
Abstract
Background
Studies have reported that a noncoding hexanucleotide repeat in C9ORF72, is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) among Caucasian population, nevertheless it is rare in Chinese population. Therefore, we aimed to investigate the mutation spectrum of Chinese ALS patients with FTD (ALS-FTD).
Methods
ALS patients with and without cognitive impairments were enrolled. Clinical features were collected including age, sex, disease duration, ALSFRS-r, family history and cognitive evaluation. Thirty-six ALS genes were screened by whole exome sequencing (WES) and repeat-primed polymerase chain reaction (PCR) were used for detection of and abnormal repeat expansions of C9ORF72.
Results
A total of 1208 patients, including 66 familial ALS (FALS) and 1142 sporadic ALS (SALS) patients were included. Twenty-three patients with sporadic ALS and one familial ALS index had concomitant FTD, which accounts for 1.99% (24/1208) of patients with ALS. In sporadic ALS-FTD, one case harboring C9ORF72 expansion variant, two cases harboring ANXA11 variants and one individual carrying CCNF variant were identified. A recurrent UBQLN2 variant was detected in a familial ALS-FTD patient. All of the ALS-FTD patients carrying variants in known causative genes manifested motor symptom onset (two bulbar onset and three limb onset) and developed cognitive impairment thereafter. It is not easy to draw a conclusion of the genotype-phenotype association in ALS-FTD with certain variants, limited by the small number of patients.
Conclusion
Our findings provide an overview of spectrum of genetic variants in Chinese ALS-FTD patients. Variants of uncertain significance in UBQLN2, ANXA11 and CCNF were identified and further studies are required for causal relations of these variants with ALS-FTD.
Similar content being viewed by others

Background
Amyotrophic lateral sclerosis (ALS) is a relentlessly progressive neurologic disease characterized by progressive paralysis and ultimately respiratory failure, with an incidence of 2.6/100 000 per year [1]. Familial ALS accounts for 5 to 10% of all ALS cases, with mutations in several genes including SOD1, TARDBP, OPTN, FUS, UBQLN2 and VCP. Frontotemporal dementia (FTD) is the second most common cause of early onset dementia, and is characterized by progressive degeneration of the frontal and temporal cortex. It occurs with an incidence of 3.5–4.1/100,000 per year in individuals under 65 [2]. A family history of dementia or psychiatric conditions is present in 40% of FTD patients. Mutations in MAPT and GRN and a hexanucleotide expansion in C9ORF72 together accounts for approximately 15% of familial FTD cohorts; mutations in other genes, including VCP, CHMP2B, and TBK1, are rare [3, 4].
ALS and FTD were traditionally considered as completely different neurological disorders with distinct clinical features. Studies over the last 30 years have demonstrated the co-occurrence of ALS and FTD. About 15% of ALS cases showing a clear diagnosis of FTD while around 15% of patients with FTD displaying ALS [5, 6]. What’s more, the description of several mutations that are common to both ALS and FTD provides a genetic link between them. Studies have reported that a noncoding hexanucleotide repeat in C9ORF72, is the most common genetic cause of ALS and FTD among Caucasian population [7, 8]. Mutations in UBQLN2 [9, 10], VCP [11], TARDBP [12], FUS [13] have also been identified in patients with ALS and FTD. Thus, ALS and FTD are now considered to be a spectrum of one overlapping diseases, commonly referred to as ALS/FTD.
Mutation spectrum of ALS varies among populations of different ethnic background. While C9ORF72 is the most prevalent mutant gene in Caucasian, it is rare in Chinese population. Few studies had explored the genetic epidemiology of ALS/FTD in Chinese populations. Therefore, we aimed to investigate the mutant condition in Chinese ALS patients with FTD (ALS-FTD) and to characterize the genotype-phenotype association of these patients.
Methods
Participants and data collection
This study included 1208 patients who were diagnosed of definite or probable ALS according to revised E1 Escorial criteria at neurology department of Peking Union Medical College Hospital (PUMCH) from September 2011 to September 2021. All patients were registered into the ALS clinical research database registration system of PUMCH and a series of clinical features were collected including age, sex, disease duration, diagnosis delay, onset site, disease evolution, revised ALS functional rating scale (ALSFRS-r), family history and cognitive evaluation. A positive family history was defined as relatives within two generations were diagnosed of ALS. Cognitive evaluation battery included: Mini-Mental State Examination score (MMSE) and the Montreal Cognitive Assessment (MoCA) and Edinburgh Cognitive and Behavioural ALS Screen (ECAS) [14]. Diagnosis of ALS with behavioral variant FTD required the individual met at least one non-overlapping supportive diagnostic features from the Neary criteria [15] for FTD. Besides, 2445 ethnicity matched healthy controls were also included in this study.
Genetic testing and bioinformation analysis
Peripheral venous blood samples were collected from the ALS patients, and DNA was isolated from mononuclear cells using a DNA isolation kit (Blood DNA Kit V2, CW2553), according to the ALS online database and previous reports [16,17,18]. Thirty-six genes identified in ALS-FTD, ALS, and FTD were screened by whole exome sequencing (WES), including the genes SOD1, ALS2, SETX, FUS, VAPB, TARDBP, OPTN, VCP, UBQLN2, SIGMAR1, FIG4, CHMP2B, PFN1, NEFH, PRPH, TFG, TAF15, GRN, CHCHD10, TUBA4A, TBK1, NEK1, GLE1, MATR3, CCNF, ANXA11, HNRNPA1, SQSTM1, ERBB4, TIA1, SPG11, KIF5A, GLT8D1, DNAJC7, TBP, CTSF and MFSD8. In brief, WES procedure comprised three standard steps: end-repair of fragmented DNA, A-tailing, and adapter ligation and amplification. Sequencing was completed by the HiSeq2000 platform (Illumina, San Diego, California, USA) as paired-end 200 bp reads. Variants with read depth ≥ 10 and genotype quality ≥ 20 were retained. Average depth and coverage were above 100X and 98.9%, respectively. The sequence was aligned to the human reference genome (UCSC hg19) using a Burrows-Wheeler Aligner and then reformatted using SAM tools (https://doi.org/10.1093/bioinformatics/btv178). Variant frequencies were determined in gnomeAD (http://gnomad-old.broadinstitute.org/), ExAC (http://exac.broadi-nstitute.org/) and 1000 Genomes (http://www.1000genomes.org) to remove the common single nucleotide polymorphisms (SNP). Only non-synonymous, splicing and frameshift variants with a minor allele frequency (MAF) of less than 0.5% or absent in population databases were selected for further analysis. SIFT (http://sift.bii.a-star.edu.sg/) and PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) were used for variant annotation. Besides, fragment-length and repeat-primed PCR was performed to detect duplications or deletions of ATXN2 and C9ORF72 in ALS/FTD patients. The criteria used to detect causative mutations were based on the recommendations of the American College of Medical Genetics and Genomics (ACMG) [19].
Verification of variants by Sanger sequencing
PCR has been performed to validate the variants identified by WES. Primers were designed using Primer 3 online software (http://bioinfo.ut.ee/primer3-0.4.0/). Amplification products were subjected to Sanger sequencing using the ABI 3730 automated DNA-sequencing system (Applied Biosystems). The sequence was aligned to the human reference genome (UCSC hg19) using CodonCode Aligner tool.
Ethics
The study was approved by the ethics committee of Peking Union Medical College Hospital. Informed consent forms were obtained from all patients or their families.
Results
Clinical information of ALS patients
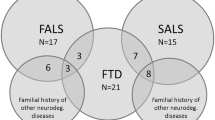
A total of 1208 patients, including 66 FALS probands and 1142 unrelated SALS patients were enrolled in current study. The male/female ratios were 1.7:1.0 (15/9). Of which, 23 SALS patients and 1 FALS index had concomitant frontotemporal lobar dementia, which accounts for 2.01% (23/1142) ALS-FTD in SALS and 1.52% (1/66) in FALS respectively. Eleven patients had bulbar-onset while fourteen patients had limb-onset. Of 24 ALS-FTD patients, nine presented motor symptoms onset, 11 presented cognitive impairment onset and four patients presented motor and cognitive deficits almost simultaneously. Cognition deteriorated in all patients over disease progression. The demographic features of patient are presented in Table 1.
Genetic results
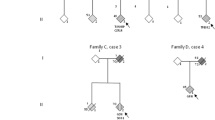
In total, one C9ORF72 expansion variant were identified in one sporadic ALS-FTD patient. The same missense variants c.107 C > G (p.P36R) in the ANXA11 (NM_145869.2) was found in two unrelated ALS-FTD patients. One heterozygous missense variant c.499G > A (p.V167M) in the CCNF (NM_001761.3) was identified in a sporadic ALS-FTD patient. A heterozygous variant c.1498 C > T (p.P500S) in the UBQLN2 (NM_013444.3) were identified in one familial ALS-FTD patient (Table 2). None of these was detected in 2445 healthy control individuals.
Based on ACMG criteria, the three detected variants were categorized as variant of uncertain significance (VUS). Genetic variants account for 12.5% (3/24) SALS-FTD patients and 100.0% (1/1) FALS-FTD. The average sequencing depth of coverage ranged from 88.2 to 142.6, with > 99% sequences mapping to the genome.
One recurrent missense variant in UBQLN2, c.1498 C > T (p.P500S) were identified in one familial ALS/FTD patient. The same genetic alteration had already been reported in ALS by Teyssou et al. [20]. This variant was absent from the exome variant server including the ExAC, gnomAD and the 1000 Genomes Project, as well as our inhouse controls.
Novel heterozygous variant in ANXA11, c.107 C > G (p.P36R) was identified in two unrelated sporadic ALS-FTD patients. The p.P36R variant was predicted to be conserved through revolution. The variant was predicted by to be ‘damaging’ by SIFT and ‘probably damaging’ by Polyphen-2 in silico analysis.
Additionally, another missense variant in CCNF, c.499G > A (p.V167M) was identified in one sporadic ALS-FTD patient. The p.V167M variant was highly conserved among different species. The p.V167M variant is predicted to be ‘tolerable’ by SIFT and ‘probably damaging’ by Polyphen-2 in silico analysis.
The patients mentioned above were negative for other common ALS causal gene mutations, including ATXN2 abnormal repeat expansions.
Clinical characteristics of patients with certain variants
All of the ALS-FTD patients carrying variants in known causative genes manifested motor symptom onset (two bulbar onset and three limb onset) and developed cognitive impairment thereafter. It is not easy to draw a conclusion of the genotype-phenotype association in ALS-FTD with certain variants, limited by the small number of patients. An additional file shows the clinical information of patients in more detail [see Additional file 1].
Discussion
Advances in sequencing technologies have shed fresh insight into the concept of ALS and FTD from two separate clinical entities into a continual disease spectrum. Identification of the genes related to ALS/FTD spectrum may facilitate our understanding of the genetic etiology and common pathogenic factors leading to neurodegeneration. In total, four nonsynonymous variants in UBQLN2, ANXA11 and CCNF, as well as one C9ORF72 expansion variant, were identified in 5 unrelated ALS-FTD patients.
Historically, comorbidity rates of FTD were documented as 3% in sporadic ALS and 15% in familial ALS [21], but recent studies [2, 6, 22] using frontal lobe–based neuropsychological measures report the vary from 28 to 48%. The comorbidity rates of FTD in SALS (2.01%) and in FALS (1.52%) in our database, which is lower than that in the Caucasian population. The difference may be owing to not only different ethnicity and diagnostic criteria, but also the limits of health care and economy in some parts of China.
Our study revealed the discrepancy of mutation spectrum between Chinese and Caucasians. C9ORF72 repeat expansion is the most common mutation in studies of pan-European and North American patient populations [23, 24], which is not as common among Chinese, Korean, and Japanese ALS patients [25,26,27]. In our previous study, 40.0% (8/20) of FALS and 18.8% (44/234) of SALS carried mutations in these genes, with a mutant frequency varying from SOD1 (FALS 30.6%, SALS 1.5%) to UBQLN2 (FALS 5%, SALS 0) [28]. One C9ORF72 repeat expansion was identified in a 65-year-old female with limb-onset ALS, with a mutant frequency of 0.39% (1/254), which is consistent with low frequency of C9ORF72 mutation as previous reported. In the ALS-FTD cohort, only one C9ORF72 repeat expansion was found. Recurrent UBQLN2 variant was found in one FALS-FTD patient with a frequency of 4.0% (1/24). ATXN2 repeat expansion and variants in VCP, TARDBP, FUS was not detected in current ALS-FTD patients.
ALS-linked variants in UBQLN2 were found to be associated with dysfunction of autophagy, neuroinflammation and formation of stress granules. These recent data surely placed UBQLN2 as an essential player in noxious protein accumulation and clearance pathways in ALS and FTD pathogenesis. The p.P500S variant in UBQLN2 was previously reported in a female European patient with ALS and had a common European founder. Weakness onset appeared in the right arm at 62 years of age. Her bulbar function rapidly worsened and she died from respiratory failure after 16 months of disease duration with no cognitive defect. In the current study, the woman with the p.P500S variant had a bulbar onset ALS at 63 years of age with cognitive impairment. She died from respiratory failure after 24 months of disease duration. It is difficult to pinpoint common features of UBQLN2-mutated patients in these one studies.
Annexin 11 encoded by ANXA11 is a member of the annexin protein family. ANXA11 protein attaches to RNP granules via its structurally disordered N-terminal domain, and to lysosomes via the C-terminal annexin repeats. The dual biophysical properties of ANXA11 protein allow it to act as a molecular tether that binds neuronal stress granules (and possibly other RNP granules) to lysosomes [29]. It is speculated that ALS-associated missense mutations in either the N-terminal LCD or in the annexin repeat domain of ANXA11 disrupt formation of the molecular tether and are associated with impaired delivery of mRNA to axon terminals for local protein synthesis [29]. However, the pathogenetic mechanism of ANXA11 variant is under investigation.
Smith et al. [30] screened 751 familial ALS patient by whole-exome sequences and identified six variants including a nonsynonymous variant p.D40G and p.D38G in ANXA11 in 13 individuals. The p.D40G mutation segregated with disease in one kindreds and was present in another two unrelated cases. They observed a unique feature of abundant Annexin A11–positive protein aggregates in spinal cord motor neurons and hippocampal neuronal axons of ALS patient carrying the p.D40G variant, along with classical pathological features of ALS. The c.107 C > G (p.P36R) variant is presented once in the ExAC East Asian population (MAF 1⁄4 0.00012). The p.P36R variant is located in the CACY binding site in the N-terminal of annexin A11 near the p. D38G and p.D40G variant and most bioinformatic tools indicate pathogenicity.
CCNF is the substrate-recognizing component of the Skp1-cullin-F-box E3 ubiquitin-ligase complex, which is responsible for tagging proteins with ubiquitin and marking them for degradation via the ubiquitin-proteasome system [31]. Neuronal cells overexpressing mutant CCNF show an increase in ubiquitin-tagged proteins, which include TDP43. CCNF was identified as a causative gene for ALS on the basis of exome sequence analysis [18]of a large family of European descent who had ALS, frontotemporal dementia, or both diseases, with an autosomal dominant pattern of inheritance. The authors reported additional, potentially pathogenic variants in CCNF in familial ALS/FTD cases with an overall mutation frequency that ranged between 0.6 and 3.3% in white populations [31]. In current study, a missense variant in CCNF, c.499G > A(V167M) was identified with a frequency of 4.0% (1/24) among ALS-FTD patients.
Conclusion
In summary, ALS-FTD patients were rare in ALS cohort. Our study provided an overview of mutation spectrum of genetic variants in Chinese ALS-FTD patients. In sporadic ALS-FTD, one case harboring C9ORF72 expansion variant, two cases harboring ANXA11 variants and one individual carrying CCNF variant were identified. A recurrent UBQLN2 variant was detected in a familial ALS-FTD patient. The results uncovered the discrepancy of mutation spectrum between Chinese and Caucasians. Variants in UBQLN2, ANXA11 and CCNF were identified and further studies are required for causal relations of these variants with ALS-FTD.
Data availability
All data generated and/or analysed during the current study are not publicly available due to personal information of the patient, but available from the corresponding author on reasonable request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis.
- FTD:
-
Frontotemporal dementia.
- ALS-FTD:
-
ALS patients with FTD.
- FALS:
-
familial ALS.
- SALS:
-
sporadic ALS.
- WES:
-
whole exome sequencing.
- PCR:
-
polymerase chain reaction.
- PUMCH:
-
Peking Union Medical College Hospital.
- MMSE:
-
Mental State Examination score.
- MoCA:
-
Montreal Cognitive Assessment.
- ECAS:
-
Edinburgh Cognitive and Behavioural ALS Screen.
- ALSFRS-r:
-
ALS functional rating scale-revised.
- MAF:
-
minor allele frequency.
- ACMG:
-
the American College of Medical Genetics and Genomics.
References
Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206.
Harvey RJ, Skelton-Robinson M, Rossor MN. The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatry. 2003;74(9):1206–9.
Sieben A, Van Langenhove T, Engelborghs S, Martin JJ, Boon P, Cras P, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012;124(3):353–72.
Mignarri A, Battistini S, Tomai Pitinca ML, Monti L, Burroni L, Ginanneschi F, et al. Double trouble? Progranulin mutation and C9ORF72 repeat expansion in a case of primary non-fluent aphasia. J Neurol Sci. 2014;341(1–2):176–8.
Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134(Pt 9):2582–94.
Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65(4):586–90.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68.
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–5.
Gellera C, Tiloca C, Del Bo R, Corrado L, Pensato V, Agostini J, et al. Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2013;84(2):183–7.
Nalbandian A, Donkervoort S, Dec E, Badadani M, Katheria V, Rana P, et al. The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol neuroscience: MN. 2011;45(3):522–31.
Borroni B, Archetti S, Del Bo R, Papetti A, Buratti E, Bonvicini C, et al. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuven Res. 2010;13(5):509–17.
Yan J, Deng HX, Siddique N, Fecto F, Chen W, Yang Y, et al. Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology. 2010;75(9):807–14.
Ye S, Ji Y, Li C, He J, Liu X, Fan D. The Edinburgh Cognitive and Behavioural ALS Screen in a Chinese Amyotrophic Lateral Sclerosis Population. PLoS ONE. 2016;11(5):e0155496.
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–54.
Abel O, Powell JF, Andersen PM, Al-Chalabi A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat. 2012;33(9):1345–51.
Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–6.
Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun. 2016;7:11253.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet medicine: official J Am Coll Med Genet. 2015;17(5):405–24.
Teyssou E, Chartier L, Amador MD, Lam R, Lautrette G, Nicol M, et al. Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol Aging. 2017;58:239. e11- e20.
Hudson AJ. Amyotrophic lateral sclerosis/parkinsonism/dementia: clinico-pathological correlations relevant to Guamanian ALS/PD. The Canadian journal of neurological sciences Le journal canadien des sciences neurologiques. 1991;18(3 Suppl):387–9.
Massman PJ, Sims J, Cooke N, Haverkamp LJ, Appel V, Appel SH. Prevalence and correlates of neuropsychological deficits in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1996;61(5):450–5.
van der Zee J, Gijselinck I, Dillen L, Van Langenhove T, Theuns J, Engelborghs S, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34(2):363–73.
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–30.
Kim EJ, Kwon JC, Park KH, Park KW, Lee JH, Choi SH, et al. Clinical and genetic analysis of MAPT, GRN, and C9orf72 genes in Korean patients with frontotemporal dementia. Neurobiol Aging. 2014;35(5):1213 e13-7.
Garcia-Redondo A, Dols-Icardo O, Rojas-Garcia R, Esteban-Perez J, Cordero-Vazquez P, Munoz-Blanco JL, et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mutat. 2013;34(1):79–82.
Ogaki K, Li Y, Atsuta N, Tomiyama H, Funayama M, Watanabe H, et al. Analysis of C9orf72 repeat expansion in 563 Japanese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(10):2527. e11-6.
Liu Q, Liu F, Cui B, Lu CX, Guo XN, Wang RR, et al. Mutation spectrum of Chinese patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(11):1272–4.
Liao YC, Fernandopulle MS, Wang G, Choi H, Hao L, Drerup CM, et al. RNA Granules Hitchhike on Lysosomes for Long-Distance Transport, Using Annexin A11 as a Molecular Tether. Cell. 2019;179(1):147–64. e20.
Smith BN, Topp SD, Fallini C, Shibata H, Chen HJ, Troakes C, et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Science translational medicine. 2017;9(388).
D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, et al. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature. 2010;466(7302):138–42.
Acknowledgements
We thank the patients and their family members for their contribution to scientific research in ALS and FTD.
Funding
The study was supported by the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS) grant (2021-1-I2M-034, 2021-1-I2M-018 and 2016-I2M-1-002), the Strategic Priority Research Program of the Chinese Academy of Sciences “Biological basis of aging and therapeutic strategies” (Grant number: XDB39040100), the National Natural Science Foundation of China (Grant number: 81971293, 81788101), and the National Key Research and Development Program of China (2016YFC0905100).
Author information
Authors and Affiliations
Contributions
XY: conceptualization, methodology, data curation, formal analysis, writing original draft; XS, LL, KZ, SL, JL, ZC, DH, DS: resources, data curation; QL and LC: conceptualization, supervision, writing-reviewing and editing; ML and XZ: conceptualization, data curation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethic approval and consent to participate
The study was approved by the ethics committee of Peking Union Medical College Hospital. Informed consent forms were obtained from all patients or their families.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, X., Sun, X., Liu, Q. et al. Mutation spectrum of chinese amyotrophic lateral sclerosis patients with frontotemporal dementia. Orphanet J Rare Dis 17, 404 (2022). https://doi.org/10.1186/s13023-022-02531-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02531-2