
Abstract
The DNA damage response (DDR) is a signaling cascade that is triggered by DNA damage, involving the halting of cell cycle progression and repair. It is a key event leading to senescence, which is characterized by irreversible cell cycle arrest and the senescence-associated secretory phenotype (SASP) that includes the expression of inflammatory cytokines. Human cytomegalovirus (HCMV) is a ubiquitous pathogen that plays an important role in the senescence process. It has been established that DDR is necessary for HCMV to replicate effectively. This paper reviews the relationship between DDR, cellular senescence, and HCMV, providing new sights for virus-induced senescence (VIS).
Similar content being viewed by others


Background
Cellular senescence was first formally described by Hayflick et al. over 50 years ago [1, 2]. Since then, it has been understood that cellular senescence is a stress-induced transformation in cellular states, including terminal cell cycle arrest and the development of senescence-associated secretory phenotypes (SASP) [3]. Senescence can be triggered by various types of cellular and environmental stresses such as telomere shortening, oncogene activation, oxidative stress, and DNA damage [4, 5]. Although many different factors lead to senescence, the DNA damage response (DDR) is a common factor in all of these mechanisms. Studies have demonstrated that senescence can be caused by persistent DDR [6, 7], a signaling cascade activated by DNA damage [5], in which cells respond to DNA damage by pausing cell cycle progression and trying to repair [7, 203].
Human cytomegalovirus (HCMV) is a β-herpesvirus that infects a variety of cell types, including fibroblasts, epithelial cells, macrophages, endothelial cells, dendritic cells, and smooth muscle cells [8]. As an enveloped, double-stranded DNA (dsDNA) virus, it has the largest genome of human viruses [9]. Herpesvirus genes are expressed in a “temporal cascade,” whereby the first set of viral genes, the immediate-early (IE) genes, drive the subsequent expression of delayed-early (DE) and late (L) genes [10,11,12,13]. During HCMV infection, the 72-kDa and 86-kDa IE1 and IE2 proteins are among the first and most widely expressed proteins. It is assumed that these proteins operate as transcriptional regulators by interacting with numerous cellular proteins that communicate with one another [14, 15].
A growing number of studies have shown that many viral infections, including HCMV [16,17,18,19], can also activate cellular senescence responses and that virus-induced senescence (VIS) has much in common with other forms of cellular senescence [20]. However, the precise regulatory mechanisms directly linking HCMV to cellular senescence remain unknown. As DDR signaling pathways are critical for the replication of HCMV [21,22,23], it would be interesting to investigate if HCMV can cause or worsen cellular senescence through DDR. In this review, we first provide a detailed explanation of how DNA damage response (DDR) begins and develops as well as how DDR contributes to the establishment of cellular senescence. We then concentrate on how HCMV influences DDR and ultimately causes cellular senescence which is characterized by the senescence-associated secretory phenotype (SASP).
DNA damage response(DDR)
DNA damage activates a signaling cascade named DNA damage response (DDR) [5], in which cells respond to DNA damage by pausing cell cycle progression and trying to repair [7, 203] (Fig. 1). This complicated network of signaling channels made up of sensors, transducers, and effectors. The sensor delivers a signal to the transducer when it locates damaged DNA, such as DNA double-strand breaks (DSBs) or single-stranded DNA (ssDNA). The transducer amplifies the signal and transmits it to the effector. The effector executes a series of cellular responses, including initiating activation of cell cycle checkpoints and mobilizing the corresponding damage repair pathways [22]. If DNA damage is repaired in time, the cell will quickly return to normal; however, if the DNA damage is particularly severe and cannot be repaired, the cell may undergo apoptosis or cellular senescence. The former is programmed cell death, a form of cellular suicide that removes damaged cells from the cell population [24]; the latter is a natural irreversible cell cycle arrest, induced by DDR. It remains unclear what determines the choice between apoptosis and senescence, but determinants may include cell type and the intensity, duration, and nature of the damage [7].
The MRE11-RAD50-NBS1 (MRN) complex and the single-stranded DNA-binding protein replication protein A (RPA) are the major sensor proteins that detect DSBs and ssDNA, respectively [24,25,26,27]. These proteins then recruit ATM (ataxia-telangiectasia mutated) and ATR (ATM- and Rad3-related), both of which are the main kinases of the DDR [25]. ATM is largely engaged in DSB repair, whereas ATR is primarily involved in the recognition of ssDNA wrapped by RPA [26] (Fig. 1). Although ATM and ATR recognize distinct forms of DNA damage, both are needed for proper checkpoint activation when DSBs are encountered [27,28,29,30]. The cis-local phosphorylation of histone H2AX (γ-H2AX) by ATM and ATR is a critical step in DDR [31]. MDC1 (mediator of DNA damage checkpoint protein 1) is hyperphosphorylated in an ATM-dependent manner, generating a phospho-specific domain that can detect γH2AX [32,33,34]. MDC1 recruitment to γH2AX amplified local ATM activity and the spreading of γH2AX along the chromatin from the DSB. This in turn raises the local concentration of many DDR components at the site of DNA damage, resulting in a positive feedback loop that amplifies ATM activity [7, 35,36,37]. Co-localization of ATRIP (ATR interaction protein) [39] and the 9-1-1 complex (composed of RAD9, RAD1, and HUS1) [40] is also required for ATR activation by RPA-coated ssDNA [38, 39]. Furthermore, topoisomerase II binding protein 1 (TOPBP1) is an ATR signaling pathway amplifier [40, 41] (Fig. 1).

The DNA damage response. Responses to DNA damage caused by double-strand breaks (DSBs) and single-stranded DNA (ssDNA). The MRN complex detects DNA DSBs and signals them by activating ATM. The accumulation of ssDNA at stalled or stressed replication forks activates ATR. Following the activation of transducer kinases, DNA damage signaling is initiated, which includes DNA repair processes (lower panel) and cell cycle checkpoints (upper panel). Direct and indirect interactions are indicated by solid and dashed arrows, respectively. This figure was modified according to the published Fig. 1 in reference [203].
Regulation of cell cycle progression by DDR
DNA damage signaling activates cell cycle checkpoints, halting cell cycle advancement and allowing time for DNA repair, preventing damaged DNA replication. The cell cycle is divided into four stages: G1, S, G2, and M, each with its own set of regulatory proteins. Cyclin D, CDK4/6, and p16INK4a are examples of G1 phase regulatory proteins, whereas cyclin E, CDK2, and p21 are examples of S phase regulatory proteins. The production of cyclin E and CDK2 complexes is required for cells to enter S phase; G2 phase regulatory proteins primarily involve cyclin B and CDK1, and the formation of a complex between the two causes cells to enter pre-M phase [42,43,44].
Activated ATM and ATR phosphorylate the activating checkpoint kinases CHK2 and CHK1, with ATR primarily activating CHK1 but also ATM [45, 46]. Activated CHK1 and CHK2 then phosphorylate the cell division cycle 25 (CDC25) phosphatase and the tumor suppressor protein p53, causing their inactivation or degradation and activation or stabilization, respectively [47,48,49,50]. Furthermore, active CHK1 in yeast stimulates Wee1 kinase, which inactivates CDK1 and CycB [51, 52]. Both eventually produce cell cycle arrest: the ATM-CHK2-P53 pathway regulates the G1 checkpoint, while the ATR-CHK1 pathway controls the S and G2/M checkpoints [42, 53], and both pathways can cause cell cycle arrest through p53 activation (Fig. 1 upper panel). p53 is a transcription factor that regulates genes involved in DNA repair, cell cycle arrest, apoptosis, and metabolism [54, 55]. Phosphorylated P53 promotes the expression of the cyclin-dependent kinase inhibitor (CDKI) p21. Both p21 and p16 cyclin-dependent kinase inhibitors are components of the tumor suppressor pathway and a major modulator of senescence-associated cell cycle arrest. CycE and CDK2 are inhibited by p21 activation. CycD and cdk4/6 cyclins are inhibited by p16 activation. The mechanism of p16 induction remains unknown [56]. Furthermore, both p21 and p16 can keep the retinoblastoma protein (pRB) hypophosphorylated and active, resulting in cellular senescence [57].
Repair mechanisms in the DNA damage response
The primary repair pathways for DSBs are non-homologous end-joining (NHEJ) and homologous recombination (HR). Non-homologous end joining (NHEJ) re-ligates a DSB without extensive processing of the DNA around the DSB and is present throughout the cell cycle, making it a relatively easier, faster, and more extensive repair mechanism of the two. HR, on the other hand, necessitates resection of the DNA at the break site to form substantial single-stranded overhangs that can invade the homologous sister strand, which is more difficult and precise and occurs only in the S/G2 phase [6, 58]. On the DSB, γH2AX progressively recruits MDC1, RNF8, and RNF168, triggering a ubiquitination cascade around the DSB [59]. Following this, the DSB repair proteins BRCA1 and 53BP1 are recruited [59, 60]. The 53BP1-RIF1 and the BRCA1-CtIP pathway are in competition with each other and their selection is regulated by the cell cycle and histone modifications [44, 61, 62]. In the G1 phase, the recruitment of 53BP1-RIF1 enhances NHEJ repair by antagonizing the recruitment of the BRCA1-CtIP complex [44]. In the S/G2 phase, CtIP cooperates with nucleases to produce extensive single-stranded overhangs by excising DNA at the break site and invading the sister homologous strand. During this process, exposed ssDNA is first bound by RPA [6], and then the recombinase RDA51 displaces RPA in the involvement of recombinant mediators BRCA1, PALB2, and BRCA2 to form RAD51-single-stranded DNA nucleoprotein filaments. This nucleoprotein filament structure is capable of facilitating multiple processes such as homology search, strand invasion, and DNA polymerization [44, 63, 64] (Fig. 1 lower panel).
The link between cellular senescence and DNA damage
Cellular senescence is a state of irreversible cell cycle arrest. Cellular senescence can be caused by a variety of factors, including telomere malfunction, DNA damage, oncogene activation, and organelle stress [5, 204]. DNA damage is likely the most powerful cause of cellular senescence, as DNA carries information about all of the proteins and RNAs produced by the cell [65, 66]. If DNA damage cannot be repaired and continues, it can result in prolonged DDR signaling and long-term proliferation arrest in the form of cellular senescence [48]. DDR foci harboring unrepaired DSBs have been reported in cultured senescent cells [49]. Inhibiting DDR signaling kinases (ATM, ATR, CHK1, and CHK2) permits senescent cells to re-enter the cell cycle [67,68,69]. Furthermore, even in the absence of physical DNA damage, alterations in DDR sensors alone can cause cell cycle arrest [70].
Cellular senescence was initially identified as the mechanism that regulates the limited replicative lifespan of cultivated cells, also known as replicative senescence (RS) [2], a type of telomere-induced cellular senescence (TIS). Telomeres shorten with each round of DNA replication due to a lack of telomere maintenance mechanisms like telomerase expression or telomere recombination. Such ends are regarded as double-strand breaks (DSBs) below a specific length, triggering a DNA damage response (DDR) [68, 71]. However, aberrant activation of the proliferative pathway can also cause cellular senescence. Oncogene-induced senescence (OIS) is characterized by substantial activation of the DDR pathway and the formation of DDR foci in senescent cells (also known as senescence-associated DNA-damage foci; SDFs) [7, 67, 72, 73]. Furthermore, mitochondrial dysfunction induces increased ROS generation in senescent cells, resulting in DNA damage and DDR activation [74, 75], which drives cellular senescence [76, 77]. Clearly, all of these senescence-inducing conditions influence DDR, which plays a critical role in cellular senescence (Fig. 2).

The relationship between cellular senescence and DNA damage. Senescence can be activated by different stimuli, including telomere shortening, DNA damage, oxidative stress, and oncogene activation. A central factor involved in all aspects of senescence is the sustained DNA damage response (DDR), which causes cell cycle arrest via the p53 and RB-dependent pathways and SASP secretion via the NF-kB and cGAS-STING pathways, ultimately inducing cellular senescence. This figure was modified according to the published Fig. 1 in reference [204].
Furthermore, cellular senescence is frequently regarded as a stress response that, in addition to the characteristic stable cell cycle arrest, involves a pro-inflammatory phenotype known as the senescence-associated secretory phenotype (SASP), which is primarily mediated by the cGAS-STING, NF-κB, and C/EBPβ signaling pathways [3, 78]. Studies have shown that the gene expression of SASP often requires sustained DDR signaling and that key DDR proteins such as ATM, NBS1, and CHK2 are involved in the activation of SASP genes [78, 79] (Fig. 3). Defective DDR signaling is a fundamental mechanism of DNA damage, cellular senescence, and aging [80].

cGAS is a key linkage between DNA damage and SASP. Exogenous and Endogenous aberrant DNA bind cyclic GMP-AMP synthase (cGAS) and activate the synthesis of 2′3′-cyclic GMP-AMP (2′3′-cGAMP), which binds to and induces oligomerization of STING (stimulator of interferon genes) in the endoplasmic reticulum and its incorporation into vesicles. When STING is activated, it attracts and activates TANK-binding kinase 1 (TBK1), which phosphorylates STING and the interferon regulatory factor IRF3, activating the NF-κB signaling cascade. The sensor kinase ataxia telangiectasia mutated (ATM) also activates TBK1, through the phosphorylation of NF-κB essential modulator (NEMO), a member of the IB kinase complex that activates NF-κB. In response to nuclear DNA damage, ATM can potentially activate STING in a non-canonical manner. PARP-1, poly (ADP-ribose) polymerase 1, is an essential DNA damage sensor. This figure was modified according to the published Fig. 3 in reference [202].
Cyclic GMP-AMP synthase (cGAS) has been found to be a key linkage between DNA damage, SASP gene expression, and cellular senescence [81]. SASP gene expression is reduced when cGAS is deleted [81]. cGAS or stimulator of interferon genes (STING) deprived cells are unable to induce senescence by DNA damage stimulation, and p16, p21, and SASP are also not increased [81,82,83]. The binding of cGAS to cytoplasmic dsDNA fragments, including double-stranded DNA from the leaky senescent nuclei and exogenous nucleic acids (viruses), initiates the cGAS-STING pathway [202, 84, 85] (Fig. 3). Activated cGAS catalyzes the formation of cyclized dinucleotides (cGAMP) from ATP and GTP. cGAMP translocates to the endoplasmic reticulum, where it binds to and activates STING [84, 86]. Activated STING translocates to the Golgi apparatus and recruits TANK-binding kinase 1 (TBK1) and IκB kinase (IKK), which activate Interferon regulatory factor 3 (IRF3) and NF-kB [86,87,88].In most unstimulated cells, NF-kB dimers are found in the cytoplasm as complexes with IkB proteins. Upon stimulation, IkB is phosphorylated by the IKK complex, ubiquitinated, and targeted for degradation, thus releasing the NF-kB subunits that translocate to the nucleus and induce transcription of inflammatory proteins like type I interferon [86,87,88,89]. Direct activation of the NF-kB signaling pathway by nuclear DNA damage necessitates the activation of ATM and PARP1 [43, 90], resulting in the phosphorylation and ubiquitination of sumoylated NEMO. PARP-1 is an essential DNA damage sensor [91]. NEMO is a regulatory subunit of the IκB kinase complex. Ubiquitinated NEMO coupled with ATM is exported into the cytoplasm, where it activates the IKK complex and then the NF-kB signaling cascade like the traditional pathway [90, 92].
Cellular senescence Induced by infection with HCMV and other viruses
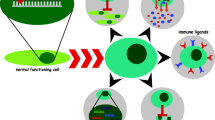
Immature myeloid lineage cells present in the bone marrow and circulating in the blood are considered as primary sites for viral latency [93,94,95,96]. Although persistent CMV infection is systemically controlled by the immune system and viral particles are detectable only in times of reactivation, life-long exposure to HCMV has been demonstrated to severely impair the T cell system. It increases the number of highly differentiated, exhausted CD4 and CD8 T cells, named terminally differentiated T Cells [97, 98]. One of the most robust markers in describing these exhausted T cells is the lack of the costimulatory molecule CD28, a member of the tumor necrosis factor receptor family that interacts with CD80 and/or CD86 expressed on activated antigen-presenting cells [99]. The age-dependent accumulation of exhausted CD28+ T cells, which preferentially produce the pro-inflammatory cytokines IFN-γ and TNF-α, is thought to contribute—together with components of the innate immune system—to the low-grade pro-inflammatory background observed in elderly persons (inflamm-aging) [100] (Fig. 4A).

The model of HCMV-induced senescence. Human cytomegalovirus (HCMV) has two modes of infection: latent and productive. (A) In the latent infection, cells from the myeloid lineage are considered as primary sites. Long exposure to HCMV enables terminal differentiation of T cells leading to accumulation of exhausted CD28 T cells, which secrete TNF-α and IFN-γ to promote inflamm-aging. (B) In the productive infection, HCMV-infected cells show senescence phenotype, including cell cycle arrest and SASP.
Cells from the myeloid lineage are thought to play a critical role in HCMV latency and reactivation but do not support productive infection [93,94,95]. Instead, this virus can infect many other cell types, in most of which virus causes a productive infection, such as macrophage, endothelial cells, fibroblasts [101, 102]. Complex mechanisms control the reactivation of the HCMV from latency. Inflammation has been shown to have the potential to cause latent HCMV to reactivate [103,104,105] (Fig. 4).
Tracking HCMV infection with single-cell transcriptomics revealed that infection outcome (productive or latent) is also based on viral gene expression levels at early stages of infection [106]. High early viral gene expression levels, particularly of immediate early (IE) genes, facilitate productive infection [106, 107]. In the productive infection, previous reports demonstrate that HCMV induces premature senescence in early passage human fibroblasts, similar to senescent cells which have reached the limit of their replicative capacity [108]. Specifically, the IE1 protein activates and interacts with p53, causing p53 accumulation [109, 110]. The IE2 protein inhibits cellular DNA synthesis, resulting in cell cycle arrest through a functional p53 pathway [111]. The interaction of IE1, IE2, and p53 above ultimately evokes the senescence phenotype in HCMV-infected cells [16, 110, 111]. Additionally, HCMV infection upregulates the expression of p16, which is necessary for ideal viral replication [112]. Furthermore, HCMV infection affects the inflammatory phenotype in addition to causing cell cycle arrest [17] (Fig. 4B).
According to recent researches, virus infections, such as measles virus, human respiratory syncytial virus and COVID-19, can prematurely stimulate cellular senescence, known as virus-induced senescence (VIS) [23, 113, 114]. Measles virus (MV) infection has been proven to induce p53 and p16-pRb pathway-dependent cellular senescence via cell [115]. Epstein–Barr virus (EBV), Kaposi sarcoma herpesvirus (KSHV) and human respiratory syncytial virus (RSV) infections can trigger DNA damage-mediated cellular senescence through replicative stress or induction of mitochondrial ROS [23, 116, 117]. Senescence markers and SASP factors have been found in tissue samples of the nasopharyngeal cavity and lungs of patients suffering from coronavirus disease 2019 (COVID-19) with severe disease progression [20]. A basic research study, assessing the occurrence of VIS, found that human diploid fibroblast models exposed to high-titer retrovirus exhibited typical senescence and the activated cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway after the fifth day of infection [118].
HCMV infection can promote cellular senescence by modulating the DDR
As mentioned above, Virus-induced senescence (VIS) has been a widespread event [20]. Viral infections generate a variety of cellular impairments, including DNA damage [23], as well as significant biological changes in host cells, such as cellular senescence [23, 115, 119]. Similarly, like activators of DNA damage, oncogenes [120,121,122] and oxidative stress [123,124,125], we speculate that DDR plays a key role in cellular senescence induced by infection with HCMV.
After penetration of the plasma membrane, components of the virion, including its 240-kb linear double-stranded DNA (dsDNA) genome (which consists of two unique coding sequences [UL and US] flanked by a series of inverted repeat, are rapidly transported to the nucleus, where viral transcription and replication take place [21, 126]. It has been proved that the entrance of the HCMV genome into the infected cell nucleus can initiate DDR during productive infection [21,22,23].
HCMV is a DNA damage-inducing factor
HCMV infection is genotoxic to host cells, and the type and quantity of damage rely on viral genome expression and the cell cycle phase at the time of viral infection [127]. Infected host cells cause particular breaks on chromosome 1, 1q42 and 1q21, during the S phase [128]. Stably transfected cells expressing HCMV UL76 develop chromosome aberrations including micronuclei and misaligned chromosomes, lagging and bridging, and activate the DNA damage signal γH2AX, causing foci formation in nuclei [129]. HCMV infection interfering with cellular replication can induce replication stress (RS) with ensuing implications for genomic integrity. In addition, expression of IE1 and IE2, driven by the viral major immediate early enhancer and promoter (MIEP), has been determined to induce RS alone [130].
Furthermore, there is accumulating evidence that viral infection can generate oxidative stress [131,132,133], which can lead to DNA damage [74, 75]. HCMV infection has been found to increase ROS generation [18] and mitochondrial biogenesis [134]. ROS promotes HCMV replication via paracrine and autocrine pathways, and N-acetylcysteine, a ubiquitous H2O2 scavenger, decreases HCMV replication activation [135]. Interestingly, HCMV appears to utilize virus-specific mechanisms to protect the cells from the harmful effects of ROS and maintain redox homeostasis [125]. There is no doubt that HCMV and ROS have a complementary relationship, and there is evidence that both HCMV and ROS can cause DNA damage, but there is still no direct evidence that HCMV-mediated increase in ROS leads to DNA damage, which would be interesting to investigate.
HCMV influences cell cycle checkpoint activation during DDR
Human cytomegalovirus (HCMV) infection activates multiple DDR proteins, including ATM and downstream effector proteins p53 and H2AX [21, 110, 136]. These proteins are also necessary for efficient HCMV replication [137, 138]. Activated p53 directly induces p21 [139], ultimately leading to cell cycle arrest in HCMV-infected fibroblasts [138, 140]. Immediate early 1 (IE1) of HCMV is an important viral protein for the induction of DDR. Its stimulation of cellular DDR was first described by Castillo et al., who showed that IE1 was sufficient to activate ATM. ATM subsequently activates the p53 pathway by phosphorylation [110]. This conclusion was later supported by additional research, which also showed that the DSB marker γH2AX is similarly activated in an IE1-dependent way [137]. In addition to the activation of ATM by IE1, HCMV infection also leads to ATM autophosphorylation [136]. Interestingly, p53 is bound by IE2 but its transactivation activity is inhibited [141, 142]。.
Efficient HCMV replication requires a host DDR that centers on the presence of ATM and E2F1 protein [137]. E2F1 is a protein in the E2F family that belongs to the RB-regulated activator class [143, 144]. It has been shown that RB inactivation and deregulation of E2F1 leads to DNA double-strand break (DSB) accumulation and cell cycle checkpoint signaling [145,146,147,148] (Fig. 5). One of the earliest impacts of HCMV infection has been identified as RB family protein inactivation [137]. IE1, IE2, pp71, and pUL97 of HCMV, all of which can inactivate RB family members [15, 149,150,151,152,153,154,155,156], lead to dysregulation of E2F1 proteins and induction of DSBs [146]. And the resulting activation of ATM and its downstream target phosphorylation, including H2AX and p53, contribute to the replication of HCMV and cell cycle arrest in the host cell [137, 157].

Model of the host DNA damage response induced by HCMV infection. Efficient replication of HCMV requires DDR centered on ATM and E2F1. HCMV infection can activate multiple DDR proteins, including ATM and the downstream effector proteins p53 and H2AX. IE1, IE2, pp71, and pUL97 of HCMV can inactivate RB family members, leading to dysregulation of E2F1 proteins and subsequent production of DSBs.
However, the role of DDR in HCMV replication has long been controversial. Although ATM is important for virus replication in cells [137], HCMV replication in cells lacking ATM has also been reported [21], Some DDR proteins have been shown to mislocate from the nucleus to the cytoplasm after infection, blocking checkpoint signaling and inhibiting host DDR. Therefore, HCMV is also able to escape some of the consequences produced by DDR [158, 159]. In conclusion, ATM and ATR control multiple pathways, and more research are needed to elucidate how HCMV targets DDR and which specific components are regulated by HCMV.
HCMV infection affects damage repair mechanisms during DDR
The replication of the human cytomegalovirus (HCMV) genome is assumed to be biphasic [160]. The initial phase of infection is characterized by origin-specific replication from the input circularized genome, which leads to single copies of the virus. Later, replication switches to a rolling loop process, resulting in the formation of huge concentric circles [157, 161]. HR happens along conjugated DNA, as indicated by the inversion of genomic sequences in contiguous monomeric units. HR occurs between freely cleaved monomeric and conjugated structures as well, with intermediate structures forming branches late in the infection period [162,163,164,165]. It has been suggested that these recombinant structures trigger a DNA damage response (DDR) in host cells during herpesvirus replication [21, 158, 166, 167]. Previous research has also revealed that viral proteins can govern HR [168,169,170].
The IE1 protein of HCMV is not only a strong activator of DDR, but it can also accelerate HR [110, 137, 168]. Further research found that the IE1 protein, in a novel way, can activate flap endonuclease 1 (FEN1), a cellular factor recently identified to be involved in HR-mediated repair of stalled replication forks by actively inducing DSBs [171], hence restarting stalled replication forks in viral replication [172]. Furthermore, IE1 binds to p53 and inhibits p53’s inhibitory impact on Rad51, enabling HR [173]. Rad51 is a key regulator of HR, and its levels are much higher in HCMV-infected human foreskin fibroblasts (HFFs) [21], but not in normal cells [174]. We hypothesize that viruses may employ the cellular HDR process to boost the efficiency and fidelity of viral genome replication [160, 175, 176].
HCMV infection regulates SASP secretion
IL-8 and IL-6 are important SASP factors that participate in HCMV infection. HCMV UL76 protein can activate the NF-kB system via the DDR, thereby inducing IL-8 expression [92] and enhancing HCMV replication [177]. As a crucial part of SASP, IL8 activates the chemokine receptor CXCR2 (IL8RB), enhancing DDR and promoting replicative senescence(RS) and oncogene-induced senescence (OIS) [178, 179]. US28, a G protein–coupled receptor encoded by HCMV promotes the production of interleukin-6 (IL-6) [180, 181], whose depletion would cause the inflammatory network to collapse and abolished senescence entry and maintenance [182].
In addition, it has been proved that cellular senescence was induced in host cells upon HCMV infection [81], which was recognized as an antiviral immune response [113, 119, 183]. Mechanistically, this induction of cellular senescence was mainly due to activation of the cGAS-STING pathway triggered by HCMV dsDNA as well as the subsequent SASP secretion [85, 184]. Interestingly, it has been demonstrated that HCMV has evolved multiple strategies to antagonize the activation of GAS-STING signaling in host cells. UL31 and UL42 interacted with cGAS respectively, inhibiting DNA binding and enzymatic activity of cGAS [185, 186]; pp65 selectively bound to cGAS and prevented its interaction with STING, thus inactivating the signaling pathway through the cGAS/STING/IRF3 axis [187]. UL82, UL94 and US9 interacted with STING respectively, disrupting the translocation of STING and impairing the TBK1 recruitment to the STING signalsome [188,189,190,191]; pUL48 had a ubiquitinating effect on STING and IE2 protein facilitated the proteasome-dependent degradation of STING, both of them inhibiting STING-induced IFN-β promoter activation [192, 193]; UL35 and UL37 × 1 downmodulated this signaling pathway at the level of the key signaling factor TBK1 [194, 195]; UL138 inhibited the pathway downstream of STING but upstream of IRF3 phosphorylation and NF-κB function [196]. Although the cGAS-STING signaling induced by HCMV dsDNA was challenged by the HCMV encoded inhibitors described above serving for the viral immune escape, this pathway remained activated [184] and subsequently induced cellular senescence [81].
Previous research has demonstrated that HCMV-infected fibroblasts can mimic senescence-associated inflammation and elicit a significant inflammatory response, potentially leading to the development of age-related inflammatory disorders [17]. As a result, we hypothesize that DDR is intrinsically connected to HCMV-induced SASP production and cellular senescence.
Conclusion
Previous studies have shown that HCMV infection triggers molecular mechanisms associated with host cell senescence [16, 109,110,111,112] as well as inflammatory responses [17, 184, 197]. However, there is little evidence to explain why HCMV can cause senescence-associated phenotypes in host cells. A growing number of studies demonstrate that HCMV might alter the DNA damage response (DDR), for example, by acting directly as a DNA damage agent, interacting with essential DDR proteins, and activating the cGAS itself as aberrant DNA [21, 110, 136]. As a result, we argue that DDR may be one of the reasons why HCMV can generate the senescence phenotype.
Interestingly, cellular senescence has been proposed as a key mechanism of viral invasion resistance [183]. Viral entrance generates major biological changes in infected host cells as a viral-triggered state shift that may lead to cellular senescence [23, 115], with varied degrees of impact on virus proliferation [119, 198]. Stable cell cycle stoppage and the release of pro-inflammatory cytokines and chemokines associated with cellular senescence may give rise to antiviral response features [119]. Leading to speculation that cellular senescence may have evolved as an antiviral defense mechanism [183, 199]. This notion is strengthened by the function of endogenous IFN-b, which is generated by DNA damage, in the induction of senescence [200]. Surprisingly, recent researches highlight a commensal-like function for HCMV in the immunosurveillance of aging cells in immunocompetent hosts: on the one hand, HCMV can be reactivated in senescent fibroblasts, but with low IE1/2 expression and the absence of productive infection, and on the other hand, CD4 CTLs are able to target HCMV-gB antigens to recognize and clear senescent cells [119, 183, 201].
In conclusion, the significance of HCMV in the aging process is receiving increased attention and is intricately related to all aspects of aging. Here we focus on the effects of HCMV on cellular senescence. As to how HCMV causes cellular senescence, there are necessarily many other mechanisms involved besides DDR, and more research is needed to demonstrate this.
Data Availability
Not applicable.
Abbreviations
- ATM:
-
Ataxia-telangiectasia mutated
- ATR:
-
ATM- and Rad3-related
- CDC25:
-
Cell division cycle 25
- cGAS:
-
Cyclic GMP-AMP synthase
- DDR:
-
The DNA damage response
- DSBs:
-
DNA double-strand breaks
- HCMV:
-
Human cytomegalovirus
- HR:
-
Homologous recombination
- IKK:
-
IκB kinase
- MDC1:
-
Mediator of DNA damage checkpoint protein 1
- MRN:
-
MRE11-RAD50-NBS1
- NHEJ:
-
Non-homologous end-joining
- RPA:
-
Replication protein A
- SASP:
-
Senescence-associated secretory phenotype
- ssDNA:
-
Single-stranded DNA
- STING:
-
Stimulator of interferon genes
- TBK1:
-
TANK-binding kinase 1
- TOPBP1:
-
Topoisomerase II binding protein 1
- VIS:
-
virus-induced senescence
References
Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621.
Hayflick L. THE LIMITED IN VITRO LIFETIME OF HUMAN DIPLOID CELL STRAINS. Exp Cell Res. 1965;37:614–36.
Schmitt CA, Tchkonia T, Niedernhofer LJ, Robbins PD, Kirkland JL, Lee S. COVID-19 and cellular senescence. Nat Rev Immunol 2022:1–13.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–35.
Di Micco R, Krizhanovsky V, Baker D. d’Adda di Fagagna F: Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75–95.
Feringa FM, Raaijmakers JA, Hadders MA, Vaarting C, Macurek L, Heitink L, Krenning L, Medema RH. Persistent repair intermediates induce senescence. Nat Commun. 2018;9:3923.
d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22.
Sinzger C, Digel M, Jahn G. Cytomegalovirus cell tropism. Curr Top Microbiol Immunol. 2008;325:63–83.
Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, et al. Genetic content of wild-type human cytomegalovirus. J Gen Virol. 2004;85:1301–12.
Britt WJ, Prichard MN. New therapies for human cytomegalovirus infections. Antiviral Res. 2018;159:153–74.
DeMarchi JM, Schmidt CA, Kaplan AS. Patterns of transcription of human cytomegalovirus in permissively infected cells. J Virol. 1980;35:277–86.
Wathen MW, Stinski MF. Temporal patterns of human cytomegalovirus transcription: mapping the viral RNAs synthesized at immediate early, early, and late times after infection. J Virol. 1982;41:462–77.
Tyl MD, Betsinger CN, Cristea IM. Virus-host protein interactions as footprints of human cytomegalovirus replication. Curr Opin Virol. 2022;52:135–47.
Fortunato EA, Sommer MH, Yoder K, Spector DH. Identification of domains within the human cytomegalovirus major immediate-early 86-kilodalton protein and the retinoblastoma protein required for physical and functional interaction with each other. J Virol. 1997;71:8176–85.
Poma EE, Kowalik TF, Zhu L, Sinclair JH, Huang ES. The human cytomegalovirus IE1-72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J Virol. 1996;70:7867–77.
Noris E, Zannetti C, Demurtas A, Sinclair J, De Andrea M, Gariglio M, Landolfo S. Cell cycle arrest by human cytomegalovirus 86-kDa IE2 protein resembles premature senescence. J Virol. 2002;76:12135–48.
Wolf J, Weinberger B, Grubeck-Loebenstein B. The immunoregulatory effects of CMV-infection in human fibroblasts and the impact on cellular senescence. Immun Ageing. 2012;9:1.
Wang S, Zhou X, He X, Ma S, Sun C, Zhang J, Xu X, Jin W, Yan J, Lin P, Mao G. Suppressive effects of pterostilbene on human cytomegalovirus (HCMV) infection and HCMV-induced cellular senescence. Virol J. 2022;19:224.
Mao G, Li H, Ding X, Meng X, Wang G, Leng SX. Suppressive effects of sirtinol on human cytomegalovirus (hCMV) infection and hCMV-induced activation of molecular mechanisms of senescence and production of reactive oxygen species. Mech Ageing Dev. 2016;158:62–9.
Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, Wyler E, Belenki D, Kaltenbrunner S, Pammer M, et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature. 2021;599:283–9.
Luo MH, Rosenke K, Czornak K, Fortunato EA. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J Virol. 2007;81:1934–50.
Xiaofei E, Kowalik TF. The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses. 2014;6:2155–85.
Martínez I, García-Carpizo V, Guijarro T, García-Gomez A, Navarro D, Aranda A, Zambrano A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence. 2016;7:427–42.
Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–41.
Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst). 2004;3:883–7.
Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–37.
Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–7.
Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008;22:297–302.
Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–56.
Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59.
Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–7.
Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–6.
Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–61.
Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–6.
Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. Embo j. 2004;23:2674–83.
Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26.
Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21:187–200.
Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–6.
Weiss RS, Matsuoka S, Elledge SJ, Leder P. Hus1 acts upstream of chk1 in a mammalian DNA damage response pathway. Curr Biol. 2002;12:73–7.
Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–55.
Liu S, Bekker-Jensen S, Mailand N, Lukas C, Bartek J, Lukas J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol. 2006;26:6056–64.
Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23.
Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–96.
Hustedt N, Durocher D. The control of DNA repair by the cell cycle. Nat Cell Biol. 2016;19:1–9.
Buscemi G, Perego P, Carenini N, Nakanishi M, Chessa L, Chen J, Khanna K, Delia D. Activation of ATM and Chk2 kinases in relation to the amount of DNA strand breaks. Oncogene. 2004;23:7691–700.
Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol. 2003;5:255–60.
Mailand N, Falck J, Lukas C, Syljuâsen RG, Welcker M, Bartek J, Lukas J. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–9.
Fumagalli M, Rossiello F, Mondello C. d’Adda di Fagagna F: stable cellular senescence is associated with persistent DDR activation. PLoS ONE. 2014;9:e110969.
Galbiati A, Beauséjour C. d’Adda di Fagagna F: a novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell. 2017;16:422–7.
Ou YH, Chung PH, Sun TP, Shieh SY. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol Biol Cell. 2005;16:1684–95.
Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell. 2001;12:551–63.
Lin AB, McNeely SC, Beckmann RP. Achieving Precision death with cell-cycle inhibitors that target DNA replication and repair. Clin Cancer Res. 2017;23:3232–40.
Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9.
Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017;36:3943–56.
Zhang Y, Xiong Y. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science. 2001;292:1910–5.
Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–77.
Campisi J. d’Adda di Fagagna F: Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40.
Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71.
Al-Hakim A, Escribano-Diaz C, Landry MC, O’Donnell L, Panier S, Szilard RK, Durocher D. The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair (Amst). 2010;9:1229–40.
Smeenk G, Mailand N. Writers, readers, and erasers of histone ubiquitylation in DNA double-strand break repair. Front Genet. 2016;7:122.
Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–83.
Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288:11135–43.
Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12:68–78.
Park JY, Zhang F, Andreassen PR. PALB2: the hub of a network of tumor suppressors involved in DNA damage responses. Biochim Biophys Acta. 2014;1846:263–75.
Ou HL, Schumacher B. DNA damage responses and p53 in the aging process. Blood. 2018;131:488–95.
Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, Yosef R, Sagiv A, Agrawal A, Shapira A, et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun. 2018;9:5435.
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42.
di d’Adda F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8.
Mallette FA, Ferbeyre G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle. 2007;6:1831–6.
Bonilla CY, Melo JA, Toczyski DP. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol Cell. 2008;30:267–76.
Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell. 2004;14:501–13.
Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7.
Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5.
Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid Med Cell Longev 2016, 2016:3565127.
Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev. 2018;170:30–6.
Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–12.
Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in Cell Senescence: is Mitophagy the Weakest Link? EBioMedicine. 2017;21:7–13.
Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory phenotype. Mol Cell. 2015;59:719–31.
Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9.
Olivieri F, Albertini MC, Orciani M, Ceka A, Cricca M, Procopio AD, Bonafè M. DNA damage response (DDR) and senescence: shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget. 2015;6:35509–21.
Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114:E4612–e4620.
Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–70.
Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–6.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91.
Kato K, Omura H, Ishitani R, Nureki O. Cyclic GMP-AMP as an endogenous second Messenger in Innate Immune Signaling by cytosolic DNA. Annu Rev Biochem. 2017;86:541–66.
Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657–74.
Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92.
Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27:96–108.
Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62.
Miyamoto S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 2011;21:116–30.
Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac-de Paillerets B, Hofman P, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 2011;25:1245–61.
Costa H, Nascimento R, Sinclair J, Parkhouse RM. Human cytomegalovirus gene UL76 induces IL-8 expression through activation of the DNA damage response. PLoS Pathog. 2013;9:e1003609.
Hahn G, Jores R, Mocarski ES. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci U S A. 1998;95:3937–42.
Kondo K, Kaneshima H, Mocarski ES. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc Natl Acad Sci U S A. 1994;91:11879–83.
Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, Sinclair JH. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol. 1991;72(Pt 9):2059–64.
Tarrant-Elorza M, Rossetto CC, Pari GS. Maintenance and replication of the human cytomegalovirus genome during latency. Cell Host Microbe. 2014;16:43–54.
Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Würzner R, Schönitzer D, Grubeck-Loebenstein B. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8 + T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005;79:3675–83.
Ouyang Q, Wagner WM, Zheng W, Wikby A, Remarque EJ, Pawelec G. Dysfunctional CMV-specific CD8(+) T cells accumulate in the elderly. Exp Gerontol. 2004;39:607–13.
Arnold CR, Wolf J, Brunner S, Herndler-Brandstetter D, Grubeck-Loebenstein B. Gain and loss of T cell subsets in Old Age—Age-Related reshaping of the T cell repertoire. J Clin Immunol. 2011;31:137–46.
Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54.
Myerson D, Hackman RC, Nelson JA, Ward DC, McDougall JK. Widespread presence of histologically occult cytomegalovirus. Hum Pathol. 1984;15:430–9.
Sinzger C, Kahl M, Laib K, Klingel K, Rieger P, Plachter B, Jahn G. Tropism of human cytomegalovirus for endothelial cells is determined by a post-entry step dependent on efficient translocation to the nucleus. J Gen Virol. 2000;81:3021–35.
Reeves M, Sissons P, Sinclair J. Reactivation of human cytomegalovirus in dendritic cells. Discov Med. 2005;5:170–4.
Söderberg-Nauclér C, Fish KN, Nelson JA. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell. 1997;91:119–26.
Söderberg-Nauclér C, Fornara O, Rahbar A. Cytomegalovirus driven immunosenescence-An immune phenotype with or without clinical impact? Mech Ageing Dev. 2016;158:3–13.
Molecular factors shaping. Whether HCMV infection is productive or latent. Nat Microbiol. 2023;8:373–4.
Schwartz M, Shnayder M, Nachshon A, Arazi T, Kitsberg Y, Levi Samia R, Lavi M, Kuint R, Tsabari R, Stern-Ginossar N. Molecular characterization of human cytomegalovirus infection with single-cell transcriptomics. Nat Microbiol. 2023;8:455–68.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602.
Hwang ES, Zhang Z, Cai H, Huang DY, Huong SM, Cha CY, Huang ES. Human cytomegalovirus IE1-72 protein interacts with p53 and inhibits p53-dependent transactivation by a mechanism different from that of IE2-86 protein. J Virol. 2009;83:12388–98.
Castillo JP, Frame FM, Rogoff HA, Pickering MT, Yurochko AD, Kowalik TF. Human cytomegalovirus IE1-72 activates ataxia telangiectasia mutated kinase and a p53/p21-mediated growth arrest response. J Virol. 2005;79:11467–75.
Song YJ, Stinski MF. Inhibition of cell division by the human cytomegalovirus IE86 protein: role of the p53 pathway or cyclin-dependent kinase 1/cyclin B1. J Virol. 2005;79:2597–603.
Zannetti C, Mondini M, De Andrea M, Caposio P, Hara E, Peters G, Gribaudo G, Gariglio M, Landolfo S. The expression of p16INK4a tumor suppressor is upregulated by human cytomegalovirus infection and required for optimal viral replication. Virology. 2006;349:79–86.
Li Z, Tian M, Wang G, Cui X, Ma J, Liu S, Shen B, Liu F, Wu K, Xiao X, Zhu C. Senotherapeutics: an emerging approach to the treatment of viral infectious diseases in the elderly. Front Cell Infect Microbiol. 2023;13:1098712.
Schulz L, Hornung F, Häder A, Radosa L, Brakhage AA, Löffler B, Deinhardt-Emmer S. Influenza Virus-Induced Paracrine Cellular Senescence of the lung contributes to enhanced viral load. Aging Dis. 2023;14:1331–48.
Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013;27:2356–66.
Koopal S, Furuhjelm JH, Järviluoma A, Jäämaa S, Pyakurel P, Pussinen C, Wirzenius M, Biberfeld P, Alitalo K, Laiho M, Ojala PM. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog. 2007;3:1348–60.
Hafez AY, Luftig MA. Characterization of the EBV-Induced persistent DNA damage response. Viruses 2017, 9.
Thangaraj A, Chivero ET, Tripathi A, Singh S, Niu F, Guo ML, Pillai P, Periyasamy P, Buch S. HIV TAT-mediated microglial senescence: role of SIRT3-dependent mitochondrial oxidative stress. Redox Biol. 2021;40:101843.
Baz-Martínez M, Da Silva-Álvarez S, Rodríguez E, Guerra J, El Motiam A, Vidal A, García-Caballero T, González-Barcia M, Sánchez L, Muñoz-Fontela C, et al. Cell senescence is an antiviral defense mechanism. Sci Rep. 2016;6:37007.
Shen Y, Zhu H, Shenk T. Human cytomagalovirus IE1 and IE2 proteins are mutagenic and mediate hit-and-run oncogenic transformation in cooperation with the adenovirus E1A proteins. Proc Natl Acad Sci U S A. 1997;94:3341–5.
Soroceanu L, Cobbs CS. Is HCMV a tumor promoter? Virus Res. 2011;157:193–203.
Geisler J, Touma J, Rahbar A, Söderberg-Nauclér C, Vetvik K. A review of the potential role of human cytomegalovirus (HCMV) infections in breast Cancer carcinogenesis and abnormal immunity. Cancers (Basel) 2019, 11.
Furukawa T, Sakuma S, Plotkin SA. Human cytomegalovirus infection of WI-38 cells stimulates mitochondrial DNA synthesis. Nature. 1976;262:414–6.
McCormick AL, Smith VL, Chow D, Mocarski ES. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J Virol. 2003;77:631–41.
Tilton C, Clippinger AJ, Maguire T, Alwine JC. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J Virol. 2011;85:12585–93.
Mocarski ES Jr. Biology and replication of cytomegalovirus. Transfus Med Rev. 1988;2:229–34.
AbuBakar S, Au WW, Legator MS, Albrecht T. Induction of chromosome aberrations and mitotic arrest by cytomegalovirus in human cells. Environ Mol Mutagen. 1988;12:409–20.
Fortunato EA, Dell’Aquila ML, Spector DH. Specific chromosome 1 breaks induced by human cytomegalovirus. Proc Natl Acad Sci U S A. 2000;97:853–8.
Siew VK, Duh CY, Wang SK. Human cytomegalovirus UL76 induces chromosome aberrations. J Biomed Sci. 2009;16:107.
Merchut-Maya JM, Bartek J Jr., Bartkova J, Galanos P, Pantalone MR, Lee M, Cui HL, Shilling PJ, Brøchner CB, Broholm H, et al. Human cytomegalovirus hijacks host stress response fueling replication stress and genome instability. Cell Death Differ. 2022;29:1639–53.
Aubert M, Chen Z, Lang R, Dang CH, Fowler C, Sloan DD, Jerome KR. The antiapoptotic herpes simplex virus glycoprotein J localizes to multiple cellular organelles and induces reactive oxygen species formation. J Virol. 2008;82:617–29.
McGuire KA, Barlan AU, Griffin TM, Wiethoff CM. Adenovirus type 5 rupture of lysosomes leads to cathepsin B-dependent mitochondrial stress and production of reactive oxygen species. J Virol. 2011;85:10806–13.
Tung WH, Hsieh HL, Lee IT, Yang CM. Enterovirus 71 induces integrin β1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: role of HO-1/CO in viral replication. J Cell Physiol. 2011;226:3316–29.
Kaarbø M, Ager-Wick E, Osenbroch P, Kilander A, Skinnes R, Müller F, Eide L. Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion. 2011;11:935–45.
Xiao J, Deng J, Lv L, Kang Q, Ma P, Yan F, Song X, Gao B, Zhang Y, Xu J. Hydrogen Peroxide Induce Human Cytomegalovirus replication through the activation of p38-MAPK signaling pathway. Viruses. 2015;7:2816–33.
Shen YH, Utama B, Wang J, Raveendran M, Senthil D, Waldman WJ, Belcher JD, Vercellotti G, Martin D, Mitchelle BM, Wang XL. Human cytomegalovirus causes endothelial injury through the ataxia telangiectasia mutant and p53 DNA damage signaling pathways. Circ Res. 2004;94:1310–7.
Pickering EX, Debatis MT, Castillo M, Lagadinos J, Wang A, Lu S, Kowalik S. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011;7:e1001342.
Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol. 2006;80:8390–401.
Castillo JP, Kowalik TF. Human cytomegalovirus immediate early proteins and cell growth control. Gene. 2002;290:19–34.
Hannemann H, Rosenke K, O’Dowd JM, Fortunato EA. The presence of p53 influences the expression of multiple human cytomegalovirus genes at early times postinfection. J Virol. 2009;83:4316–25.
Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finkel T, Epstein SE. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science. 1994;265:391–4.
Tanaka K, Zou JP, Takeda K, Ferrans VJ, Sandford GR, Johnson TM, Finkel T, Epstein SE. Effects of human cytomegalovirus immediate-early proteins on p53-mediated apoptosis in coronary artery smooth muscle cells. Circulation. 1999;99:1656–9.
Iaquinta PJ, Lees JA. Life and death decisions by the E2F transcription factors. Curr Opin Cell Biol. 2007;19:649–57.
Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16:245–56.
Rogoff HA, Pickering MT, Frame FM, Debatis ME, Sanchez Y, Jones S, Kowalik TF. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol. 2004;24:2968–77.
Pickering MT, Kowalik TF. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene. 2006;25:746–55.
Frame FM, Rogoff HA, Pickering MT, Cress WD, Kowalik TF. E2F1 induces MRN foci formation and a cell cycle checkpoint response in human fibroblasts. Oncogene. 2006;25:3258–66.
Powers JT, Hong S, Mayhew CN, Rogers PM, Knudsen ES, Johnson DG. E2F1 uses the ATM signaling pathway to induce p53 and Chk2 phosphorylation and apoptosis. Mol Cancer Res. 2004;2:203–14.
Kalejta RF, Bechtel JT, Shenk T. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol. 2003;23:1885–95.
Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science. 2008;320:797–9.
Prichard MN, Sztul E, Daily SL, Perry AL, Frederick SL, Gill RB, Hartline CB, Streblow DN, Varnum SM, Smith RD, Kern ER. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J Virol. 2008;82:5054–67.
Zhang Z, Huong SM, Wang X, Huang DY, Huang ES. Interactions between human cytomegalovirus IE1-72 and cellular p107: functional domains and mechanisms of up-regulation of cyclin E/cdk2 kinase activity. J Virol. 2003;77:12660–70.
Hagemeier C, Caswell R, Hayhurst G, Sinclair J, Kouzarides T. Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. Embo j. 1994;13:2897–903.
Kalejta RF. Human cytomegalovirus pp71: a new viral tool to probe the mechanisms of cell cycle progression and oncogenesis controlled by the retinoblastoma family of tumor suppressors. J Cell Biochem. 2004;93:37–45.
Kalejta RF, Shenk T. Proteasome-dependent, ubiquitin-independent degradation of the rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc Natl Acad Sci U S A. 2003;100:3263–8.
Pajovic S, Wong EL, Black AR, Azizkhan JC. Identification of a viral kinase that phosphorylates specific E2Fs and pocket proteins. Mol Cell Biol. 1997;17:6459–64.
Skaliter R, Lehman IR. Rolling circle DNA replication in vitro by a complex of herpes simplex virus type 1-encoded enzymes. Proc Natl Acad Sci U S A. 1994;91:10665–9.
Gaspar M, Shenk T. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc Natl Acad Sci U S A. 2006;103:2821–6.
O’Dowd JM, Zavala AG, Brown CJ, Mori T, Fortunato EA. HCMV-infected cells maintain efficient nucleotide excision repair of the viral genome while abrogating repair of the host genome. PLoS Pathog. 2012;8:e1003038.
Lehman IR, Boehmer PE. Replication of herpes simplex virus DNA. J Biol Chem. 1999;274:28059–62.
Skaliter R, Makhov AM, Griffith JD, Lehman IR. Rolling circle DNA replication by extracts of herpes simplex virus type 1-infected human cells. J Virol. 1996;70:1132–6.
Courcelle CT, Courcelle J, Prichard MN, Mocarski ES. Requirement for uracil-DNA glycosylase during the transition to late-phase cytomegalovirus DNA replication. J Virol. 2001;75:7592–601.
Severini A, Scraba DG, Tyrrell DL. Branched structures in the intracellular DNA of herpes simplex virus type 1. J Virol. 1996;70:3169–75.
Severini A, Sevenhuysen C, Garbutt M, Tipples GA. Structure of replicating intermediates of human herpesvirus type 6. Virology. 2003;314:443–50.
Strang BL, Stow ND. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J Virol. 2005;79:12487–94.
Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A. 2005;102:5844–9.
Wilkinson DE, Weller SK. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol. 2004;78:4783–96.
Kulkarni AS, Fortunato EA. Stimulation of homology-directed repair at I-SceI-induced DNA breaks during the permissive life cycle of human cytomegalovirus. J Virol. 2011;85:6049–54.
Nakai-Murakami C, Shimura M, Kinomoto M, Takizawa Y, Tokunaga K, Taguchi T, Hoshino S, Miyagawa K, Sata T, Kurumizaka H, et al. HIV-1 Vpr induces ATM-dependent cellular signal with enhanced homologous recombination. Oncogene. 2007;26:477–86.
Trojanek J, Croul S, Ho T, Wang JY, Darbinyan A, Nowicki M, Del Valle L, Skorski T, Khalili K, Reiss K. T-antigen of the human polyomavirus JC attenuates faithful DNA repair by forcing nuclear interaction between IRS-1 and Rad51. J Cell Physiol. 2006;206:35–46.
Fehrmann RS, Karjalainen JM, Krajewska M, Westra HJ, Maloney D, Simeonov A, Pers TH, Hirschhorn JN, Jansen RC, Schultes EA, et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat Genet. 2015;47:115–25.
Schilling EM, Scherer M, Rothemund F, Stamminger T. Functional regulation of the structure-specific endonuclease FEN1 by the human cytomegalovirus protein IE1 suggests a role for the re-initiation of stalled viral replication forks. PLoS Pathog. 2021;17:e1009460.
Kulkarni AS, Fortunato EA. Modulation of homology-directed repair in T98G glioblastoma cells due to interactions between wildtype p53, Rad51 and HCMV IE1-72. Viruses. 2014;6:968–85.
Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci U S A. 1995;92:2298–302.
Boichuk S, Hu L, Hein J, Gjoerup OV. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol. 2010;84:8007–20.
Kudoh A, Iwahori S, Sato Y, Nakayama S, Isomura H, Murata T, Tsurumi T. Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in Epstein-Barr virus replication compartments. J Virol. 2009;83:6641–51.
Murayama T, Kuno K, Jisaki F, Obuchi M, Sakamuro D, Furukawa T, Mukaida N, Matsushima K. Enhancement human cytomegalovirus replication in a human lung fibroblast cell line by interleukin-8. J Virol. 1994;68:7582–5.
Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–55.
Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18.
Halwachs-Baumann G, Weihrauch G, Gruber HJ, Desoye G, Sinzger C. hCMV induced IL-6 release in trophoblast and trophoblast like cells. J Clin Virol. 2006;37:91–7.
Rahbar A, Boström L, Lagerstedt U, Magnusson I, Söderberg-Naucler C, Sundqvist VA. Evidence of active cytomegalovirus infection and increased production of IL-6 in tissue specimens obtained from patients with inflammatory bowel diseases. Inflamm Bowel Dis. 2003;9:154–61.
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31.
Reddel RR. Senescence: an antiviral defense that is tumor suppressive? Carcinogenesis. 2010;31:19–26.
Paijo J, Döring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, Witte G, Messerle M, Hornung V, Kaever V, Kalinke U. cGAS senses human cytomegalovirus and induces type I Interferon responses in human monocyte-derived cells. PLoS Pathog. 2016;12:e1005546.
Huang ZF, Zou HM, Liao BW, Zhang HY, Yang Y, Fu YZ, Wang SY, Luo MH, Wang YY. Human cytomegalovirus protein UL31 inhibits DNA sensing of cGAS to Mediate Immune Evasion. Cell Host Microbe. 2018;24:69–80e64.
Fu YZ, Guo Y, Zou HM, Su S, Wang SY, Yang Q, Luo MH, Wang YY. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS Pathog. 2019;15:e1007691.
Biolatti M, Dell’Oste V, Pautasso S, Gugliesi F, von Einem J, Krapp C, Jakobsen MR, Borgogna C, Gariglio M, De Andrea M, Landolfo S. Human cytomegalovirus tegument protein pp65 (pUL83) dampens type I Interferon Production by inactivating the DNA sensor cGAS without affecting STING. J Virol 2018, 92.
Fu YZ, Su S, Gao YQ, Wang PP, Huang ZF, Hu MM, Luo WW, Li S, Luo MH, Wang YY, Shu HB. Human cytomegalovirus tegument protein UL82 inhibits STING-Mediated signaling to evade antiviral immunity. Cell Host Microbe. 2017;21:231–43.
Nukui M, Roche KL, Jia J, Fox PL, Murphy EA. Protein S-Nitrosylation of human cytomegalovirus pp71 inhibits its ability to limit STING antiviral responses. J Virol 2020, 94.
Zou HM, Huang ZF, Yang Y, Luo WW, Wang SY, Luo MH, Fu YZ, Wang YY. Human cytomegalovirus protein UL94 targets MITA to evade the antiviral Immune response. J Virol 2020, 94.
Choi HJ, Park A, Kang S, Lee E, Lee TA, Ra EA, Lee J, Lee S, Park B. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat Commun. 2018;9:125.
Kumari P, Saha I, Narayanan A, Narayanan S, Takaoka A, Kumar NS, Tailor P, Kumar H. Essential role of HCMV deubiquitinase in promoting oncogenesis by targeting anti-viral innate immune signaling pathways. Cell Death Dis. 2017;8:e3078.
Kim JE, Kim YE, Stinski MF, Ahn JH, Song YJ. Human cytomegalovirus IE2 86 kDa protein induces STING degradation and inhibits cGAMP-Mediated IFN-beta induction. Front Microbiol. 2017;8:1854.
Fabits M, Goncalves Magalhaes V, Chan B, Girault V, Elbasani E, Rossetti E, Saeland E, Messerle M, Pichlmair A, Lisnic VJ, Brinkmann MM. The Cytomegalovirus tegument protein UL35 antagonizes pattern recognition receptor-mediated type I IFN transcription. Microorganisms; 2020. p. 8.
Ren Y, Wang A, Wu D, Wang C, Huang M, Xiong X, Jin L, Zhou W, Qiu Y, Zhou X. Dual inhibition of innate immunity and apoptosis by human cytomegalovirus protein UL37x1 enables efficient virus replication. Nat Microbiol. 2022;7:1041–53.
Albright ER, Mickelson CK, Kalejta RF. Human cytomegalovirus UL138 protein inhibits the STING pathway and reduces Interferon Beta mRNA Accumulation during Lytic and latent infections. mBio. 2021;12:e0226721.
Bianco C, Mohr I. Restriction of human cytomegalovirus replication by ISG15, a host Effector regulated by cGAS-STING double-Stranded-DNA sensing. J Virol 2017, 91.
Hsieh TH, Tsai TT, Chen CL, Shen TJ, Jhan MK, Tseng PC, Lin CF. Senescence in Monocytes facilitates Dengue Virus infection by increasing infectivity. Front Cell Infect Microbiol. 2020;10:375.
Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell. 2006;17:1583–92.
Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, Guha M, Li N, Chen Q, Yang T, et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015;11:785–97.
Hasegawa T, Oka T, Son HG, Oliver-García VS, Azin M, Eisenhaure TM, Lieb DJ, Hacohen N, Demehri S. Cytotoxic CD4(+) T cells eliminate senescent cells by targeting cytomegalovirus antigen. Cell. 2023;186:1417–1431e1420.
Antonio, Pezone Fabiola, Olivieri Maria Vittoria, Napoli Antonio, Procopio Enrico Vittorio, Avvedimento Armando, Gabrielli Inflammation and DNA damage: cause effect or both Nature Reviews Rheumatology 2023;19(4):200–211. https://doi.org/10.1038/s41584-022-00905-1
Virus DNA Replication and the Host DNA Damage Response Annual Review of Virology 2018;5(1):141–164. https://doi.org/10.1146/virology2018.5.issue-1, https://doi.org/10.1146/annurev-virology-092917-043534.
Shmulevich R, Krizhanovsky V: Cell Senescence, DNA Damage, and Metabolism. Antioxid Redox Signal. 2021;34:324–334.
Funding
This work was supported by Chinese Traditional Medicine Science and Technology Projects of Zhejiang Province (GZY-ZJ-KJ-24055, 2018ZA004, 2018ZB002 and 2019ZB005), the Health Bureau of Zhejiang Province (2020KY387 and 2022KY009).
Author information
Authors and Affiliations
Contributions
MGX designed the study. WXN prepared figures. WXN and ZXQ wrote the original draft. MGX and WSY were responsible for review and editing of the final draft. MGX and WSY contributed to the project administration and supervision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved the manuscript for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, X., Zhou, X., Wang, S. et al. DNA damage response(DDR): a link between cellular senescence and human cytomegalovirus. Virol J 20, 250 (2023). https://doi.org/10.1186/s12985-023-02203-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-023-02203-y