
Abstract
Background
While simple Audit & Feedback (A&F) has shown modest effectiveness in reducing low-value care, there is a knowledge gap on the effectiveness of multifaceted interventions to support de-implementation efforts. Given the need to make rapid decisions in a context of multiple diagnostic and therapeutic options, trauma is a high-risk setting for low-value care. Furthermore, trauma systems are a favorable setting for de-implementation interventions as they have quality improvement teams with medical leadership, routinely collected clinical data, and performance-linked to accreditation. We aim to evaluate the effectiveness of a multifaceted intervention for reducing low-value clinical practices in acute adult trauma care.
Methods
We will conduct a pragmatic cluster randomized controlled trial (cRCT) embedded in a Canadian provincial quality assurance program. Level I–III trauma centers (n = 30) will be randomized (1:1) to receive simple A&F (control) or a multifaceted intervention (intervention). The intervention, developed using extensive background work and UK Medical Research Council guidelines, includes an A&F report, educational meetings, and facilitation visits. The primary outcome will be the use of low-value initial diagnostic imaging, assessed at the patient level using routinely collected trauma registry data. Secondary outcomes will be low-value specialist consultation, low-value repeat imaging after a patient transfer, unintended consequences, determinants for successful implementation, and incremental cost-effectiveness ratios.
Discussion
On completion of the cRCT, if the intervention is effective and cost-effective, the multifaceted intervention will be integrated into trauma systems across Canada. Medium and long-term benefits may include a reduction in adverse events for patients and an increase in resource availability. The proposed intervention targets a problem identified by stakeholders, is based on extensive background work, was developed using a partnership approach, is low-cost, and is linked to accreditation. There will be no attrition, identification, or recruitment bias as the intervention is mandatory in line with trauma center designation requirements, and all outcomes will be assessed with routinely collected data. However, investigators cannot be blinded to group allocation and there is a possibility of contamination bias that will be minimized by conducting intervention refinement only with participants in the intervention arm.
Trial registration
This protocol has been registered on ClinicalTrials.gov (February 24, 2023, #NCT05744154).
Similar content being viewed by others


Background
Low-value clinical practices are tests or treatments that are not supported by evidence and may cause unnecessary harm [1]. They expose patients to adverse events such as avoidable irradiation, postoperative complications, medication or transfusion side effects, unnecessary interventions on incidental findings, and direct and indirect expenses [2,3,4,5]. They are also a major barrier to timely access to appropriate care and threaten the sustainability of modern healthcare systems [2,3,4]. Stakeholders in high-income countries have expressed the urgent need to develop strategies to address the burden of low-value care with interventions targeting de-implementation [6,7,8,9]. De-implementation is defined here as discontinuing or abandoning practices that are not proven to be effective, are less effective or less cost-effective than an alternative practice, or are potentially harmful [10, 11].
Audit & Feedback (A&F), a summary of the clinical performance of healthcare provided over a specified period of time, has been shown to have a modest effect on the de-implementation of low-value practices [12]. Organizations such as National Audit Commissioners advocate for enhancing A&F to support clinicians in their use of feedback data [13, 14]. Theory and evidence, largely based on the implementation of high-value practices, suggest that multifaceted interventions addressing determinants for success (barriers and facilitators) may be more effective and cost-effective than simple A&F [12, 15, 16]. However, the mechanisms for change, barriers, and facilitators for de-implementation differ from those for implementation [16, 17]. As such, studies on the effectiveness of interventions designed to increase high-value care may not be generalizable to the reduction of low-value practices. There is therefore a need for research on the incremental effectiveness of multifaceted interventions over simple A&F for de-implementation [18]. Additionally, although the barriers and facilitators of de-implementation have been well documented [17, 19], experts acknowledge the critical need for research on the effectiveness of interventions tailored to these determinants for success to support the de-implementation of low-value practices [17, 20,21,22,23].
Injury is the leading cause of productive life years lost and is second only to heart and stroke disease for in-patient costs [24]. Given the need to make rapid decisions in a context of multiple competing diagnostic and therapeutic options, trauma care is a high-risk setting for low-value care. Trauma quality assurance programs in place in Canada [25,26,27,28,29] and worldwide [30,31,32] have been associated with improvements in quality of care [33,34,35], but they are based on simple A&F and exclusively target adherence to high-value clinical practices. Low-value practices in trauma care are frequent, subject to significant inter-hospital variation [36, 37], and are associated with increased complications, length of stay, and in-patient costs [36]. Trauma systems are a favorable setting for de-implementation interventions as they possess many documented facilitators including quality improvement teams with medical leadership, routinely collected clinical data, and performance linked to accreditation [19]. Furthermore, potential gains are huge due to the resource-intensive nature of trauma care. Trauma systems are thus the ideal setting to advance knowledge on de-implementation of low-value care.
Our primary objective is to assess the effectiveness of a multifaceted intervention embedded in a provincial quality assurance program compared to simple A&F to reduce low-value adult acute trauma care. Secondary objectives are to (i) identify the mechanisms associated with a successful implementation of the intervention, (ii) evaluate whether intervention effectiveness changes over time, (iii) assess the effect of the intervention on clinical outcomes and resource use, (iv) assess unintended consequences, and (v) evaluate cost-effectiveness.
Methods
This protocol is reported according to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) statement (Additional file 1) [38] and the Consolidated Standards of Reporting Trials (CONSORT) extension for cluster trials (Additional File 2) [39].
Trial design
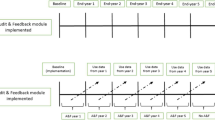
We will conduct a parallel arm, pragmatic, superiority cluster randomized trial (cRCT). The trial is pragmatic as it is embedded in a Canadian provincial trauma quality assurance program. The trial scored 5/5 on 6 of the 9 domains of the PRagmatic Explanatory Continuum Indicator Summary (PRECIS 2) tool, 3/5 for organization intervention and 4/5 for the outcome, indicating a high level of pragmatism (Additional file 3: Table S1) [40]. As the trial will be based on routinely collected data that are available at no extra cost, we will use a baseline observation period pre-randomization to increase study power [41]. We will randomize at the trauma center level because quality programs operate at the local trauma committee level in each trauma center. A stepped wedge design will not be used as the minimum 1-year rollout period is unacceptably long for stakeholders and parallel arm cRCTs have fewer risks of bias than stepped wedge cRCTs [42].
Setting
The trial will be embedded in the Québec Trauma Care Continuum, a provincial regionalized trauma system comprising 57 adult trauma centers of which 3 are level I (highly specialized urban centers), 5 are level II (similar capacity to level I but in smaller cities), 22 are level III (hospitals in small towns transferring most major trauma to level I/II centers after stabilization), and 27 are level IV (rural community hospitals). All centers undergo periodic verification in line with designation, conducted by the Institut national d’excellence en santé et services sociaux (INESSS) and overseen by the Ministry of Health and Social Services [43]. Verification includes simple A&F on adherence to high-value care and risk-adjusted outcomes and submission of an action plan within 6 months of reception of the A&F report. Local trauma committees in each center are required to ensure the quality of the trauma program according to designation requirements. Committees include the program medical director (chair), the program manager, heads of critical care, emergency and surgical departments, and multidisciplinary services, and a hospital administrator. Quality improvement activities include trimestral committee meetings with chart review and discussions with clinical and administrative leads locally and at referring centers to identify improvement strategies such as the development of local care protocols. Formal letters of agreement are signed by heads of clinical departments to operate changes in their services when required.
Inclusion/exclusion criteria and recruitment
We will include all adult level I–III trauma centers in the Québec Trauma Care Continuum (n= 30). All centers will be recruited as the trial will be embedded in the 2023 evaluation cycle of the provincial trauma quality assurance program, which is mandatory [43]. Level IV centers will not be included as most see fewer than 20 trauma patients per year. Patient-level inclusion criteria will be all adult (> 16 years of age) admissions with a primary diagnosis of injury to any of these trauma centers. Admissions with a primary diagnosis of thermal injuries, foreign bodies, drowning, or late effects of injuries will be excluded as diagnostic and therapeutic approaches for these diagnoses are distinct [44].
Intervention development
The multifaceted intervention was developed using extensive background work, conducted according to recommended steps for de-implementation [16, 45, 46]. First, we identified 63 potentially low-value practices through a scoping review and clinician survey [47]. Second, we synthesized evidence on the benefits, harms, and cost-effectiveness of these practices [48,49,50,51]. Third, we measured their incidence using trauma registry data [37]. Fourth, two panels of international experts (76% participation) and local stakeholders (94% participation) and three patient–partners (75% participation) reached an evidence-informed consensus on 16 practices that should be targeted for de-implementation [52]. Fifth, we derived quality indicators for 12 of these practices with trauma registry data and showed that 6 had moderate to high validity [36]. These quality indicators target (i) initial diagnostic imaging (head, cervical spine, or whole-body computed tomography [CT] in low-risk patients), (ii) specialist consultation (neurosurgical consult for mild traumatic brain injury and spine service consult for isolated transverse process fractures), and (iii) repeat imaging for transfers (repeat scan in patients with no disease progression and no additional details needed). Based on this work, we developed a multifaceted intervention with clinical and implementation science experts, patient–partners, and accreditation professionals using UK Medical Research Council guidelines for the Development of Complex Interventions (Additional file 3: Figure S1) [53, 54]. We matched frequently reported barriers and facilitators reported by experts in our consensus study [52] and documented in a knowledge synthesis [19] to implementation strategies using the Consolidated Framework for Implementation Research (CFIR)-Experts Recommendations for Implementing Change (ERIC) tool (Additional file 3: Table S2) [45, 55, 56].
Trial interventions
Trial interventions will be delivered by research team members twice over two evaluation cycles (12 months per cycle with a refinement phase in the first cycle; Additional file 3: Fig. S1, Table S3).
The intervention group will receive the multifaceted intervention (Additional file 3: Table S4, Figures S2.1 & S2.2), which includes (1) refinement with end users; (2) an A&F report sent to local governing authorities presenting the following: performance compared to peers (simple A&F), a summary message indicating if action is required and a list of potential actions; (3) educational materials (a clinical vignette; consequences of the practice; links to practice guidelines, clinical decision rules and shared decision-making tools; a case review tool); (4) virtual educational meetings with the local trauma medical director, trauma program manager and data analyst on how to use the report and the case review tool and how to assess their barriers and facilitators to prepare their own action plan with the CFIR-ERIC matching tool [57]; and (5) two virtual facilitation visits 2 and 4 months after the transmission of the report (or at more appropriate times depending on needs) to support committees in preparing their action plan.
In line with recommendations [54] and to avoid contamination, we have embedded the refinement phase in the intervention (Additional file 3: Figure S1, Table S3, Fig. 1). We will first conduct four 60-min focus groups with 8–10 local trauma committee members [58] in trauma centers randomized to the intervention arm (stratified by designation level), with additional focus groups to obtain informational redundancy if required [59]. We will use a semi-structured interview guide (Additional file 3: Table S5) to gather reactions to the prototype, generate ideas on potential modifications, and identify other potential barriers and facilitators using the CFIR[60]. Second, we will recruit 5–8 end users [61] in the intervention arm and not involved in the previous phases to test intervention usability using a Think Aloud Protocol [62, 63]. We will use a one-to-one approach asking participants to articulate their reactions and their understanding of the information while going through the A&F report and the educational materials. We will modify the intervention in the presence of problems considered important by end users (e.g., difficulty in interpreting quality indicators) or in the presence of more minor problems (e.g., aesthetic details regarding the presentation of materials) reported frequently (by ≥ 5/8 end users). We will repeat this process with 5–8 additional end users until no major or frequent problems remain [61]. The intervention will be refined iteratively across focus group and usability testing rounds. We will strive for equal representation of males and females for focus groups and usability testing. Participation in this stage will also be balanced for clinical experience (i.e., < 2, 2–10, > 10 years) and disciplines (e.g., surgery, emergency medicine, radiology, critical care, nursing, management).

Implementation over two evaluation cycles
The control group will receive the quality improvement intervention currently in place in the Québec Trauma Care Continuum (i.e., simple A&F report presenting their performance compared to peers on quality indicators measuring adherence to high-value care and risk-adjusted outcomes) with the addition of quality indicators on low-value care (already planned for the 2023 evaluation cycle). Simple A&F was chosen for the control because it is standard practice in the Québec Trauma Care Continuum and in most integrated trauma systems and the effectiveness of simple A&F for de-implementation has been studied [12].
Randomization, allocation, blinding, and adherence
Centers will be randomized to simple A&F (control) or the multifaceted intervention (intervention) in a 1:1 ratio. An independent statistician at the Ottawa Methods Center (outside of the province in which the trial will be conducted and fully independent from the sites) will generate the allocation sequence using the covariate-constrained allocation technique [64] to ensure that study arms are balanced for designation level (I/II and III), cluster size, and the pre-intervention measure of the primary outcome. The allocation sequence will only be revealed on trial initiation. An independent INESSS professional will allocate centers to their randomization group. Local trauma committees will not be told explicitly what aspect of the intervention is randomized; they will only be aware that there are two variations of the intervention. Data extractors and data analysts will be blinded to group allocation. Due to the nature of the intervention, it will not be possible to blind the investigators to allocation groups. To minimize non-adherence and contamination, only participants in the intervention arm will participate in the refinement phase. We do not envisage major problems with adherence because quality assurance activities are mandatory in line with designation requirements [43].
Outcome measures
Our primary outcome is low-value initial diagnostic imaging, calculated as the proportion of low-risk patients who receive head, cervical spine, or whole-body CT in the emergency department (Additional file 3: Table S6). This indicator was selected on consultation with our advisory committee as it applies to the most patients, is considered to have the greatest consequences in terms of adverse effects (unnecessary radiation, delays in care, incidental findings) and resource use (availability of CT scans, staff workload, costs), and is a good indicator of global overuse in a trauma system [36]. Secondary outcomes are low-value specialist consultation (neurosurgical and spine) and repeat imaging for transfers. Other secondary outcomes are mortality, complications, resource use (intensive care unit [ICU] and hospital length of stay), and unintended consequences (unplanned readmission, missed injuries); determinants for successful implementation of the intervention (process evaluation); and incremental cost-effectiveness ratios (economic evaluation).
Follow-up will be based on continuous data collection at each site, which is mandatory in line with designation requirements. Sites will be followed up over the 18-month interval (6 to 24 months) after implementation over two evaluation cycles using these data (Fig. 1, Additional file 3: Table S3). Our advisory committee recommended evaluation over two cycles as identifying problems, implementing solutions and operating change takes place over more than one cycle [65]. We will allow for a 6-month lag period post-implementation in the first cycle, corresponding to the time allotted to centers to submit an action plan. Because data collection is mandatory in all centers, we do not anticipate any loss to follow-up.
Study data
Data on low-value practices, mortality, complications, unplanned readmissions, ICU and hospital length of stay, and missed injuries will be extracted from the provincial trauma registry, used successfully in recent work [36]. Data collection is mandatory for all transfers and/or admissions with a primary diagnosis of injury in all provincial trauma centers [66]. Chart data are extracted in each center using coding protocols and are centralized at the Ministry of Health where they are submitted to data quality protocols. Supervision by a data manager, on-going training, an electronic forum, and quarterly meetings with stakeholders are used to ensure data quality. A recent re-abstraction of a random sample of 80 patient charts stratified by injury type and severity suggested 98% concordance on fields used for A&F (unpublished data).
Process evaluation
We will assess fidelity, reach, and contextual factors using Medical Research Council guidance on process evaluation of complex interventions [67]. To assess fidelity, we will monitor the ability to deliver the multifaceted intervention and simple A&F as intended. The research team member delivering the intervention will document whether intervention components were delivered as planned and describe the challenges encountered. We will also evaluate the download of educational materials (e.g., case revision tool, clinical decision rules, shared decision-making tools) using Google Analytics [68] and examine submitted action plans to document whether they address feedback and describe strategies to facilitate de-implementation based on a local assessment of barriers and facilitators [69]. To assess reach, we will conduct 20 individual interviews (recommended sample size for informational redundancy) [70] with medical directors and trauma program managers in 20 participating centers (10 in each arm, stratified by designation level), 1 to 2 months before the end of each evaluation cycle (Fig. 1, Additional file 3: Table S3). They will be asked about strategies used to disseminate feedback to clinicians involved in trauma care, challenges in implementing the action plan, and unintended consequences of the intervention (e.g., difficulty planning transfer management without scans). We will also evaluate contextual influences on feedback responses, including elements within (e.g., competing priorities, support and commitment of decision makers, available resources) and outside the trauma center (e.g., national initiatives, policies, discussions with clinicians in other centers) [67]. Finally, at the end of the trial (months 27 to 30; Fig. 1, Additional file 3: Table S3), we will conduct 20 individual interviews (10 in each arm) with clinicians working in participating centers (stratified by designation level) to determine their recall and understanding of feedback received and the actions taken to change the practices in their organization. These clinicians will be identified through trauma program managers who will not be given any information about the purpose of the interviews to minimize selection bias. We will strive for an equal representation of males and females and of clinicians with different clinical experiences and disciplines for interviews.
Sample size calculations
As all level I–III trauma centers will participate in this trial, we calculated the statistical power to detect a minimum important difference with a fixed number of clusters and patients per cluster (Additional file 3: Table S6). All sample size parameter values were accurately estimated by historical data from four 18-month observation periods between April 1, 2017, and March 31, 2020, using trauma registry data from the 30 level I–III trauma centers in the Québec Trauma Care Continuum [36]. Based on stakeholder input, a 25% relative difference in the primary outcome (proportion of patients with low-value initial diagnostic imaging) would be considered a minimum important difference. With 15 clusters in arm 1 and 15 clusters in arm 2 and an average of 285 patients per cluster period in a parallel arm cRCT with a baseline observation period, we will achieve 90% power to detect a 25% relative difference between the intervention and control arms (0.14 in the control arm versus 0.105 in the intervention arm) at a two-sided 5% level of significance [71]. We assumed a within-period intra-cluster correlation coefficient of 0.044, and a cluster autocorrelation coefficient of 0.984 (correlation from before to after). Given that the median observed absolute difference for interventions targeting de-implementation in the most recent meta-analysis on the topic was 10.5% [12], an absolute difference of 3.5% is considered plausible. For our secondary outcomes, we will have 90% power to detect absolute differences of 7.4% and 16.0% in neurosurgical consultation and repeat imaging for transfers, respectively [71].
Data analysis plan
All statistical analyses will be based on an intention-to-treat principle and will be conducted by a statistician blinded to group assignment. Analysis of the primary outcome will be conducted at the individual patient level using weighted independence estimating equations with robust standard errors to provide valid statistical inference for the cluster-average target estimand [72]. A small sample correction will be applied to control for the small number of clusters [73]. To produce the intervention effect as a relative risk with a 95% confidence interval, we will use modified robust Poisson regression for clustered data [74]. To obtain correct type I error rates, variables used in the covariate-constrained allocation will be included as covariates (volume, designation level, and baseline proportion), and to improve power and precision [75], the analysis will adjust for prespecified patient risk factors: age, comorbidities, and injury severity (anatomical and physiological parameters). Secondary outcomes measured as binary variables will be analyzed using a similar approach. Secondary outcomes measured as continuous variables (i.e., ICU and hospital length of stay) will be analyzed using linear regression after log transformation. To address the secondary objective of evaluating trends over time, we will model repeated outcomes over 6-month intervals and include time and group-by-time interactions. We will use least square mean differences between the arms to determine whether outcomes have changed over time [76]. There are missing data on covariables in the registry, which will be handled with multiple imputation, shown to lead to accurate effect estimates for trauma quality indicators in simulations [77,78,79].
We will synthesize qualitative data from transcribed focus groups and individual interviews through thematic analysis [80] using QDA miner software (Provalis Research, Montréal, Canada) [81]. We chose thematic analysis because it allows for data organization and reduction, which facilitates a comprehensible description and interpretation of relevant elements [82]. To ensure credibility and reliability, peer debriefing will be carried out by experienced researchers at different analysis stages [83, 84]. We will ensure transferability using purposive sampling to maximize the chances of obtaining information that is relevant to the research objectives [83]. Credibility and trustworthiness will be reinforced by involving a coder in qualitative data analysis with no knowledge of the subject and by independent validation of the entire data analysis by a member of the research team [80]. We will use descriptive statistics to report on the sex and gender, experience, and discipline of participants and to assess intervention fidelity. To assess adherence and contamination, we will analyze process data for external influences and the use of implementation strategies in control centers. We will triangulate data on intervention effectiveness and process evaluation using a joint display to explain findings (i.e., why the intervention was effective or not) [58, 85, 86].
Effectiveness data will only be analyzed at the end of the trial. Process evaluation data collected at the end of the first cycle will be analyzed to inform intervention refinements for the second cycle, and data collected at the end of the trial will be used to refine the intervention prior to upscaling.
Subgroup analyses
We have planned subgroup analyses by designation level, baseline proportion of low-value initial imaging, patient sex (information on patient gender is not available), and patient age for effectiveness; and by clinician gender, age, years in practice, and discipline for the refinement of intervention and for process evaluation.
Sensitivity analysis
If contamination is present, we will use an instrumental variable adjustment method [87] in sensitivity analyses. To evaluate the influence of non-adherence, we will conduct per-protocol analyses. To assess the influence of multiple imputation, we will conduct complete case analyses. Fine and Grey models will be used to assess the influence of competing risks of mortality for analyses on hospital and ICU length of stay.
Economic evaluation
We are currently conducting an early economic evaluation examining the potential benefit of the multifaceted intervention, which has informed intervention development. This includes a systematic review of the cost-effectiveness of A&F interventions [15], a budget impact analysis of low-value practices and a simulation-based early economic evaluation to estimate the potential cost-effectiveness of a hypothetical multifaceted intervention targeting low-value trauma care (initial results suggest the intervention is always dominant). Following the trial, we will update the simulation model with trial data to evaluate the cost-effectiveness of the true intervention. The economic evaluation will be conducted according to Canadian Agency Guidelines for Economic Evaluation of Health Technologies [88] and results will be reported according to the Consolidated Health Economic Evaluation Reporting Standards 2022 statement [89].
Trial management
The main coordinating center will be at the CHU de Québec—Université Laval Research Center. Patient-level data from the trauma registry is hosted at the Ministry of Health and Social Services, and an extraction of this database with no patient-identifiable information will be accessed by the trial analyst (blinded to group allocation) through a password-protected portal at INESSS offices. Qualitative data from intervention refinement and process evaluations will be kept in password-protected computers in locked offices at the CHU de Québec-UL research center. Our trial coordinator will work with an INESSS professional to coordinate trial initiation and recruitment/randomization, implement the intervention, and obtain trauma registry data. They will also be responsible for liaising with trauma program managers in each center and coordinating the collection of qualitative data for the process evaluation. The trial steering committee, co-led by the two principal investigators (LM, MB), includes a representative of INESSS (CT), trauma medical directors of the three regional trauma committees (CM, TR, SS), patient partners (GP, PR) and equity, diversity, and inclusion champions (HW, NY). The committee will oversee trial conduct and knowledge translation activities and ensure we meet project deliverables according to timelines. A data monitoring committee will not be used as data for effectiveness analysis is not collected within the trial and the risk to participants is considered very low. Any trial modifications will be submitted to the trial steering committee for approval, recorded in our registered protocol, and reported in final manuscripts.
Stakeholder, patient, and public involvement
Our primary stakeholder partner is the provincial paragovernmental organization responsible for overseeing healthcare quality (INESSS). Our research team has been working in collaboration with this organization since 2010. They have been involved in designing the trial so it can be embedded in their 2023 evaluation cycle, and they will be involved in all phases of trial conduct. The trial has also been designed in collaboration with the Trauma Association of Canada (association of clinicians involved in trauma care across Canada) and Health Services Organisation (responsible for hospital accreditation programs in Canada). The design of this cRCT has also been informed by national research collaboratives, the Canadian Traumatic brain injury Research Consortium and the Canadian Critical Care Trials Group, and national knowledge translation networks, Choosing Wisely Canada, and the A&F Metalab. To ensure trial results are relevant to other high-income countries with similar trauma care infrastructures, we have partnered with the UK Trauma Audit Research Network and Victoria State Trauma Outcomes Registry and Monitoring groupin Australia. Three patients (PR, GP, MA) with varying injury profiles were integrated in the consensus process to select low-value practices targeted in this trial [52]. We first conducted a preparatory meeting to explore their perceptions on low-value trauma care, establish their priorities, and prepare for their integration. Patient perceptions and priorities were presented by a patient at the consensus meeting. All patients participated in the meeting and voted on practices to be retained. Patients will not be involved in intervention refinement as they are not end users [90]. However, two patients (PR, GP) are on the trial steering committee. No members of the general public are involved.
Ethics and dissemination
This trial has been approved by the CHU-de-Québec – Université Laval research ethics board (#113,664). Our dissemination plan was designed using published guidelines [91] and includes (i) one-page visual summaries on partner organization websites, (ii) peer-reviewed publications, (iii) presentations at clinical and academic (de/implementation science) conferences and to end users (patient support groups, clinicians, accreditation authorities), (iv) media channels and policy briefs, and (v) half-day workshop with implementation science and accreditation stakeholders on the development multifaceted interventions targeting de-implementation.
Discussion
The intervention has a high probability of success because it targets a problem identified by stakeholders, is based on extensive background work, will be refined with end users considering barriers and facilitators, is low-cost (embedded in an existing quality assurance platform, based on routinely collected data), and is linked to accreditation. This setting provides a unique opportunity to develop a cost-efficient, acceptable, sustainable intervention that can be integrated into existing trauma quality assurance platforms. Bias is anticipated to be minimal (no attrition, identification, or recruitment bias). However, there may be differences in engagement that will be explored through process evaluation. Furthermore, investigators cannot be blinded to group allocation and there is a possibility of contamination bias that will be minimized by conducting intervention refinement only with participants in the intervention arm and will be assessed in process evaluations.
This project will advance de-implementation science through the application of knowledge on barriers and facilitators to intervention design and on the incremental effectiveness of a multifaceted de-implementation intervention over simple A&F in a pragmatic setting. If the intervention is effective and cost-effective, we will upscale across Canada with national and provincial trauma authorities and promote uptake in other healthcare domains. We will also explore international uptake through collaborating organizations in the UK, Australia, and the USA. This intervention has the potential to reduce the adverse effects and indirect expenses of low-value trauma care for patients and families. It could also free up resources, reduce delays to care, and reduce clinician workload, ultimately improving efficiency at a time of unprecedented strain on healthcare resources.
Availability of data and materials
Individual patient data are held by provincial authorities and cannot be transmitted by study investigators. Supporting material will be made available through the research program website. Data from intervention refinement and process evaluations will be made available on request.
Abbreviations
- A&F:
-
Audit & feedback
- CFIR-ERIC:
-
Consolidated Framework for Implementation Research-Experts Recommendations for Implementing Change
- CHU:
-
Centre Hospitalier Universitaire
- CONSORT:
-
Consolidated Standards of Reporting Trials
- cRCT:
-
Cluster randomized controlled trial
- CT:
-
Computed tomography
- ICU:
-
Intensive care unit
- INESSS:
-
Institut d’Excellence en Santé et en Services Sociaux
- PRECIS-2:
-
Pragmatic Explanatory Continuum Indicator Summary
- QDA:
-
Qualitative data analysis
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Trials
References
Choosing Wisely Canada. https://choosingwiselycanada.org/about/. Accessed September 13th, 2022.
Berwick DM, Hackbarth AD. Eliminating waste in US health care. JAMA. 2012;307(14):1513–6.
Boat TF, Chao SM, O’Neill PH. From waste to value in health care. JAMA. 2008;299(5):568–71.
Reilly BM, Evans AT. Much ado about (doing) nothing. Ann Intern Med. 2009;150(4):270–1.
Brownlee S, Chalkidou K, Doust J, et al. Evidence for overuse of medical services around the world. Lancet. 2017;390(10090):156–68.
Association médicale du Québec. Plus de cinq milliards mal dépensés dans le système de santé. AMQ. https://www.amq.ca/fr/publications/nos-communiques-de-presse/communiques-2013/item/502-plus-de-cinq-milliards-mal-depenses-dans-le-systeme-de-sante. Accessed 17 Sept 2018.
Canadian Institute for Health Information. Unnecessary Care in Canada. CIHI. https://www.cihi.ca/sites/default/files/document/choosing-wisely-baseline-report-en-web.pdf. Accessed 17 Sept 2018.
Choosing Wisely Canada. https://choosingwiselycanada.org/. Accessed 22 Aug 2022.
Institut de la pertinence des actes médicaux (IPAM). https://ipam.ca/. Accessed 22 Aug 2022.
McKay VR, Morshed AB, Brownson RC, Proctor EK, Prusaczyk B. Letting go: conceptualizing intervention de-implementation in public health and social service settings. Am J Community Psychol. 2018;62(1–2):189–202.
Prasad V, Ioannidis JP. Evidence-based de-implementation for contradicted, unproven, and aspiring healthcare practices. Implement Sci. 2014;9:1.
Ivers N, Jamtvedt G, Flottorp S, et al. Audit and feedback: effects on professional practice and healthcare outcomes. Cochrane Database Syst Rev. 2012;6:Cd000259.
Healthcare Quality improvement Partnership (HQIP). Maximising the quality improvement potential of the National Clinical Audit and Patient Outcomes Programme. https://www.hqip.org.uk/wp-content/uploads/2021/11/Maximising-the-QI-potential-of-the-national-clinical-audit-and-outcome-review-programme.pdf. Accessed 22 Aug 2022.
Health Service Executive. National Review of Clinical Audit. https://www.hse.ie/eng/services/publications/national-review-of-clinical-audit-report-2019.pdf. Accessed 22 Aug 2022.
Moore L, Guertin JR, Tardif PA, et al. Economic evaluations of audit and feedback interventions: a systematic review. BMJ Qual Saf. 2022;31(10):754–67.
Niven DJ, Mrklas KJ, Holodinsky JK, et al. Towards understanding the de-adoption of low-value clinical practices: a scoping review. BMC Med. 2015;13:255.
Norton WE, Chambers DA. Unpacking the complexities of de-implementing inappropriate health interventions. Implement Sci. 2020;15(1):2.
Squires JE, Sullivan K, Eccles MP, Worswick J, Grimshaw JM. Are multifaceted interventions more effective than single-component interventions in changing health-care professionals’ behaviours? An overview of systematic reviews. Implement Sci. 2014;9:152.
van Dulmen SA, Naaktgeboren CA, Heus P, et al. Barriers and facilitators to reduce low-value care: a qualitative evidence synthesis. BMJ Open. 2020;10(10):e040025.
Dyson J, Cowdell F. How is the theoretical domains framework applied in designing interventions to support healthcare practitioner behaviour change? A systematic review. Int J Qual Health Care. 2021;33(3):mzab106.
Powell BJ, Fernandez ME, Williams NJ, et al. Enhancing the impact of implementation strategies in healthcare: a research agenda. Front Public Health. 2019;7:3.
Burton CR, Williams L, Bucknall T, et al. In: Theory and practical guidance for effective de-implementation of practices across health and care services: a realist synthesis. Southampton: NIHR Journals Library; 2021.
Norton WE, Kennedy AE, Chambers DA. Studying de-implementation in health: an analysis of funded research grants. Implement Sci. 2017;12(1):144.
Parachute. The cost of injury in Canada. Parachute: Toronto, ON. http://www.parachutecanada.org/downloads/research/Cost_of_Injury-2015.pdf. Accessed 22 Aug 2022.
Alberta Health Services. Providing care to trauma patients in Alberta. https://www.albertahealthservices.ca/info/page13243.aspx. Accessed 26 Aug 2022.
Critical Care Services Ontario. Regional trauma network development. https://criticalcareontario.ca/wp-content/uploads/2020/10/Regional-Trauma-Network-Development-Guide.pdf. Accessed 26 Aug 2022.
Institut National d'excellence en santé et en services sociaux. Évaluation du réseau. https://www.inesss.qc.ca/thematiques/sante/traumatologie/continuum-de-services-en-traumatologie-cst/evaluation-du-reseau.html. Accessed 26 Aug 2022.
Provincial Health Services Authority. Trauma Services BC. http://www.phsa.ca/our-services/programs-services/trauma-services-bc. Accessed 26 Aug 2022.
Trauma NB. Injury prevention and trauma care for all. https://nbtrauma.ca/. Accessed 26 Aug 2022.
American College of Surgeons. Trauma quality improvement program. https://www.facs.org/quality-programs/trauma/quality/trauma-quality-improvement-program/. Accessed 22 Aug 2022.
Monash University. Victorian State Trauma Outcomes Registry and Monitoring Group (VSTORM). https://www.monash.edu/medicine/sphpm/vstorm/home. Accessed 22 Aug 2022.
The Trauma Audit & Research Network. https://www.tarn.ac.uk/. Accessed 22 Aug 2022.
Batomen B, Moore L, Strumpf E, Champion H, Nandi A. Impact of trauma centre accreditation on mortality and complications in a Canadian trauma system: an interrupted time series analysis. BMJ Qual Saf. 2021;30(11):853–66.
Batomen B, Moore L, Strumpf E, Yanchar NL, Thakore J, Nandi A. Trauma system accreditation and patient outcomes in British Columbia: an interrupted time series analysis. Int J Qual Health Care. 2020;32(10):677–84.
Moore L, Lavoie A, Bourgeois G, Lapointe J. Donabedian’s structure-process-outcome quality of care model: validation in an integrated trauma system. J Trauma Acute Care Surg. 2015;78(6):1168–75.
Moore L, Bérubé M, Tardif P-A, et al. Validation of quality indicators targeting low-value trauma care: a multicenter retrospective cohort study. JAMA Surg (In Press). 2022;157(11):1008–16.
Soltana K, Moore L, Bouderba S, et al. Adherence to clinical practice guideline recommendations on low-value injury care: a multicenter retrospective cohort study. Value Health. 2021;24(12):1728–36.
Chan AW, Tetzlaff JM, Altman DG, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7.
Campbell MK, Piaggio G, Elbourne DR, Altman DG, Group C. Consort 2010 statement: extension to cluster randomised trials. BMJ. 2012;345:e5661.
Loudon K, Treweek S, Sullivan F, Donnan P, Thorpe KE, Zwarenstein M. The PRECIS-2 tool: designing trials that are fit for purpose. BMJ. 2015;350:h2147.
Copas AJ, Hooper R. Cluster randomised trials with different numbers of measurements at baseline and endline: sample size and optimal allocation. Clin Trials. 2020;17(1):69–76.
Hemming K, Taljaard M. Reflection on modern methods: when is a stepped-wedge cluster randomized trial a good study design choice? Int J Epidemiol. 2020;49(3):1043–52.
Ministère de la santé et des services sociaux. Trajectoire - Traumatologie. https://www.msss.gouv.qc.ca/professionnels/soins-et-services/guide-urgences-trajectoire-traumatologie/. Accessed 13 Sept 2022.
Institut National d'excellence en santé et en services sociaux. Portrait du réseau québécois de traumatologie adulte: 2013 à 2016. https://www.connexiontccqc.ca/wp-content/uploads/INESSS_Reseau_traumatologie_adulte-3.pdf. Accessed 3 March 2023.
Bonafide CP, Keren R. Negative studies and the science of deimplementation. JAMA Pediatr. 2018;172(9):807–9.
Grimshaw JM, Patey AM, Kirkham KR, et al. De-implementing wisely: developing the evidence base to reduce low-value care. BMJ Qual Saf. 2020;29(5):409–17.
Moore L, Lauzier F, Tardif PA, et al. Low-value clinical practices in injury care: a scoping review and expert consultation survey. J Trauma Acute Care Surg. 2019;86(6):983–93.
Berube M, Moore L, Tardif PA, et al. Low-value injury care in the adult orthopaedic trauma population: a systematic review. Int J Clin Pract. 2021;75(12):e15009.
Conombo B, Guertin JR, Tardif PA, et al. Economic evaluation of in-hospital clinical practices in acute injury care: a systematic review. Value Health. 2022;25(5):844–54.
Malloum Boukar K, Moore L, Tardif PA, et al. Value of repeat CT for nonoperative management of patients with blunt liver and spleen injury: a systematic review. Eur J Trauma Emerg Surg. 2021;47(6):1753–61.
Moore L, Tardif PA, Lauzier F, et al. Low-value clinical practices in adult traumatic brain injury: an umbrella review. J Neurotrauma. 2020;37(24):2605–15.
Moore L, Berube M, Tardif PA, et al. Quality indicators targeting low-value clinical practices in trauma care. JAMA Surg. 2022;157(6):507–14.
O’Cathain A, Croot L, Duncan E, et al. Guidance on how to develop complex interventions to improve health and healthcare. BMJ Open. 2019;9(8):e029954.
Skivington K, Matthews L, Simpson SA, et al. A new framework for developing and evaluating complex interventions: update of Medical Research Council guidance. BMJ. 2021;374:n2061.
Waltz TJ, Powell BJ, Fernandez ME, Abadie B, Damschroder LJ. Choosing implementation strategies to address contextual barriers: diversity in recommendations and future directions. Implement Sci. 2019;14(1):42.
Michie S, van Stralen MM, West R. The behaviour change wheel: a new method for characterising and designing behaviour change interventions. Implement Sci. 2011;6:42.
Consolidated Framework for Implementation Research. Strategy Design. https://cfirguide.org/choosing-strategies/. Accessed 22 Aug 2022.
Creswell J, Creswell J. Research design: qualitative, quantitative, and mixed methods approaches. Newbury Park: SAGE Publications; 2017.
Guest G, Namey E, McKenna K. How many focus groups are enough? Building an evidence base for nonprobability sample sizes. Field Methods. 2017;29(1):3–22.
Damschroder LJ, Aron DC, Keith RE, Kirsh SR, Alexander JA, Lowery JC. Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4:50.
U.S. Department of Health and Human Services. Research-based web design & usability guidelines. https://www.usability.gov/sites/default/files/documents/guidelines_book.pdf. Accessed 1 Sept 2022.
Jaspers MW, Steen T, van den Bos C, Geenen M. The think aloud method: a guide to user interface design. Int J Med Inform. 2004;73(11–12):781–95.
Bainbridge L. Verbal protocol analysis. Philadelphia, PA, US: Taylor & Francis; 1990.
Moulton LH. Covariate-based constrained randomization of group-randomized trials. Clin Trials. 2004;1(3):297–305.
Stelfox HT, Straus SE. Measuring quality of care: considering measurement frameworks and needs assessment to guide quality indicator development. J Clin Epidemiol. 2013;66(12):1320–7.
Ministère du Travail de l'Emploi et de la Solidarité sociale. S-4.2, r. 27 - Règlement sur la transmission de renseignements concernant les usagers victimes de traumatismes majeurs. https://www.legisquebec.gouv.qc.ca/fr/document/rc/S-4.2,%20r.%2027. Accessed 13 Sept 2022.
Moore GF, Audrey S, Barker M, et al. Process evaluation of complex interventions: Medical Research Council guidance. BMJ. 2015;350:h1258.
DataDriven. How to track downloads in Google Analytics - Complete guide. https://www.datadrivenu.com/track-downloads-google-analytics/. Published 2022. Accessed 13 Sept 2022.
Lorencatto F, Gould NJ, McIntyre SA, et al. A multidimensional approach to assessing intervention fidelity in a process evaluation of audit and feedback interventions to reduce unnecessary blood transfusions: a study protocol. Implement Sci. 2016;11(1):163.
Vasileiou K, Barnett J, Thorpe S, Young T. Characterising and justifying sample size sufficiency in interview-based studies: systematic analysis of qualitative health research over a 15-year period. BMC Med Res Methodol. 2018;18(1):148.
Hemming K, Kasza J, Hooper R, Forbes A, Taljaard M. A tutorial on sample size calculation for multiple-period cluster randomized parallel, cross-over and stepped-wedge trials using the Shiny CRT Calculator. Int J Epidemiol. 2020;49(3):979–95.
Kahan BC, Li F, Copas AJ, Harhay MO. Estimands in cluster-randomized trials: choosing analyses that answer the right question. Int J Epidemiol. 2022;52(1):107–18.
Ford WP, Westgate PM. A comparison of bias-corrected empirical covariance estimators with generalized estimating equations in small-sample longitudinal study settings. Stat Med. 2018;37(28):4318–29.
Zou GY, Donner A. Extension of the modified Poisson regression model to prospective studies with correlated binary data. Stat Methods Med Res. 2013;22(6):661–70.
European Medicines Agency. Guideline on adjustment for baseline covariates in clinical trials. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-adjustment-baseline-covariates-clinical-trials_en.pdf. Accessed 22 Aug 2022.
Hooper R, Forbes A, Hemming K, Takeda A, Beresford L. Analysis of cluster randomised trials with an assessment of outcome at baseline. BMJ. 2018;360:k1121.
Moore L, Hanley JA, Lavoie A, Turgeon A. Evaluating the validity of multiple imputation for missing physiological data in the national trauma data bank. J Emerg Trauma Shock. 2009;2(2):73–9.
Moore L, Hanley JA, Turgeon AF, Lavoie A, Emond M. A multiple imputation model for imputing missing physiologic data in the national trauma data bank. J Am Coll Surg. 2009;209(5):572–9.
Moore L, Lavoie A, LeSage N, et al. Multiple imputation of the Glasgow Coma Score. J Trauma. 2005;59(3):698–704.
Braun V, Clark V, Hayfield N, Terry G. Thematic analysis. In: springer, ed. Handbook of research methods in health social sciences. Singapour: Springer Nature; 2019.
Provalis Research. QDA Miner. https://provalisresearch.com/products/qualitative-data-analysis-software/. Accessed August 22th, 2022.
Braun V, Clarke V. Using thematic analysis in psychology. Qual Res Psychol. 2006;3(2):77–101.
Anney V. Ensuring the quality of the findings of qualitative research: looking at trustworthiness criteria. JETERAPS. 2015;5(2):272–81.
Carnevale FA. Authentic qualitative research and the quest for methodological rigour. Can J Nurs Res. 2002;34(2):121–8.
Guetterman TC, Fetters MD, Creswell JW. Integrating quantitative and qualitative results in health science mixed methods research through joint displays. Ann Fam Med. 2015;13(6):554–61.
Younas A, Pedersen M, Durante A. Characteristics of joint displays illustrating data integration in mixed-methods nursing studies. J Adv Nurs. 2020;76(2):676–86.
Sussman JB, Hayward RA. An IV for the RCT: using instrumental variables to adjust for treatment contamination in randomised controlled trials. BMJ. 2010;340:c2073.
Canada's Drug and Health Technology Agency. Guidelines for the economic evaluation of health technologies: Canada. https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-0. Accessed 29 Feb 2023.
Husereau D, Drummond M, Augustovski F, et al. Consolidated Health Economic Evaluation Reporting Standards 2022 (CHEERS 2022) statement: updated reporting guidance for health economic evaluations. Value Health. 2022;25(1):3–9.
Gray-Burrows KA, Willis TA, Foy R, et al. Role of patient and public involvement in implementation research: a consensus study. BMJ Qual Saf. 2018;27(10):858–64.
Canadian Institutes of Health Research (CIHR). Ethics of Health Research Involving First Nations, Inuit and Métis People. https://cihr-irsc.gc.ca/e/29339.html. Accessed 29 March 2021.
Acknowledgements
The authors would like to thank Xavier Neveu for his help with the statistical analysis plan.
Funding
This work is supported by the Canadian Institutes of Health Research (#186093, https://webapps.cihr-irsc.gc.ca/decisions/p/project_details.html?applId=467919&lang=en).
Author information
Authors and Affiliations
Consortia
Contributions
LM and MB developed the overarching scientific aims, designed the project, and secured funding. AFT, RF, and NY. AB, MT, HTS, PA, NI, JG, YO, and FL contributed to the design of the randomized controlled trial. AB, AFT, NY, EM, JP, HTS, SB, BH, PLB, NI, JG, MS, BG, and FL contributed to the design of the intervention. MT and YO designed the randomization strategy and the statistical analysis plan. MB and HW ensured consideration of equity, diversity, and inclusion in the study design. MB and HW co-designed usability testing. MB, AL, and MS co-designed intervention refinement and process evaluations. JRG designed the economic evaluation. All authors contributed to the development, drafting, or review of the manuscript. The authors approved the final version of the manuscript.
Ethics declarations
Ethics approval and consent to participate
We have obtained approval from the CHU de Québec-UL Research Ethics Board (#113664). Informed consent is not required as the trial is embedded within a mandatory quality assurance process linked to accreditation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
SPIRIT 2013 Checklist: Recommended items to address in a clinical trial protocol and related documents*.
Additional file 2.
CONSORT 2010 checklist of information to include when reporting a cluster randomised trial.
Additional file 3:
Table S1. PRECIS-2 scores for trial domains with rationale. Figure S1. Integration of the project within UK Medical Research Council guidelines for the Development of Complex Interventions. Table S2. Matching of barriers with implementation strategies according to the CFIR-ERIC tool. Table S3. Schedule of enrolment, interventions, and assessments. Table 4. Intervention prototype as per the TIDieR checklist. Figure S2.1. Example of a page of the A&F report for one quality indicator (intervention arm). Figure S2.2. Example of output from patient chart revision tool† (intervention arm). Table S5. Questions for the semi-structured interviews for focus groups (intervention refinement). Table S6. Sample size calculation for primary and selected secondary outcomes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Moore, L., Bérubé, M., Belcaid, A. et al. Evaluating the effectiveness of a multifaceted intervention to reduce low-value care in adults hospitalized following trauma: a protocol for a pragmatic cluster randomized controlled trial. Implementation Sci 18, 27 (2023). https://doi.org/10.1186/s13012-023-01279-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13012-023-01279-y