand. Dis Chest. 1957;32(6):687–9.
The UC. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45(D1):D158–69.
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–61.
Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA. Genenames.org: the HGNC and VGNC resources in 2017. Nucleic Acids Res. 2017;45(D1):D619–25.
Traebert M, Hattenhauer O, Murer H, Kaissling B, Biber J. Expression of type II Na-P(i) cotransporter in alveolar type II cells. Am J Physiol. 1999;277(5 Pt 1):L868–73.
Frei P, Gao B, Hagenbuch B, Mate A, Biber J, Murer H, et al. Identification and localization of sodium-phosphate cotransporters in hepatocytes and cholangiocytes of rat liver. Am J Physiol Gastrointest Liver Physiol. 2005;288(4):G771–8.
Huber K, Muscher A, Breves G. Sodium-dependent phosphate transport across the apical membrane of alveolar epithelium in caprine mammary gland. Comp Biochem Physiol A Mol Integr Physiol. 2007;146(2):215–22.
Sabbagh Y, O’Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20(11):2348–58.
Xu Y, Yeung CH, Setiawan I, Avram C, Biber J, Wagenfeld A, et al. Sodium-inorganic phosphate cotransporter NaPi-IIb in the epididymis and its potential role in male fertility studied in a transgenic mouse model. Biol Reprod. 2003;69(4):1135–41.
Yin BW, Kiyamova R, Chua R, Caballero OL, Gout I, Gryshkova V, et al. Monoclonal antibody MX35 detects the membrane transporter NaPi2b (SLC34A2) in human carcinomas. Cancer Immun. 2008;8:3.
Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18(17):4570–9.
Xie J, Zhu XY, Liu LM, Meng ZQ. Solute carrier transporters: potential targets for digestive system neoplasms. Cancer Manag Res. 2018;10:153–66.
National Center for Biotechnology Information (NCBI) [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; [1988] – [cited 2022 Sep 25]. Available from: https://www.ncbi.nlm.nih.gov/.
Forster IC. The molecular mechanism of SLC34 proteins: insights from two decades of transport assays and structure-function studies. Pflugers Arch. 2018.
Ghezzi C, Meinild AK, Murer H, Forster IC. Voltage- and substrate-dependent interactions between sites in putative re-entrant domains of a Na(+)-coupled phosphate cotransporter. Pflugers Arch. 2011;461(6):645–63.
Werner A, Kinne RK. Evolution of the Na-P(i) cotransport systems. Am J Physiol Regul Integr Comp Physiol. 2001;280(2):R301–12.
Forster IC, Hernando N, Biber J, Murer H. Phosphate transport kinetics and structure-function relationships of SLC34 and SLC20 proteins. Curr Top Membr. 2012;70:313–56.
Fenollar-Ferrer C, Patti M, Knopfel T, Werner A, Forster IC, Forrest LR. Structural fold and binding sites of the human Na(+)-phosphate cotransporter NaPi-II. Biophys J. 2014;106(6):1268–79.
Fenollar-Ferrer C, Forster IC, Patti M, Knoepfel T, Werner A, Forrest LR. Identification of the first sodium binding site of the phosphate cotransporter NaPi-IIa (SLC34A1). Biophys J. 2015;108(10):2465–80.
Hernando N, Wagner CA. Mechanisms and regulation of intestinal phosphate absorption. Compr Physiol. 2018;8(3):1065–90.
Hattenhauer O, Traebert M, Murer H, Biber J. Regulation of small intestinal Na-P(i) type IIb cotransporter by dietary phosphate intake. Am J Physiol. 1999;277(4 Pt 1):G756–62.
Radanovic T, Wagner CA, Murer H, Biber J. Regulation of intestinal phosphate transport. I. Segmental expression and adaptation to low-P(i) diet of the type IIb Na(+)-P(i) cotransporter in mouse small intestine. Am J Physiol Gastrointest Liver Physiol. 2005;288(3):G496-500.
Xu H, Uno JK, Inouye M, Xu L, Drees JB, Collins JF, et al. Regulation of intestinal NaPi-IIb cotransporter gene expression by estrogen. Am J Physiol Gastrointest Liver Physiol. 2003;285(6):G1317–24.
Stauber A, Radanovic T, Stange G, Murer H, Wagner CA, Biber J. Regulation of intestinal phosphate transport. II. Metabolic acidosis stimulates Na(+)-dependent phosphate absorption and expression of the Na(+)-P(i) cotransporter NaPi-IIb in small intestine. Am J Physiol Gastrointest Liver Physiol. 2005;288(3):G501–6.
Arima K, Hines ER, Kiela PR, Drees JB, Collins JF, Ghishan FK. Glucocorticoid regulation and glycosylation of mouse intestinal type IIb Na-P(i) cotransporter during ontogeny. Am J Physiol Gastrointest Liver Physiol. 2002;283(2):G426–34.
Xu H, Collins JF, Bai L, Kiela PR, Ghishan FK. Regulation of the human sodium-phosphate cotransporter NaP(i)-IIb gene promoter by epidermal growth factor. Am J Physiol Cell Physiol. 2001;280(3):C628–36.
Segawa H, Kaneko I, Yamanaka S, Ito M, Kuwahata M, Inoue Y, et al. Intestinal Na-P(i) cotransporter adaptation to dietary P(i) content in vitamin D receptor null mice. Am J Physiol Renal Physiol. 2004;287(1):F39-47.
Jin C, Zoidis E, Ghirlanda C, Schmid C. Dexamethasone and cyclic AMP regulate sodium phosphate cotransporter (NaPi-IIb and Pit-1) mRNA and phosphate uptake in rat alveolar type II epithelial cells. Lung. 2010;188(1):51–61.
Brown AJ, Zhang F, Ritter CS. The vitamin D analog ED-71 is a potent regulator of intestinal phosphate absorption and NaPi-IIb. Endocrinology. 2012;153(11):5150–6.
Ishihara Y, Hagiwara K, Zen K, Huqun, Hosokawa Y, Natsuhara A. A case of pulmonary alveolar microlithiasis with an intragenetic deletion in SLC34A2 detected by a genome-wide SNP study. Thorax. 2009;64(4):365–7.
Vismara MF, Colao E, Fabiani F, Bombardiere F, Tamburrini O, Alessio C, et al. The sodium-phosphate co-transporter SLC34A2, and pulmonary alveolar microlithiasis: presentation of an inbred family and a novel truncating mutation in exon 3. Respir Med Case Rep. 2015;16:77–80.
Mehta K, Dell S, Birken C, Al-Saleh S. Pulmonary Alveolar Microlithiasis. Can Respir J. 2016;2016:4938632.
Ozbudak IH, Bassorgun CI, Ozbilim G, Luleci G, Sarper A, Erdogan A, et al. Pulmonary alveolar microlithiasis with homozygous c.316G>C (p.G106R) mutation: a case report. Turk Patoloji Derg. 2012;28(3):282–5.
Gaber K, Najem S, Bader O, Bendardaf A. New mutation of SCL34A2 Gene in a family with pulmonary alveolar microlithiasis in Libya. Chest. 2012;142(4):439A.
Alastal AYH. Pulmonary alveolar microlithiasis caused by two homozygous mutations. Am J Respir Crit Care Med. 2017;195:A3438.
Izumi H, Kurai J, Kodani M, Watanabe M, Yamamoto A, Nanba E, et al. A novel SLC34A2 mutation in a patient with pulmonary alveolar microlithiasis. Hum Genome Var. 2017;4:16047.
Yin X, Wang H, Wu D, Zhao G, Shao J, Dai Y. SLC34A2 Gene mutation of pulmonary alveolar microlithiasis: report of four cases and review of literatures. Respir Med. 2013;107(2):217–22.
Wang H, Yin X, Wu D, Jiang X. SLC34A2 gene compound heterozygous mutation identification in a patient with pulmonary alveolar microlithiasis and computational 3D protein structure prediction. Meta Gene. 2014;2:557–64.
Proesmans M, Boon M, Verbeken E, Ozcelik U, Kiper N, Van de Casseye W, et al. Pulmonary alveolar microlithiasis: a case report and review of the literature. Eur J Pediatr. 2012;171(7):1069–72.
Zhong YQ, Hu CP, Cai XD, Nie HP. A novel mutation of the SLC34A2 gene in a Chinese pedigree with pulmonary alveolar microlithiasis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2009;26(4):365–8.
Stokman L, Nossent EJ, Grunberg K, Meijboom L, Yakicier MC, Voorhoeve E, et al. A case of pulmonary alveolar microlithiasis associated with a homozygous 195 kb deletion encompassing the entire SLC34A2 gene. Clin Case Rep. 2016;4(4):412–5.
Ellison J. Novel human pathological mutations. Gene symbol: SLC34A2. Disease: pulmonary alveolar microlithiasis. Hum Genet. 2009;125(3):333.
Dandan S, Yuqin C, Wei L, Ziheng P, Dapeng Z, Jianzhu Y, et al. Novel deletion of SLC34A2 in Chinese patients of PAM shares mutation hot spot with fusion gene SLC34A2-ROS1 in lung cancer. J Genet. 2018;97(4):939–44.
Simon CT, Lewis TC, Neemuchwala F, Arteta M, Rabah R. Pulmonary alveolar microlithiasis: a case report with a novel mutation in the SLC34A2 gene and review of the literature. Human Pathol: Case Rep. 2018;13:33–5.
Zhang D, Xiao K, Guan W, Hu X, Yan P, Xie L. An inbred family with pulmonary alveolar microlithiasis in China: a genome-wide SNP study. Int J Clin Exp Pathol. 2017;10(1):446–52.
Pascual-González YCE, Luburich P, Vicens-Zygmunt V. Pulmonary alveolar microlithiasis: genetic disorder and concomitant altered calcium metabolism. Eur Respir J. 2020;56(Suppl. 64):3490.
Yazdani MF, Morris-Rosendahl D, Chen L, Devaraj A, Chua F. Images of the month 1: cough before the storm: A case of pulmonary alveolar microlithiasis. Clin Med (Lond). 2020;20(1):110–1.
Sigur E, Roditis L, Labouret G, Bieth E, Simon S, Martin-Blondel A, et al. Pulmonary alveolar microlithiasis in children less than 5 years of age. J Pediatr. 2020;217(158–64): e1.
de Oliveira MA, Schwerk N, Schutz K, Schramm L, Dreissig A, Grewendorf S, et al. Pulmonary alveolar microlithiasis: a novel patient and brief review of the literature. Klin Padiatr. 2022;234(5):317–9.
Panjwani A, Kadhem H, Alwedaie SMJ, Alawadhi AMT. A novel genetic variant of pulmonary alveolar microlithiasis. Lung India. 2022;39(2):212–3.
Yi S, Li M, Yang Q, Zhang X, Chen F, Qin Z, et al. Novel SLC12A1 mutations cause Bartter syndrome in two patients with different prognoses. Clin Chim Acta. 2022;531:120–5.
Gaber K NS, Twair A, Dharrat A. Pulmonary alveolar microlitiasis in living adult monozygotic twin; CT scan versus CXR family screening results. Am J Respirat Crit Care Med. 2012;185(MeetingAbstracts A5427).
Virkki LV, Biber J, Murer H, Forster IC. Phosphate transporters: a tale of two solute carrier families. Am J Physiol Renal Physiol. 2007;293(3):F643–54.
Steinhaus R, Proft S, Schuelke M, Cooper DN, Schwarz JM, Seelow D. MutationTaster2021. Nucleic Acids Res. 2021;49(W1):W446–51.
Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou LP, Mi H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022;31(1):8–22.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–7.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9): e67.
Dogan OT, Ozsahin SL, Gul E, Arslan S, Koksal B, Berk S, et al. A frame-shift mutation in the SLC34A2 gene in three patients with pulmonary alveolar microlithiasis in an inbred family. Intern Med. 2010;49(1):45–9.
Olauson H, Brandenburg V, Larsson TE. Mutation analysis and serum FGF23 level in a patient with pulmonary alveolar microlithiasis. Endocrine. 2010;37(2):244–8.
Yang Y, Qiao JH, An JH, Zhang Y, Yu T, Jia B, et al. Detection of SLC34A2 in patients with pulmonary alveolar microlithiasis and the effect of SLC34A2 on transportation of calcium and phosphate in human alveolar epithelial cells. Zhonghua Jie He He Hu Xi Za Zhi. 2008;31(12):908–11.
Zhang XD, Gao JM, Luo JM, Zhao Y. Pulmonary alveolar microlithiasis: a case report and review of the literature. Exp Ther Med. 2018;15(1):831–7.
Castillo S, Hamblin MJ. A caucasian woman with pam due to a rare mutation. Am J Respirat Crit Care Med. 2017;195:A3439.
Werner A, Patti M, Zinad HS, Fearn A, Laude A, Forster I. Molecular determinants of transport function in zebrafish Slc34a Na-phosphate transporters. Am J Physiol Regul Integr Comp Physiol. 2016;311(6):R1213–22.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
Wang H, Yin X, Wu D. Novel human pathological mutations. SLC34A2. Disease: pulmonary alveolar microlithiasis. Hum Genet. 2010;127(4):471.
Senyigit A, Yaramis A, Gurkan F, Kirbas G, Buyukbayram H, Nazaroglu H, et al. Pulmonary alveolar microlithiasis: a rare familial inheritance with report of six cases in a family. Contribution of six new cases to the number of case reports in Turkey. Respiration. 2001;68(2):204–9.
Gocmen A, Toppare MF, Kiper N, Buyukpamukcu N. Treatment of pulmonary alveolar microlithiasis with a diphosphonate–preliminary results of a case. Respiration. 1992;59(4):250–2.
Ishida T, Matsumura Y, Miyake A. A case of pulmonary alveolar microlithiasis showing respiratory failure after 33 years and revealing microliths in bronchoalveolar lavage. Nihon Kyobu Shikkan Gakkai Zasshi. 1991;29(5):627–31.
Oka S, Shiraishi K, Ogata K, Goto Y, Yasuda T, Yanagihara H. Pulmonary alveolar microlithiasis. Report of three cases. Am Rev Respir Dis. 1966;93(4):612–6.
Jonsson ALM, Hernando N, Knopfel T, Mogensen S, Bendstrup E, Hilberg O, et al. Impaired phosphate transport in SLC34A2 variants in patients with pulmonary alveolar microlithiasis. Hum Genom. 2022;16(1):13.
Lambert G, Forster IC, Stange G, Kohler K, Biber J, Murer H. Cysteine mutagenesis reveals novel structure-function features within the predicted third extracellular loop of the type IIa Na(+)/P(i) cotransporter. J Gen Physiol. 2001;117(6):533–46.
Ma T, Qu D, Yan B, Zhang Q, Ren J, Hu Y. Effect of SLC34A2 gene mutation on extracellular phosphorus transport in PAM alveolar epithelial cells. Exp Ther Med. 2018;15(1):310–4.
Hernando N, Myakala K, Simona F, Knopfel T, Thomas L, Murer H, et al. Intestinal depletion of NaPi-IIb/Slc34a2 in mice: renal and hormonal adaptation. J Bone Miner Res. 2015;30(10):1925–37.
Saito A, Nikolaidis NM, Amlal H, Uehara Y, Gardner JC, LaSance K, et al. Modeling pulmonary alveolar microlithiasis by epithelial deletion of the Npt2b sodium phosphate cotransporter reveals putative biomarkers and strategies for treatment. Sci Transl Med. 2015;7(313):313ra181.
Acknowledgements
Not applicable.
Funding
Open access funding provided by the Royal Danish Library. The authors received no funding for the preparation of this review article.
Author information
Authors and Affiliations
Contributions
All the authors conceived the manuscript. ÅLMJ, EB, OH, US and JHC designed the review. ÅLMJ performed the literature search, selected the studies for inclusion, extracted and analyzed data, drafted and revised the manuscript. All authors contributed to the manuscript editing and critical revision for important intellectual/scientific content. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jönsson, Å.L.M., Hilberg, O., Simonsen, U. et al. New insights in the genetic variant spectrum of SLC34A2 in pulmonary alveolar microlithiasis; a systematic review. Orphanet J Rare Dis 18, 130 (2023). https://doi.org/10.1186/s13023-023-02712-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-023-02712-7