
Abstract
Radiotherapy (RT) is a highly effective anti-cancer therapy delivered to around 50–60% of patients. It is part of therapy for around 40% of cancer patients who are cured of their disease. Until recently, the focus of this anti-tumour efficacy has been on the direct tumour cytotoxicity and RT-induced DNA damage. Recently, the immunomodulatory effects of RT on the tumour microenvironment have increasingly been recognized. There is now intense interest in potentially using RT to induce an anti-tumour immune response, which has led to rethinking into how the efficacy of RT could be further enhanced. Following the breakthrough of immune check point inhibitors (ICIs), a new era of immuno-oncology (IO) agents has emerged and established immunotherapy as a routine part of cancer treatment. Despite ICI improving outcomes in many cancer types, overall durable responses occur in only a minority of patients. The immunostimulatory effects of RT make combinations with ICI attractive to potentially amplify anti-tumour immunity resulting in increased tumour responses and improved outcomes. In contrast, tumours with profoundly immunosuppressive tumour microenvironments, dominated by myeloid-derived cell populations, remain a greater clinical challenge and RT may potentially further enhance the immunosuppression. To harness the full potential of RT and IO agent combinations, further insights are required to enhance our understanding of the role these immunosuppressive myeloid populations play, how RT influences these populations and how they may be therapeutically manipulated in combination with RT to improve outcomes further. These are exciting times with increasing numbers of IO targets being discovered and IO agents undergoing clinical evaluation. Multidisciplinary research collaborations will be required to establish the optimal parameters for delivering RT (target volume, dose and fractionation) in combination with IO agents, including scheduling to achieve maximal therapeutic efficacy.
Similar content being viewed by others
Background
Radiotherapy (RT) is a highly effective anti-cancer therapy known to induce direct DNA damage to tumour cells. More recently, the immunomodulatory effects of RT on the tumour microenvironment (TME) has encouraged investigations into how RT efficacy might be enhanced. RT is able to induce a local anti-tumour immune response, potentially leading to systemic anti-tumour immunity and contributing to tumour regression outside of the local radiation field, termed the “abscopal effect” [1]. Though reports of abscopal effects date back to 1953, systemic responses remain an extremely rare clinical occurrence [2]. Therefore, the research focus has been on understanding the mechanisms of RT induced anti-tumour immunity and potentially manipulating this further with therapeutic immuno-oncology (IO) agents to increase the frequency of systemic responses. There has been a surge of interest in IO agents following the clinical success of Ipilimumab in the treatment of metastatic melanoma [3]. Ipilimumab is a monoclonal antibody (mAb) that targets Cytotoxic T Lymphocyte Associated Protein 4 (CTLA-4), inhibiting its actions as a suppressive immune checkpoint and thereby facilitating an anti-tumour immune response. This success has led to the development of other immune checkpoint inhibitors (ICIs), with durable remissions observed using mAbs against Programmed Cell Death Protein/Ligand 1 (anti-PD1/PD-L1) in numerous disease groups and improved survival in metastatic disease. Despite ICIs being recognized as breakthrough therapies, only the minority of cancer patients respond to such treatment [4,5,6].
The immunomodulatory potential of both RT and ICIs has provided rationale for combining RT and IO agents to further improve overall response rates and the duration of responses. Rare abscopal effects are now increasingly reported as case reports in patients receiving combined treatment, but they are still the rare minority and further prospective clinical trials are required [7,8,9,10,11]. While many pre-clinical studies investigating RT and IO combinations demonstrate proof of principle with long-term tumour control in murine tumour models, translating this into clear clinical benefit has proved challenging [12,13,14,15]. In a study of metastatic lung cancer patients, combining anti-CTLA4 mAb with RT induced systemic responses where anti-CTLA4 alone had failed; however, disease control was only achieved in 31% (12/39) of patients with only two complete responses [16]. Further, in the TONIC trial, no benefit was observed from combining single-site RT with anti-PD1 [17].
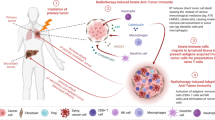
The immune contexture of the TME is now understood to be a significant predictive biomarker of response to immunotherapies. Tumours with an abundance of infiltrating T-cells appear to be most likely to respond to ICI, whereas tumours with an abundance of immunosuppressive myeloid cells and few infiltrating T-cells fail to exhibit a durable response [18,19,20,21]. RT may enhance the ability of T-cell rich tumours to respond to ICI but to compound matters further, RT may also modulate the TME to support myeloid populations [22]. These discoveries have encouraged investigations into stimulatory IO agents which induce T-cell infiltration and activation, or which reprogram myeloid populations in tumours where RT drives immunosuppression (Fig. 1) [12, 23].

The choice of therapeutic agent in combination with radiotherapy may depend on the TME. Radiotherapy can drive the expansion and activation of T-cells in tumours with a T-cell rich TME. This leads to upregulation of PD-L1 and these tumours respond to ICIs. Conversely, tumours with a myeloid rich TME do not respond to ICI, RT drives further expansion and suppressive myeloid cells and may need to be combined with IO agents which re-programme myeloid cells and/or enhance T-cell priming and infiltration. ICI immune checkpoint inhibitor, IO immuno-oncology, M2 M2-polarised macrophage, mAb monoclonal antibody, MDSC myeloid-derived suppressor cell, RT radiotherapy, TME tumour microenvironment
This review will therefore describe our current understanding of the complex interaction of RT with the TME, how IO agents may modulate this and the challenges of translating combined therapies into the clinic.
The effects of RT on the induction of systemic immunity
T-cell priming and antigen-specific immune response
The therapeutic success of RT has been shown to be dependent on effector T-cells and their ability to respond to tumour antigens [24]. Irradiation triggers immunogenic cell death (ICD) by inducing DNA damage and the subsequent release of damage-associated molecular patterns (DAMPs) from tumour cells, turning tumour cells into an “in situ vaccine” [25]. These effects promote dendritic cell (DC) antigen presentation, and the differentiation of naïve T-cells towards an effector phenotype.
Irradiated cells release DAMP signals which enhance the function of DCs. The release of adenosine triphosphate recruits DCs to the tumour. Calreticulin is translocated to the surface of dying cells where it is recognized by DCs, promoting phagocytosis. The passive release of HMGB1 enables DCs to efficiently process and cross-present antigens through toll-like receptor (TLR) 4 dependent signaling, inducing an effective T-cell mediated immune response [26,27,28]. Therefore, through stimulating DCs, RT is an effective adjuvant for immunotherapies.
Depletion studies in murine lymphoma models indicate that DCs—not B cells or macrophages—are indeed the major antigen presenting cell (APC) required for durable anti-tumour immunity, demonstrated when RT is combined with stimulatory CD40 mAb [29]. CD40 agonists are known to enhance DC function through increased surface expression of major histocompatibility complex (MHC) molecules and the production of pro-inflammatory cytokines [23]. Exploring novel strategies to augment DC function may improve therapeutic outcomes post-RT.
The presence of cytosolic DNA and micronuclei in irradiated tumour cells also activates cGas/STING signalling pathways which stimulate downstream production of immunogenic type I interferons (IFN), responsible for the maturation of DCs amongst other immunostimulatory events [30,31,32,33,34]. This pathway also activates CD8+ T-cells and has been shown to potentiate the effects of PD-L1 blockade [30, 35]. The cGas/STING cascade is negatively regulated by protective DNA damage response (DDR) pathways. Targeting DDR pathways via PARP or CHK1 inhibitors significantly increases surface expression of PD-L1 and augments cytotoxic T-cell infiltration in in vivo models of small cell lung carcinoma [36]. The PARP inhibitor Olaparib is therefore undergoing clinical trial evaluation in combination with anti-PD1 mAbs to assess clinical efficacy [31, 37]. Enhancing the effects of RT-induced DNA damage through DDR inhibitors is a logical approach to improving tumour response and is currently being extensively investigated; the addition of an ICI such as anti-PD1 to overcome tumour-induced immunosuppression is an exciting prospective approach [38].
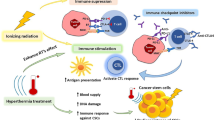
RT has also been described to activate mTOR signalling as part of DDR pathways [39, 40]. This may lead to an increase in peptide presentation by tumours and recognition by effector T-cells [40]. Upon activation, T-cells produce IFNγ which increases antigen spread and further enhances MHC expression on tumour cells, augmenting immune recognition [41]. When cytotoxic CD8+ T-cells induce tumour cell death, they release new tumour antigens which further strengthens the immune response. This has led to the concept of RT-induced “in situ anti-tumour vaccination”, whereby the enhanced immunogenicity of the irradiated tumour might augment systemic responses, as shown in Fig. 2, panel A [42,43,44].

Radiotherapy induces a local anti-tumour immune response (a). Radiotherapy delivered to multiple sites may augment systemic responses (b). RT-induced immunogenic cell death stimulates the release of DAMPs and type 1 IFNs, which enhance antigen uptake and presentation by DCs. DCs present tumour antigens to T cell receptors, priming naïve T-cells to an effector phenotype. These T-cells migrate into the local tumour to exert their cytotoxic effects, or re-enter the circulation and migrate to distal, non-irradiated tumour sites (Panel A). At distal tumour sites, DCs may also activate T-cells against novel tumour antigens. RT delivered to multiple sites may therefore increase the quantity and diversity of migrating T-cells, enhancing the potential for systemic immune responses at non-irradiated sites (b). DAMPs damage-associated molecular patterns, DC dendritic cell, IFN interferon, MHC I major histocompatibility complex class I, RT radiotherapy, TCR T-cell receptor
T-cell trafficking: recruitment and migration
Both resident and infiltrating T-cell populations appear to be required for effective control of irradiated tumours and distal tumour sites following RT and anti-PD1 combinations [45]. Local RT may initially induce cell death in radiosensitive tumour-residing T lymphocytes, but it may also stimulate T-cell tumour infiltration.
T-cell recruitment to the TME is mediated by adaptive and innate immune responses induced by RT. Once matured, DCs migrate to tumour draining lymph nodes (LNs) where they prime naïve T-cells towards an effector phenotype. These effector T-cells, along with CD4 + T helper cells, are trafficked to the tumour site via chemokine gradients [46, 47]. RT damage also induces increased expression of adhesion molecules ICAM and VCAM1 on tumour and endothelial cells, which attracts effector T-cells. Blocking infiltrating T-cells from binding to these adhesion molecules impedes T-cell mediated tumour rejection, which highlights the significance of leukocyte recruitment in establishing anti-tumour immunity [48].
Work from our own laboratory investigating T-cell receptor clonality has suggested that following RT and anti-PD1 combinations, the immune response in the local TME is dominated by polyclonal expansion of pre-existing T-cell clones [45]. The eradication of secondary tumours outside of the local radiation field was only observed in mice receiving the RT and anti-PD1 combination therapy. It is unclear whether this systemic response was dependent on the migration of pre-existing T-cells clones or de novo activation of T-cells at the secondary site, but both resident and infiltrating T-cells were required for primary response. On-going research aims to elucidate the mechanism of action of T-cell trafficking to better understand how combination approaches succeed in targeting distal metastases.
Pre-clinical studies of RT-anti-PD1 combinations suggest that irradiating draining LNs impacts T-cell infiltration into the primary tumour, modifies intra-tumoural chemokine expression and reduces overall survival [49]. Since the lymphatic system plays a crucial role in the migration of DCs and T-cells, it is likely that irradiating LNs impedes cell migration away from the primary tumour towards distal sites. Understanding the impact of LN irradiation on systemic immunity may shape clinical decisions for elective nodal irradiation.
Further studies are also needed to clarify the significance of T-cell trafficking in phenotypically immunosuppressive tumours, such as prostate cancers. These may benefit from IO agents which stimulate leukocyte trafficking, such as anti-CD40. The administration of agonistic CD40 alone or in combination with RT leads to a significant increase in CD8+ and CD4 + T-cells infiltrating the tumour, and the combined approach may successfully induce T-cell dependent immunity [50,51,52].
The infiltration of T-cells with a memory phenotype (CD8+ , CD103 +) into the TME correlates with improved overall survival in lung and ovarian tumours [53, 54]. Resident memory T-cells (Trm) are a recently identified subset of T-cells which reside in the tissue without recirculating and are linked to enhanced tumour control. In mouse models of melanoma, anti-PD1 treatment enhances infiltration of plastic circulating memory T-cells, which give rise to Trm cells [55]. Both cell subsets express PD1 and the presence of either memory T-cell is sufficient to induce an immune response, though Trm cells do so with a stronger efficacy. In another study, combining RT-anti-PD1 with an additional mAb against the macrophage-specific phagocytic receptor Mer-TK delayed abscopal tumour growth through the retention of Trm cells in the secondary TME [56]. Further studies are needed to better understand the impact of RT on Trm populations and the significance of these cells in inducing abscopal responses.
The impact of myeloid cell populations on RT and immunotherapy combinations
Tumours with a high proportion of myeloid and other suppressive effector cells within the TME can hinder anti-tumour immune responses. These tumours are radioresistant and lack clinical responsiveness to ICI [18, 57,58,59,60]. Despite the immunostimulatory effects discussed above, RT can encourage an immunosuppressive TME through the recruitment of myeloid-derived suppressor cells (MDSC) populations and the repolarization of macrophages into an M2 phenotype [61].
Immediately following RT, there is a relative increase in MDSCs, tumour associated macrophages (TAMs) and regulatory T-cells (Tregs) within the TME, as these appear to be more radioresistant than T lymphocytes [62]. MDSCs and TAMs drive tumour growth and angiogenesis whilst negatively regulating T-cells [58, 63, 64]. Tregs contribute to immunosuppression partially via CTLA4 checkpoint signaling which inhibits cytotoxic T-cell activation [65, 66]. Irradiated tumour cells release oxygen and nitrogen radicals, which encourage the polarization of macrophages from an inflammatory M1 phenotype into a tumour-supporting M2 phenotype. These M2 TAMs secrete cytokines and matrix metalloproteinases which assist tumour immune evasion [67]. Repopulation of the TME with T-cells following RT will therefore be vital for anti-tumour immune responses.
RT stimulates the recruitment of suppressive cells to the TME by increasing the expression of inflammatory chemokines such as CXCL5 and CXCL2; these appear important in recruiting MDSCs and Tregs via STING-activated CCR2 pathways, as well as stimulating the production of TGFβ [68, 69]. TGFβ is well known for its role in supporting tumour progression and has recently been shown to impede anti-tumour immunity post-RT [70, 71]. Within the TME, TGFβ suppresses the effector functions of T-cells and natural killer cells, and inhibits DC maturation. TGFβ also promotes M2 macrophage polarity and favours the conversion of CD4 + T-cells into immunosuppressive Tregs. In murine models of colorectal cancer (CRC), mice receiving a TGFβ inhibitor were resistant to metastatic formation [72]. However, utilizing TGFβ inhibitors to improve RT efficacy has not consistently induced tumour control due to the complex role played by this cytokine as both a tumour promotor and tumour suppressor [68, 73]. RT also stimulates production of chemokines CCL2 and CCL5, which recruit inflammatory monocytes that differentiate into immunosuppressive TAMs in the TME. Dual CCL2/CCL5 antagonists which target these monocytic populations improve RT efficacy and reduce tumour metastases in poorly immunogenic breast and pancreatic tumour models, highlighting the significance of myeloid populations [74,75,76].
MDSC have been shown to contribute to patient resistance to ICI [77,78,79]. RT and anti-PD1 combinations fail to induce long-term tumour clearance in tumours with high populations of myeloid cells residing in the TME [57]. Circulating myeloid populations also appear to influence therapeutic response, as high peripheral levels of monocytic MDSC are associated with greater tumour burden, lower numbers of antigen-specific T-cells and resistance to ICIs [19]. In the treatment of melanoma with Ipilimumab, non-responders demonstrated significantly higher proportions of MDSC, neutrophils and monocytes in the TME [77]. MDSC populations in the TME have also been negatively correlated with the efficacy of DC-based immunotherapies in combination with RT [58, 80].
The utilization and development of therapeutic agents which manipulate or reprogram myeloid populations provide another exciting opportunity to improve clinical responses to RT-IO combinations. Stimulatory IO agents employed to activate anti-tumour T-cell responses can also reprogram suppressive cell populations. CD40 agonists manipulate macrophages to acquire an M1 phenotype, thereby upregulating pathways associated with effector T-cell priming [81]. Macrophage-specific MerTK is also a therapeutic target of interest, with improved survival rates seen with inhibitory mAbs combined with RT-anti-PD1 in lung adenocarcinomas, and tumour regression when combined with RT and TGFβ blockade in poorly immunogenic tumour models [56, 82]. TLR signalling is central to several stages of T-cell activation, making TLRs an attractive therapeutic target [83, 84]; TLR agonists may also repolarise macrophages to an M1 phenotype and convert MDSCs into APCs, stimulating T-cell responses [85,86,87]. In preclinical studies, RT potentiates the effects of TLR7/8 agonists, inducing durable anti-tumour immune responses and reduced metastases in several disease subsets [12, 88,89,90].
Targeting TGFβ represents another potential strategy to enhance combination therapies. TGFβ inhibitors in combination with anti-PD1 mAbs demonstrated CD8+ T-cell infiltration and improved tumour control in a subset of urothelial cancer patients and poorly immunogenic breast cancer models [71, 91]. Promising results were also seen in metastatic CRC models in which TGFβ inhibition sensitized previously unresponsive tumours to anti-PD1 [92]. RT-induced TGFβ was shown to inhibit abscopal responses even in combination with anti-PD-1 and anti-CD137 mAbs; this was overcome by TGFβ blockade [93]. TGFβ blockade has also demonstrated promising results in combination with an agonistic OX40 mAb in metastatic breast cancer, where there was a synergistic T-cell dependent response [94]. Clinical trials are under development to study triple-combinations of TGFβ inhibitors, RT and ICIs in non-small cell lung carcinoma (NSCLC), hepatocellular carcinoma and pancreatic tumours [95, 96].
Phosphodiesterase-5 (PDE5) inhibitors are currently approved for non-malignant conditions and have been shown to reduce the immunosuppressive capacity of MDSCs in murine tumour models. PDE5 inhibitors increase T-cell infiltration and activation through Arg-1 and NOS down-regulation resulting in improved efficacy of adoptive T-cell therapies [97]. In clinical trials of HNSCC and metastatic melanoma, the PDE5 inhibitor Tadalafil improved clinical outcomes and augmented immune responses by reducing MDSC function [98]. This provides rational for trialing PDE5 inhibitors in RT and immunotherapy combinations. There are currently several clinical trials underway investigating the therapeutic manipulation of myeloid populations in conjunction with RT and ICI [18]. However, further studies are needed to fully elucidate the impact of these populations on the efficacy of treatments and determine whether their reprogramming may overcome resistance to RT-ICI combinations.
Hypoxia has long been established as a significant factor in radioresistance [99]. More recently, the effect of oxygen-deficient TMEs on immunosuppressive cell populations has emerged as potentially important [61]. RT is able to support hypoxic environments by disrupting tumour vasculature and the generation of reactive oxygen species [100,101,102,103]. Hypoxic tumours produce potent T-cell suppressor adenosine, recruit MDSCs and TAMs via CSF1 signalling, and further support immunosuppression via TGFβ signalling [61, 104]. The expression of hypoxia-inducible factor-1 (HIF-1) stimulates secretion of stromal-derived factor-1 which further recruits MDSCs via CXCR4 binding [61]. Through HIF-1, hypoxia also upregulates PD-L1 expression on MDSCs and tumour cells. Patients with hypoxic tumours may therefore benefit from anti-PD1 mAbs [104]. However, disrupted vasculature in hypoxic tumours may hinder the delivery of IO agents to the tumour site. The addition of a therapeutic agent to re-oxygenate tumours, such as nitrous oxide, is therefore being considered to increase the frequency of responders to combination therapies [105, 106].
Clinical considerations for the delivery of RT and immunotherapy combinations
Due to the complex immunological interplay between RT and IO agents (Table 1), there are many uncertainties regarding how the delivery of RT may impact immunotherapy efficacy. Establishing the optimal RT dose, fractionation and target volume along with the optimal scheduling of IO agents are just some factors required for successful clinical translation.
The dose and number of fractions of RT will likely play a critical role in the immunomodulation of the TME. High-doses of 12–16 Gy delivered in a single fraction induce protective DDR pathways within the tumour which hinder T-cell response, whereas lower doses have been shown to optimally induce the production of IFNβ required for DC activation [33]. It is currently unclear whether single high-doses or fractionated low-doses would better complement ICIs., High-dose RT (12 or 20 Gy) has been shown to increase PD-L1 expression on tumour cells, where anti-PD1 treatment can induce successful tumour control [13, 14, 107, 108]. Conversely, lower doses given in fractions (18 × 2 Gy or 5 × 2 Gy) have also been shown to increase PD-L1 expression and may result in earlier expression, suggesting that further studies into optimal RT doses are required [109, 110]. Though higher doses may induce cell death in lymphocytes, it is likely that different T-cell populations exhibit differing sensitivities to RT, as Trm population have been demonstrated to increase in proportion following local irradiation of solid tumours [111, 112]. IFNγ, which is found at greater levels within the irradiated tumour than at secondary sites, can also mediated T-cell survival post RT, suggesting targeting multiple tumour sites could increase the efficacy of immune responses [111, 113, 114].
Currently the majority of clinical trials investigating RT-IO combinations employ single site irradiation, which may not be optimal. In the situation of multiple metastases, multi-site irradiation may improve therapeutic outcomes by reducing disease burden and also by increasing RT-induced immune stimulation, as shown in Fig. 2, panel B [115, 116]. In this situation, RT delivery to a single tumour may not liberate enough tumour antigens to generate a sufficiently robust systemic anti-tumour immune response. Increasing RT target volume to reduce disease burden and increase immune infiltration may therefore enhance the efficacy of IO agents. However, consideration must also be given to the potential increase in patient toxicity associated with large volume fields and multiple sites of RT, as well as the potential for RT to induce cell death in radiosensitive immune cells within the tumour and in local LNs [49]. Further research is required to address these important questions.
Scheduling of the IO agent relative to the delivery of RT is also likely to affect the generation of systemic immune responses. The optimal schedule is likely to depend on disease group, tumour site and the RT-IO combination employed. Research from our laboratory in murine models of CRC evaluated three regimes of RT with anti-PD1: concurrent delivery at the start of RT cycle; concurrent delivery at the end of RT cycle; sequential delivery 7 days after RT completion. Acquired resistance to radiotherapy was overcome by concurrent delivery of anti-PD1 with effective anti-tumour immunity and tumour control; sequential delivery was less effective [13]. However, the recently published PACIFIC trial of NSCLC patients demonstrated improved progression-free survival with sequential anti-PD1 delivery, with the greatest benefit seen in patients who began anti-PD1 in the shortest timeframes to completing RT [117]. This emerging data highlights the importance of scheduling RT with IO agents and the requirement for further study across different tumour types.
Further investigations are also needed to identify biomarkers which inform on RT-induced immunological changes, which will guide decision making in the clinic. Predictive biomarkers to identify the most appropriate IO agent for each patient would increase the frequency of responders. PD-L1 expression has emerged as a potential prognostic biomarker correlated with improved survival after RT, though its value as a predictive biomarker for RT-IO agent combinations remains to be seen [118,119,120]. There has been some success using tumour immune infiltrates—the tumour ‘immunoscore’—to predict RT outcomes, though again further studies should investigate its predictive value for combination therapies [121,122,123,124]. Due to the complex interplay between RT and the anti-tumour immune response, surveillance of patients throughout treatment using dynamic biomarkers would allow for real-time decision making to improve treatment efficacy and prevent toxicities. Therefore, there is an urgent need for studies to monitor RT-induced changes to the immune microenvironment throughout treatment and across tumour types, and for clinical trials to include predictive biomarker discovery in their study outcomes.
Conclusion
The ability of RT to reprogram the TME has complex local and systemic consequences. The immunostimulatory effects of RT make combinations with IO agents attractive to amplify effective anti-tumour immunity and improve outcomes. In contrast, immunosuppressive TMEs dominated by MDSC populations remain a greater clinical challenge, as RT may potentially enhance immunosuppression. To harness the full potential of RT-IO agent combinations, further insights are required to understand the role of these immunosuppressive myeloid populations, how RT influences them and the optimal ways to therapeutically manipulate them to improve clinical outcomes. These are exciting times with increasing numbers of IO targets being discovered and undergoing clinical evaluation [125,126,127]. The experience with ICI informs us that it is unlikely that a single IO agent will be sufficient to induce durable anti-tumour immunity in all patients, and so combination approaches will be required. Using multiple agents will create further challenges related to toxicity and adverse effects. Multidisciplinary research collaborations will be required to establish the optimal target volume, dose and fractionation to deliver RT in combination with IO agents to achieve maximal therapeutic efficacy. Discovering dynamic RT-related biomarkers will also be critical for translation. Prognostic and predictive immune biomarkers will enable clinicians to assess the immune microenvironment throughout RT, predict patient benefit from therapeutic agents and monitor their response, enabling patient treatment plans to be personalised.
Availability of data and materials
Not applicable.
Abbreviations
- APC:
-
Antigen presenting cell
- CRC:
-
Colorectal cancer
- CTLA4:
-
Cytotoxic T lymphocyte associated protein 4
- DAMP:
-
Damage associated molecular patterns
- DC:
-
Dendritic cell
- DDR:
-
DNA damage response
- HIF1:
-
Hypoxia inducible factor 1
- HMGB1:
-
High mobility group box protein 1
- HNSCC:
-
Head and neck squamous cell carcinoma
- ICAM:
-
Intracellular adhesion molecule
- ICD:
-
Immunogenic cell death
- ICI:
-
Immune checkpoint inhibitor
- IFN:
-
Interferon
- IO:
-
Immuno-oncological
- LN:
-
Lymph node
- mAb:
-
Monoclonal antibody
- MDSC:
-
Myeloid derived suppressor cell
- MHC:
-
Major histocompatibility complex/class 1
- mTOR:
-
Mammalian target of rapamycin
- NSCLC:
-
Non small cell lung carcinoma
- PARP:
-
Poly (ADP-ribose) polymerase
- PD-1/PD-L1:
-
Programmed cell death protein/ligand 1
- PDE-5:
-
Phosphodiesterase-5
- RT:
-
Radiotherapy
- STING:
-
Stimulator of interferon genes
- TAM:
-
Tumour associated macrophages
- TGFβ:
-
Transforming growth factor beta
- TLR:
-
Toll-like receptor
- TME:
-
Tumour microenvironment
- Tregs:
-
Regulatory T-cells
- Trm:
-
Resident memory T-cells
- VCAM:
-
Vascular cell adhesion molecule
References
Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10(7):718–26.
Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol. 1953;26(305):234–41.
Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Vol. 11, Nature Reviews Cancer. NIH Public Access; 2011. p. 805–12.
Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389(10064):67–76.
Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–46.
Powles T, O’Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: Updated results from a phase 1/2 open-label study. JAMA Oncol. 2017;3(9):e172411.
Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res. 2013;1(6):365–72.
Grimaldi AM, Simeone E, Giannarelli D, Muto P, Falivene S, Borzillo V, et al. Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. Oncoimmunology. 2014;3(5):e28780.
Qin Q, Nan X, Miller T, Fisher R, Teh B, Pandita S, et al. Complete local and abscopal responses from a combination of radiation and nivolumab in refractory Hodgkin’s lymphoma. Radiat Res. 2018;190(3):322–9.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15(17):5379–88.
Dagoglu N, Karaman S, Caglar HB, Oral EN. Abscopal effect of radiotherapy in the immunotherapy era: systematic review of reported cases. Cureus. 2019;11(2):e4103.
Dovedi SJ, Melis MHM, Wilkinson RW, Adlard AL, Stratford IJ, Honeychurch J, et al. Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood. 2013;121(2):251–9.
Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74(19):5458–68.
Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124(2):687–95.
Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11(2Pt1):728–34.
Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. 2018;24(12):1845–51.
Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920–8.
Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front Immunol. 2018;9(6):1310.
Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247–57.
Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caracò C, Curvietto M, et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014;63(7):675–83.
Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61(7):1019–31.
Gough MJ, Young K, Crittenden M. The impact of the myeloid response to radiation therapy. Clin Dev Immunol. 2013;2013:281958.
Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–43.
Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589–95.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31(1):51–72.
Golden EB, Frances D, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3:e28518.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61.
Dovedi SJ, Lipowska-Bhalla G, Beers SA, Cheadle EJ, Mu L, Glennie MJ, et al. Antitumor efficacy of radiation plus immunotherapy depends upon dendritic cell activation of effector cd8 T cells. Cancer Immunol Res. 2016;4(7):621–30.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–52.
McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer. 2020;20(4):203–17.
Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71(7):2488–96.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8(1):15618.
MacKenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. CGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461–5.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–20.
Sen T, Rodriguez BL, Chen L, Della Corte CM, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9(5):646–61.
Dillon MT, Boylan Z, Smith D, Guevara J, Mohammed K, Peckitt C, et al. PATRIOT: A phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid. Clin Transl Radiat Oncol. 2018;12:16–20.
Fulton B, Short SC, James A, Nowicki S, McBain C, Jefferies S, et al. PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin Transl Radiat Oncol. 2018;8:12–6.
Ma Y, Vassetzky Y, Dokudovskaya S. mTORC1 pathway in DNA damage response. Biochim Biophys Acta - Mol Cell Res. 2018;1865(9):1293–311.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–71.
Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-γ production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180(5):3132–9.
Chakraborty M, Abrams SI, Camphausen K, Liu K, Scott T, Coleman CN, et al. Irradiation of tumor cells up-regulates fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol. 2003;170(12):6338–47.
Vanpouille-Box C, Pilones KA, Wennerberg E, Formenti SC, Demaria S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine. 2015;33(51):7415–22.
Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862–70.
Dovedi SJ, Cheadle EJ, Popple AL, Poon E, Morrow M, Stewart R, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin Cancer Res. 2017;23(18):5514–26.
Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181(5):3099–107.
Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711-723.e4.
Riegler J, Gill H, Ogasawara A, Hedehus M, Javinal V, Oeh J, et al. VCAM-1 density and tumor perfusion predict T-cell infiltration and treatment response in preclinical models. Neoplasia (US). 2019;21(10):1036–50.
Marciscano AE, Ghasemzadeh A, Nirschl TR, Theodros D, Kochel CM, Francica BJ, et al. Elective nodal irradiation attenuates the combinatorial efficacy of stereotactic radiation therapy and immunotherapy. Clin Cancer Res. 2018;24(20):5058–71.
Honeychurch J, Glennie MJ, Johnson PWM, Illidge TM. Anti-CD40 monoclonal antibody therapy in combination with irradiation results in a CD8 T-cell-dependent immunity to B-cell lymphoma. Blood. 2003;102(4):1449–57.
Sonpavde G, McMannis JD, Bai Y, Seethammagari MR, Bull JMC, Hawkins V, et al. Phase I trial of antigen-targeted autologous dendritic cell-based vaccine with in vivo activation of inducible CD40 for advanced prostate cancer. Cancer Immunol Immunother. 2017;66(10):1345–57.
Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, Victor CT, et al. Radiotherapy and CD40 activation separately augment immunity to checkpoint blockade in cancer. Cancer Res. 2018;78(15):4282–91.
Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpréville V, et al. CD8+ CD103 + tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. 2015;194(7):3475–86.
Webb JR, Milne K, Watson P, DeLeeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker cd103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. 2014;20(2):434–44.
Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martýnez-Cano S, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat Commun. 2017;8:16073.
Caetano MS, Younes AI, Barsoumian HB, Quigley M, Menon H, Gao C, et al. Triple therapy with MeRTK and PD1 inhibition plus radiotherapy promotes abscopal antitumor immune responses. Clin Cancer Res. 2019;25(24):7576–84.
Deng L, Liang H, Burnette B, Weichselbaum RR, Fu YX. Radiation and anti-PD-L1 antibody combinatorial therapy induces T cell-mediated depletion of myeloid-derived suppressor cells and tumor regression. Oncoimmunology. 2014;3:e28499.
Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222(1):162–79.
Wang D, An G, Xie S, Yao Y, Feng G. The clinical and prognostic significance of CD14+HLA-DR−/low myeloid-derived suppressor cells in hepatocellular carcinoma patients receiving radiotherapy. Tumor Biol. 2016;37(8):10427–33.
Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–34.
Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol. 2015;25(1):18–27.
Kachikwu EL, Iwamoto KS, Liao YP, Demarco JJ, Agazaryan N, Economou JS, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys. 2011;81(4):1128–35.
Shojaei F, Zhong C, Wu X, Yu L, Ferrara N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol. 2008;18(8):372–8.
Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13(3):193–205.
Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression—implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16(6):356–71.
Liyanage UK, Moore TT, Joo H-G, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169(5):2756–61.
Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222(1):155–61.
Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35:S185–98.
Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8(1):1736.
Yang L, Pang Y, Moses HL. TGF-β and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31(6):220–7.
Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, et al. TGFβ is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015;75(11):2232–42.
Calon A, Espinet E, Palomo-Ponce S, Tauriello DVF, Iglesias M, Céspedes MV, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571–84.
Sow H, Ren J, Camps M, Ossendorp F, ten Dijke P. Combined inhibition of TGF-β signaling and the PD-L1 immune checkpoint is differentially effective in tumor models. Cells. 2019;8(4):320.
Connolly KA, Belt BA, Figueroa NM, Murthy A, Patel A, Kim M, et al. Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget. 2016;7(52):86522–35.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–62.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5.
Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res. 2015;21(24):5453–9.
Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, Chevriaux A, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 2016;76(18):5241–52.
Hansen GL, Gaudernack G, Brunsvig PF, Cvancarova M, Kyte JA. Immunological factors influencing clinical outcome in lung cancer patients after telomerase peptide vaccination. Cancer Immunol Immunother. 2015;64(12):1609–21.
Finkelstein SE, Iclozan C, Bui MM, Cotter MJ, Ramakrishnan R, Ahmed J, et al. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo-adjuvant treatment of high-risk soft tissue sarcoma patients. Int J Radiat Oncol Biol Phys. 2012;82(2):924–32.
Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. CD40 ligation activates murine macrophages via an IFN-γ-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol. 2005;174(10):6013–22.
Crittenden MR, Baird J, Friedman D, Savage T, Uhde L, Alice A, et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget. 2016;7(48):78653–66.
Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299(5609):1033–6.
Kobold S, Wiedemann G, Rothenfußer S, Endres S. Modes of action of TLR7 agonists in cancer therapy. Vol. 6, Immunotherapy. Future Medicine Ltd.; 2014. p. 1085–95.
Wang J, Shirota Y, Bayik D, Shirota H, Tross D, Gulley JL, et al. Effect of TLR agonists on the differentiation and function of human monocytic myeloid-derived suppressor cells. J Immunol. 2015;194(9):4215–21.
Spinetti T, Spagnuolo L, Mottas I, Secondini C, Treinies M, Rüegg C, et al. TLR7-based cancer immunotherapy decreases intratumoral myeloid-derived suppressor cells and blocks their immunosuppressive function. Oncoimmunology. 2016;5(11):e1230578.
Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, et al. Toll-like receptor 3 signaling converts tumorsupporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci U S A. 2012;109(6):2066–71.
Dovedi SJ, Adlard AL, Ota Y, Murata M, Sugaru E, Koga-Yamakawa E, et al. Intravenous administration of the selective toll-like receptor 7 agonist DSR-29133 leads to anti-tumor efficacy in murine solid tumor models which can be potentiated by combination with fractionated radiotherapy. Oncotarget. 2016;7(13):17035–46.
Schölch S, Rauber C, Tietz A, Rahbari NN, Bork U, Schmidt T, et al. Radiotherapy combined with TLR7/8 activation induces strong immune responses against gastrointestinal tumors. Oncotarget. 2015;6(7):4663–76.
Dewan MZ, Vanpouille-Box C, Kawashima N, DiNapoli S, Babb JS, Formenti SC, et al. Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clin Cancer Res. 2012;18(24):6668–78.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–8.
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–43.
Rodríguez-Ruiz ME, Rodríguez I, Mayorga L, Labiano T, Barbes B, Etxeberria I, et al. TGFb blockade enhances radiotherapy abscopal efficacy effects in combination with anti-PD1 and anti-CD137 immunostimulatory monoclonal antibodies. Mol Cancer Ther. 2019;18(3):621–31.
Garrison K, Hahn T, Lee WC, Ling LE, Weinberg AD, Akporiaye ET. The small molecule TGF-β signaling inhibitor SM16 synergizes with agonistic OX40 antibody to suppress established mammary tumors and reduce spontaneous metastasis. Cancer Immunol Immunother. 2012;61(4):511–21.
Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFb, in advanced solid tumors. Clin Cancer Res. 2018;24(6):1287–95.
Dahmani A, Delisle JS. TGF-β in T cell biology: Implications for cancer immunotherapy. Cancers (Basel). 2018;10(6):194.
Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691–702.
Hassel JC, Jiang H, Bender C, Winkler J, Sevko A, Shevchenko I, et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology. 2017;6(9):e1326440.
Horsman MR, Overgaard J. The impact of hypoxia and its modification of the outcome of radiotherapy significance of hypoxia. J Radiat Res. 2016;57(S1):90–8.
Chen FH, Chiang CS, Wang CC, Tsai CS, Jung SM, Lee CC, et al. Radiotherapy decreases vascular density and causes hypoxia with macrophage aggregation in TRAMP-C1 prostate tumors. Clin Cancer Res. 2009;15(5):1721–9.
Tsai JH, Makonnen S, Feldman M, Sehgal CM, Maity A, Lee WMF. Ionizing radiation inhibits tumor neovascularization by inducing ineffective angiogenesis. Cancer Biol Ther. 2005;4(12):1395–400.
Tsai CS, Chen FH, Wang CC, Huang HL, Jung SM, Wu CJ, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys. 2007;68(2):499–507.
Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5(5):429–41.
Eckert F, Zwirner K, Boeke S, Thorwarth D, Zips D, Huber SM. Rationale for combining radiotherapy and immune checkpoint inhibition for patients with hypoxic tumors. Front Immunol. 2019;10:407.
Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74(3):665–74.
Graham K, Unger E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int J Nanomed Dovepress. 2018;13:6049–58.
Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, et al. Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol Res. 2015;3(4):345–55.
Patel KR, Martinez A, Stahl JM, Logan SJ, Perricone AJ, Ferris MJ, et al. Increase in PD-L1 expression after pre-operative radiotherapy for soft tissue sarcoma. Oncoimmunology. 2018;7(7):e1442168.
Derer A, Spiljar M, Bäumler M, Hecht M, Fietkau R, Frey B, et al. Chemoradiation increases PD-L1 expression in certain melanoma and glioblastoma cells. Front Immunol. 2016;7:610.
Grapin M, Richard C, Limagne E, Boidot R, Morgand V, Bertaut A, et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: a promising new combination. J Immunother Cancer. 2019;7(1):1–12.
Arina A, Beckett M, Fernandez C, Zheng W, Pitroda S, Chmura SJ, et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat Commun. 2019;10(1):3959.
Grayson JM, Harrington LE, Lanier JG, Wherry EJ, Ahmed R. Differential sensitivity of naive and memory CD8+ T cells to apoptosis in vivo. J Immunol. 2002;169(7):3760–70.
Gerber SA, Sedlacek AL, Cron KR, Murphy SP, Frelinger JG, Lord EM. IFN-γ mediates the antitumor effects of radiation therapy in a murine colon tumor. Am J Pathol. 2013;182(6):2345–54.
Zimmerman M, Yang D, Hu X, Liu F, Singh N, Browning D, et al. IFN-γ upregulates Survivin and ifi202 expression to induce survival and proliferation of tumor-specific T cells. PLoS One. 2010;5(11):e14076.
Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545(7652):60–5.
Luke JJ, Lemons JM, Karrison TG, Pitroda SP, Melotek JM, Zha Y, et al. Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. J Clin Oncol. 2018;36(16):1611–8.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N Engl J Med. 2017;377(20):1919–29.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379(24):2342–50.
Lee VHF, Lo AWI, Leung CY, Shek WH, Kwong DLW, Lam KO, et al. Correlation of PD-L1 expression of tumor cells with survival outcomes after radical intensity-modulated radiation therapy for non-metastatic nasopharyngeal carcinoma. PLoS ONE. 2016;11(6):e0157969–e0157969.
Iijima M, Okonogi N, Nakajima NI, Morokoshi Y, Kanda H, Yamada T, et al. Significance of PD-L1 expression in carbon-ion radiotherapy for uterine cervical adeno/adenosquamous carcinoma. J Gynecol Oncol. 2020;31(2):e19.
Angell H, Galon J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr Opin Immunol. 2013;25:261–7.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4.
Hagland HR, Lea D, Watson MMC, Søreide K. Correlation of circulating T-cells in pre-operative blood to intratumoral density and location of CD3+ and CD8+ T-cells in colorectal cancer: a potential for an immunoscore by liquid biopsy? Eur J Cancer. 2017;72:S49-50.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71.
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94.
Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci USA. 2010;107(17):7875–80.
Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73(9):2782–94.
Acknowledgements
Not applicable.
Funding
Eleanor J Cheadle and Timothy M Illidge are supported by the NIHR Manchester Biomedical Research Centre. Madyson Colton is supported by the Christie Charitable Fund.
Author information
Authors and Affiliations
Contributions
MC wrote and edited the manuscript, and designed Fig. 2. JH designed the first figure. EC, JH and TI edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declares that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Colton, M., Cheadle, E.J., Honeychurch, J. et al. Reprogramming the tumour microenvironment by radiotherapy: implications for radiotherapy and immunotherapy combinations. Radiat Oncol 15, 254 (2020). https://doi.org/10.1186/s13014-020-01678-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13014-020-01678-1