
Abstract
Background
Developmental and epileptic encephalopathies (DEEs) are a heterogeneous group of severe disorders that are characterized by early-onset, refractory seizures and developmental slowing or regression. Genetic variations are significant causes of these changes. De novo variants in an increasing number of candidate genes have been found to be causal. The YWHAG gene is one such gene that has been reported to cause developmental and epileptic encephalopathy 56 (DEE56). Here, we report a heterozygous missense variant, c.170G > A (p.R57H), in the YWHAG gene that caused early-onset epilepsy and developmental delay in a Chinese family.
Methods
We described the clinical manifestations of the proband and his mother in detail. Then, we use trio-based whole-exome sequencing to search the etiology of this family.
Results
Both the proband and his mother exhibited early-onset seizures, intellectual disability, and developmental delay. While the proband attained seizure control with sodium valproate, his mother's seizures were not well controlled. Trio-based whole-exome sequencing revealed a heterozygous missense variant, c.170G > A (p.R57H), in the YWHAG gene, which was considered as the cause of early-onset epilepsy and developmental delay in this family.
Conclusions
Our report further confirmed that YWHAG haploinsufficiency results in developmental and epileptic encephalopathy 56.
Similar content being viewed by others


Background
Developmental and epileptic encephalopathies (DEEs) are a heterogeneous group of severe disorders that are characterized by early-onset, refractory seizures and developmental slowing or regression. Genetic variations are significant causes of DEEs [1, 2]. With the wide application of whole-genome and whole-exome sequencing, an increasing number of candidate genes have been found to be causal, but most of these are discovered in only a small proportion of cases [3]. YWHAG (* 605356) is one such gene that has been recently related to DEEs.
The YWHAG gene, located on 7q11.23, which encodes YWHAG, is a member of the 14-3-3 protein family and is highly expressed in the brain, skeletal muscle, and heart [4]. Eleven variants in the YWHAG gene have been reported to cause developmental and epileptic encephalopathy 56 (DEE56) (# 617665) or autism [3, 5,6,7,8]. The affected patients exhibit early-onset epilepsy, intellectual disability, motor developmental delay, speech impairment, and sometimes behavioral problems [3, 5]. However, the disorder has great clinical heterogeneity, as some patients may have a mild phenotype, with normal delivery and normal development, and only epilepsy is observed [9]. Here, we report a missense variant c.170G > A (p.R57H) in exon 2 of YWHAG, which caused early-onset epilepsy and developmental delay in a Chinese family.
Methods
Ethical approval and participants
Written informed consent for participation and whole-exome sequencing and subsequent Sanger sequencing as a part of the diagnostic process (approved by the Medical Ethical Committee of Affiliated Hospital of Qingdao University) was obtained from the proband’s parents and grandparents. Detailed clinical manifestations from the proband and his mother were then collected.
Whole-exome sequencing
To explore the etiology of the early-onset seizures and developmental delay in this family, we performed trio-based whole-exome sequencing on the proband and his parents. Peripheral blood samples (2 ml) were collected from the boy and his family members. Then, the samples were sent to MyGenostics, Beijing, for trio-whole exon sequencing using the Illumina HiSeq X Ten system. Following sequencing, raw image files were processed using Bcl2Fastq software (Bcl2Fastq 2.18.0.12, Illumina, Inc.) for base calling and raw data generation. Then the clean reads were aligned to the reference human genome (hg19). The final identified variants were evaluated using three algorithms, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant [SIFT; (http://sift.jcvi.org/)], and MutationTaster (http://www.mutationtaster.org/), to predict pathogenicity. Variant interpretation was performed according to the American College of Medical Genetics (ACMG) guidelines [10]. Sanger sequencing was utilized for further validation of variants and variant detection in other relatives.
Results
The proband
The patient (III.1, Fig. 1 A) is a boy who was born at full-term via cesarean section. His parents are nonconsanguineous. His father is healthy. His mother (II.2, Fig. 1 A) has seizures and developmental delay. He was the first child of his parents, and his mother had no history of miscarriages. His birth weight was 3200 g (p50), birth length was 50 cm (p50), and birth head circumference was 34 cm (p50). There was no history of asphyxia and anoxia. He has no unusual features. His gross motor development has been delayed since birth. He could raise his head at the age of 4 months, turn over at the age of 6 months, sit at the age of 8 months and walk without assistance at the age of 1 year and 6 months. He is now 3 years and 10 months and cannot jump. His language development is seriously delayed. Now, he can say only “papa, mama, no” or express his needs by pointing to the object. He can understand simple commands. He is very irritable. He had his first seizure at the age of 1 year and 11 months, manifested as upward gaze of the eyes, lip cyanosis, and limb trembling, which is considered a generalized tonic–clonic seizure (GTCS). EEG at that time did not reveal a seizure and showed no abnormalities in the background. Neuroimaging was unremarkable. Sodium valproate treatment was initiated, and he was seizure free after 4 months of treatment. Interval EEG and brain MRI performed again at 3 years and 10 months showed no abnormalities.

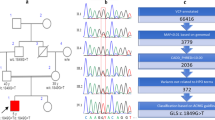
A: The family genogram, in which the black arrow indicates the proband. The proband is represented by a black box, and the affected mother is represented by a black circle. B: Sanger sequencing results of the family. The red arrow points to the variant site. The proband and his mother carry the variant, but the father (II.1), the grandfather (I.1) and the grandmother (II.2) do not carry the variant c.170G > A (p.R57H). C: crystal structure of YWHAG (PDB:3UZD). Left: Dimeric YWHAG is shown as bottle green ribbons, and the phosphopeptide ligand is shown as an orange stick. Right: close-up view of the binding groove and side chains of the residues crucial for phosphopeptide binding. The conserved triad of two arginines and a tyrosine residue (Arg-57, Arg-132, and Tyr-133), which form the positively charged patch, are shown in green. D: Partial sequence alignment of YWHAG orthologs and different human 14-3-3 proteins surrounding the variant. Identical residues across all proteins are shown in black
The proband’s mother
The proband’s mother (II.2, Fig. 1A) had her first seizure at the age of 2 years, also manifested as GTCS. Her relatives did not report other types of seizures. Interval EEG was unremarkable. She was treated with antiepileptic drugs, but it is not known what they were, and she had a 5- to 6-year remission of epilepsy. During pregnancy, she stopped taking drugs without the guidance of a doctor, and the seizures returned. She is currently being treated with carbamazepine, phenytoin sodium and sodium valproate, but the seizures are not completely controlled. The mother also had global developmental delay. She sat at the age of 8 months and walked without support at the age of 1 year and 5 months. She did not do well in school. She can cook, but she cannot shop. At examination, she could understand some things but could not express herself very well. She refused to take intelligence tests and refused further examination with EEG and brain MRI.
Whole-exome sequencing results
We performed trio-based whole-exome sequencing on the proband and his parents. We identified a heterozygous variant c.170G > A (p.R57H) in exon 2 of YWHAG (NM_012479), which was inherited from his mother (Fig. 1B). Further investigation with Sanger sequencing found that the grandmother and grandfather did not carry the variant (Fig. 1B). This is a heterozygous variant that causes the arginine at position 57 to be replaced by histidine. PolyPhen-2 predicted this variant to be damaging, with a score of 1.000. The variant was also predicted to be disease causing by MutationTaster and damaging by SIFT (with a score of 0.000). According to ACMG guideline, the p.R57H variant in YWHAG gene meets the criteria to be classified as likely pathogenic (PS2 + PM2 + PP1 + PP3 + PP4) [10]. This site is highly conserved between species and subtypes of the protein family, and the arginine at position 57 is part of the highly conserved triad of two arginines and a tyrosine (Arg132-Arg57-Tyr133) that normally form a positively charged patch within a binding groove for interacting phosphopeptides [3, 11]. This ability of the protein to bind phosphopeptides is potentially affected by substitution at this site (Fig. 1C and D).
Discussion
Functional study of YWHAG
The YWHAG gene is located on 7q11.23, which encodes YWHAG, a member of the 14-3-3 protein family and is highly expressed in the brain, skeletal muscle, and heart [4]. 14-3-3 proteins function in vital cellular processes, such as metabolism, protein trafficking, signal transduction, apoptosis and cell cycle regulation [12]. 14-3-3 proteins exist in monomeric and dimeric states as homo- and heterodimers, respectively, but YWHAG is almost entirely dimeric. Each monomer consists of a bundle of nine α-helices (αA to αI), of which helices αC, αE, αG, and αI form a conserved peptide-binding groove, which has a positively charged patch on one side and a hydrophobic patch on the other (Fig. 1 C). The positively charged patch is formed by a conserved triad of two arginines and a tyrosine residue (Arg-57, Arg-132, and Tyr-133), which bind the phosphate group of the interacting phosphopeptide/protein [3, 11, 13]. In previous study, de novo variants in the YWHAG gene were predicted to impair dimerization (p.E15A) and phosphopeptide binding (p.R132C)[3]. In the same way, the replacement of arginine by histidine at position 57 was predicted to affect the ability of the protein to bind phosphopeptides, thus affecting the biological function downstream. Previous functional experiments carried out by knocking down ywhag1 in zebrafish revealed reduced brain size and increased diameter of the heart tube in zebrafish [14]. Other researchers found that both the overexpression and the knockdown of Ywhag in mice disrupt neuronal migration of pyramidal neurons, which indicates that a balance of Ywhag expression is required during cortical development to prevent delays in neuronal migration [15, 16]. Kim et al. found that Ywhag homozygous knockout mice was prenatally lethal, and heterozygous mice showed developmental delay. In addition, behavioral analyses found that heterozygous mice display hyperactive and depressive-like behavior along with more sensitive responses to acute stress than littermate control mice [17]. In summary, we hypothesize that the variant affecting the ability of the protein to bind phosphopeptides may also result in delays in neuronal migration, thus leading to developmental delay, epilepsy and so forth.
Clinical manifestations and diagnosis
Until now, fourteen variants in 25 patients were found to result in YWHAG deficiency (including our patients) [3, 5,6,7,8, 18,19,20]. The cases of fourteen of the patients were described with detailed clinical manifestations (Table 1). Affected patients mainly manifested as early-onset epilepsy, mild-moderate intellectual disability, motor developmental delay, speech impairment, and sometimes behavioral problems. Some of them have facial dysmorphism such as prominent forehead, long palpebral fissures, bulbous nose, and absent Cupid's bow. However, a mild phenotype was also observed, as one patient was reported to have normal development, and only epilepsy was observed[9]. This suggests the great clinical heterogeneity of this disease. The age of seizure onset is usually less than 2 years. The most common seizure types are GTCS, absence seizures and myoclonic seizures. One of the patients reported by Kanani et al. experienced only a single generalized tonic–clonic seizure, although most of the patients had multiple seizure types and multiple seizures. Most of the patients had normal interictal EEG and cranial MRI results (Table 1) [3, 5]. Both of our patients had mild-moderate ID, motor developmental delay, and speech impairment. They experienced multiple GTCSs but no other types of seizures. The proband was irritable. Although quite a few patients had facial dysmorphism, no facial dysmorphism were observed in our patients. A recent paper reported a patient carrying the same variant as our patient, whose clinical presentation was roughly the same as ours. But, some of his EEG recordings showed generalized or bifrontal spikes and SW complexes [19]. A total of fourteen variants have been found so far. Except for the variants shown in Table 1, a de novo p.D129E variant was reported in an individual with Lennox-Gastaut syndrome (LGS) [6]. A p.K50Q de novo variant was identified in a subject with autism [7]. Very recently, two novel variants (p.R42Ter and p.K125E) were reported in two unrelated families with childhood myoclonic epilepsy and FS. They all have normal intelligent and motor development [20]. And another variant p. R132H was reported in a patient with DEE56 in a cohort study [18].Among these patients, there seem to be hot spot variants. p.R132C was found in 4/15 patients, and p.Y133S was found in 2/15 patients (Table 1). In addition to the patient whose case is summarized in Table 1, the p.Y133S variant was reported in a patient severely affected by a neurodevelopmental disorder who had no detailed clinical description[8]. If a patient has a phenotype similar to that described above, the diagnosis can be established by genetic analysis to identify a pathogenic variant in the YWHAG gene.
Treatment and prognosis
The seizures of most patients are sensitive to antiepileptic drugs. Usually, seizure control can be achieved by sodium valproate and levetiracetam and ethosuximide and stiripentol. One patient was found to have no response to lamotrigine [5]. The proband in our study attained seizure control after 4 months of treatment with sodium valproate. However, his mother attained only partial control with phenytoin sodium, carbamazepine, and sodium valproate. This may be partly due to the sudden withdrawal of drugs during pregnancy and the subsequent irregular use of drugs after pregnancy. Motor and speech rehabilitation may be useful to patients; however, due to the small number of cases, there are no data to support this. In light of the reported patients and our patients, most patients with DEE56 seem to have a relatively good prognosis. They may achieve self-care ability, and the seizures can be controlled with standard antiepileptic drugs [5].
Conclusion
YWHAG haploinsufficiency is now a recognized etiology of DEE56. From the reported patients and our cases, we can see that although patients with DEE56 have early-onset seizures and global developmental delay, most of them seem to have a relatively good prognosis. We report a missense variant in the YWHAG gene in a Chinese family, which will expand the variant spectrum and further enrich the genotype–phenotype relationships of DEE56. Since some patients may get married and have children due to the relatively good prognosis, we also emphasize the importance of genetic diagnosis and prenatal diagnosis for females who have mild-moderate intellectual disability, developmental delay and especially epilepsy.
Availability of data and materials
The variant is available in the ClinVar repository [https://www.ncbi.nlm.nih.gov/clinvar/]. the accession number is SCV002064257. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive[21] in National Genomics Data Center [22], China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA for human: HRA002140) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human/. Please refer to the following website for details https://ngdc.cncb.ac.cn/gsa-human/browse/HRA002140.
Abbreviations
- DEE:
-
Developmental and epileptic encephalopathies
- GTCS:
-
Generalized tonic-clonic seizure
- LGS:
-
Lennox-Gastaut syndrome
- ACMG:
-
American college of medical genetics
References
Helbig I, Tayoun AA. Understanding genotypes and phenotypes in epileptic encephalopathies. Mol Syndromol. 2016;7(4):172–81.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):512–21.
Guella I, McKenzie MB, Evans DM, Buerki SE, Toyota EB, Van Allen MI, Suri M, Elmslie F, Simon MEH, van Gassen KLI, et al. De novo mutations in YWHAG cause early-onset epilepsy. Am J Hum Genet. 2017;101(2):300–10.
Horie M, Suzuki M, Takahashi E, Tanigami A. Cloning, expression, and chromosomal mapping of the human 14-3-3gamma gene (YWHAG) to 7q11.23. Genomics. 1999;60(2):241–3.
Kanani F, Titheradge H, Cooper N, Elmslie F, Lees MM, Juusola J, Pisani L, McKenna C, Mignot C, Valence S, et al. Expanding the genotype-phenotype correlation of de novo heterozygous missense variants in YWHAG as a cause of developmental and epileptic encephalopathy. Am J Med Genet A. 2020;182(4):713–20.
Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501(7466):217–21.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–15.
Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542(7642):433–438.
Stern T, Orenstein N, Fellner A, Lev-El Halabi N, Shuldiner AR, Gonzaga-Jauregui C, Lidzbarsky G, Basel-Salmon L, Goldberg-Stern H. Epilepsy and electroencephalogram evolution in YWHAG gene mutation: a new phenotype and review of the literature. Am J Med Genet A. 2021;185(3):901–8.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Gene Med. 2015;17(5):405–24.
Yang X, Lee WH, Sobott F, Papagrigoriou E, Robinson CV, Grossmann JG, Sundström M, Doyle DA, Elkins JM. Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc Natl Acad Sci USA. 2006;103(46):17237–42.
Morrison DK. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009;19(1):16–23.
Xiao B, Smerdon SJ, Jones DH, Dodson GG, Soneji Y, Aitken A, Gamblin SJ. Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature. 1995;376(6536):188–91.
Komoike Y, Fujii K, Nishimura A, Hiraki Y, Hayashidani M, Shimojima K, Nishizawa T, Higashi K, Yasukawa K, Saitsu H, et al. Zebrafish gene knockdowns imply roles for human YWHAG in infantile spasms and cardiomegaly. Genesis. 2010;48(4):233–43.
Cornell B, Wachi T, Zhukarev V, Toyo-Oka K. Overexpression of the 14-3-3gamma protein in embryonic mice results in neuronal migration delay in the developing cerebral cortex. Neurosci Lett. 2016;628:40–6.
Wachi T, Cornell B, Marshall C, Zhukarev V, Baas PW, Toyo-oka K. Ablation of the 14-3-3gamma protein results in neuronal migration delay and morphological defects in the developing cerebral cortex. Dev Neurobiol. 2016;76(6):600–14.
Kim DE, Cho CH, Sim KM, Kwon O, Hwang EM, Kim HW, Park JY. 14-3-3γ haploinsufficient mice display hyperactive and stress-sensitive behaviors. Experimental neurobiology. 2019;28(1):43–53.
Brunet T, Jech R, Brugger M, Kovacs R, Alhaddad B, Leszinski G. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin Genet. 2021;100(1):14–28.
Sedláčková L, Štěrbová K, Vlčková M. A novel variant in YWHAG further supports phenotype of developmental and epileptic encephalopathy. Am J Med Genet A. 2021;185(5):1363–5.
Ye XG, Liu ZG, Wang J, Dai JM, Qiao PX, Gao PM, Liao WP. YWHAG mutations cause childhood myoclonic epilepsy and febrile seizures: molecular sub-regional effect and mechanism. Front Genet. 2021;12: 632466.
Chen T, Chen X, Zhang S, Zhu J, Tang B, Wang A, Dong L, Zhang Z, Yu C, Sun Y, et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteomics Bioinform. 2021. https://doi.org/10.1016/j.gpb.2021.08.001.
Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic acids research 2022, 50(D1):D27-d38.
Acknowledgements
We thank for the child and his family for cooperation.
Funding
This study is supported by the Taishan Scholars Program of Shandong Province, (NO. tsqn201909191). In detail, the fund pays for the polish fee and publication charge of the article.
Author information
Authors and Affiliations
Contributions
ZY: Conceptualization, Validation, Writing-Original Draft; ZF S: Formal analysis, Analysis of data; JX: Writing-review, Funding; CQ Y: Validation, Writing-review; FL: Investigation, Analysis of data; HP1: Acquisition, analysis of data; XF: Validation, Acquisition of data; YZ: Conceptualization, interpretation of data, revision of the draft; HP2: Writing-review & Editing; All authors critically reviewed the manuscript, participated in its revision and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Study approval and ethical clearance was obtained from the Medical Ethical Committee of Affiliated Hospital of Qingdao University. Written informed consent for participation was obtained from all of participants in this study. For the proband who was under the age of 16, written informed consent for participation was obtained from his parents prior to data collection.
Consent for publication
The written informed consent for publication of clinical details and/or clinical images was obtained from the proband’s parents and their grandparents.
Competing interests
All authors declare that they do not have any conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yi, Z., Song, Z., Xue, J. et al. A heterozygous missense variant in the YWHAG gene causing developmental and epileptic encephalopathy 56 in a Chinese family. BMC Med Genomics 15, 216 (2022). https://doi.org/10.1186/s12920-022-01377-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01377-8