
Abstract
Background
De novo heterozygous mutations in the GNAO1 gene, encoding the Gα o subunit of G-proteins, are the cause of a severe neurodevelopmental disorder, featuring early infantile seizures, profound cognitive dysfunction and, occasionally, movement disorder (early infantile epileptic encephalopathy-17).
Methods
We report a further case of this association in a 20 month-old Spanish girl with neonatal-onset refractory seizures, progressive microcephaly, oral-lingual dyskinesia and nearly absent psychomotor development. We performed whole-exome sequencing, a computational structural analysis of the novel gene variant identified and reviewed the previously reported cases.
Results
Trio whole-exome-sequencing uncovered a de novo p.Leu199Pro GNAO1 mutation. Computational structural analysis indicates this novel variant adversely affects the stability of the G-protein heterotrimeric complex as a whole. Of note, our patient showed a sustained seizure reduction while on a ketogenic diet.
Conclusions
With this observation, a total of twelve patients with GNAO1 encephalopathy have been reported. Oral-lingual dyskinesia and responsiveness of seizures to ketogenic diet are novel features. The distorted sex ratio (12/12 females) of the condition remains unexplained; a differential gender effect of the disruption of G-protein- mediated signal transduction on the developing brain can be hypothesized.
Similar content being viewed by others


Background
Epileptic encephalopathy is a group of neurological disorders characterized by severe and progressive cognitive and behavioral impairments, which are most probably caused or worsened by epileptic activity [1]. Heterozygous mutations in the GNAO1 gene (MIM*139311), encoding the alpha subunit of the heterotrimeric guanine nucleotide-binding proteins (G proteins), were first described as a cause of early infantile epileptic encephalopathy (EIEE) in 2013 [2]. Subsequent reports have broadened the spectrum of clinical presentation [3–5] which includes prominent dyskinesia and intellectual disability with few or no seizures (4) and the condition is currently classified as EIEE17 (MIM #615473). Interestingly, all the cases reported to date involve female subjects despite the disease-causing gene maps to chromosome 16q13.
The GNAO1 gene product is the alpha subunit of Go, a member of the G-protein family, involved in cellular signal transduction. Three functional G-protein subtypes are defined, namely inhibitory G-proteins (Gi), stimulatory G-proteins (Gs) and “other” (Go) the latter being abundantly expressed in the brain [6]. Typically, G-proteins are characterized by their alpha subunit, which binds and hydrolyzes GTP and interacts with specific GPCRs (G-protein coupled receptors) or effector molecules, such as adenylate cyclase. Relevant to neuronal excitability, Go proteins modulate neurotransmitter release by mediating the presynaptic auto-inhibitory effect of several neurotransmitters on their receptors, including M2/M4 muscarinic, alpha(2) adrenergic, mu/delta opioid and GABA-B receptors [7].
We here report the first Spanish girl with this disorder carrying a de novo GNAO1 heterozygous mutation. State-of-the-art computational analyses suggest that this novel variant causes significant defects in protein structural stability.
Methods
Clinical report
The patient is a 21 month-old girl with a severe encephalopathy presenting with neonatal seizures. She is the second child of unrelated, healthy parents. A 4 year-old sister is healthy. Pregnancy and delivery were uneventful. Neonatal physical examination was reportedly normal, with head circumference of 34 cm (35th percentile). Seizures appeared on the third day of life and were tonic generalized or manifested as facial congestion, drooling, or motor phenomena such as sucking, grimacing or winking. They lasted from seconds to a few minutes, recurred up to 40 times per day and were refractory to treatment, including a pyridoxal phosphate-containing vitamin cocktail.
She was referred for study at the age of 3 months. The physical examination revealed a head growth deceleration (head circumference at 38,8 cm, 3rd percentile), low-set ears without other dysmorphic features, drowsiness with lack of visual fixation when awake, decreased muscle tone, reduced spontaneous movements and occasional startle. Interictal EEG recordings showed diffuse background slowing, and multifocal high-voltaged sharp waves and spike and slow-wave complexes, with predominance over both central-parietal areas. An extensive metabolic workup, including CSF lactate, amino acids and neurotransmitter metabolites, array CGH, fundoscopic examination and visual evoked potentials were normal. A previous brain MRI study, performed at 18 days of life, showed signs consistent with mild cerebral atrophy, thin corpus callosum and delayed myelination (Fig. 1).

Brain MRI at age 18 days. Sagittal T1WI view showing thinning of the corpus callosum (arrow), with anterior predominance, and some degree of frontal lobe volume reduction (a). Axial T2WI view showing asymmetric enlargement of temporal subarachnoidal spaces (arrow) and mildly prominent sulci over the cerebral convexity, suggesting incipient atrophy. Myelination is delayed with minimal signals observed at the posterior limb of the internal capsule (arrowhead) and corpus callosum (b)
On follow-up, EEG’s continued to show a slowed and high-voltaged background activity with poor differentiation and absent sleep elements plus very abundant, almost constant, bilateral sharp wave elements, consistent with severe epileptic encephalopathy. Seizures persisted unchanged despite the use of multiple antiepileptic drugs, including phenobarbital, valproate, levetiracetam, vigabatrin, carbamazepine, zonisamide, clobazam, topiramate and lacosamide in different combinations as well as a short trial with hydroaltesone. Although a slight improvement in seizure frequency was eventually obtained with a carbamazepine, topiramate and clobazam at age 11 months she was still suffering 15–20 seizures per day, mostly tonic generalized with upgaze deviation. With the institution of a 1:3 ketogenic diet (KD) seizure frequency progressively diminished and she remained seizure-free for almost two months. However, non-compliance with the KD resulted in recurrence of seizures. She currently averages 1–4 seizures per day. At age 19 months an EEG performed during sleep captured two seizures with, respectively, right and left mesial frontal origin. At age 21 months, the patient is severely disabled; she has not attained cephalic control or visual fixation and does not respond to any social cues. On examination, head circumference is 43 cm (<−3SD), height 86 cm (33th percentile) and weight 14 Kg (96th percentile). There is severe axial hypotonia, spastic tetraparesis, brisk deep tendon reflexes and paucity of voluntary movements. She has recently begun to display dyskinetic buccolingual movements, but without evidence of adventitious movements involving the trunk or limbs.
Genetic studies and whole-exome sequencing
Peripheral blood samples were collected from the trio (patient and both progenitors). Genomic DNA was isolated using a standard salting-out method. The study was approved by the Vall d'Hebron University Hospital Institutional Review Board and informed consent was obtained from the patient’s parents according to the Helsinki declaration.
Exome sequencing of DNA samples from the trio was performed at the Centre Nacional d’Anàlisi Genòmica (CNAG) in Barcelona. 1 μg of genomic DNA was fragmented with an ultra sound device (Covaris, Woburn, MA, USA), denatured and hybridized with capture oligos (NimbleGen SeqCap EZ Exome v3.0 exome enrichment kit, Roche, Madison, WI, USA) which enriches for ~44 Mb of the human exonic regions. Captured sequences were enriched with streptavidin-conjugated paramagnetic beads. End repair, A-tailing, Illumina adapter ligation and post-capture amplification were done with TruSeq PE Cluster Kit v3, Illumina Corp, La Jolla, CA). Each captured exome was sequenced in one HiSeq2000 lane using version 3 chemistry. Coverage over 30X -was achieved for more than 80 % of the target. Alignment and variant calling were carried out following the GATK best practices for whole-exome sequencing. Sequencing reads were aligned to b37-decoy reference using BWA (http://bio-bwa.sourceforge.net/), then we marked duplicates, performed local realignment around indels, recalibrated the base quality scores, called variants with HaplotypeCaller and filtered them using Variant Quality Score Recalibration (VQSR) and hard filters. Refinement was done considering the pedigree, the population frequencies from Exome aggregation consortium (http://exac.broadinstitute.org/) [8], NHLBI Exome Sequencig Project (http://evs.gs.washington.edu/EVS/) [9], 1000 genomes project (http://www.1000genomes.org/) [10] and dbsnp (http://www.ncbi.nlm.nih.gov/SNP/) [11] and finally, the functional annotation (SnpEff, SnpSift) and the disease. In a first tier analysis (diagnostic exome sequencing) we investigated any variants within genes previously associated with epilepsy. This was followed by consideration of variants in any other gene covered by WES (research exome sequencing). Both, de novo mutations and transmitted alleles were analyzed. All candidate selected de novo mutations were absent in ExAC, and passed visual inspection of alignment quality using the Integrative Genomics Viewer (IGV).
Sanger sequencing was performed to confirm the GNAO1 de novo missense variant identified through whole-exome sequencing. To that end, exon six from the proband and progenitors was amplified by Polymerase-Chain-Reaction (PCR), purified and sequenced using the BigDye Terminator cycle sequencing kit v3.1 and an automated sequencer ABI PRISM 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA) (primer sequences and PCR conditions available upon request).
Structural modeling and free-energy calculations
Homology modeling human Gαo protein
The human WT Gαo protein was homology modeled from the crystal structure of mouse Gαo (PDB id: 3C7K) with MODELLER [12] (residues 22–346, see Additional file 1: Table S1 for sequence alignment). The sequence identity between mouse and human protein is 98 %. During homology modeling, conserved residues were maintained in their original crystal structure rotameric orientation. The co-crystallized ligands GDP and MG were directly transferred from the crystal structure into the homology model. The WT homology model was energy-minimized in the AMBER14SB force-field [13] with 1000 steps of steepest descent and 100 steps of conjugate gradients in CHIMERA [14]. The p.Leu199Pro mutation was inserted into the WT homology model with CHIMERA by applying the most appropriate side-chain rotamer and then energy-minimizing the mutant protein as performed previously in the AMBER14SB force-field.
Calculation of free-energy change
The predicted free-energy change (ddG) of the p.Leu199Pro mutation in human Gαo was made using ROSETTA [15] with a conversion factor of 0.73 employed to convert Rosetta Energy Units to kcal/mol [16]. The program was executed in “high-resolution mode” with full protein flexibility in side-chains and backbone (without bound GDP) and with the following flags: “iterations” set to 50, “local_opt_only” set to false, “sc_min_only” set to false, “opt_radius” set to 12.0, and “fa_max_dis” set to 9.0.
Molecular dynamics simulations
Both Gαo WT and Gαo Leu199Pro molecular dynamics (MD) systems were built using Amber LEap from AmberTools14 [13]. Ions were added to neutralize both systems and salt was added at a concentration of 0.2 M in each. Each system was then equilibrated with MD using ACEMD (Accelerating Biomolecular Dynamics in the Microsecond Time Scale) [17] at 300° and a pressure of 1 atm (NPT) in the AMBER14SB force-field for a total of 7.0 ns with harmonic restraints on the protein progressively relaxed from 10 kcal mol−1 Å−2 to 0 kcal mol−1 Å−2. Production run MD simulations of 1000 ns were then performed for both WT and mutant Leu199Pro proteins at 300° (NVT) in the AMBER14SB force-field using ACEMD [17]. VMD 1.9.2 [18] was used for RMSD (root-mean-square deviation) analyses of MD simulations, including heat-map generation.
Results
Whole-exome sequencing
Whole exome sequencing from the patient and her parents (trio-based variant analysis) identified a single de novo variant within the epilepsy candidate genes, a heterozygous mutation GRCh37/hg19:chr16:56370645, NM_020988.2: c.596 T > C in GNAO1, producing the amino acid change p.Leu199Pro (NP_066268.1) in the proband. Read depth at the variant position was 38X, 42X and 50X for the proband and progenitors, respectively. The mutation was confirmed by PCR and Sanger sequencing (Fig. 2). This variant has not been previously described as related to the phenotype and is not present in any of the public genomic databases listed above. In silico prediction sofwares (SIFT [19], Mutation Taster [20] andPolyPhen-2 [21]) classified the novel variant as damaging. Despite the location of the variant in close proximity to a splice junction, the ESEfinder [22, 23] and Netgene2 [24, 25] scores did not indicate an effect on splicing (data not shown).

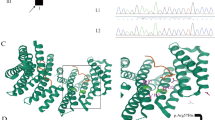
a Left, pedigree of the affected patient, showing the carrier of the heterozygous of the de novo mutation (filled symbol) and her unaffected sister and parents. Right, validation of the identified mutation by means of PCR and Sanger sequencing: electropherograms show the wild-type sequence in the progenitors (top) and the novel GNAO1 c.596 T > C (NM_020988.2) variant in the proband (bottom), producing the amino acid change p.Leu199Pro (NP_066268.1). b Multiple sequence alignment of the GNAO1 protein region containing Leucine 199 (NP_066268.1) illustrating the high degree of evolutionary conservation of the affected residue
Computational structural analysis
The human WT Gαo protein with bound GDP was homology modeled from the crystal structure of mouse Gαo (PDB id: 3C7K) using MODELLER [8]. As these proteins share 98 % sequence identity, homology modeling accuracy can be considered very high (see Methods for details and Supplementary Information). The Leu199Pro mutation was introduced with CHIMERA [14]. The Leu199Pro mutation site is located in the GTPase domain of Gαo, specifically residing in the interior of a β-sheet that is packed between the N- and C-termini and interacting with adjacent α-helices. According to the homology model of human Gαo, Leu199 is not involved in GDP/GTP binding so its mutation to proline is not predicted to directly affect ligand binding or hydrolysis (see Fig. 3a).

a Homology model of human WT Gαo. The mutated Leu199 is highlighted in yellow space-fill, protein backbone with secondary structure in beige, GDP in red/orange space-fill, magnesium in green. b Structural superposition of mutant Leu199Pro Gαo protein after 1 μ-second of MD simulation (in pink with mutation Leu199Pro shown as red space-fill) and heterotrimeric guanine-nucleotide-binding protein Gi (PDB id: 1GG2, Giα in beige, Giβ in cyan, Giγ in green). c GTPase domain of mutant Leu199Pro Gαo (in pink) compared to WT (in green) after 1 μ-second MD simulations (mutation Leu199Pro shown in red space-fill). d Plot of RMSD (a) for Cα atoms of WT Gαo protein and Leu199pro mutant during 1 μ-second MD simulations (RMSD comparisons made against initial structure)
The WT and mutant forms of human Gαo were analysed with ROSETTA [15] to calculate the predicted free-energy change (ddG) of the mutation. Using a conversion factor of 0.73 to convert from Rosetta Energy Units to kcal/mol [12], the predicted ddG of Leu199Pro is +6.1 kcal/mol. This suggests the mutation has a destabilizing effect on the protein and could possibly cause defects in its folding. To investigate this effect in more detail, both the WT protein and Leu199Pro mutant were subjected to a 1 μ-second MD simulation using ACEMD [13]. During these simulations, the WT protein remains stable in terms of both its secondary and tertiary structure with an RMSD that fluctuates between 1.0 and 2.0 Å (Fig. 3d). However the Leu199Pro mutant displays significant structural change in its GTPase domain, with multiple H-bonding in its central B-sheet broken, causing the creation of a large flexible loop region and increased positional fluctuation (Fig. 3c), with a protein RMSD of 1.5–3.0 Å across the simulation (Fig. 3d). In addition, the mutation also affects other areas of the domain, in particular α-helices: 208–217 and 257–262, both which lose their structural integrity and experience greater positional fluctuation during the simulation (Fig. 3c).
In the context of the protein as a whole, heat-map representations of positional fluctuation reveal structural disturbances across both domains, GTPase and helical, in the mutant Leu199Pro protein (see Fig. 4). These disturbances occur in areas already described in the GTPase domain but also in several loop regions throughout the protein.

a RMSD per residue heat-map (scale 0–15, from low/blue to high/red) for WT Gαo throughout a 1 μ-second MD simulation. b RMSD per residue heat-map for mutant Leu199Pro Gαo. RMSD comparisons made against respective initial structures
Discussion
Early infantile epileptic encephalopathy-17 is a severe neurological disorder characterized by onset of intractable seizures in the first weeks or months of life, usually associated with EEG abnormalities, caused by heterozygous mutations in GNAO1. Neurological outcome is uniformly poor in all the infants described thus far. Imaging may disclose indistinct brain abnormalities, such as cerebral atrophy, delayed myelination, and/or or thin corpus callosum.
Including the present report, twelve patients with GNAO1 encephalopathy have been described (Table 1). Early-onset epileptic encephalopathy has been the most frequent presentation (n = 9), but two out of the eight described Japanese patients, showed either few, late-onset seizures or none at all [4]. Clinical-EEG patterns are variable, and patients are referred to as suffering from Ohtahara, malignant migrating partial epilepsy of infancy, West syndrome or other forms of early epileptic encephalopathy phenotype [2–5]. Some patients show involuntary movements, including chorea, dystonia, athetosis or stereotypies [2, 4].
Refractoriness to antiepileptic drugs appears a common feature in all patients displaying the EIEE phenotype. In our patient a complete cessation of seizures was noted one month after the institution of a ketogenic diet and the patient remains seizure-free five months into the diet. Considering the widely admitted deleterious effect of seizures on the developing brain, institution of this therapeutic modality early in the evolution of GNAO1 encephalopathy seems advisable. Based on the loss of calcium-current inhibition found in vitro in GNAO1 mutants, resulting from altered Gαo -mediated signaling induced by norepinephrine, Nakamura et al. suggested the use of selective calcium-channel blockers or the use of the high-voltage activated calcium channels modulator topiramate to alleviate epilepsy in patients with GNAO1 mutations [2]. Our patient, at variance with previous cases where treatment was specified, did receive topiramate since the age of 8 months, but without noticeable improvement in seizures or changes in her developmental profile.
All twelve patients described carried heterozygous de novo missense mutations [2–5]. Indeed, the p.Leu199Pro is predicted to cause protein dysfunction in both of the described GNAO1 isoforms, according to the used in silico prediction tools. Furthermore, the application of the recently published ACMG guidelines for interpretation of sequence variants [26] to the p.Leu199Pro variant classified it as pathogenic by virtue of fulfilling one strong (PS2) plus two moderate (PM1, PM2) and two supporting (PP2,PP3) pathogenic criteria.
In vitro functional studies showed that three out of four analyzed missense GNAO1 mutations resulted in abnormal, cytoplasmic localization of the protein, while electrophysiological studies proved that all four variants produced impaired N-type calcium channel current inhibition after norepinephrine application [2]. Interestingly, a mouse model harboring a heterozygous (p.Gly184Ser) Gnao1 missense mutation displayed a severe and lethal epileptic phenotype that was absent in the Gnao1 hemizygous knock-out mouse, suggesting that pathogenic GNAO1 mutations may act through a gain-of-function, dominant effect [27].
In our present work, we investigated the effect of the novel p. Leu199Pro mutation in the structure of the human Gαo protein using state-of-the-art computational tools. These provide evidence that the mutation causes significant defects in protein stability. The observed disturbances occur locally in the GTPase domain where the mutation is situated but also in several loop regions throughout the protein. This suggests the p.Leu199Pro mutation decreases overall structural stability and perhaps folding of the Gαo protein. From a functional point of view, the location of the p.Leu199Pro mutation may be particularly significant in terms of interactions Gαo makes with its binding partner Gβϒ and others. With respect to the crystal structure of the complete heterotrimeric guanine-nucleotide-binding protein Gi (PDB id: 1GG2), when the Gαo subunit is superimposed, the mutation p.Leu199Pro is in direct contact with Gβϒ (see Fig. 1b). Therefore any introduced structural instability in this interface would likely adversely affect the stability of the Gαβϒ complex as a whole. From the observed results, it is certainly conceivable that the p.Leu199Pro mutation could generate this level of instability. The issue of a female gender bias in this disorder deserves special consideration. A distorted sex ratio can be found in many complex disorders, for instance autism spectrum disorder [28] and a male-biased effect has been described for 16p13.11 copy number variants as predisposing factor to a range of neurodevelopmental disorders [29]. Possible causes include hormonal factors, sexual dimorphic brain differences, imprinting or genetic modifiers, with a net effect of greater protection of the female brain. It is conceivable that the penetrance of GNAO1 mutation is higher in the less resilient male brain thus producing a more severe phenotype. While female bias in EIEE-17 remains unexplained, it could be posited that disruption of G-protein signal transduction may lead to prenatal lethality in males.
Conclusions
We describe a further female patient with GNAO1-associated neonatal-onset epilepsy and a severe neurodevelopmental disorder. Seizures were amenable to treatment with KD. Structural analysis of the novel mutation suggests it introduces significant structural instability into both the Gαo subunit and the whole G-protein heterotrimeric complex. The resulting defective cellular signal transduction is in all probability deleterious during brain development and putatively fatal in the male embryo.
Abbreviations
- ACEMD:
-
accelerating biomolecular dynamics in the microsecond time scale
- BWA:
-
Burrows-wheeler aligner
- CGH:
-
comparative genomic hybridization
- CSF:
-
cerebrospinal fluid
- ddG:
-
predicted free-energy change
- EEG:
-
electroencephalography
- EEIE:
-
early infantile epileptic encephalopathy
- GABA:
-
gamma-aminobutyric acid
- GATK:
-
genome analysis toolkit
- GDP:
-
guanosine diphosphate
- Gi:
-
inhibitory G-proteins
- Go:
-
“other” G-proteins
- GPCRs:
-
G-protein coupled receptors
- Gs:
-
stimulatory G-proteins
- GTP:
-
guanosine triphosphate
- IGV:
-
integrative genome viewer
- KD:
-
ketogenic diet
- MD:
-
molecular dynamics
- MG:
-
magnesium
- MRI:
-
magnetic resonance imaging
- PCR:
-
polymerase-chain-reaction
- PDB:
-
protein data bank
- RMSD:
-
root-mean-square deviation
- VQSR:
-
variant quality score recalibration
- WT:
-
wildtype
References
Dulac O. Epileptic encephalopathy. Epilepsia. 2001;42 Suppl 3:23–6.
Nakamura K, Kodera H, Akita T, Shiina M, Kato M, Hoshino H, et al. De novo mutations in GNAO1, encoding a G-alpha-o subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet. 2013;93:496–505.
EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. 2014;95:360–70.
Saitsu H, Fukai R, Ben-Zeev B, Sakai Y, Mimaki M, Okamoto N, et al. Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur J Hum Genet. 2016;24:129–34.
Talvik I, Moller R, Vaher M, Vaher U, Larsen LHG, Dahl H, et al. Clinical phenotype of De novo GNAO1 mutation: case report and review of literature. Child Neurology Open. 2015. doi:10.1177/2329048X15583717.
Strathmann M, Wilkie TM, Simon MI. Alternative splicing produces transcripts encoding two forms of the alpha subunit of GTP-binding protein Go. Proc Natl Acad Sci U S A. 1990;87:6477–81.
Brown DA, Sihra TS. Presynaptic signaling by heterotrimeric G-proteins. Handb Exp Pharmacol. 2008;184:207–60.
Exome Agregation Consortium, Lek M, Karczewski KJ, Minkel EV, Samocha KE, Banks E et al. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv preprint first posted online Oct. 30, 2015; doi: 10.1101/030338.
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/) (accessed January, 2016)
A global reference for human genetic variation, The 1000 Genomes Project Consortium, Nature 526, 68–74 (01 October 2015) doi:10.1038/nature15393.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Sali A. Comparative protein modeling by satisfaction of spatial restraints. Mol Med Today. 1995;1:270–7.
Case DA, Cheatham 3rd TE, Darden T, Gohlke H, Luo R, Merz Jr KM, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–88.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–25.
Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins. 2011;79:830–8.
Harvey MJ, Giupponi G, De Fabritiis G. ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput. 2009;5:1632–9.
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7, e46688.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. Nat Methods. 2010;7:248–9.
Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–508.
Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucl Acids Res. 2003;31:3568–71.
Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site prediction in Arabidopsis thaliana DNA by combining local and global sequence information. Nucl Acids Res. 1996;24:3439–52.
Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220:49–65.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Kehrl JM, Sahaya K, Dalton HM, Charbeneau RA, Kohut KT, Gilbert K, et al. Gain-of-function mutation in Gnao1: a murine model of epileptiform encephalopathy (EIEE17)? Mamm Genome. 2014;25:202–10.
Whiteley P, Todd L, Carr K, Shattock P. Gender ratios in Autism, Asperger syndrome and Autism spectrum disorder. Autism Insights. 2010;2:17–24.
Tropeano M, Ahn JW, Dobson RJ, Breen G, Rucker J, Dixit A, et al. Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders. PLoS One. 2013;18:e61365.
Acknowledgements
This work was funded by grants from Instituto de Salud Carlos III Madrid, Spain (PI12/01005), Ministerio de Economía y Competitividad, Spain (SAF2014-58396-R) and AGAUR, Barcelona, Spain (SGR2009/0078); AM-G is the recipient of a scholarship from Vall d’Hebron Institut de Recerca (Barcelona, Spain); EC-L supported by a Beatriu de Pinós post-doctoral grant from AGAUR (Barcelona, Spain).
The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison (a full list of contributing groups can be found at http://exac.broadinstitute.org/about) and the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
AMG carried out the molecular genetic studies and validated the findings, JD performed the computational structural analyses, JLP provided the clinical and neuroradiological data, MCGJ and LMG carried out metabolic studies related to treatment and helped to draft the manuscript, ECL set up the bioinformatic analysis pipeline, JG devised the computational approach to mutation analysis and interpreted the data, AM conceived the study and drafted the manuscript. All authors have read and approved the final manuscript.
Additional file
Additional file 1: Table S1.
Sequence alignment as calculated by NCBI-BLAST between human Gαo (Query) and mouse Gαo (Sbjct, PDB id: 3C7K). (DOC 69 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Marcé-Grau, A., Dalton, J., López-Pisón, J. et al. GNAO1 encephalopathy: further delineation of a severe neurodevelopmental syndrome affecting females. Orphanet J Rare Dis 11, 38 (2016). https://doi.org/10.1186/s13023-016-0416-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-016-0416-0