
Abstract
Objective
Alström syndrome (ALMS) is a rare autosomal recessive genetic disorder that is caused by homozygous or compound heterozygous mutation in the ALMS1 gene. Dilated cardiomyopathy (DCM) is one of the well-recognized features of the syndrome ranging from sudden-onset infantile DCM to adult-onset cardiomyopathy, sometimes of the restrictive hypertrophic form with a poor prognosis. We aimed to evaluate severe cardiomyopathy in Alström syndrome in infancy and display susceptible specific mutations of the disease, which may be linked to severe DCM. Secondarily we reviewed published mutations in ALMS1 with cardiomyopathies in the literature.
Method
We represent new mutagenic alleles related to severe cardiomyopathy and cardiac outcome in this patient cohort. We evaluated echocardiographic studies of nine Turkish patients diagnosed with Alström syndrome (between 2014 and 2020, at age two weeks to twenty years). Thus, we examined the cardiac manifestations of a single-centre prospective series of nine children with specific ALMS mutations and multisystem involvement. All patients underwent genetic and biochemical testing, electrocardiograms, and echocardiographic imaging to evaluate systolic strain with speckle tracking.
Results
Four of the patients died from cardiomyopathy. Three patients (including three of the four fatalities) with the same mutation (c.7911dupC [p.Asn2638Glnfs*24]) had cardiomyopathy with intra-familial variability in the severity of cardiomyopathy. Global longitudinal strain, a measure of systolic contractile function, was abnormal in all patients that can be measured.
Conclusion
Cardiac function in ALMS patients with infantile cardiomyopathy appears to have different clinical spectrums depending on the mutagenic allele. The c.7911dupC (p. Asn2638Glnfs*24) mutation can be related to severe cardiomyopathy. Parents can be informed and consulted about the progression of severe cardiomyopathy in a child carrying this mutagenic allele.
Similar content being viewed by others


Introduction
Alström syndrome (ALMS) (OMIM# 203800) is a rare autosomal recessive genetic disorder [1]. About 800 individuals diagnosed with Alström syndrome have been identified worldwide. It is caused by mutations in the ALMS1 gene located on chromosome 2p13, which encodes a predicted 461.2-kDa protein of 4169 amino acids. The functions of this protein include microtubule organization, particularly the formation and maintenance of cilia [2]. The metabolic abnormalities include severe insulin resistance, type 2 diabetes (T2D), hypertriglyceridemia, thyroid, and hepatic dysfunction.
Dilated cardiomyopathy occurs in over 60% of patients with ALMS at different life periods. The onset and progression of the cardiomyopathy show intra-familial variability. More than 40% of infants with ALMS present severe dilated cardiomyopathy, although this is reported as improving after infancy [3, 4]. In about 20% of Alström syndrome cases, restrictive cardiomyopathy with fibrosis and pulmonary hypertension develops during adolescence or adulthood. Diffuse interstitial myocardial fibrosis in ALMS is associated with left ventricular (LV) systolic function [5,6,7,8].
Hence, ALMS is a complex multisystem disorder; particular mutations may explain the variability of clinical presentations. The mutations related to the severity of cardiomyopathy have not been defined yet. Here, we represent a family with multiple affected members with ALMS with a highly deleterious mutation associated with severe infantile cardiomyopathy and cardiac outcome in Alström syndrome patients. We aimed to evaluate severe cardiomyopathy in Alström syndrome in infancy and to display susceptible specific mutations of the disease, which may be linked to severe DCM. Secondarily we reviewed published mutations of ALMS with cardiomyopathies in the literature.
Materials and methods
Between March 2014 and November 2020, we evaluated the echocardiographic studies of nine Turkish patients diagnosed with ALMS (Table 1, Additional file 1: Table S1). All the patients had mutations in the ALMS1 gene (Fig. 1. Pedigree X and Y).

Pedigree of the patients, Sanger sequencing confirmation of mutation on ALMS1 gene
We measured kidney and liver function tests, haemoglobin A1c (HbA1c), and lipid panels for all the patients and performed standard 12-lead electrocardiograms (ECGs). Cardiac measurements were performed according to the American Society of Echocardiography (ASE) guidelines [9].
We calculated LV ejection and shortness fractions (LVEF and LVSF, respectively) using the Teicholz formula [9]. We analyzed right ventricular (RV) functions with RV fractional area change (FAC) and Tricuspid annular plane systolic excursion (TAPSE).
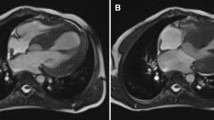
Further analysis of global myocardial function was performed with Speckle tracking echocardiography (GLS). Cardiac magnetic resonance imaging (CMR) imaging studies were performed on two patients (Patients A and C) to assess cardiac anatomy.
The ALMS1 gene was sequenced for each patient after the clinical diagnosis, including all exons and exon–intron boundaries, using next-generation sequencing on the Illumina Miseq platform. Lipid panel, blood glucose, insulin, and thyroid function tests were collected. ECG verified eye findings. Tympanometry and distortion product otoacoustic emissions (DPOE) testing evaluated the hearing situation (Table 2).
Results
The demographic and clinical characteristics of the patients are displayed in Table 2. Eight of the patients (8/9) had been diagnosed with cardiomyopathy. Six patients had infancy onset cardiomyopathy, while the other two had been diagnosed after infancy/ adolescence or early adulthood. Two patients of the six infantile-onset cardiomyopathies with the same mutation died before two years old, at 16 months and 22 months, respectively. Three patients with adolescence-onset CMP responded partially to heart failure medications. However, their recent ejection fractions were still at the lower end of the normal range in two and deteriorated in one patient. In 2 families with multiple affected children (Families X, Y), the siblings had similar spectrums for cardiomyopathy; in Family X, the older patient had mild cardiomyopathy while his sister, who was monitored by echocardiograms biannually since birth, displayed severe infantile cardiomyopathy until the age of two then improved with heart failure treatment to a mild form of cardiomyopathy.
Five patients were clinically stable at the evaluation time, without symptoms or signs of heart failure. There was no significant resting tachycardia and normal blood pressure. All patients received beta-blockers and angiotensin-converting enzyme inhibitors. The ECG was normal in 3 patients and abnormal in 2 patients. Abnormalities included left ventricular hypertrophy (n = 1), left axis deviation (n = 3), non-specific T wave changes (n = 3), right axis deviation (n = 1), second-degree atrioventricular block (n = 1). and the ECG was otherwise normal.
Among all patients, we found left ventricular dilatation or systolic dysfunction in all patients (Table 1). Valvar involvement was also noted in two patients with thickened mitral valve leaflets and moderate mitral regurgitation. One patient had restrictive cardiomyopathy, and another showed prominent LV trabeculations with deep intertrabecular recesses filled with blood from the ventricular cavity. Hypertrabeculation with multiple crypts at the apical postero-lateral aspects of the left ventricle. Patients' detailed individual echocardiographic values were presented in Additional file 1: Table S1.
CMR was performed on two children and had evidence of decreased left ventricular function. Strain parameters of the left ventricle, including GLS, were calculated in three available patients. GLS of the left ventricle was abnormal in three children (Fig. 2).

Left ventricular dilatation of patients with Alström syndrome on echocardiography. a Patient C, b Patient B, c Patient C, d Patient E, e Patient F
Genetic testing
ALMS1 whole gene sequencing was performed for all nine patients in this study (Table 3, Fig. 2). We detected a homozygous single nucleotide duplication on exon 10 of the ALMS1 gene (LRG_741t1[A1] c.7911dupC (p. Asn2638Glnfs*24) as previously reported by Marshall et al. [8]. This mutation is predicted to form a truncated protein, thus being highly deleterious and possibly responsible for the severe phenotype. A parental segregation study by Sanger sequencing confirmed the mutation (Table 4).
The outcome of patients with c.7911dupC (p. Asn2638Glnfs*24) mutation
Patients in Pedigree X had similar onset, progression, and outcome of the disease processes of Alström syndrome (Fig. 1). The dilated cardiomyopathy was reported in infancy, although it was evident soon after birth and progressed to death in two cases, with severe dilated and worsening cardiomyopathy, despite the anti-congestive medication. Several months later, the other four patients with the same mutation manifested dilated cardiomyopathy but survived beyond infancy. The seven-year-old and five-year-old girls had systolic dysfunction on echocardiography. One of them was determined to have had cardiomyopathy during infancy since he had systolic dysfunction (Fig. 2).
Discussion
Here, we have represented severe infantile-onset cardiomyopathy in a family with a recently recognized mutation of ALMS1 [8]. Although genotype–phenotype correlation has not been demonstrated for a specific clinical spectrum for the individual mutations in ALMS1 in all reported articles on cardiomyopathies, we think that the c.7911dupC (p. Asn2638Glnfs*24) mutation may be one of the significant determinants of the severe cardiomyopathy.
The cohort of patients with ALMS has demonstrated two major clinical spectrums related to cardiomyopathy: infantile-onset (43%) and those with later onset (18%). Cardiomyopathy has not been diagnosed in the remaining patients (39%, ages 2–33 years). Patients with infantile-onset cardiomyopathy have had apparent recovery of cardiac function within three years [4]. Previous reports have suggested the complete recovery of infantile-onset cardiomyopathy in Alström syndrome patients [4].
Bond et al. observed seven families in which heart failure due to dilated cardiomyopathy had been presented as a symptom within the first three months. In that study, all patients responded to conventional supportive therapy, and the cardiomyopathy resolved spontaneously within six months. The authors suggested that dilated cardiomyopathy in ALMS presenting in the first year of life can regress completely [10].
Brofferio et al. suggested that cardiac involvement in ALMS is more common than previously reported. A third (13 of 38) of patients in their study had infantile cardiomyopathy, with EF results ranging from 40 to 66%. The authors proposed longitudinal follow-up studies to determine whether this mild cardiomyopathy progresses into a more significant disease in all Alström syndrome patients [11].
Due to the reported outcomes of cardiomyopathy in ALMS, most series report cardiac function with a history of infantile cardiomyopathy that improves within the first 2–3 years of life [12, 13]. However, we suggest that recovery of cardiac function is not characterized by time; instead, it is a matter of the type and characterization of the mutation. The severity of cardiomyopathy depends on the truncated protein, accordingly to the mutation. In our patient series with the ALMS, c.7911dupC (p. Asn2638Glnfs*24) mutation, cardiomyopathy was partially resolved in two cases. In contrast, two patients with the same mutation died despite maximal drug support. Similar to our results, Mahamid et al. [14] reported two brothers, 2 and 3 years of age, diagnosed with Alström syndrome during febrile respiratory infection, initially presenting in infancy with severe dilated cardiomyopathy. The disease course in the older sibling has mainly resolved while cardiomyopathy in the younger sibling deteriorated despite maximal support with heart failure medications.
Our findings of LV systolic and diastolic dysfunctions in all the children correspond with previous reports [15]. Phenotypic variation, such as differences in severity of cardiomyopathy among siblings with the same mutation, suggests that besides any variability due to the mutation itself, there is an interplay between multitudes of potential genetic modifiers, with environmental factors leading to the range in severity of the Alström syndrome phenotypic spectrum [16].
In genotype–phenotype correlation studies, researchers have found no association between the location or type of ALMS1 mutations and T2D, body mass index (BMI), or dilated cardiomyopathy [10, 17]. However, in another study, Ichihara et al. did identify a link between the polyglutamine repeat in ALMS and early onset myocardial infarct [18].
Despite advances in our knowledge of the spectrum of ALMS1 mutations, we still do not have enough evidence for prognostic predictions based on genotype. Histopathology of affected patients has shown diffuse interstitial fibrosis affecting the myocardium, later confirmed by cardiac MRI [5, 6]. Dilated cardiomyopathy (DCM) can occur suddenly in infancy (in the first months of life) due to aberrant differentiation of cardiomyocytes [19, 20]. Histologic findings of infantile cardiomyopathy have not been presented yet, but there are studies in which mitogenic cardiomyopathy is described in patients with Alström syndrome [21, 22].
Mitogenic cardiomyopathy is a very rare human phenotype. Shenje et al. showed homozygous or compound heterozygous mutations among six infants with Alström syndrome, suggesting mitogenic cardiomyopathy is the leading cause of lethal cardiomyopathy. They showed that ALMS1 is a key for cell cycle regulation in perinatal cardiomyocytes. They removed the hearts of each individual they studied, either at the time of transplantation due to end-stage heart failure or after death from heart failure [21]. Although we did not perform autopsies, mitogenic cardiomyopathy may be the cause of death of our patient with severe infantile cardiomyopathy. Restrictive cardiomyopathy develops slowly in adolescents and adults [23]. In line with previous reports, our first adolescent case (Patient A) died because of restrictive cardiomyopathy.
Although intra-familial differences observed in the disease presentation of our patients carrying the same mutation (Patients B, C, D, E, and F) complicated our notion about this recently recognized mutation, we suggest that there are mutations that tend to severe cardiomyopathy. The phenotypic differences observed among individuals within the same family may result from gene–gene, protein–protein interactions or functional polymorphisms [24]. Some authors have suggested that environmental and unknown genetic modifiers probably interact with ALMS1 [13] children growing up in the same family with similar environmental factors to support expression patterns of genotype. Further to the evidence from our patients, Hollander et al. showed that the clinical course of Alström syndrome might vary concerning cardiac disease manifestations, even in monozygotic twins [25].
In a literature review of 44 patients from our country by Ozanturk et al., the authors reported death of two ALMS patients with an unknown genetic mutation might be linked to CMP [26]. Our patients had not been displayed in that study. In our ALMS cohort, cardiomyopathy appeared to be a primary manifestation.
In a study by Brofferio et al., there were subclinical strain echocardiographic abnormalities in nearly all patients in their cohort, suggesting the presence of myocardial disease in patients without overt cardiomyopathy, including those with recovered from infantile cardiomyopathy [11]. However, the individuals in the current cohort also displayed significant metabolic disturbances, including T2D, vision and hearing loss, elevated triglyceride and cholesterol levels, and obesity. These metabolic disturbances can themselves be substantial cardiac risk factors over time. Our findings of abnormal GLS with LV systolic dysfunction and restrictive cardiomyopathy of both ventricles support their results.
In another study, while mild interstitial myocardial fibrosis was present in patients without a prior history of dilated cardiomyopathy, moderate-to-severe interstitial fibrosis was found in ALMS patients with dilated cardiomyopathy [4]. Progressive fibrosis and cardiac risk factors (obesity, T2DM) can explain the restrictive cardiomyopathy of our case (Patient A), diagnosed in adolescence with restrictive cardiomyopathy.
Conclusion
Early diagnosis of ALMS and genetic testing provide important prognostic information to the paediatric cardiologist and enable the family to consider likely outcomes. First, appreciation of the aetiology of cardiomyopathy can guide appropriate management plans. Second, determining the exact causal mutation(s) in the index patient can clarify the inheritance pattern. Thus, families can be consulted on the recurrence risk in future offspring and seek prenatal testing in subsequent pregnancies.
Our comprehensive evaluation of nine children with Alström syndrome suggests that cardiac function in ALMS patients with infantile cardiomyopathy has different clinical spectrums depending on the mutagenic allele. We recommend that the c.7911dupC (p. Asn2638Glnfs*24) mutation can be related to severe cardiomyopathy.
Availability of data and materials
Please contact author for data requests.
References
Bond J, Flintoff K, Higgins J, et al. The importance of seeking ALMS1 mutations in infants with dilated cardiomyopathy. J Med Genet. 1997;42(2):e10.
Brofferio A, Sachdev V, Hannoush H, et al. Characteristics of cardiomyopathy in Alström syndrome: prospective single-center data on 38 patients. Mol Genet Metab. 2017;121:336–43.
Chang KT, Taylor GP, Meschino WS, Kantor PF, Cutz E. Mitogenic cardiomyopathy: a lethal neonatal familial dilated cardiomyopathy characterized by myocyte hyperplasia and proliferation. Hum Pathol. 2010;41(7):1002e8.
Collin GB, Marshall JD, Ikeda A, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31(1):74–8.
Edwards NC, Moody WE, Yuan M, et al. Diffuse LV interstitial fibrosis is associated with sub-clinical myocardial dysfunction in Alström Syndrome: an observational study. Orphanet J Rare Dis. 2005;10:83.
Hearn T, Renforth GL, Spalluto C, et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat Genet. 2002;31(1):79–83.
Hollander SA, Alsaleh N, Ruzhnikov M, Jensen K, Rosenthal D, Stevenson D, Manning M. Variable clinical course of identical twin neonates with Alström syndrome presenting coincidentally with dilated Cardiomyopathy. Am J Med Genet. 2017;173A:1687–9.
Ichihara S, Yamamoto K, Asano H, et al. Identification of a glutamic acid repeat polymorphism of ALMS1 as a novel genetic risk marker for early-onset myocardial infarction by genome-wide linkage analysis. Circ Cardiovasc Genet. 2013;6(6):569–78.
Lopez L, Colan ST, Frommelt PC, et al. Recommendations for quantification methods during the performance of a pediatric echocardiogram: a report from the pediatric measurements writing group of the American society of echocardiography pediatric and congenital heart disease council. J Am Soc Echocardiogr. 2010;23:465–95.
Loudon MA, Bellenger NG, Carey CM, Paisey RB. Cardiac magnetic resonance imaging in Alström syndrome. Orphanet J Rare Dis. 2009;4:14.
Louw JJ, Corveleyn A, Jia Y, et al. Homozygous loss-of-function mutation in ALMS1 causes the lethal disorder mitogenic cardiomyopathy in two siblings. Eur J Med Genet. 2014;57(9):532–5.
Mahamid J, Lorber A, Horovitz Y, Shalev SA, Collin GB, Naggert JK, et al. Extreme clinical variability of dilated cardiomyopathy in two siblings with Alström syndrome. Pediatr Cardiol. 2013;34(2):455–8.
Marshall JD, Maffei P, Collin GB. Naggert JK Alström syndrome: genetics and clinical overview. Curr Genom. 2011;12(3):225–35.
Marshall JD, Bronson RT, Collin GB, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165(6):675–83.
Marshall JD, Ludman MD, Shea SE, et al. Genealogy, natural history and phenotype of Alström syndrome in a large Acadian kindred and three additional families. Am J Genet. 1997;73:150–61.
Marshall JD, Muller J, Collin GB, et al. Alström syndrome: mutation spectrum of ALMS1. Hum Mutat. 2015;36(7):660–8.
Michaud J, Hon E, Guilbert F, Weill J, Puech B, Benson L, et al. Natural history of Alstr6m syndrome early childhood: onset with dilated cardiomyopathy. J Pediatr. 1996;128(2):225–9.
Nerakh G, Ranganath R. Alström syndrome presenting as isolated dilated cardiomyopathy. Indian J of Pediatr. 2019;86(3):296–8.
Ozantürk A, Marshall JD, Collin GB, et al. The phenotypic and molecular genetic spectrum of Alström syndrome in 44 Turkish kindreds and a literature review of Alström syndrome in Turkey. J Hum Genet. 2015;60:1–9.
Richard J, Paisey B, Steeds R, et al. Alström syndrome. In: Ardinger HH, Pagon RA, Wallace SE, editors., et al., GeneReviews (R). Seattle WA: University of Washington; 1993.
Shenje LT, Andersen P, Halushka MK, et al. Mutations in Alström protein impair terminal differentiation of cardiomyocytes. Nat Commun. 2014;5:3416.
Smith JC, McDonnell B, Retallick C, et al. Is arterial stiffening in Alström Syndrome linked to the development of cardiomyopathy? Eur J Clin Invest. 2007;37:99–105.
T’Hart LM, Dekker JM, Heine RJ, Maassen JA. Lack of association between gene variants in the ALMS1 gene and Type 2 diabetes mellitus. Diabetologia. 2003;46:1023–4.
Nussinov R, Tsaid CJ, Jang H. Protein ensembles link genotype to phenotype. PLoS Comput Biol. 2019;15(6): e1006648.
Toulany A, Shea S. Warren AE Doppler tissue, strain, and strain rate imaging in pediatric patients with Alström syndrome: are there regional functional abnormalities? J Am Soc Echocardiogr. 2006;19(1):14–20.
Zerbini C, Weinberg DS. Perez-Atayde AR DNA ploidy analysis of myocardial hyperplasia. Hum Pathol. 1992;23(12):1427e30.
Acknowledgements
Not applicable
Funding
No funding was used during this study.
Author information
Authors and Affiliations
Contributions
SD, ED, AG take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation. SD designed the study, planned the concept, and prepared and edited manuscript. RD, ED, FO, GY, SD had roles in data acquisition and analysis. RD had a role in the literature overview and data analysis. All of the authors gave final approval of this version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by Istanbul University-Cerrahpasa Institutional Review Board (November 3, 2020; 29430533–604.01–01-139311). Detailed informed consent was obtained for publication from all patients or their parents/legally authorized representatives.
Competing interests
The authors declare that they have no competing interests, including specific financial interests, relationships and/or affiliations relevant to the subject matter or materials included.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1.
Detailed individual echocardiographic studies of nine Turkish patients diagnosed with ALMS.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dedeoglu, S., Dede, E., Oztunc, F. et al. Mutation identification and prediction for severe cardiomyopathy in Alström syndrome, and review of the literature for cardiomyopathy. Orphanet J Rare Dis 17, 359 (2022). https://doi.org/10.1186/s13023-022-02483-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02483-7