
Abstract
Background
Echovirus 9 (E9) is associated with a wide variety of diseases and medical conditions, and the clinical symptoms of sporadic cases caused by E9 often are severe. With a high global prevalence, E9 has caused multiple outbreaks worldwide. However, little is known about the genetic and geographic population dynamics of E9.
Method
A total of 131 VP1 gene sequences, including15 generated in this study and 116 obtained from GenBank, were used to coestimate time-resolved phylogenies to infer viral evolution and transmission in worldwide. Overlapping fragments representing whole genomes were amplified by reverse transcription polymerase chain reaction (RT-PCR) using specific primers. Then, we reported the genetic characteristics of fifteen E9 strains in the Chinese Mainland. Similarity plots and bootscanning analysis were used to determine recombination patterns of E9.
Results
The estimated mean evolutionary rate of global E9 VP1 gene was 4.278 × 10−3 substitutions per site per year (95% confidence interval [CI], 3.822 × 10−3/site/year to 4.710 × 10−3/site/year), and the common ancestor of E9 likely emerged around 1868 (95% CI, 1840 to 1892). The full-length genomic sequences of the fifteen E9 strains showed 76.9–79.6% nucleotide identity and 95.3–95.9% amino acid identity with E9 Barty strain. 11 of 15 E9 whole genome sequence present four recombination patterns, and E9 recombinants have extensive genetic exchanges in the 2C and P3 regions with other Enterovirus B (EV-B) circulated in China. Four of six E9 strains were temperature sensitive, and two were temperature resistant, and a comparative genomics analysis suggested that 411, 865 and 867 amino acid substitution in the P1 region was related to temperature sensitivity.
Conclusion
This study highlights a persistent transmission network of E9 in worldwide, provides valuable information regarding the molecular epidemiology of E9.
Similar content being viewed by others


Background
E9, which belongs to the species EV-B of the Picornaviridae family, is a positive sense, single-stranded RNA virus [1]. E9 has been frequently associated with aseptic meningitis (AM) [2], hand, foot, and mouth disease (HFMD) [3], and acute flaccid paralysis (AFP) [4]. Less frequently, E9 is also associated with multiorgan infection that can rapidly lead to critical conditions, such as acute rhabdomyolysis [5], acute liver failure [6], acute renal failure [7], acute onset of type I diabetes mellitus [8]. Hence, E9 is associated with a wide variety of diseases and conditions, and the clinical symptoms of sporadic cases caused by E9 often are severe.
E9 has caused multiple outbreaks worldwide, including Asia (Japan [9] [10], China [11], et al.), Europe (Russia [12], England [13], Belgium [14], Spain [15]), Oceania (Australia [16]), South America (Brazil [17]), North America (Mexico [15], America [18]). The detection rate of E9 was found to be high in environmental monitoring [19]. Moreover, the susceptible population of E9 was preschool children [20], and nurseries are high-risk places for outbreaks [21], so the consequences are serious once an outbreak occurs. Therefore, it is necessary to strengthen the monitoring and research on E9, and guard against the re-outbreak of E9.
Despite its clinical impact and its high global prevalence, little is known about the genetic and geographic population dynamics of E9. Continuous molecular epidemiological surveillance is important to help identify newly emerging strains and to better understand trends in viral circulation [22]. In our previous study, we divided E9 into A-G genotypes and investigated the distribution of genotypes [23]. This study analyzed the population dynamics of E9 in viral evolution and transmission in global basing on the former study [24]. Recombination patterns of E9 were analysed in our study, and temperature sensitivity tests were used to determine the environmental transmission capacity.
Materials and methods
Ethics statement and sample collection
This study did not involve human participants or human experimentation. Only specimens (stool samples, throat swab samples) were collected from HFMD patients for public health purposes at the urging of the Ministry of Health, P. R. of China. Written informed consent for the use of their clinical samples was obtained from the parents of the children whose samples were analyzed. This study was approved by the second session of the Ethics Review Committee of the National Institute for Viral Disease Control and Prevention (NIVDC), Chinese Center for Disease Control and Prevention, all experimental protocols were approved by NIVDC, and the procedures were in compliant with the approved protocol.
Based on national HFMD surveillance program, clinical specimens (throat swabs, rectal swabs, herpes swabs, or stools) were collected from HFMD patients from thirty-one provinces or municipalities in mainland of China. All clinical specimens from HFMD patients were collected according to applicable standards and previously described protocols [25].
Virus isolation and molecular typing
Viruses were isolated from original clinical specimens by propagation in human rhabdo-myosarcoma (RD) and human larynx carcinoma (HEp-2) cells by conventional methods. The cell lines were provided by the WHO Global Poliovirus Specialized Laboratory in the USA and were originally purchased from the American Type Culture Collection (Manassas, VA, USA). After complete EV-like CPE were observed, we harvested the infected cell cultures.
Viral RNA was extracted from the cell culture using a QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany). RT-PCR was performed to amplify the partial VP1 coding region using the PrimeScript One Step RT-PCR Kit Ver.2 (TaKaRa, Dalian, China) with primers 490 and 492 (EV-B universal primers). The EV Genotyping Tool (a BLAST server) based on entire VP1 region was used for enterovirus serotyping. Finally, fifteen E9 strains were extracted from the specimen during the years 2013–2019 located in six provinces of China (Table 1) and include in this study.
Full-length genome sequencing
The full-length viral genome sequences were amplified by “primer-walking” strategy to close the gaps as necessary. Briefly, overlapping fragments representing whole genomes were amplified by RT-PCR using specific primers. The 5′end of the genome was amplified based on the manufacturer’s instruction with 5′-Full RACE Kit (Takara, Shiga, Japan). The 3′end of the genome was amplified using an oligo-dT primer (7500A) described in a previous study [26]. The primer pairs used for each step are listed in Table 2.
Phylodynamic and recombination analysis
131 entire VP1 sequences of E9 (including 116 E9 entire VP1 sequences downloaded from GenBank and 15 sequences obtained from this study) were used to construct Bayesian inference of phylogeny using Markov Chain Monte Carlo (MCMC). GTR (General Time Reversible) + G4 (Gamma distributed rate with four rate catagories) was selected as the best-fitting nucleotide substitution model calculated by JModeltest v2.0.1 software. Phylodynamic analyses were performed in the GTR + G model of nucleotide substitutions under the strict clock: Uncorrelated Log-normal setting for 80 million MCMC by the BEAST v1.8.4 software. After the maximum clade credibility tree had been constructed, convergence was assessed with effective sample size (ESS) values higher than 200 using TRACER v1.6 software. The analysis was sampled at every 10,000 states. Posterior probabilities were calculated with a burn-in of 10 million states. The analysis of collected data was conducted by Tracer v1.6, and Tree Annotator program was employed to output the results of the maximum clade credibility (MCC) tree model. FigTree program was then used to plot the MCC molecular evolutionary tree.
The P2 and P3 coding region sequences of the fifteen E9 strains were analyzed using the BLAST server to compare their identity with sequences from GenBank. Enterovirus genome sequences with a similarity higher than 85% were selected as potential recombination parents. Similarity plots and bootscanning analysis were performed by the SimPlot v3.5.1 with a 200-nucleotide window moving in 20-nucleotide steps. According to the distribution of informative sites, we determined recombination breakpoints.
Assay of temperature sensitivity
According to the spatiotemporal separation distribution and genotypes, we selected six representative E9 stains (SaX-2013-18, HeB-2013-65, HeN-2013-321, HuN-2014-103, JX-2019-806, X57) to test the temperature sensitivity of the E9 strains. The temperature sensitivity of six E9 strains and two selected control strains (HTYT-ARL-AFP02F/XJ/CHN/2011, showing no temperature sensitivity and KS-MGTH90F/XJ/CHN/2011, showing temperature sensitivity) were assayed on monolayer RD cells in 24-well plates. The 24-well plates were inoculated with 50μL of undiluted virus stock solution. The plates were incubated at different temperature conditions, i.e. the optimum temperature of 36 °C and the supra-optimal temperature of 39.5 °C for virus propagation. After adsorption at 36 °C or 39.5 °C for 1 h, the unabsorbed virus inoculum was removed and 100 μL of maintenance medium was added to each well. The plates were continuously incubated at 36 or 39.5 °C and harvested respectively at five time points after infection (4, 8, 16, 24, and 48 h). The CCID50 was calculated by end-point dilution method on monolayer RD cells in 96-well plates at 36 °C. Virus isolates showing more than 2-logarithm reduction in titer at different temperatures were considered temperature-sensitive.
Results
Origin and evolution analysis and population dynamics of E9
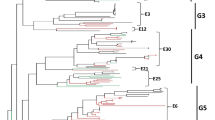
Bayesian phylogenetic analysis is to reconstruct time‐scaled phylogenies which can better reflect the actual virus origin and evolution. Bayesian phylogenetic analysis was performed based on E9 VP1 sequence alignment (N = 131). The results showed that the estimated mean evolutionary rate was 4.278 × 10−3 substitutions per site per year (95% CI, 3.822 × 10−3/site/year to 4.710 × 10−3/site/year). The common ancestor of E9 likely emerged around 1868 (95% CI, 1840 to 1892) (Fig. 1). Genotype A only includes the prototype Hill strain, whose origin can be traced back to 1895. After half a century of transmission, it was first isolated from the feces of healthy children in 1953. The origin of genotypes B, C, D, E and F can be traced back to 1953, they have the same ancestor with genotype A. It is speculated that genotypes B–F were evolved from genotype A. The origin of genotype C could date back to 1987. Significantly, all E9 strains isolated from Russia were clustered into genotype C, and the origin of these E9 strains could date back to 1989, which suggested E9 had been circulating in Russia for about 20 years before the outbreak of aseptic meningitis caused by E9 [12]. Genotype D was the most common genotype in worldwide, the origin of genotype D could date back to 1993. In 2003 and 2000, D2 and D3 sub-genotypes were formed respectively. D2 and D3 sub-genotypes are the dominant genotypes of E9 in Chinese mainland. In D2 sub-genotype, the Taiwan strain of E9 was firstly isolated in 2008, the rest were all isolated from Yunnan, China. It is suggested that D2 sub-genotype of E9 may be originated from Taiwan, China, and transmitted in Yunnan, China. Only one strain of E9 was isolated from Thailand, others were Chinese mainland strains in D3 sub-genotype. The time of origin of the D3 sub-genotype in mainland China was earlier than that in the Thailand strain, we speculated that the Thailand strain of E9 was imported from China and was not transmitted in Thailand. The origin of genotype E and F can be traced back to 2001 and 1992. Genotype G was formed by the evolution of ancient ancestors in 1995.

The MCC phylogenetic tree generated using the MCMC method based on the complete VP1 sequences of 121 E9 variants and the Chinese isolates was marked in red. The scale bar represents time in years. The tree was node-labelled with inferred dates of lineage splits. Each genotype is noted on the right
Bayesian skyline plot analyses were performed to reconstruct the past population history of E9 by measuring the dynamics of VP1 effective population size over time. There was no obvious amplification in the population of E9 before 1990s, which maintained at a stable level. A burst of the skyline plot was observed in 1990s-2010s, probably because of faster and increasing branch of evolution of E9 at that time which were accordance with repeated E9 outbreaks reports in that time [9, 12, 18]. And after 2010s, the effective population size is to a certain level and remained stable up to now (Fig. 2).

Bayesian Skyline Plots of E9
Full-length genome analysis of the fifteen Chinese E9 strains
The whole genome sequence of the fifteen Chinese E9 strains was determined to be 7454–7456nt long. The ORF of the fifteen Chinese E9 strains was 6609nt in length, encoding a polypeptide of 2203 amino acids, with a 741–743nt 5’-UTR and a 103-105nt 3’-UTR. The overall nucleotide composition of the fifteen strains was as follows: 27.8–28.5%A, 23.6–24.2%T, 23.0–23.6%C, and 24.2–25.0%G.
The prototype strain Hill of E9 (E9/Hill) was isolated from the rectal swab of a healthy child in 1953 and shown to be nonpathogenic in newborn mice. In contrast, strain Barty (E9/Barty) isolated from a child suffering from aseptic meningitis, are highly virulent in newborn mice [28]. Unlike Hill strain, Barty strain contains a C-terminal extension to the capsid protein VP1 with a argnine-glycine-aspartic acid (RGD) motif [29]. In previous studies, researcher considered that RGD motif was a significant factor affecting pathogenicity of E9 strains [28]. Alignment of the fifteen E9 strains in this study with strain Barty and the prototype Hill strain found that all isolates exhibited the VP1 extension with a RGD motif. We concluded that the pathogenicity of the fifteen E9 strains in this study was similar to that of strain Barty.
Then we aligned the fifteen E9 strains in this study with E9 Barty strains (Barty/X92886). The full-length genomic sequences of the fifteen E9 strains showed 76.9–79.6% nucleotide identity and 95.3–95.9% amino acid identity with E9 Barty strains. The nucleotide sequence and amino acid sequence similarities of the P1 region with Barty strains was 79.2–80.6% and 95.1–95.7%, respectively. The nucleotide sequence and amino acid sequence similarities of the fifteen E9 strains with Barty strains were 74.0–77.2% and 75.8–78.5%, 72.4–78.3% and 74.2–79.8% in the P2 and P3 region, respectively. However, fifteen E9 strains have higher homology with other EV-B prototype strains than E9, suggesting that recombination might occur in these coding regions.
Phylogenetic analysis of the fifteen E9 strains compared with other EV-B genomes
Maximum likelihood phylogenetic trees were constructed based on the VP1, P1, P2, and P3 coding region nucleotide sequences of the prototype sequence of all EV-B in the GenBank database and the fifteen Chinese E9 strains in this study (Fig. 3). The VP1 and P1 phylogenetic trees indicated that the fifteen E9 strains with the prototype of E9 (Barty strains) were clustered in single clade as expected. Unlike the VP1 and P1 phylogenetic trees, the phylogenetic trees based on the P2 and P3 coding regions showed that the fifteen E9 strains did not cluster in single clade and shared higher similarity with the prototype sequence of other EV-B strains than Barty strains. In P2 coding region, JX19-806 shared the highest similarity with the prototype sequence of E1. In P3 coding region, HeB13-65, TJ16-30, HuN14-103, SaX13-18, X57 and SaX13-97 were clustered with EV86, and JX19-806 was clustered with E1, EV-B84, EV101. These results suggest that the fifteen Chinese E9 strains may experience different recombination patterns in the evolutionary process.

Maximum likelihood phylogenetic trees were illustrated in (a–d) based on the VP1, P1, P2, and P3 coding regions of the prototype sequence of all EV-B in the GenBank database and the fifteen Chinese E9 strains in this study. Strains isolated from this study were marked with black circles. The scale bars indicate the substitution per site per year
Four recombination patterns of the fifteen E9 strains
Potential evidence of recombination in the genome of the fifteen E9 strains was investigated via similarity plot and boot scanning analysis. The fifteen E9 strains had no significant recombination with prototype strains of other EV-B. Then, the P2 and P3 regions of the fifteen E9 strains were used to screen closely sequences available which have a strong possibility to occur recombination event in GenBank using BLAST from NCBI. All closely sequences which showing over 85% similarity with the query sequence were downloaded from GenBank.
Similarity plot and boot scanning analysis showed that the other 11 E9 strains had recombined except for HeN-2013-44, HeN-2013-91, HeN-2013-321 and JX-2019-806. We found four recombination patterns of the fifteen E9 strains named respectively RF1-RF4 via. To definitively illustrate the recombination, four groups were defined as A-D groups respectively based on the recombination forms RF1-RF4 used as the query sequence.
In the 5’-UTR and P1 region, all the fifteen E9 strains shared the highest similarity with the E9 strain isolated from Yunnan of China in 2010(KM812-JN596587). However, in the 2C-3D coding region, the query strains shared the highest identity with other serotype strains. In the 2C coding region, RF1 possibly recombined with the CVB5 strain (P727/CHN/2013/KP289438), RF2 and RF3 recombined with the E3 strain (123R2/CHN/2018/MK791150) with a high supporting value, RF4 possibly recombined with the EV-B86 strain (BAN00-10,354/USA/AY843304). In the P3 regions, RF1 and RF4 recombined with the CVB5 strain (P727/CHN/2013/KP289438) with a higher probability. RF2 and RF3 recombined with the E3 (123R2/CHN/2018/MK791150) strain in the 3A-3C regions and recombined with E11(520 k/CHN/YN/2010/KP294524) and EV-B106(KS-MGTH90F/CHN/XJ/2011/KX171337) respectively in the 3D region (Fig. 4).

Similarity plot and bootscanning analysis of 4 types of recombination forms of the fifteen E9 strains
Environmental tolerance of the six China E9 strains
We selected six E9 representative stains from the fifteen E9 according to the Spatiotemporal separation distribution and genotypes to test the tolerance to the environment (Fig. 5). The results indicated that four strains (SaX-2013-18, HeB-2013-65, HeN-2013-321 and HuN-2014-103) were temperature sensitive and two others (JX19-806 and X57) were temperature resistant. The results showed that JX19-806 and X57 had higher tolerance to temperature compared to other strains.

Temperature sensitivity test curves of the six representative Chinese E9 strains. Blue and orange lines represent the growth trends of the viruses on RD cells at 36 ℃ and 39.5 ℃, respectively. The Xinjiang EV-B85 strain (HTYT-ARL-AFP02F/XJ/CHN/2011, showing no temperature sensitivity) and the EV-B106 strain (KS-MGTH90F/XJ/CHN/2011, showing temperature sensitivity) were used as experimental controls
The P1 region encodes the structural protein (vpl—vp4) of the virus and constitutes the capsid of the virus. In previous studies, capsid genes were shown to involve in determining the optimal growth temperature [30]. To investigate the possible mechanism underlying the difference in temperature sensitivity among these strains, we aligned nucleotide and amino acid substitutions in the P1 region. A total of 40 amino acid substitutions in the P1 region were summarized in Table 3. Comparison of amino acid substitutions showed that the temperature resistant strains have more amino acid substitutions than the temperature sensitive strains. Significantly, the amino acids at position 411, 865 and 867 in the temperature resistant strains had same mutations, whereas in the three temperature sensitive strains, the amino acid was same.
Discussion
E9 virus has been circulating in worldwide for half a century, which endangers human health and increases the burden of disease. However, the molecular epidemiological study of E9 is very limited. We analyzed all available E9 sequence data in the word, including fifteen E9 genome-wide data obtained from the mainland of China in 2013–2019 in this study. Analysis of the origin and evolution of E9 indicated that the estimated mean evolutionary rate was 4.278 × 10−3 substitutions per site per year. In previous study, the mean evolutionary rate of EV71 was estimated to be 4.6 × 10−3 substitutions per site per year during its propagation process [31]. The mean evolutionary rate of E9 in worldwide was approximate to that of EV71, which hints the evolution of E9 is active. A burst of effective population size was observed in 1990s–2010s, then the effective population size is to a certain higher level than before and remained stable. Hence, we should be alert to the outbreak of E9 in the future.
D genotype of E9 is the dominant gene in Chinese mainland. The earliest strain of E9 D2 sub-genotype was isolated from Taiwan, China, we speculate that D2 sub-genotype of E9 circulating in mainland of Chinese was imported from Taiwan. Except for one strain isolated from Thailand, the remaining D3 sub-genotype of E9 was isolated from mainland of Chinese, we speculate that the Thailand strain of E9 was imported from mainland of Chinese. JX19-806 was the only C2 sub-genotype strain isolated from mainland of Chinese and its nucleotide sequence shows the highest similarities with the French strain isolated in 2012, which hints the E9 transmission along the border. As a lesson learned from these cases, in addition to the crucial route, domestic transmission, we should pay attention to the prevention of epidemics along the border.
Recombination is an important form of the evolution of enterovirus, and P2 and P3 were the main regions of recombination [32]. Analysis of the complete genome sequence of the fifteen E9 sequence data shows that E9 circulated in mainland of China had four recombination patterns. Recombination forms have no obvious time and regional characteristics. Recombination of E9 is characterized by universality and polymorphism.
It is very interesting that, among the six tested E9 strains, four (SaX-13-18, HeB-13-65, HeN-13-321 and HuN-14-103) were temperature sensitive and the other two (JX19-806 and X57) were temperature resistant. We aligned nucleotide and amino acid substitutions in the P1 region. According to the comparison of amino acid substitutions, we speculate that the amino acid substitutions at 411, 865 and 867 positions are related to changes in temperature-sensitivity. However, additional evidence is needed to support this hypothesis.
In conclusion, we analyzed the origin and evolution of E9 in worldwide. It is indicated that more attention should be paid for the prevention of epidemics along the border and the outbreak of E9 in the future. We reported the full-length genome sequences of fifteen E9 strains isolated from the mainland of China in 2013–2019. Sequence analysis suggested that these fifteen E9 strains have high genetic diversity compared with Barty strain, suggesting recombination within the non-structural protein-encoding region, and extensive genetic exchange with other EV-B serotypes, such as CVB5, E3, E11, EV-B106, and EV-B86. Some of the isolated E9 strains were temperature resistant. Hence, E9 has the potential to become a more common strain. This study provides valuable information regarding the molecular epidemiology of E9.
Although this study has included all E9 available sequences worldwide, this study is still lack of sequences isolated in certain regions and time periods. This may bias the bioinformatics analysis. With the coverage of the E9 sequence, the bioinformatics analysis will include sequences with a larger time and regional distribution span, and the results will be more accurate and representative.
Conclusion
This study highlights a persistent transmission network of E9 in worldwide, provides valuable information regarding the molecular epidemiology of E9.
Availability of data and materials
Condensed anonymized data are available from the corresponding author on reasonable request.
Abbreviations
- E9:
-
Echovirus 9
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- CI:
-
Confidence interval
- EV-B:
-
Enterovirus B
- AM:
-
Aseptic meningitis
- HFMD:
-
Hand, foot, and mouth disease
- AFP:
-
Acute flaccid paralysis
- NIVDC:
-
National Institute for Viral Disease Control and Prevention
- RD:
-
Rhabdo-myosarcoma
- Hep-2:
-
Human larynx carcinoma
- MCMC:
-
Markov Chain Monte Carlo
- GTR:
-
General Time Reversible
- G4:
-
Gamma distributed rate with four rate catagories
- ESS:
-
Effective sample size
- MCC:
-
Maximum clade credibility
- RGD:
-
Argnine-glycine-aspartic acid
References
Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Dempsey DM, Dutilh BE, et al. Changes to virus taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2019). Arch Virol. 2019;164(9):2417–29.
Takeshima S, Yoshimoto T, Shiga Y, Kanaya Y, Neshige S, Himeno T, et al. Clinical, epidemiological and etiological studies of adult aseptic meningitis: report of 13 cases with mumps meningitis. Rinsho Shinkeigaku. 2015;55(9):630–6.
Ma S, Zhang Y, Du C, Yang T, Liu Q, Pan Y, et al. Dynamic Constitution of the pathogens inducing encephalitis in hand, foot and mouth disease in kunming, 2009–2011. Jpn J Infect Dis. 2015;68(6):504–10.
Shaukat S, Angez M, Alam MM, Jebbink MF, Deijs M, Canuti M, et al. Identification and characterization of unrecognized viruses in stool samples of non-polio acute flaccid paralysis children by simplified VIDISCA. Virol J. 2014;11(1):146.
Josselson J, Pula T, Sadler JH. Acute rhabdomyolysis associated with an echovirus 9 infection. Arch Intern Med. 1980;140(12):1671–2.
Bajema KL, Simonson PD, Greninger AL, Çoruh B, Pottinger PS, Bhattacharya R, et al. Acute liver failure due to echovirus 9 associated with persistent B-Cell depletion from rituximab. Open Forum Infect Dis. 2017;4(3):ofx174.
Tanaka T, Takada T, Takagi D, Takeyama N, Kitazawa Y. Acute renal failure due to rhabdomyolysis associated with echovirus 9 infection: a case report and review of literature. Jpn J Med. 1989;28(2):237–42.
Vreugdenhil GR, Schloot NC, Hoorens A, Rongen C, Pipeleers DG, Melchers WJ, et al. Acute onset of type I diabetes mellitus after severe echovirus 9 infection: putative pathogenic pathways. Clin Infect Dis. 2000;31(4):1025–31.
Gondo K, Kusuhara K, Take H, Ueda K. Echovirus type 9 epidemic in Kagoshima, southern Japan: seroepidemiology and clinical observation of aseptic meningitis. Pediatr Infect Dis J. 1995;14(9):787–91.
Akasu Y. Outbreak of aseptic meningitis due to ECHO-9 in northern Kyushu island in the summer of 1997. Kurume Med J. 1999;46(2):97–104.
Ma YL, He K, Li QF, Zhao LJ. Investigation of first echo 9 enterovirus encephalitis outbreak in Baoshan. Yunnan CTM. 2018;18(1):69–71.
Golitsyna LN, Fomina SG, Novikova NA, Epifanova NV, Parfenova OV, Lukovnikova LB, et al. Molecular genetic echovirus 9 variants identified in patients with aseptic meningitis in Russia in 2007–2009. Vopr Virusol. 2011;56(6):37–42.
Hobson D, Hoskins JM, Horner J, Clarke AH, Wood FB. Clinical, epidemiological, and virological aspects of echovirus type 9 infection in Sheffield, 1960. Br J Prev Soc Med. 1962;16:84–92.
Nihoul E, Quersin-Thiry L, Weynants A. ECHO virus type 9 as the agent responsible for an important outbreak of aseptic meningitis in Belgium. Am J Hyg. 1957;66(1):102–18.
Foreman RE, Gutierrez A. Aseptic meningitis associated with echoviral type 6 and 9 infections in Carlsbad, New Mexico. A report on nine patients. Rocky Mt Med J. 1978;75(4):209–13.
Ashwell MJ, Smith DW, Phillips PA, Rouse IL. Viral meningitis due to echovirus types 6 and 9: epidemiological data from Western Australia. Epidemiol Infect. 1996;117(3):507–12.
Nogueira RM, Nascimento JP, Alencar A. Pathogenicity, for newborn mice, of echovirus type 9 samples isolated from cases of meningitis during an outbreak in Rio de Janeiro. Mem Inst Oswaldo Cruz. 1983;78(2):193–8.
Centers for Disease Control and Prevention (CDC). Outbreaks of aseptic meningitis associated with echoviruses 9 and 30 and preliminary surveillance reports on enterovirus activity—United States, 2013. MMWR Morb Mortal Wkly Rep. 2003;52(32):761–4.
Tiwari S, Dhole TN. Assessment of enteroviruses from sewage water and clinical samples during eradication phase of polio in North India. Virol J. 2018;15(1):157.
Horbowska H, Grodzicka-Królak H, Przestalska-Malkin H, Wielopolska H. Echo 9 type virus meningitis epidemics in preschool children. Pediatr Pol. 1978;53(6):753–7.
Toth M, Osvath P, Galambos M, Voltay B. Kindergarten outbreak of an exanthematous disease caused by echovirus type 9. Acta Paediatr Acad Sci Hung. 1964;5:235–9.
Lema C, Torres C, Van der Sanden S, Cisterna D, Freire MC, Gómez RM. Global phylodynamics of echovirus 30 revealed differential behavior among viral lineages. Virology. 2019;531:79–92.
Si FF, Wei HY, Guan LY, Ji TJ, Wang DY, Zhang Y, Zhu SL, Song Y, Han ZZ, Li JH, Xu WB, Yan DM. Molecular epidemiological study of ECHO-9 associated with hand, foot, and mouth disease in China, 2013–2019. Chin J Virol. 2021;37(1):115–22.
Grenfell BT, Pybus OG, Gog JR, Wood JLN, Daly JM, Mumford JA, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303(5656):327–32.
Xu W, Zhang Y. Isolation and characterization of vaccine-derived polioviruses, relevance for the global polio eradication initiative. Methods Mol Biol. 2016;1387:213–26.
Lo S-H, Huang Y-C, Huang C-G, Tsao K-C, Li W-C, Hsieh Y-C, et al. Clinical and epidemiologic features of Coxsackievirus A6 infection in children in northern Taiwan between 2004 and 2009. J Microbiol Immunol Infect. 2011;44(4):252–7.
Zhu YJ, Pan Y, Chen JY, Liu YL, Shi HJ, Liao HW, et al. Complete genome sequence characteristics of human echovirus 9 strain isolated in Yunnan China. Chin J Virol. 2013;29(002):132–6.
Zimmermann H, Eggers HJ, Nelsen-Salz B. Cell attachment and mouse virulence of echovirus 9 correlate with an RGD motif in the capsid protein VP1. Virology. 1997;233(1):149–56.
Jung YT, Kim GR, Paik SY. Molecular biological characterization of enterovirus variant isolated from patients with aseptic meningitis. Exp Mol Med. 1998;30(2):101–7.
Royston L, Essaidi-Laziosi M, Pérez-Rodríguez FJ, Piuz I, Geiser J, Krause K-H, et al. Viral chimeras decrypt the role of enterovirus capsid proteins in viral tropism, acid sensitivity and optimal growth temperature. PLoS Pathog. 2018;14(4):e1006962.
Tan X, Huang X, Zhu S, Chen H, Yu Q, Wang H, et al. The persistent circulation of enterovirus 71 in People’s Republic of China: causing emerging nationwide epidemics since 2008. PLoS ONE. 2011;6(9):e25662.
Kyriakopoulou Z, Pliaka V, Amoutzias GD, Markoulatos P. Recombination among human non-polio enteroviruses: implications for epidemiology and evolution. Virus Genes. 2015;50(2):177–88.
Acknowledgements
We thank the National Institute for Viral Disease Control and Prevention, for the unidentifed isolate from AFP surveillance. We also thank the support of provincial disease control and prevention center.
Funding
This study was supported by the National Key Technology R&D Program of China (Project No. 2021YFF0703800). We also acknowledge the funding received from National key R & D plan (2021YFC0863000), and the Municipal Natural Science Foundation of Beijing of China (L192014). The funding body was not involved in the study design, clinical sample collection, data analysis, and interpretation or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization, FFS, DMY; Formal analysis, FFS, TJJ, DYW, YZ, SLZ, JHL, WBX, DMY; Writing—original draft preparation, FFS; Writing—review and editing, DMY; Project administration, DMY; Funding acquisition, DMY. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethical standards
Animal and Human Rights Statement This study did not involve human experimentation. Biosafety evaluations were approved by the National Institute for Viral Disease Control and Prevention, China CDC. All experimental operations were handled following the Standard Operational Protocol approved by China CDC.
Consent for publication
Written informed consent for specimen collection, testing and publication was obtained from all individuals (or their parents) in the study population.
Competing interests
The authors declare that no competing interests exist.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Si, F., Ji, T., Wang, D. et al. Origin and evolution analysis and genetic characteristics of echovirus 9 in China. Virol J 19, 98 (2022). https://doi.org/10.1186/s12985-022-01820-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-022-01820-3