
Abstract
The emergence of a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and more recently, the independent evolution of multiple SARS-CoV-2 variants has generated renewed interest in virus evolution and cross-species transmission. While all known human coronaviruses (HCoVs) are speculated to have originated in animals, very little is known about their evolutionary history and factors that enable some CoVs to co-exist with humans as low pathogenic and endemic infections (HCoV-229E, HCoV-NL63, HCoV-OC43, HCoV-HKU1), while others, such as SARS-CoV, MERS-CoV and SARS-CoV-2 have evolved to cause severe disease. In this review, we highlight the origins of all known HCoVs and map positively selected for mutations within HCoV proteins to discuss the evolutionary trajectory of SARS-CoV-2. Furthermore, we discuss emerging mutations within SARS-CoV-2 and variants of concern (VOC), along with highlighting the demonstrated or speculated impact of these mutations on virus transmission, pathogenicity, and neutralization by natural or vaccine-mediated immunity.
Similar content being viewed by others

Background
Coronaviruses (CoVs) can infect humans and animals to cause mild to severe disease, including death [1]. CoVs are divided into four genera: alpha- and beta-CoVs predominantly originate in bats and infect other mammals, while gamma- and delta-CoVs originate in and largely infect avian species [2]. CoV infection in animals is generally associated with gastric symptoms [3], such as acute diarrhea in young pigs that are infected with porcine epidemic diarrhea virus (PEDV) and swine acute diarrhea syndrome coronavirus (SADS-CoV) [4, 5]. While CoVs mainly circulate in animals, such as pigs, camels, cats, and bats [6], there have been at least seven documented instances where these viruses have spilled over into humans [7]. These events have led to the emergence of human coronaviruses (HCoVs) that are low and high pathogenic. The origin of the most recently emerged human coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is speculated to be associated with Rhinolophus bats, but the zoonotic transmission pathway remains unknown.
HCoV-229E, HCoV-OC43, HCoV-NL63 and HCoV-HKU1 represent endemic and low pathogenic HCoVs, and are responsible for one-third of common cold symptoms [8]. High pathogenic HCoVs such as severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2 cause or have caused severe disease in humans with case-fatality rates of 10.9%, 34.3%, and 2.1%, respectively [9,10,11]. SARS-CoV, MERS-CoV and SARS-CoV-2 are beta-CoVs [12, 13]. MERS-CoV belongs to the Merbecovirus subgenus, while SARS-CoV and SARS-CoV-2 belong to the SARS-related coronavirus (SARSr-CoV) species within the Sarbecovirus subgenus [14]. It remains unclear why most HCoVs evolved to largely cause minor illness while MERS-CoV continues to cause severe disease [15,16,17]. In this review, we have highlighted the origins of HCoVs and mapped positively selected for mutations within HCoV proteins to discuss the evolutionary trajectory of SARS-CoV-2. We have also discussed emerging mutations within SARS-CoV-2 and variants of concern (VOC), along with highlighting the demonstrated or speculated impact of these mutations on virus transmission, pathogenicity, and neutralization by natural or vaccine-mediated immunity.
Origin of human coronaviruses
All known HCoVs are speculated to have an evolutionary origin in bats or rodents [1, 3, 18] (Fig. 1), with five of seven HCoVs originating in bats [3, 19,20,21] (Table 1). Bats are speculated to be primordial hosts for all CoV lineages due to ubiquitous detection of diverse CoVs and constant CoV population growth, which contrasts epidemic-like growths observed in other animals [22]. Although bats and alpacas can serve as MERS-CoV reservoirs [23, 24], dromedary camels are the major reservoir host and primary contributor to human infections [25,26,27,28] (Fig. 1). The full extent of wildlife or intermediate animal reservoirs of SARS-CoV-2 is currently unknown.

Speculated animal origins of known human coronaviruses. HCoV species are organized chronologically (top to bottom) by their speculated dates of spill over into humans. Intermediate hosts (top to bottom) shown are alpacas, cattle, civet cats, dromedary camels, pangolins, and unknown (denoted as a question mark). Genome similarity to humans (A) indicates percentage similarity of CoV genomes detected in reservoir species with corresponding human CoV. Genome similarity to humans (B) indicates percentage similarity of CoV genomes detected in intermediate species with corresponding human CoV. Non-human CoVs that are highly pathogenic in animals, such as PEDV and SADS-CoV, are not shown here. Genomic percentage similarities were extracted from existing primary studies [20, 21, 32, 56, 60, 277,278,279,280,281,282,283]
SARS-CoV-2 is believed to have originated in a seafood market in Wuhan, Hubei Province, China [29], although limited contact-tracing at the beginning of the pandemic does not allow for definitive characterization of the exact events that led to the first human-to-human transmission, including the index patient or initial animal contact. Nonetheless, it is speculated that the natural reservoirs of SARS-CoV-2 are Rhinolophus bats (Table 1) since diverse SARSr-CoVs have been detected in multiple Rhinolophus species [22, 30, 31], including RaTG13 in R. affinis [32]. RaTG13 is 96.2% identical to SARS-CoV-2 at the whole genome level [32]. Moreover, SARS-CoV-2 contains a polybasic furin-like cleavage site between S1 and S2 spike (S) protein subunits, similar to Rhinolophus CoV RmYN02 [33, 34], which shares 93.3% whole genome nucleotide identity with SARS-CoV-2 [34]. However, the receptor binding domain (RBD) of SARS-CoV-2 is only 85% and 61.3% identical to those of RaTG13 and RmYN02, respectively [34,35,36]. RaTG13 and RmYN02 were discovered in bats of China’s Yunnan province, over 1500 km away from Wuhan [34, 35]; however, this does not preclude the possibility of virus spill over as bats can fly long distances. Virus transmission and transport by susceptible intermediate reservoirs or humans is also possible.
Phylogenetic analyses have identified a possible recombination-mediated origin for SARS-CoV-2 [37,38,39]. Neutralizing antibodies to SARS-CoV and SARS-CoV-2 have been detected in Malayan pangolins (Manis javanica), suggesting that SARSr-CoVs have been circulating in pangolins since 2003 [40]. Recombination of CoVs within Malayan pangolins has been suggested given the 97.4% amino acid similarity within the RBDs of pangolin SARSr-CoVs and SARS-CoV-2 [35, 41], including conservation of all critical residues required for successful human ACE2 (hACE2)-mediated cellular entry [35, 39, 41, 42] and the detection of pangolin SARSr-CoVs that bind to hACE2 [43]. Additionally, bats and pangolins may share underground caves [44], facilitating ecological contact in high density areas. However, the lack of robust evidence of direct SARS-CoV-2 emergence from a pangolin CoV precursor [45], along with the reported high pathogenicity of SARSr-CoVs in infected pangolins [41, 42, 45] makes it unlikely that pangolins are intermediate reservoirs of SARSr-CoVs.
The nucleotide percentage similarity of CoVs detected in reservoir species is generally lower than CoVs detected in intermediate species. Adaptive evolution of CoVs in intermediate species facilitates successful spill over into humans (Fig. 1). Since SARS-CoV-2 is more closely related to bat SARSr-CoVs than to pangolin SARSr-CoVs (Fig. 1), it seems unlikely that pangolins are intermediate hosts, unless we haven’t yet detected the full range of SARSr-CoVs in pangolins. It is uncertain whether an unknown intermediate host provided an opportunistic amplifying role or a stable reservoir for the zoonotic transmission of SARS-CoV-2.
While direct human infection with bat SARSr-CoVs has not been reported [46], it is possible that the majority of adaptive evolution of SARSr-CoVs occurs in bats, prior to spill over into humans [47]. Some notable adaptations include carrying the lowest level of CpG dinucleotides among known beta-CoV genomes [48], similar to a mechanism of escaping innate immunity observed in camel MERS-related CoVs strains [49, 50]. The relatively few SARSr-CoVs detected in the Hubei Province [35] are phylogenetically distant from SARS-CoV-2 [51]. Indeed, if SARS-CoV-2 did transmit from animals to humans, further sampling in Hubei Province may identify more closely related SARSr-CoVs in archived animal specimens. Investigating the possibility of an infected person travelling to Wuhan and unwittingly spreading the virus will be more difficult in the absence of archived samples and records of travel history.
Despite the abundance of SARSr-CoVs and beta-CoVs in bat species [52, 53], it is likely that additional reservoirs and intermediate hosts remain undetected [54]. Pigs, alpacas, and dromedary camels also maintain a variety of CoVs with the potential to transmit to humans [3, 12, 20, 55,56,57]. Independent insertions within RBDs of SARS-CoV, MERS-CoV, and SARS-CoV-2 suggest convergent evolution, which will likely lead to emergence of more pathogenic HCoVs [58]. Further sampling of bats, pangolins, and other species that share an ecological niche with bats may help piece together the puzzle surrounding the spill over of SARS-CoV-2 into humans [59] and also help discover other CoVs with potential to infect humans.
Aside from consistent spill over of MERS-CoV from camels [60], HCoVs have emerged through limited spill over events, followed by human-to-human transmission [3, 61]. While challenging to predict, future spill over events are likely, due to the long history of CoV host shifting [62,63,64,65]. Anthropogenic factors such as urbanization and deforestation increase habitat overlap of humans and animals, providing increased zoonotic transmission opportunities [57, 66]. Areas of high contact between humans, wildlife, and domesticated animals, such as live animal wet markets provide opportunity for viral recombination and adaptation to a broader range of animal species prior to transmission to humans [57]. Identifying existing CoV diversity in such areas will enhance our understanding of ecological opportunities for zoonosis and will help us better predict and prevent the emergence of future HCoVs.
Evolution of SARS-CoV-2 and its variants
Co-evolution of CoVs with their hosts is driven by genetic diversity that is selected through evolutionary pressures. CoV genetic diversity is made possible by a large genome (26.4–31.7 kb) [67], high mutation rate due to a low fidelity viral polymerase (~ 10–4 substitutions per site per year) [68, 69], and high recombination frequency (up to 25% for the entire genome in vivo) [70, 71]. Mutations that confer greater fitness are selected for, leading to antigenic drift. Ratios of the rates of non-synonymous/synonymous mutations (dN/dS) greater than one, less than one and equal to one indicate positive selection, negative (purifying) selection and neutral evolution, respectively [72]. SARS-CoV-2 genomes are currently under purifying selection [73, 74]. Despite observing little viral diversity at the beginning of the COVID-19 pandemic [75, 76], positive selection with presumed advantages such as increased transmission rates has now been documented [77,78,79] (Fig. 2, Table 2). However, functional characterization of these mutations remains under-investigated.

Mutations identified in human coronaviruses. Red dots within the genomes correspond to specific amino acid residues that have been strongly positively selected for such that a specific mutation has become dominant in the region where it emerged [74, 78, 83,84,85,86,87,88,89,90,91, 94,95,96, 99,100,101, 104, 111, 116, 117, 121, 123,124,125, 129, 131, 132, 135, 138,139,140, 146, 151,152,153,154, 158, 162, 278, 284,285,286,287,288,289,290,291,292,293]. Genomic regions highlighted by red bars correspond to deletions that have been selected for, while purple bars correspond to regions with significant polymorphisms within a CoV species. Beta-CoV Lineage B (Sarbecovirus) is represented within the blue shaded area, beta-CoV Lineage C (Merbecovirus) is represented within the yellow shaded area, beta-CoV Lineage A (Embecovirus) is represented within the red shaded area, and alpha-CoVs are represented within the green shaded area. Genome length in kilobases (kb) is noted on top. See Table 2 for more details
Antigenic drift is most frequently observed in viral surface proteins that are highly exposed to selection pressures of the immune system, such as neutralizing antibodies [80]. Indeed, CoV spike genes, particularly the S1 and RBD coding regions, have the highest detected non-synonymous mutation rates [81, 82], a trend observed across the majority of HCoVs (Fig. 2). For low pathogenic and endemic HCoVs, multiple positively selected for residues and polymorphic sites are found in the N-terminal domain (NTD) of S [83,84,85,86,87,88]. A notable exception is HCoV-HKU1, for which there is a shortage of sequencing data outside of the hemagglutinin esterase (HE) gene. Emerging data suggest that positively selected for and homoplastic sites have been observed within the SARS-CoV-2 NTD as well [78, 89,90,91]. Given the observations with other HCoVs (Fig. 2) and the detection of neutralizing epitopes within the SARS-CoV-2 NTD [91, 92], we speculate that with continued circulation, vaccination and convalescent sera therapy, further positively selected for mutations in the NTD are likely to occur. Further retrospective research on the evolution of endemic HCoVs may help predict the likely evolutionary trajectory of SARS-CoV-2.
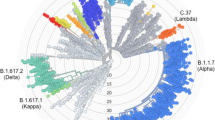
CoV genomic mutations give rise to virus variants, and closely related variants are grouped into clades. SARS-CoV-2 variants have been clustered into nine clades: L, V, S, G, GH, GR, GV, GRY and O [93, 94] (Table 3), named after their most representative mutations [95]. Clade L dominated the beginning of the pandemic [38], prior to the appearances of clade S and the less defined clade O in early January, 2020 [73, 93, 96]. Clades V and G appeared in mid-January, followed by clades GH and GR at the end of February, clade GV at the end of June, and clade GRY in September, 2020 [94, 97, 98]. Clades L and V are likely extinct, while clades G, GH, GR, and GRY comprise the majority of global SARS-CoV-2 sequences currently [97, 98]. Clade S has also been declining since the emergence of clade G [93]. Following rapid dissemination of clade G and its derivatives, such as B.1.1.7, B.1.351, P.1, and B.1.617.2 variants (Table 5), we may see the rise of other variants, selected by mounting population-level immunity and other yet unidentified factors [89, 99,100,101], highlighting the need for international genome surveillance efforts and global data sharing via the established GISAID resource [102].
Clade G is characterized in part by the single nucleotide polymorphism (SNP) A23403G within subdomain 2 of the S1 gene, resulting in amino acid mutation D614G [103, 104] (Fig. 2, Table 2). D614G is now detected globally in B.1.1.7, B,1,351, P.1, B.1.617.2 and other variants [97, 104, 105] and increases the infectivity of SARS-CoV-2 by increasing respiratory viral load [106, 107], possibly due to increased S openness [108, 109] or cleavability [110], causing this mutation to become dominant upon emergence [93, 111, 112]. There is also an epidemiological correlation between D614G and anosmia (loss of smell) [109], potentially due to greater viral loads in the olfactory epithelium. Preliminary evidence suggests that D614G increases viral susceptibility to neutralization [113], with uncertain impacts on disease severity [104, 114, 115].
D614G is usually accompanied by three other mutations which represent clade G [104, 116, 117] (Table 3). Of these mutations, P323L in the RNA-dependent RNA polymerase (RdRp), encoded by Nsp12 (Fig. 2, Table 2), is particularly interesting as CoV RdRp tends to be highly conserved by purifying selection given its critical role in viral genome replication [118, 119] (Table 4). P323 falls outside of the RdRp catalytic site and within a relatively uncharacterized interface domain that may interact with proteins that regulate viral polymerase function [120]. The correlation of this mutation with increased point mutations [121] elsewhere in the genome raises an intriguing hypothesis that P323L diminishes RdRp proofreading ability, leading to increased mutation rates. Moreover, P323L downregulates the association of Nsp12 with the Nsp8 primase subunit (Table 4), reducing polymerase activity and viral replication [122]. Decreased replication could decrease symptomology, leading to reduced COVID-19 detection and greater population-level spread. It is important to characterize the cumulative effect of all mutations, as any reduction in transmission due to P323L could be compensated for by the co-existing D614G mutation. Multiple factors may contribute to the success of clade G and its derivatives via rapid spread with low detection in human populations [104].
Positively selected for residues within SARS-CoV-2 Nsp6 [74, 123,124,125,126] are intriguing since Nsp6 is relatively conserved in other coronaviruses [126] (Fig. 2, Table 2). SARS-CoV-2 Nsp6 inhibits IFN-1 responses [127] and may reduce delivery of viral factors to host lysosomes similar to its SARS-CoV ortholog [128] (Table 4). The Nsp6 L37F mutation may impair Nsp6 function [129], decreasing viral replication and causing increased asymptomatic infections [130]. A similar homoplasy occurs in MERS-CoV Nsp6 [74, 131] (Fig. 2), although the outcome of this mutation is unknown. The associated clade V mutation (Table 3) in ORF3a (G251V) reduces viral replication through decreased SARS-CoV-2 intraviral ORF3a-S and ORF3a-membrane protein (M) binding affinity [132]. Nsp6 (L37F) and ORF3a (G251V) mutations were likely selected to decrease pathogenicity and disease severity. A separate positively selected ORF3a mutation (Q57H) [111] characteristic of clade GH variants (Table 3) is speculated to increase ORF3a-S and ORF3a-M binding affinity, promoting virus replication [132]. The ORF3a viroporin is essential for SARS-CoV-2 pathogenesis [133] and limits apoptosis in infected cells relative to its SARS-CoV ortholog [134], potentially contributing to less severe disease outcomes.
Another mutation of interest (L84S) lies within ORF8 [123, 124, 135], a protein implicated in evasion of host immune responses [136, 137] (Table 4). ORF8 was under strong directional selection at the beginning of both SARS-CoV-2 [124] and SARS-CoV outbreaks [138], supporting the theory that it facilitates zoonotic transmission and adaptation in alternate hosts [139, 140]. However, the over-representation of ORF8 deletions in SARS-CoV with no apparent effect on viral survival [138] suggests that ORF8 may be dispensable in humans [139], and L84S mutations may not be significant. While L84S may be important in SARS-CoV-2 virulence and pathogenesis given ORF8’s role in attenuation of host immunity (Table 4), the continued decline of L84S representation among global SARS-CoV-2 sequences [93] suggests otherwise.
Mutations RG203KR within SARS-CoV-2 nucleoprotein (N) have become dominant and characteristic of clade GR [123]. RG203KR alters N protein morphology, resulting in increased intraviral protein binding affinity [132]. N-M interactions are necessary for CoV viral assembly [141, 142], while N-envelope (E) interactions potentially increase production of virus-like particles [143]. Therefore, increased intraviral N protein binding affinities could contribute to increased viral replication. RG203KR may also confer immune evasion properties to SARS-CoV-2 considering the rapid expansion of clade GR and the role of N protein in antagonizing human antiviral immune responses [144, 145] (Table 4). The global prevalence of variant B.1.1.7 has generated clade GRY from clade GR [146].
Clade GV is associated with the European variant 20A.EU1 containing spike NTD mutation A222V [105, 147]. A222 is located within a speculated B lymphocyte epitope [148] that may impact neutralization by human antibodies, consistent with observed SARS-CoV-2 re-infection with a clade GV variant [149]. The rise in prevalence of variant 20A.EU1 and clade GV is most likely associated with the relaxing of travel-associated restrictions across Europe near the end of the summer of 2020 considering the rapid decline in prevalence of global clade GV sequences in 2021 [97, 150].
Ongoing SARS-CoV-2 evolution and the rise of variants of concern
An aforementioned trend across HCoVs is positively selected residues within RBD [84, 85, 88, 138, 139, 151,152,153,154] (Fig. 2, Table 2), which facilitates interactions with host cellular proteins, providing a crucial target for the host immune response [155]. Accordingly, SARS-CoV-2 RBD is rapidly evolving, leading to novel variants [156, 157] (Fig. 2, Table 2). SARS-CoV-2 variants associated with greater transmissibility, altered virulence, or the ability to escape natural infection- and vaccine-mediated immunity or current diagnostic tests are called Variants of Concern (VOC; Table 5).
Early data suggest that RBD mutation N501Y emerged recurrently in multiple regions due to increased transmissibility, and is associated with multiple VOCs [89, 99, 100, 158] (Table 5). SARS-CoV-2 N501 serves as one of six critical S residues required for binding to ACE2 [159] and N501Y increases viral infectivity through greater S-hACE2 binding affinity, likely due to stronger interactions with ACE2 residues Y41 and K353 [160]. Other critical residues within the SARS-CoV-2 RBD (L455, F486, Q493, S494, Y505) [73] should be closely monitored as mutations may increase SARS-CoV-2 transmission in humans and facilitate zooanthroponotic transfer to other species.
Early studies of the highly transmissible B.1.1.7 variant [77, 161] originating in the United Kingdom described 17 co-occurring non-synonymous mutations or deletions [89], which are more than expected since the mutation rate of SARS-CoV-2 is estimated to be around 2.4 × 10–3 per site per year [135]. In addition to N501Y, spike 69-70del, Y144del, and P681H mutations are speculated to be of functional significance [78, 162] (Table 5). Spike NTD 69-70del variants have shown significant transmission expansion, with speculated increased resistance to antibody-mediated neutralization [92] likely associated with sequestration of a protruding spike loop [78]. Y144del confers antibody resistance due to loss of a negative surface charge [163, 164]. Spike P681 is located in a known CoV mutational hotspot [83, 101] directly adjacent to the SARS-CoV-2 S1/S2 furin cleavage site (aa 681–684) [89, 165, 166] which promotes virus entry into host cells [167]; mutation in this region may increase cleavability and membrane fusion to enhance infectivity. P681 is also within an antigenic epitope recognized by B and T lymphocytes, implicating host immune response alterations [168]. P681H may therefore represent adaptive evolution to evade host immunity, although confirmatory studies are required. Another speculated B.1.1.7 mutation at ORF8 (Q27stop) causes early protein termination [89]. Truncated ORF8 has been associated with milder symptoms [169], although increased mortality is also associated with the B.1.1.7 variant [79, 170]. Emerging mutations in B.1.1.7 must be monitored and investigated, such as the sub-lineage VOC202102/02 that contains the RBD mutation E484K, which is associated with antibody resistance [171,172,173].
Another variant containing N501Y is B.1.351, which was first detected in South Africa in December, 2020 [100], but likely originated in October, 2020 [101]. This variant contains eight non-synonymous mutations in S, including three within the RBD (K417N, E484K, N501Y) and three in the NTD that may contribute to increased transmissibility [100, 101]. Both N501Y and E484K are located within the receptor binding motif (RBM) of the RBD. E484 interacts with residue K31 on hACE2 [174], one of two critical hACE2 RBD-interacting residues [159, 175], suggesting that E484K may affect the binding affinity of SARS-CoV-2 with hACE2. However, preliminary studies demonstrate contradictory binding affinity observations [176, 177]; further studies are required. In addition, E484K confers some resistance to antibody-mediated neutralization of SARS-CoV-2 in vitro [91, 154, 178,179,180,181], consistent with the observation that E484 is an important recognition site for neutralizing antibodies [181, 182], and raising concerns about E484K being an immune escape mutation appearing in multiple independent SARS-CoV-2 lineages [172, 183,184,185,186]. Similarly, spike K417 is within a neutralizing antibody epitope [100]. Preliminary evidence suggests K417N reduces recognition by human antibodies [187]. K417N may impact RBD-hACE2 binding affinity and stabilize E484K, though these effects remain uncertain [91, 177, 187, 188].
Mutations within the RBD (K417T, E484K, N501Y) have also been observed in the P.1 variant (Table 5) that likely originated in Brazil and has since spread to other countries [101, 189,190,191]. In contrast, the P.2 variant only contains E484K, likely acquired through convergent evolution with P.1 [186, 192]. Little is known about the P.1 variant, but based on emerging data [193], we speculate that the RBD mutations likely affect antibody-mediated neutralization and contribute to increased transmission as observed with B.1.351. Mutations shared between the B.1.1.7, B.1.351, and P.1 variants are speculated to have arisen independently, indicating convergent evolution [194] (Table 5). These variants also share Nsp6 3675-3677del, with unknown functional significance [194, 195].
VOC B.1.617.2 was first identified in India in late 2020 and contains positively selected for mutations within the spike protein, namely, L452R, T478K, and P681R, along with the D614G mutation [196] (Table 5). Mutation of the uncharged and hydrophobic leucine (L) residue into the positively charged and hydrophilic arginine (R) residue at spike position 452 allows for an increased electrostatic interaction with negatively charged ACE2 residues E35, E37, and D38, likely leading to the observed increase in S-hACE2 complex stability, viral infectivity, and virus replication [196, 197]. Furthermore, abolition of the hydrophobic surface patch through the L452R mutation led to reduced antibody-mediated neutralization and cellular immune recognition [196,197,198]. Spike mutation T478K has also been shown to increase electrostatic interactions in the S-hACE2 complex and may increase binding affinity similar to the S477N mutation [199]. The mutation T478K is within a neutralizing epitope close to the immune evasion mutation E484K/Q that is present in multiple SARS-CoV-2 variants, including the ancestral B.1.617 lineage and current sub-lineages B.1.617.1 and B.1.617.2 [181, 200, 201]. T478K in combination with L452R may contribute to increased resistance to neutralization by monoclonal antibodies, convalescent sera, and vaccinated sera [201, 202]. B.1.617.2 has increased replication efficiency in human airway systems relative to the B.1.1.7 lineage due to enhanced spike cleavability, which is likely augmented by the P681R mutation [201, 203]. P681R is known to increase cell-to-cell fusion in the respiratory tract, potentially increasing transmissibility and pathogenicity in infected individuals [201, 203]. B.1.617.2 may thus represent a VOC with similar resistance to antibody neutralization as B.1.351 and transmissibility beyond B.1.1.7 [200]. Recently discovered B.1.617.2 sequences containing the K417N mutation (AY.1/AY.2 lineages) must be monitored for altered antibody resistance and increased transmissibility [204].
Circulating variants containing an N439K mutation (e.g. B.1.141 and B.1.258) also show some degree of neutralization evasion [91, 198, 205], raising speculations about SARS-CoV-2 variants escaping vaccine-mediated immunity. Emerging data suggest that antibodies elicited by mRNA vaccines (BNT162b2 and mRNA-1273) have 20% and 16.7% reduced neutralization capacity, respectively, against the B.1.1.7 variant [206, 207] and 67% and 84% reduced neutralization capacity, respectively, against the B.1.351 variant [208, 209]. Neutralization capacity of sera from BNT162b2 and mRNA-1273 vaccinated individuals have 87% and 52% reduced neutralization capacity, respectively, against the B.1.617.2 variant [200, 201, 210]. The emergence of B.1.1.7 sub-lineages containing the E484K RBD mutation (e.g. VOC202102/02) pose additional challenges for vaccine-mediated immunity [171, 173, 183]. While complete vaccine failure is unlikely [206, 207, 211,212,213,214,215], immune escape variants may create a need to update current SARS-CoV-2 vaccines. Monitoring the emergence of novel SARS-CoV-2 variants is especially important as vaccine-mediated immunity provides stronger selective pressure for SARS-CoV-2 evolution.
Other variants of interest
Multiple emerging SARS-CoV-2 lineages are not considered VOCs but are still of interest and may become VOCs in the future. One variant, B.1.525, was first detected in December, 2020, in the United Kingdom and Nigeria and has since spread internationally. B.1.525 contains spike mutations 69-70del, E484K, Q677H, and F888L. Q677P/H has emerged in disparate variants and may affect spike cleavability similar to P681H [158, 216,217,218]. F888L lies between the fusion peptide and heptad repeat region of the S2 subunit [219] and may impact host cellular entry, similar to the impact of heptad repeat mutations in MERS-CoV [139, 220].
Variant B.1.526 from New York contains spike mutations D253G, D614G, and A701V, along with either E484K or S477N, creating two major B.1.526 sub-lineages. NTD mutation D253G reduces antibody-mediated neutralization [163]. A701V, shared by variant B.1.351 [100], is in the S2 subunit adjacent to the furin cleavage site [219] and may impact SARS-CoV-2 cleavability and infectivity. S477N, also found in variant 20A.EU2, increases binding to hACE2 [221, 222] and reduces antibody-mediated neutralization [178, 223], likely due to its position within a neutralizing epitope [224]. D614G and E484K are shared with multiple other variants (Table 5) and likely play a role in B.1.526 expansion.
P681H found in variant B.1.1.207 from Nigeria [162] may enhance infectivity and modulate host immunity as speculated for B.1.1.7. Similar effects are expected for P681R in variant A.23.1 that emerged in Uganda [183, 225]. The UK A.23.1 sub-lineage VUI-202102/01 also contains immune escape mutation E484K [171, 183]. Preliminary data show increased ACE2 binding affinity and reduced antibody-mediated neutralization for the P.3 variant from Brazil, which contains the spike mutations E484K, N501Y, and P681H [164]. Data also suggest increased ACE2 binding affinity and reduced neutralization profile for the B.1.620 variant from Central Africa, which contains spike mutations E484K, S477N, D614G, and P681H [226]. Other notable variants include N440K variants from India [227] that have increased transmissibility, and the R.1 variant from Japan which contains potential immune escape mutations W152L and E484K [228].
B.1.427/B.1.429 are two emerging lineages that originated in California in May 2020 [229], however, circulating B.1.427/B.1.429 variants are now being replaced by more transmissible variants, such as B.1.1.7 and B.1.617.2 [97, 230]. B.1.427/B.1.429 contains multiple positively selected for mutations within the S protein, such as S13I, W152C, and L452R, all of which contribute to some degree of resistance to antibody-mediated neutralization [229]. L452R has convergently evolved in the B.1.617 lineage and contributed to enhanced SARS-CoV-2 infectivity [196,197,198] (Table 5). Spike mutation L452Q was detected in the recently emerged C.37 lineage from Peru and is expected to have similar impacts on virus infectivity as the L452R mutation [231]. C.37 also shares Nsp6 3675-3677del with B.1.1.7, B.1.351. and P.1 variants [231], and contains the spike RBD mutation F490S that has been associated with reduced antibody-mediated neutralization [91, 178]. These variants need to be monitored for transmission expansion and convergent evolution.
Multiple factors will determine the evolutionary trajectory of SARS-CoV-2 and the COVID-19 pandemic
The future of SARS-CoV-2 and COVID-19 remains uncertain. Many virological, immunological, and social factors will influence the epidemiological trajectory of this virus. One particularly intriguing question that remains unanswered is whether SARS-CoV-2 will become endemic in the human population, like HCoVs NL63, OC43, HKU1, and 229E [232,233,234].
Currently, endemic HCoVs cause seasonal outbreaks [235], with increased circulation observed in the winter in temperate regions [232]. Cold temperatures are favourable for enveloped viruses [236], as lower temperatures enhance lipid ordering of the viral envelope, allowing the virus to remain protected outside the host for longer periods of time [237, 238]. Low temperatures also enhance aerosol transmission of respiratory viruses by allowing virions to remain suspended in the air for a longer duration [239]. Furthermore, cold and dry environments can have immunosuppressive effects on a potential host, further increasing the chances of infection [240,241,242]. Evidence suggests decreased transmission of SARS-CoV-2 in warmer climates [243,244,245,246], likely due to degeneration of viral structural stability with increasing temperatures [247]. Decreased transmission of SARS-CoV-2 was not observed during the summer of 2020 [11, 248] likely because of the sheer number of cases and an immunologically naïve population. For seasonality to have an observable impact on SARS-CoV-2 transmission, the basic reproduction number (R0) must drop from its current estimate of around 2.5 to less than 1 [249]. In theory, SARS-CoV-2 R0 should drop substantially when population herd immunity is reached through natural infection and vaccination, allowing for meteorological factors to influence viral transmission, leading to seasonal fluctuations. Other intervention mechanisms such as effective social distancing, quarantine, and contact-tracing will contribute towards reducing the R0 for SARS-CoV-2 [250, 251].
Multiple studies have demonstrated short-lasting immunity to endemic HCoVs, with waning of protective immunity and re-infections common within 80 days [85] to one year [252,253,254,255]. There is no observable association between endemic HCoV re-infection and infection severity [254]. Waning of humoral immunity within a year [256,257,258,259,260] and re-infection of immunocompetent patients [149] have been demonstrated for SARS-CoV-2, suggesting the possibility of annual outbreaks [233, 261]. A weaker initial immune response and sharper decline of antibody levels have been reported in individuals with asymptomatic SARS-CoV-2 infections [257, 258]. Thus, multiple exposures to SARS-CoV-2 may be required to develop sufficient immunity to prevent future re-infections, which may also be influenced by adaptive evolution of SARS-CoV-2 in the human population (Table 5). The duration of protection through vaccination and natural exposures is being closely monitored, along with antigenic evolution of SARS-CoV-2 that may lead to immune escape. Indeed, the evolutionary trajectories of endemic HCoVs suggest that SARS-CoV-2 will evolve to co-exist with the human population. However, with roll-out of the first ever HCoV vaccines, predicting the evolutionary trajectory of SARS-CoV-2 remains challenging.
An important factor that may influence ongoing SARS-CoV-2 transmission is the potential for cross-protection by humoral and cellular immune responses induced by related endemic HCoVs. There is evidence of cross-protection within the same genera of HCoVs [233, 262, 263], but not between genera [264]. Thus, immunity against beta-CoVs HCoV-OC43 and HCoV-HKU1 may provide some protection against COVID-19 [265,266,267,268], while immunity against alpha-CoVs HCoV-229E and HCoV-NL63 will likely provide little to no protection. Antibody-dependent enhancement has not been observed for SARS-CoV-2 [269, 270], ruling out the possibility of increased disease severity by cross-reactive antibodies generated against endemic HCoVs. The high frequency of CoV recombination during co-infections raises the additional concern that SARS-CoV-2 recombination with seasonal HCoVs could generate novel CoVs [131, 271, 272]. The role of HCoV co-infection has not been reported or extensively studied and will be especially important for immunocompromised and elderly individuals.
Conclusions
SARS-CoV-2 continues to evolve and adapt to the human population as highlighted by the emergence of novel variants. Mutations within the spike protein of SARS-CoV-2 variants confer increased transmissibility and some degree of resistance to antibody-mediated neutralization. However, recurrent attenuating mutations, such as P323L, L37F, G251V, and Q27stop have also been identified and are speculated to reduce disease severity. The appearance of attenuating mutations suggests that SARS-CoV-2 is evolving to become less pathogenic in humans. The current SARS-CoV-2 pandemic is driven by asymptomatic, pre-symptomatic, or otherwise unrecognized cases [273,274,275]. Reduced pathogenicity of SARS-CoV-2 combined with mounting population-level immunity will likely cause a reduction of severe cases of COVID-19, leading to an apparent abatement of the pandemic, followed by endemic circulation of low pathogenic SARS-CoV-2 variants. A similar evolutionary trajectory may have led to the establishment of current low-pathogenic endemic HCoVs [276].
Monitoring future emerging variants of SARS-CoV-2 is critical to determine control measures for the COVID-19 pandemic. Mutations speculated to reduce immune recognition, such as within the spike protein (S13I, 69-70del, W152L, A222V, K417N, N439K, S477N, T478K, E484K/Q, F490S, P681H/R) and nucleoprotein (RG203KR) should be studied for reduced sensitivity to natural or vaccine-induced immunity. Other factors, such as zoonotic and zooanthroponotic transmission of SARS-CoV-2, cross-protection through immunity against endemic HCoVs, and the possible creation of novel animal reservoirs through zooanthroponosis should continue to be investigated as they may have significant implications on the evolutionary trajectory of SARS-CoV-2 and the COVID-19 pandemic.
Availability of data and materials
Not applicable.
Abbreviations
- CoV:
-
Coronavirus
- dN :
-
Rate of nonsynonymous mutations
- dS :
-
Rate of synonymous mutations
- E:
-
Envelope protein
- hACE2:
-
Human ACE2
- HCoV:
-
Human coronavirus
- HE:
-
Hemagglutinin esterase
- M:
-
Membrane protein
- MERS-CoV:
-
Middle East respiratory syndrome coronavirus
- N:
-
Nucleocapsid protein
- S:
-
Spike glycoprotein
- NTD:
-
N-terminal domain
- PEDV:
-
Porcine epidemic diarrhea virus
- RdRp:
-
RNA-dependent RNA polymerase
- RBD:
-
Receptor binding domain
- RBM:
-
Receptor binding motif
- R0 :
-
Basic reproduction number
- SADS-CoV:
-
Swine acute diarrhea syndrome coronavirus
- SARS-CoV:
-
Severe acute respiratory syndrome coronavirus
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SARSr-CoV:
-
Severe acute respiratory syndrome related coronavirus
- SNP:
-
Single nucleotide polymorphism
- VOC:
-
Variant of concern
References
Su S, Wong G, Shi W, Liu J, Lai ACK, Zhou J, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24(6):490–502.
Woo PCY, Lau SKP, Lam CSF, Lau CCY, Tsang AKL, Lau JHN, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86(7):3995–4008.
Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181–92.
Lin C-M, Saif LJ, Marthaler D, Wang Q. Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Res. 2016;226:20–39.
Zhou P, Fan H, Lan T, Yang X-L, Shi W-F, Zhang W, et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature. 2018;556(7700):255–8.
MacLachlan NJ, Dubovi EJ. Coronaviridae. In: Maclachlan NJ, Dubovi EJ, editors. Fenner’s veterinary virology. Elsevier; 2017. p. 435–61.
Chen B, Tian E-K, He B, Tian L, Han R, Wang S, et al. Overview of lethal human coronaviruses. Sig Transduct Target Ther. 2020;5(1):89.
van der Hoek L. Human coronaviruses: what do they cause? Antivir Ther (Lond). 2007;12(4 Pt B):651–8.
Chan-Yeung M, Xu R-H. SARS: epidemiology. Respirology. 2003;8(s1):S9-14.
Memish ZA, Perlman S, Van Kerkhove MD, Zumla A. Middle East respiratory syndrome. Lancet. 2020;395(10229):1063–77.
ArcGIS. . COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Johns Hopkins University; 2020.
Lim Y, Ng Y, Tam J, Liu D. Human coronaviruses: a review of virus–host interactions. Diseases. 2016;4(4):26.
Touma M. COVID-19: molecular diagnostics overview. J Mol Med. 2020;98(7):947–54.
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–74.
European Centre for Disease Prevention and Control. MERS-CoV worldwide overview. ECDC; 2021.
Mehand MS, Al-Shorbaji F, Millett P, Murgue B. The WHO R&D Blueprint: 2018 review of emerging infectious diseases requiring urgent research and development efforts. Antivir Res. 2018;159:63–7.
Alshukairi AN, Zheng J, Zhao J, Nehdi A, Baharoon SA, Layqah L, et al. High prevalence of MERS-CoV infection in camel workers in Saudi Arabia. MBio. 2018;9(5):e01985-e2018.
Li X, Song Y, Wong G, Cui J. Bat origin of a new human coronavirus: there and back again. Sci China Life Sci. 2020;63(3):461–2.
Frieman M, Baric R. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. MMBR. 2008;72(4):672–85.
Tao Y, Shi M, Chommanard C, Queen K, Zhang J, Markotter W, et al. Surveillance of bat coronaviruses in Kenya identifies relatives of human coronaviruses NL63 and 229E and their recombination history. J Virol. 2017;91(5):e01953-e2016.
Pfefferle S, Oppong S, Drexler JF, Gloza-Rausch F, Ipsen A, Seebens A, et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats. Ghana Emerg Infect Dis. 2009;15(9):1377–84.
Vijaykrishna D, Smith GJD, Zhang JX, Peiris JSM, Chen H, Guan Y. Evolutionary insights into the ecology of coronaviruses. J Virol. 2007;81(8):4012–20.
Adney DR, Bielefeldt-Ohmann H, Hartwig AE, Bowen RA. Infection, replication, and transmission of middle east respiratory syndrome coronavirus in alpacas. Emerg Infect Dis J. 2016;22(6):1031–7.
Munster VJ, Adney DR, van Doremalen N, Brown VR, Miazgowicz KL, Milne-Price S, et al. Replication and shedding of MERS-CoV in Jamaican fruit bats (Artibeus jamaicensis). Sci Rep. 2016;6(1):21878.
Mohd HA, Al-Tawfiq JA, Memish ZA. Middle east respiratory syndrome coronavirus (MERS-CoV) origin and animal reservoir. Virol J. 2016;13(1):87.
Alraddadi BM, Watson JT, Almarashi A, Abedi GR, Turkistani A, Sadran M, et al. Risk factors for primary middle east respiratory syndrome coronavirus illness in humans, Saudi Arabia. Emerg Infect Dis. 2016;22(1):49–55.
Farag E, Sikkema RS, Mohamedani AA, de Bruin E, Munnink BBO, Chandler F, et al. MERS-CoV in camels but not camel handlers, Sudan, 2015 and 2017. Emerg Infect Dis J. 2019;25(12):2333–5.
Reusken C, Ababneh M, Raj V, Meyer B, Eljarah A, Abutarbush S, et al. Middle East Respiratory Syndrome coronavirus (MERS-CoV) serology in major livestock species in an affected region in Jordan, June to September 2013. Eurosurveillance. 2013;18(50):20662.
Ralph R, Lew J, Zeng T, Francis M, Xue B, Roux M, et al. 2019-nCoV (Wuhan virus), a novel coronavirus: human-to-human transmission, travel-related cases, and vaccine readiness. J Infect Dev Ctries. 2020;14(1):3–17.
Banerjee A, Kulcsar K, Misra V, Frieman M, Mossman K. Bats and coronaviruses. Viruses. 2019;11(1):41.
Menachery VD, Yount BL Jr, Debbink K, Agnihothram S, Gralinski LE, Plante JA, et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med. 2015;21:1508–13.
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3.
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020;176:104742.
Zhou H, Chen X, Hu T, Li J, Song H, Liu Y, et al. A novel bat coronavirus reveals natural insertions at the S1/S2 cleavage site of the Spike protein and a possible recombinant origin of HCoV-19. bioRxiv. 2020. https://doi.org/10.1101/2020.03.02.974139.
Zhang Y-Z, Holmes EC. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. 2020;181(2):223–7.
Day T, Gandon S, Lion S, Otto SP. On the evolutionary epidemiology of SARS-CoV-2. Curr Biol. 2020;30(15):R849–57.
Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–9.
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020;26:1–3.
Wong MC, Cregeen SJJ, Ajami NJ, Petrosino JF. Evidence of recombination in coronaviruses implicating pangolin origins of nCoV-2019. bioRxiv. 2020. https://doi.org/10.1101/2020.02.07.939207.
Wacharapluesadee S, Tan CW, Maneeorn P, Duengkae P, Zhu F, Joyjinda Y, et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat Commun. 2021;12(1):972.
Lam TT-Y, Jia N, Zhang Y-W, Shum MH-H, Jiang J-F, Zhu H-C, et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020;583(7815):282–5.
Xiao K, Zhai J, Feng Y, Zhou N, Zhang X, Zou J-J, et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. 2020;583(7815):286–9.
Wrobel AG, Benton DJ, Xu P, Borg A, Roustan C, Martin SR, et al. Structure and binding properties of Pangolin-CoV Spike glycoprotein inform the evolution of SARS-CoV-2. Nat Commun. 2020;12:837.
Lehmann D, Halbwax ML, Makaga L, Whytock R, Ndindiwe Malata L, Bombenda Mouele W, et al. Pangolins and bats living together in underground burrows in Lopé National Park, Gabon. Afr J Ecol. 2020;58(3):540–2.
Liu P, Jiang J-Z, Wan X-F, Hua Y, Li L, Zhou J, et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 2020;16(5):e1008421.
Zhao X, Ding Y, Du J, Fan Y. 2020 update on human coronaviruses: one health, one world. Med Nov Technol Devices. 2020;8:100043.
Zhao J, Cui W, Tian B. The potential intermediate hosts for SARS-CoV-2. Front Microbiol. 2020;11:580137. https://doi.org/10.3389/fmicb.2020.580137/full.
Novaes Rocha V. Viral replication of SARS-CoV-2 could be self-limitative—the role of the renin-angiotensin system on COVID-19 pathophysiology. Med Hypotheses. 2020;145:110330.
Krieg AM. The role of CpG motifs in innate immunity. Curr Opin Immunol. 2000;12(1):35–43.
Alnazawi M, Altaher A, Kandeel M. Comparative genomic analysis MERS CoV isolated from humans and camels with special reference to virus encoded helicase. Biol Pharm Bull. 2017;40(8):1289–98.
Lin X-D, Wang W, Hao Z-Y, Wang Z-X, Guo W-P, Guan X-Q, et al. Extensive diversity of coronaviruses in bats from China. Virology. 2017;507:1–10.
Yang X-L, Hu B, Wang B, Wang M-N, Zhang Q, Zhang W, et al. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of severe acute respiratory syndrome coronavirus. J Virol. 2016;90(6):3253–6.
Ge X-Y, Li J-L, Yang X-L, Chmura AA, Zhu G, Epstein JH, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503(7477):535–8.
Becker DJ, Albery GF, Sjodin AR, Poisot T, Dallas TA, Eskew EA, et al. Predicting wildlife hosts of betacoronaviruses for SARS-CoV-2 sampling prioritization. Ecology. 2020. https://doi.org/10.1101/2020.05.22.111344.
Corman VM, Eckerle I, Memish ZA, Liljander AM, Dijkman R, Jonsdottir H, et al. Link of a ubiquitous human coronavirus to dromedary camels. PNAS. 2016;113(35):9864–9.
Crossley BM, Mock RE, Callison SA, Hietala SK. Identification and characterization of a novel alpaca respiratory coronavirus most closely related to the human coronavirus 229E. Viruses. 2012;4(12):3689–700.
Perveen N, Muzaffar SB, Al-Deeb MA. Exploring human–animal host interactions and emergence of COVID-19: evolutionary and ecological dynamics. Saudi J Biol Sci. 2021;28(2):1417–25.
Gussow AB, Auslander N, Faure G, Wolf YI, Zhang F, Koonin EV. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. PNAS. 2020;117(26):15193–9.
Banerjee A, Doxey AC, Mossman K, Irving AT. Unraveling the zoonotic origin and transmission of SARS-CoV-2. Trends Ecol Evol. 2020;36:180–4.
Sabir JSM, Lam TT-Y, Ahmed MMM, Li L, Shen Y, Abo-Aba SEM, et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science. 2016;351(6268):81–4.
Corman VM, Muth D, Niemeyer D, Drosten C. Hosts and sources of endemic human coronaviruses. Adv Virus Res. 2018;100:163–88.
Rest JS, Mindell DP. SARS associated coronavirus has a recombinant polymerase and coronaviruses have a history of host-shifting. Infect Genet Evol. 2003;3(3):219–25.
Bolles M, Donaldson E, Baric R. SARS-CoV and emergent coronaviruses: viral determinants of interspecies transmission. Curr Opin Virol. 2011;1(6):624–34.
Lorusso A, Desario C, Mari V, Campolo M, Lorusso E, Elia G, et al. Molecular characterization of a canine respiratory coronavirus strain detected in Italy. Virus Res. 2009;141(1):96–100.
Alekseev KP, Vlasova AN, Jung K, Hasoksuz M, Zhang X, Halpin R, et al. Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses, based on complete genomic sequences. J Virol. 2008;82(24):12422–31.
Chan JF-W, To KK-W, Tse H, Jin D-Y, Yuen K-Y. Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol. 2013;21(10):544–55.
Woo PCY, Huang Y, Lau SKP, Yuen K-Y. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2(8):1804–20.
Vijgen L, Lemey P, Keyaerts E, Van Ranst M, St-Jean JR, Jacomy H, et al. Genetic variability of human respiratory coronavirus OC43. J Virol. 2005;79(5):3223–5.
Sánchez CM, Gebauer F, Suñé C, Mendez A, Dopazo J, Enjuanes L. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology. 1992;190(1):92–105.
Baric RS, Fu K, Schaad MC, Stohlman SA. Establishing a genetic recombination map for murine coronavirus strain A59 complementation groups. Virology. 1990;177(2):646–56.
Banerjee A, Doxey AC, Tremblay BJ-M, Mansfield MJ, Subudhi S, Hirota JA, et al. Predicting the recombination potential of severe acute respiratory syndrome coronavirus 2 and Middle East respiratory syndrome coronavirus. J Gen Virol. 2020;101(12):1251–60.
Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4(12):e1000304.
Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020;7(6):1012–23.
van Dorp L, Acman M, Richard D, Shaw LP, Ford CE, Ormond L, et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect Genet Evol. 2020;83:104351.
Simmonds P. Rampant C→U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: causes and consequences for their short- and long-term evolutionary trajectories. mSphere. 2020;5(3):e00408-e420.
Mavian C, Marini S, Manes C, Capua I, Prosperi M, Salemi M. Regaining perspective on SARS-CoV-2 molecular tracing and its implications. medRxiv. 2020. https://doi.org/10.1101/2020.03.16.20034470.
Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: insights from linking epidemiological and genetic data. Infect Dis. 2021. https://doi.org/10.1101/2020.12.30.20249034.
Kemp SA, Harvey WT, Lytras S, Consortium TC-19 GU (COG-U), Carabelli AM, Robertson DL, et al. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion H69/V70. bioRxiv. 2021. https://doi.org/10.1101/2020.12.14.422555.
Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday J, et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B117 in England. Science. 2021;372(6538):eabg3055.
Bush RM. Predicting adaptive evolution. Nat Rev Genet. 2001;2(5):387–92.
Gallaher WR. A palindromic RNA sequence as a common breakpoint contributor to copy-choice recombination in SARS-COV-2. Arch Virol. 2020;165:1–8.
Tortorici MA, Veesler D. Structural insights into coronavirus entry, Chapter chapter 4. In: Rey FA, editor. Advances in virus research. Complementary strategies to understand virus structure and function, vol. 105. Academic Press; 2019. p. 93–116.
Chibo D, Birch C. Analysis of human coronavirus 229E spike and nucleoprotein genes demonstrates genetic drift between chronologically distinct strains. J Gen Virol. 2006;87(5):1203–8.
Ren L, Zhang Y, Li J, Xiao Y, Zhang J, Wang Y, et al. Genetic drift of human coronavirus OC43 spike gene during adaptive evolution. Sci Rep. 2015;5(1):11451.
Kiyuka PK, Agoti CN, Munywoki PK, Njeru R, Bett A, Otieno JR, et al. Human coronavirus NL63 molecular epidemiology and evolutionary patterns in rural coastal Kenya. J Infect Dis. 2018;217(11):1728–39.
Dominguez SR, Sims GE, Wentworth DE, Halpin RA, Robinson CC, Town CD, et al. Genomic analysis of 16 Colorado human NL63 coronaviruses identifies a new genotype, high sequence diversity in the N-terminal domain of the spike gene and evidence of recombination. J Gen Virol. 2012;93(11):2387–98.
Pyrc K, Dijkman R, Deng L, Jebbink MF, Ross HA, Berkhout B, et al. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J Mol Biol. 2006;364(5):964–73.
Wang Y, Li X, Liu W, Gan M, Zhang L, Wang J, et al. Discovery of a subgenotype of human coronavirus NL63 associated with severe lower respiratory tract infection in China, 2018. Emerg Microbes Infect. 2020;9(1):246–55.
Rambaut A, Loman N, Pybus O, Barclay W, Barrett J, Carabelli A, et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Virological. 2020. https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563/1.
McCarthy KR, Rennick LJ, Nambulli S, Robinson-McCarthy LR, Bain WG, Haidar G, et al. Natural deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. bioRxiv. 2020. https://doi.org/10.1101/2020.11.19.389916.
Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC, et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 2020;9:e61312.
Kemp SA, Collier DA, Datir R, Ferreira I, Gayed S, Jahun A, et al. Neutralising antibodies in Spike mediated SARS-CoV-2 adaptation. medRxiv. 2020. https://doi.org/10.1101/2020.12.05.20241927.
GISAID. hCoV-19 analysis update. 2021. https://www.gisaid.org/hcov-19-analysis-update/.
Mercatelli D, Giorgi FM. Geographic and genomic distribution of SARS-CoV-2 mutations. Front Microbiol. 2020;11:1800. https://doi.org/10.3389/fmicb.2020.01800/full.
Guan Q, Sadykov M, Mfarrej S, Hala S, Naeem R, Nugmanova R, et al. A genetic barcode of SARS-CoV-2 for monitoring global distribution of different clades during the COVID-19 pandemic. Int J Infect Dis. 2020;100:216–23.
Ceraolo C, Giorgi FM. Genomic variance of the 2019-nCoV coronavirus. J Med Virol. 2020;92(5):522–8.
GISAID. Global phylogeny updated by NextStrain. GISAID; 2021.
Alam ASMRU, Islam OK, Hasan MS, Islam MR, Mahmud S, AlEmran HM, et al. Evolving infection paradox of SARS-CoV-2: fitness costs virulence? Infect Dis. 2021. https://doi.org/10.1101/2021.02.21.21252137.
European Centre for Disease Prevention and Control. Rapid increase of a SARS-CoV-2 variant with multiple spike protein mutations observed in the United Kingdom. ECDC; 2020.
Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. Epidemiology. 2020. https://doi.org/10.1101/2020.12.21.20248640.
Centers for Disease Control and Prevention. Emerging SARS-CoV-2 variants. COVID-19. 2021. https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html.
Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data—from vision to reality. Euro Surveill. 2017;22(13):30494.
Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD, et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun. 2020;11(1):6013.
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182(4):812-827.e19.
Lauring AS, Hodcroft EB. Genetic variants of SARS-CoV-2—what do they mean? JAMA. 2021;325:529–31.
Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2020;325:1–6.
Hou YJ, Chiba S, Halfmann P, Ehre C, Kuroda M, Dinnon KH, et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science. 2020;370(6523):1464–8.
Mansbach RA, Chakraborty S, Nguyen K, Montefiori DC, Korber B, Gnanakaran S. The SARS-CoV-2 spike variant D614G favors an open conformational state. bioRxiv. 2020. https://doi.org/10.1101/2020.07.26.219741.
Butowt R, Bilinska K, Von Bartheld CS. Chemosensory dysfunction in COVID-19: integration of genetic and epidemiological data points to D614G spike protein variant as a contributing factor. ACS Chem Neurosci. 2020;11(20):3180–4.
Eaaswarkhanth M, Madhoun AA, Al-Mulla F. Could the D614G substitution in the SARS-CoV-2 spike (S) protein be associated with higher COVID-19 mortality? Int J Infect Dis. 2020;96:459–60.
Omotoso OE. Contributory role of SARS-CoV-2 genomic variations and life expectancy in COVID-19 transmission and low fatality rate in Africa. Egypt J Med Hum Genet. 2020;21(1):72.
Yurkovetskiy L, Wang X, Pascal KE, Tomkins-Tinch C, Nyalile TP, Wang Y, et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell. 2020;183(3):739-751.e8.
Weissman D, Alameh M-G, de Silva T, Collini P, Hornsby H, Brown R, et al. D614G spike mutation increases SARS CoV-2 susceptibility to neutralization. medRxiv. 2020. https://doi.org/10.1101/2020.07.22.20159905.
Becerra-Flores M, Cardozo T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract. 2020;74(8):e13525.
Lorenzo-Redondo R, Nam HH, Roberts SC, Simons LM, Jennings LJ, Qi C, et al. A unique clade of SARS-CoV-2 viruses is associated with lower viral loads in patient upper airways. medRxiv. 2020. https://doi.org/10.1101/2020.05.19.20107144.
Nguyen TT, Pham TN, Van TD, Nguyen TT, Nguyen DTN, Le HNM, et al. Genetic diversity of SARS-CoV-2 and clinical, epidemiological characteristics of COVID-19 patients in Hanoi, Vietnam. PLoS ONE. 2020;15(11):e0242537.
Koyama T, Platt D, Parida L. Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ. 2020;98(7):495–504.
Min JS, Kim G-W, Kwon S, Jin Y-H. A cell-based reporter assay for screening inhibitors of MERS coronavirus RNA-dependent RNA polymerase activity. JCM. 2020;9(8):2399.
Wang Y, Anirudhan V, Du R, Cui Q, Rong L. RNA-dependent RNA polymerase of SARS-CoV-2 as a therapeutic target. J Med Virol. 2021;93(1):300–10.
Kirchdoerfer RN, Ward AB. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun. 2019;10(1):2342.
Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18:179.
Ilmjärv S, Abdul F, Acosta-Gutiérrez S, Estarellas C, Galdadas I, Casimir M, et al. Epidemiologically most successful SARS-CoV-2 variant: concurrent mutations in RNA-dependent RNA polymerase and spike protein. medRxiv. 2020. https://doi.org/10.1101/2020.08.23.20180281.
Tomaszewski T, DeVries RS, Dong M, Bhatia G, Norsworthy MD, Zheng X, et al. New pathways of mutational change in SARS-CoV-2 proteomes involve regions of intrinsic disorder important for virus replication and release. Evol Bioinform Online. 2020;16:1176934320965149.
Velazquez-Salinas L, Zarate S, Eberl S, Gladue DP, Novella I, Borca MV. Positive selection of ORF1ab, ORF3a, and ORF8 genes drives the early evolutionary trends of SARS-CoV-2 during the 2020 COVID-19 pandemic. Front Microbiol. 2020;11:550674.
Tonkin-Hill G, Martincorena I, Amato R, Lawson ARJ, Gerstung M, Johnston I, et al. Patterns of within-host genetic diversity in SARS-CoV-2. bioRxiv. 2020. https://doi.org/10.1101/2020.12.23.424229.
Cárdenas-Conejo Y, Liñan-Rico A, García-Rodríguez DA, Centeno-Leija S, Serrano-Posada H. An exclusive 42 amino acid signature in pp1ab protein provides insights into the evolutive history of the 2019 novel human-pathogenic coronavirus (SARS-CoV-2). J Med Virol. 2020;92(6):688–92.
Xia H, Cao Z, Xie X, Zhang X, Chen JY-C, Wang H, et al. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020;33(1):108234.
Cottam EM, Whelband MC, Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy. 2014;10(8):1426–41.
Benvenuto D, Angeletti S, Giovanetti M, Bianchi M, Pascarella S, Cauda R, et al. Evolutionary analysis of SARS-CoV-2: how mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J Infect. 2020;81(1):e24–7.
Wang R, Hozumi Y, Yin C, Wei G-W. Decoding asymptomatic COVID-19 infection and transmission. arXiv200701344. 2020. hp://arxivorg/abs/2007.01344.
Dudas G, Rambaut A. MERS-CoV recombination: implications about the reservoir and potential for adaptation. Virus Evol. 2016. https://doi.org/10.1093/ve/vev023.
Wu S, Tian C, Liu P, Guo D, Zheng W, Huang X, et al. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein–protein interactions. J Med Virol. 2020;93(4):2132–40. https://doi.org/10.1002/jmv.26597.
Issa E, Merhi G, Panossian B, Salloum T, Tokajian S. SARS-CoV-2 and ORF3a: nonsynonymous mutations, functional domains, and viral pathogenesis. mSystems. 2020;5(3):e00266-e320.
Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol. 2020;17:1–3.
Chaw S-M, Tai J-H, Chen S-L, Hsieh C-H, Chang S-Y, Yeh S-H, et al. The origin and underlying driving forces of the SARS-CoV-2 outbreak. J Biomed Sci. 2020;27(1):73.
Zhang Y, Zhang J, Chen Y, Luo B, Yuan Y, Huang F, et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through potently downregulating MHC-I. bioRxiv. 2020. https://doi.org/10.1101/2020.05.24.111823.
Flower TG, Buffalo CZ, Hooy RM, Allaire M, Ren X, Hurley JH. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. PNAS. 2021;118(2):e2021785118.
The Chinese SARS Molecular Epidemiology Consortium. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303(5664):1666–9.
Forni D, Cagliani R, Clerici M, Sironi M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017;25(1):35–48.
Lau SKP, Feng Y, Chen H, Luk HKH, Yang W-H, Li KSM, et al. Severe acute respiratory syndrome (SARS) coronavirus ORF8 Protein Is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J Virol. 2015;89(20):10532–47.
He R, Leeson A, Ballantine M, Andonov A, Baker L, Dobie F, et al. Characterization of protein–protein interactions between the nucleocapsid protein and membrane protein of the SARS coronavirus. Virus Res. 2004;105(2):121–5.
Masters PS. The molecular biology of coronaviruses. In: Luisa BG, editor. Advances in virus research. Elsevier; 2006. p. 193–292.
Siu YL, Teoh KT, Lo J, Chan CM, Kien F, Escriou N, et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J Virol. 2008;82(22):11318–30.
Li J-Y, Liao C-H, Wang Q, Tan Y-J, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020;286:198074.
Mu J, Xu J, Zhang L, Shu T, Wu D, Huang M, et al. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci China Life Sci. 2020;63(9):1–4.
GISAID. Clade and lineage nomenclature aids in genomic epidemiology studies of active hCoV-19 viruses. Clade and lineage nomenclature, March 2, 2021. 2021.
Bartolini B, Rueca M, Gruber CEM, Messina F, Giombini E, Ippolito G, et al. The newly introduced SARS-CoV-2 variant A222V is rapidly spreading in Lazio region, Italy. medRxiv. 2020. https://doi.org/10.1101/2020.11.28.20237016.
Zhang B, Hu Y, Chen L, Yau T, Tong Y, Hu J, et al. Mining of epitopes on spike protein of SARS-CoV-2 from COVID-19 patients. Cell Res. 2020;30(8):702–4.
To KK-W, Hung IF-N, Ip JD, Chu AW-H, Chan W-M, Tam AR, et al. Coronavirus disease 2019 (COVID-19) re-infection by a phylogenetically distinct severe acute respiratory syndrome coronavirus 2 strain confirmed by whole genome sequencing. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa1275ciaa1275.
Hodcroft EB, Zuber M, Nadeau S, Crawford KHD, Bloom JD, Veesler D, et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv. 2020. https://doi.org/10.1101/2020.10.25.20219063.
Wong AHM, Tomlinson ACA, Zhou D, Satkunarajah M, Chen K, Sharon C, et al. Receptor-binding loops in alphacoronavirus adaptation and evolution. Nat Commun. 2017;8(1):1735.
Kim Y, Cheon S, Min C-K, Sohn KM, Kang YJ, Cha Y-J, et al. Spread of mutant middle east respiratory syndrome coronavirus with reduced affinity to human CD26 during the South Korean Outbreak. MBio. 2016;7(2):e00019.
Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, Luo S, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–43.
Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN, et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2021;29(1):44-57.e9.
Piccoli L, Park Y-J, Tortorici MA, Czudnochowski N, Walls AC, Beltramello M, et al. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell. 2020;183(4):1024-1042.e21.
Wu A, Wang L, Zhou H-Y, Ji C-Y, Xia SZ, Cao Y, et al. One year of SARS-CoV-2 evolution. Cell Host Microbe. 2021;29:503–7.
Chen J, Wang R, Wang M, Wei G-W. Mutations strengthened SARS-CoV-2 infectivity. J Mol Biol. 2020;432(19):5212–26.
Tu H, Avenarius MR, Kubatko L, Hunt M, Pan X, Ru P, et al. Distinct patterns of emergence of SARS-CoV-2 spike variants including N501Y in clinical samples in Columbus Ohio. Genomics. 2021. https://doi.org/10.1101/2021.01.12.426407.
Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94(7):e00127-e220.
Santos JC, Passos GA. The high infectivity of SARS-CoV-2 B.1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation. bioRxiv. 2021. https://doi.org/10.1101/2020.12.29.424708.
Grabowski F, Preibisch G, Kochańczyk M, Lipniacki T. SARS-CoV-2 variant under investigation 202012/01 has more than twofold replicative advantage. Epidemiology. 2021. https://doi.org/10.1101/2020.12.28.20248906.
Oluniyi P. Detection of SARS-CoV-2 P681H spike protein variant in Nigeria. Virological. 2020]. https://virological.org/t/detection-of-sars-cov-2-p681h-spike-protein-variant-in-nigeria/567.
McCallum M, Marco AD, Lempp F, Tortorici MA, Pinto D, Walls AC, et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Immunology. 2021. https://doi.org/10.1101/2021.01.14.426475.
Bascos NAD, Mirano-Bascos D, Saloma CP. Structural analysis of spike protein mutations in an emergent SARS-CoV-2 variant from the Philippines. Biophysics. 2021. https://doi.org/10.1101/2021.03.06.434059.
Xia S, Lan Q, Su S, Wang X, Xu W, Liu Z, et al. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther. 2020;5(1):1–3.
Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020;78(4):779-784.e5.
Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The furin cleavage site of SARS-CoV-2 spike protein is a key determinant for transmission due to enhanced replication in airway cells. bioRxiv. 2020. https://doi.org/10.1101/2020.09.30.318311.
Yarmarkovich M, Warrington JM, Farrel A, Maris JM. Identification of SARS-CoV-2 vaccine epitopes predicted to induce long-term population-scale immunity. CR Med. 2020;1(3):100036.
Zinzula L. Lost in deletion: the enigmatic ORF8 protein of SARS-CoV-2. Biochem Biophys Res Commun. 2020;538:116–24.
Horby P, Huntley C, Davies N, Edmunds J, Ferguson N, Medley G, et al. NERVTAG paper on COVID-19 variant of concern B.1.1.7: paper from the New and Emerging Respiratory Virus Threats Advisory Group (NERVTAG) on new coronavirus (COVID-19) variant B.1.1.7. New and Emerging Respiratory Virus Threats Advisory Group; 2021.
Public Health England. PHE statement on variant of concern and new variant under investigation. Gov.UK; 2021. https://www.gov.uk/government/news/phe-statement-on-variant-of-concern-and-new-variant-under-investigation.
Wise J. Covid-19: The E484K mutation and the risks it poses. BMJ. 2021;372:n359.
Collier DA, Marco AD, Ferreira IATM, Meng B, Datir R, Walls AC, et al. SARS-CoV-2 B.1.1.7 escape from mRNA vaccine-elicited neutralizing antibodies. medRxiv. 2021. https://doi.org/10.1101/2021.01.19.21249840.
Lim H, Baek A, Kim J, Kim MS, Liu J, Nam K-Y, et al. Hot spot profiles of SARS-CoV-2 and human ACE2 receptor protein protein interaction obtained by density functional tight binding fragment molecular orbital method. Sci Rep. 2020;10(1):16862.
Luan J, Lu Y, Jin X, Zhang L. Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem Biophys Res Commun. 2020;526(1):165–9.
Nelson G, Buzko O, Spilman P, Niazi K, Rabizadeh S, Soon-Shiong P. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv. 2021. https://doi.org/10.1101/2021.01.13.426558.
Cheng MH, Krieger JM, Kaynak B, Arditi M, Bahar I. Impact of South African 501V2 variant on SARS-CoV-2 spike infectivity and neutralization: a structure-based computational assessment. bioRxiv. 2021. https://doi.org/10.1101/2021.01.10.426143.
Liu Z, VanBlargan LA, Bloyet L-M, Rothlauf PW, Chen RE, Stumpf S, et al. Landscape analysis of escape variants identifies SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. bioRxiv. 2021. https://doi.org/10.1101/2020.11.06.372037.
Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, et al. Evolution of antibody immunity to SARS-CoV-2. Nature. 2021;591:639–44.
Andreano E, Piccini G, Licastro D, Casalino L, Johnson NV, Paciello I, et al. SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv. 2020. https://doi.org/10.1101/2020.12.28.424451.
Barnes CO, Jette CA, Abernathy ME, Dam KM-A, Esswein SR, Gristick HB, et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588(7839):682–7.
Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369(6506):1014–8.
Public Health England. Variants: distribution of cases data. Gov.UK; 2021. https://www.gov.uk/government/publications/covid-19-variants-genomically-confirmed-case-numbers/variants-distribution-of-cases-data.
Gröhs Ferrareze PA, Franceschi VB, de Menezes MA, Caldana GD, Zimerman RA, Thompson CE. E484K as an innovative phylogenetic event for viral evolution: genomic analysis of the E484K spike mutation in SARS-CoV-2 lineages from Brazil. Evol Biol. 2021. https://doi.org/10.1101/2021.01.27.426895.
West AP, Barnes CO, Yang Z, Bjorkman PJ. SARS-CoV-2 lineage B.1.526 emerging in the New York region detected by software utility created to query the spike mutational landscape. Bioinformatics. 2021. https://doi.org/10.1101/2021.02.14.431043.
Sabino EC, Buss LF, Carvalho MPS, Prete CA, Crispim MAE, Fraiji NA, et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet. 2021;397(10273):452–5.
Fratev F. The N501Y and K417N mutations in the spike protein of SARS-CoV-2 alter the interactions with both hACE2 and human derived antibody: a free energy of perturbation study. Mol Biol. 2020. https://doi.org/10.1101/2020.12.23.424283.
Villoutreix BO, Calvez V, Marcelin A-G, Khatib A-M. In silico investigation of the new UK (B117) and South African (501YV2) SARS-CoV-2 variants with a focus at the ACE2–spike RBD interface. IJMS. 2021;22(4):1695.
National Institute of Infectious Diseases. Brief report: new variant strain of SARS-CoV-2 identified in travelers from Brazil. National Institute of Infectious Diseases; 2021. p. 1.
European Centre for Disease Prevention and Control. Risk related to the spread of new SARS-CoV-2 variants of concern in the EU/EEA—first update. ECDC; 2021.
Toovey OTR, Harvey KN, Bird PW, Tang JW-TW-T. Introduction of Brazilian SARS-CoV-2 484K.V2 related variants into the UK. J Infect. 2021;82:e23–4.
Voloch CM, da Silva FR, de Almeida LGP, Cardoso CC, Brustolini OJ, Gerber AL, et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. Genet Genomic Med. 2020. https://doi.org/10.1101/2020.12.23.20248598.
Vasques Nonaka CK, Miranda Franco M, Gräf T, Almeida Mendes AV, Santana de Aguiar R, Giovanetti M, et al. Genomic evidence of a Sars-Cov-2 reinfection case with E484K spike mutation in Brazil. Life Sci. 2021;27:1522–4.
Faria NR, Morales Claro I, Candido D, Moyses Franco LA, Andrade PS, Coletti TM, et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings—SARS-CoV-2 coronavirus/nCoV-2019 genomic epidemiology. Virological. 2021. https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586.
Vogels CBF, Breban MI, Ott IM, Alpert T, Petrone ME, Watkins AE, et al. Multiplex qPCR discriminates variants of concern to enhance global surveillance of SARS-CoV-2. PLoS Biol. 2021;19(5):e3001236.
Cherian S, Potdar V, Jadhav S, Yadav P, Gupta N, Das M, et al. Convergent evolution of SARS-CoV-2 spike mutations, L452R, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Mol Biol. 2021. https://doi.org/10.1101/2021.04.22.440932.
Motozono C, Toyoda M, Zahradnik J, Ikeda T, Saito A, Tan TS, et al. An emerging SARS-CoV-2 mutant evading cellular immunity and increasing viral infectivity. Microbiology. 2021. https://doi.org/10.1101/2021.04.02.438288.
Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell. 2020;182(5):1284-1294.e9.
Di Giacomo S, Mercatelli D, Rakhimov A, Giorgi FM. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J Med Virol. 2021;93(9):5638–43.
Wall EC, Wu M, Harvey R, Kelly G, Warchal S, Sawyer C, et al. Neutralising antibody activity against SARS-CoV-2 VOCs B.1.617.2 and B.1.351 by BNT162b2 vaccination. Lancet. 2021;397(10292):2331–3.
Mlcochova P, Kemp S, Dhar MS, Papa G, Meng B, Mishra S, et al. SARS-CoV-2 B.1.617.2 Delta variant emergence, replication and sensitivity to neutralising antibodies. Microbiology. 2021. https://doi.org/10.1101/2021.05.08.443253.
Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021.
Peacock TP, Sheppard CM, Brown JC, Goonawardane N, Zhou J, Whiteley M, et al. The SARS-CoV-2 variants associated with infections in India, B.1.617, show enhanced spike cleavage by furin. Microbiology. 2021. https://doi.org/10.1101/2021.05.28.446163.
Public Health England. SARS-CoV-2 variants of concern and variants under investigation in England. Gov.UK; 2021.
Thomson EC, Rosen LE, Shepherd JG, Spreafico R, Filipe AS, Wojcechowskyj JA, et al. Circulating SARS-CoV-2 spike variant N439K maintains fitness while evading antibody-mediated immunity. Cell. 2021;184:1171–87.
Edara VV, Floyd K, Lai L, Gardner M, Hudson W, Piantadosi A, et al. Infection and mRNA-1273 vaccine antibodies neutralize SARS-CoV-2 UK variant. medRxiv. 2021. https://doi.org/10.1101/2021.02.02.21250799.
Muik A, Wallisch A-K, Sänger B, Swanson KA, Mühl J, Chen W, et al. Neutralization of SARS-CoV-2 lineage B117 pseudovirus by BNT162b2 vaccine—elicited human sera. Science. 2021;371:1152–3.
Liu Y, Liu J, Xia H, Zhang X, Fontes-Garfias CR, Swanson KA, et al. Neutralizing activity of BNT162b2-elicited serum—preliminary report. N Engl J Med. 2021;384:1466–8.
Wu K, Werner AP, Moliva JI, Koch M, Choi A, Stewart-Jones GBE, et al. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. bioRxiv. 2021. https://doi.org/10.1101/2021.01.25.427948.
Choi A, Koch M, Wu K, Dixon G, Oestreicher J, Legault H, et al. Serum neutralizing activity of mRNA-1273 against SARS-CoV-2 variants. Microbiology. 2021. https://doi.org/10.1101/2021.06.28.449914.
Moderna. Moderna COVID-19 vaccine retains neutralizing activity against emerging variants first identified in the U.K. and the Republic of South Africa. Moderna; 2021. https://investors.modernatx.com/news-releases/news-release-details/moderna-covid-19-vaccine-retains-neutralizing-activity-against.
BioNTech. Pfizer and BioNTech publish results of study showing COVID-19 vaccine elicits antibodies that neutralize pseudovirus bearing the SARS-CoV-2 U.K. strain spike protein in cell culture. BioNTech. 2021. https://investors.biontech.de/news-releases/news-release-details/pfizer-and-biontech-publish-results-study-showing-covid-19.
Xie X, Liu Y, Liu J, Zhang X, Zou J, Fontes-Garfias CR, et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat Med. 2021;27:1–2.
Wang Z, Schmidt F, Weisblum Y, Muecksch F, Barnes CO, Finkin S, et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. bioRxiv. 2021. https://doi.org/10.1101/2021.01.15.426911.
Lopez Bernal J, Andrews N, Gower C, Gallagher E, Simmons R, Thelwall S, et al. Effectiveness of covid-19 vaccines against the B16172 (Delta) variant. N Engl J Med. 2021. https://doi.org/10.1056/NEJMoa2108891.
Hodcroft EB, Domman DB, Snyder DJ, Oguntuyo K, Van Diest M, Densmore KH, et al. Emergence in late 2020 of multiple lineages of SARS-CoV-2 spike protein variants affecting amino acid position 677. Infect Dis. 2021. https://doi.org/10.1101/2021.02.12.21251658.
Kim J-S, Jang J-H, Kim J-M, Chung Y-S, Yoo C-K, Han M-G. Genome-wide identification and characterization of point mutations in the SARS-CoV-2 genome. Osong Public Health Res Perspect. 2020;11(3):101–11.
Sarkar R, Mitra S, Chandra P, Saha P, Banerjee A, Dutta S, et al. Comprehensive analysis of genomic diversity of SARS-CoV-2 in different geographic regions of India: an endeavour to classify Indian SARS-CoV-2 strains on the basis of co-existing mutations. Arch Virol. 2021;166(3):801–12.
Xia S, Zhu Y, Liu M, Lan Q, Xu W, Wu Y, et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. 2020;17(7):765–7.
Forni D, Filippi G, Cagliani R, De Gioia L, Pozzoli U, Al-Daghri N, et al. The heptad repeat region is a major selection target in MERS-CoV and related coronaviruses. Sci Rep. 2015;5(1):14480.
Mejdani M, Haddadi K, Pham C, Mahadevan R. SARS-CoV-2 receptor binding mutations and antibody mediated immunity. Bioinformatics. 2021. https://doi.org/10.1101/2021.01.25.427846.
Singh A, Steinkellner G, Köchl K, Gruber K, Gruber CC. Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Res Square. 2020;11:4320.
Tea F, Stella AO, Aggarwal A, Darley DR, Pilli D, Vitale D, et al. SARS-CoV-2 neutralizing antibodies; longevity, breadth, and evasion by emerging viral variants. Infect Dis. 2020. https://doi.org/10.1101/2020.12.19.20248567.
Wang D, Mai J, Zhou W, Yu W, Zhan Y, Wang N, et al. Immunoinformatic analysis of T- and B-cell epitopes for SARS-CoV-2 vaccine design. Vaccines. 2020;3(8):355.
Bugembe DL, Phan MVT, Ssewanyana I, Semanda P, Nansumba H, Dhaala B, et al. A SARS-CoV-2 lineage A variant (A.23.1) with altered spike has emerged and is dominating the current Uganda epidemic. Infect Dis. 2021. https://doi.org/10.1101/2021.02.08.21251393.
Dudas G, Hong SL, Potter B, Calvignac-Spencer S, Niatou-Singa FS, Tombolomako TB, et al. Travel-driven emergence and spread of SARS-CoV-2 lineage B.1.620 with multiple VOC-like mutations and deletions in Europe. Epidemiology. 2021. https://doi.org/10.1101/2021.05.04.21256637.
Tandel D, Gupta D, Sah V, Harinivas Harshan K. N440K variant of SARS-CoV-2 has higher infectious fitness. Microbiology. 2021. https://doi.org/10.1101/2021.04.30.441434.
Hirotsu Y, Omata M. Detection of R1 lineage severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with spike protein W152L/E484K/G769V mutations in Japan. PLoS Pathog. 2021;17(6):e1009619.
McCallum M, Bassi J, Marco AD, Chen A, Walls AC, Iulio JD, et al. SARS-CoV-2 immune evasion by variant B.1.427/B.1.429. Immunology. 2021. https://doi.org/10.1101/2021.03.31.437925.
World Health Organization. Tracking SARS-CoV-2 variants. WHO; 2021.
Romero PE, Dávila-Barclay A, Salvatierra G, González L, Cuicapuza D, Solis L, et al. The emergence of SARS-CoV-2 variant lambda (C.37) in South America. Epidemiology. 2021. https://doi.org/10.1101/2021.06.26.21259487.
Audi A, AlIbrahim M, Kaddoura M, Hijazi G, Yassine HM, Zaraket H. Seasonality of respiratory viral infections: will COVID-19 follow suit? Front Public Health. 2020. https://doi.org/10.3389/fpubh.2020.567184/full.
Kissler SM, Tedijanto C, Goldstein E, Grad YH, Lipsitch M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science. 2020;368(6493):860–8.
Kanzawa M, Spindler H, Anglemyer A, Rutherford GW. Will coronavirus disease 2019 become seasonal? J Infect Dis. 2020;222(5):719–21.
Monto AS, DeJonge PM, Callear AP, Bazzi LA, Capriola SB, Malosh RE, et al. Coronavirus occurrence and transmission over 8 years in the HIVE cohort of households in Michigan. J Infect Dis. 2020;222(1):9–16.
Price RHM, Graham C, Ramalingam S. Association between viral seasonality and meteorological factors. Sci Rep. 2019;9(1):929.
Polozov IV, Bezrukov L, Gawrisch K, Zimmerberg J. Progressive ordering with decreasing temperature of the phospholipids of influenza virus. Nat Chem Biol. 2008;4(4):248–55.
Paynter S. Humidity and respiratory virus transmission in tropical and temperate settings. Epidemiol Infect. 2015;143(6):1110–8.
Harper GJ. Airborne micro-organisms: survival tests with four viruses. J Hyg (Lond). 1961;59(4):479–86.
Shephard RJ, Shek PN. Cold exposure and immune function. Can J Physiol Pharmacol. 1998;76(9):828–36.
Eccles R. An explanation for the seasonality of acute upper respiratory tract viral infections. Acta Otolaryngol. 2002;122(2):183–91.
Kudo E, Song E, Yockey LJ, Rakib T, Wong PW, Homer RJ, et al. Low ambient humidity impairs barrier function and innate resistance against influenza infection. Proc Natl Acad Sci USA. 2019;116(22):10905–10.
Sajadi MM, Habibzadeh P, Vintzileos A, Shokouhi S, Miralles-Wilhelm F, Amoroso A. Temperature and latitude analysis to predict potential spread and seasonality for COVID-19. SSRN J. 2020. https://www.ssrn.com/abstract=3550308.
Bukhari Q, Jameel Y. Will coronavirus pandemic diminish by summer? Social Science Research Network; 2020. Report No.: ID 3556998. https://papers.ssrn.com/abstract=3556998.
Wang J, Tang K, Feng K, Lv W. High temperature and high humidity reduce the transmission of COVID-19. SSRN J. 2020; https://www.ssrn.com/abstract=3551767.
Chen B, Liang H, Yuan X, Hu Y, Xu M, Zhao Y, et al. Predicting the local COVID-19 outbreak around the world with meteorological conditions: a model-based qualitative study. BMJ Open. 2020;10(11):e041397.
Sharma A, Preece B, Swann H, Fan X, McKenney RJ, Ori-McKenney KM, et al. Structural stability of SARS-CoV-2 virus like particles degrades with temperature. Biochem Biophys Res Commun. 2021;1(534):343–6.
Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20(5):533–4.
Petersen E, Koopmans M, Go U, Hamer DH, Petrosillo N, Castelli F, et al. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect Dis. 2020;20(9):e238–44.
Sanche S, Lin YT, Xu C, Romero-Severson E, Hengartner N, Ke R. High contagiousness and rapid spread of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis. 2020;26(7):1470–7.
Poirier C, Luo W, Majumder MS, Liu D, Mandl KD, Mooring TA, et al. The role of environmental factors on transmission rates of the COVID-19 outbreak: an initial assessment in two spatial scales. Sci Rep. 2020;10(1):17002.
Edridge AWD, Kaczorowska J, Hoste ACR, Bakker M, Klein M, Loens K, et al. Seasonal coronavirus protective immunity is short-lasting. Nat Med. 2020;26(11):1691–3.
Aldridge RW, Lewer D, Beale S, Johnson AM, Zambon M, Hayward AC, et al. Seasonality and immunity to laboratory-confirmed seasonal coronaviruses (HCoV-NL63, HCoV-OC43, and HCoV-229E): results from the Flu Watch cohort study. Wellcome Open Res. 2020;5:52.
Galanti M, Shaman J. Direct observation of repeated infections with endemic coronaviruses. J Infect Dis. 2020;223:409–15.
Callow KA, Parry HF, Sergeant M, Tyrrell DAJ. The time course of the immune response to experimental coronavirus infection of man. Epidemiol Infect. 1990;105(2):435–46.
Galipeau Y, Greig M, Liu G, Driedger M, Langlois M-A. Humoral responses and serological assays in SARS-CoV-2 infections. Front Immunol. 2020;11:610688. https://doi.org/10.3389/fimmu.2020.610688/full#B23.
Long Q-X, Tang X-J, Shi Q-L, Li Q, Deng H-J, Yuan J, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med. 2020;26(8):1200–4.
Ibarrondo FJ, Fulcher JA, Goodman-Meza D, Elliott J, Hofmann C, Hausner MA, et al. Rapid decay of anti-SARS-CoV-2 antibodies in persons with mild covid-19. N Engl J Med. 2020;383(11):1085–7.
Seow J, Graham C, Merrick B, Acors S, Pickering S, Steel KJA, et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat Microbiol. 2020;5(12):1598–607.
Choe PG, Kang CK, Suh HJ, Jung J, Song K-H, Bang JH, et al. Waning antibody responses in asymptomatic and symptomatic SARS-CoV-2 infection. Emerg Infect Dis. 2021;27(1):327–9.
Shaman J, Galanti M. Will SARS-CoV-2 become endemic? Science. 2020;370(6516):527–9.
Chan K-H, Chan JF-W, Tse H, Chen H, Lau CC-Y, Cai J-P, et al. Cross-reactive antibodies in convalescent SARS patients’ sera against the emerging novel human coronavirus EMC (2012) by both immunofluorescent and neutralizing antibody tests. J Infect. 2013;67(2):130–40.
Patrick DM, Petric M, Skowronski DM, Guasparini R, Booth TF, Krajden M, et al. An outbreak of human coronavirus OC43 infection and serological cross-reactivity with SARS coronavirus. Can J Infect Dis Med Microbiol. 2006;17(6):330–6.
Kellam P, Barclay W. The dynamics of humoral immune responses following SARS-CoV-2 infection and the potential for reinfection. J Gen Virol. 2020;101(8):791–7.
Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181(7):1489-1501.e15.
Mateus J, Grifoni A, Tarke A, Sidney J, Ramirez SI, Dan JM, et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science. 2020;370(6512):89–94.
Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584(7821):457–62.
Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature. 2020;587(7833):270–4.
Gao Q, Bao L, Mao H, Wang L, Xu K, Yang M, et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science. 2020;369(6499):77–81.
Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. PNAS. 2020;117(17):9490–6.
Wardeh M, Baylis M, Blagrove MSC. Predicting mammalian hosts in which novel coronaviruses can be generated. Nat Commun. 2021;12(1):780.
Goldstein SA, Brown J, Pedersen BS, Quinlan AR, Elde NC. Extensive recombination-driven coronavirus diversification expands the pool of potential pandemic pathogens. Evol Biol. 2021. https://doi.org/10.1101/2021.02.03.429646.
Nikolai LA, Meyer CG, Kremsner PG, Velavan TP. Asymptomatic SARS coronavirus 2 infection: invisible yet invincible. Int J Infect Dis. 2020;100:112–6.
Gandhi M, Yokoe DS, Havlir DV. Asymptomatic transmission, the achilles’ heel of current strategies to control covid-19. N Engl J Med. 2020;382(22):2158–60.
Ye Z-W, Yuan S, Yuen K-S, Fung S-Y, Chan C-P, Jin D-Y. Zoonotic origins of human coronaviruses. Int J Biol Sci. 2020;16(10):1686–97.
Vijgen L, Keyaerts E, Moës E, Thoelen I, Wollants E, Lemey P, et al. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. JVI. 2005;79(3):1595–604.
St. Jean JR, Jacomy H, Desforges M, Vabret A, Freymuth F, Talbot PJ. Human respiratory coronavirus OC43: genetic stability and neuroinvasion. J Virol. 2004;78(16):8824–34.
Bakkers MJG, Lang Y, Feitsma LJ, Hulswit RJG, de Poot SAH, van Vliet ALW, et al. Betacoronavirus adaptation to humans involved progressive loss of hemagglutinin-esterase lectin activity. Cell Host Microbe. 2017;21(3):356–66.
Woo PCY, Lau SKP, Chu C, Chan K, Tsoi H, Huang Y, et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79(2):884–95.
Hu B, Zeng L-P, Yang X-L, Ge X-Y, Zhang W, Li B, et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017;13(11):e1006698.
Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science. 2003;302(5643):276–8.
Corman VM, Ithete NL, Richards LR, Schoeman MC, Preiser W, Drosten C, et al. Rooting the phylogenetic tree of middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J Virol. 2014;88(19):11297–303.
Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. 2020;30(7):1346-1351.e2.
Lee J-S, Kim SY, Kim TS, Hong KH, Ryoo N-H, Lee J, et al. Evidence of severe acute respiratory syndrome coronavirus 2 reinfection after recovery from mild coronavirus disease 2019. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa1421.
Leao JC, Gusmao TPL, Zarzar AM, Filho JCL, de Faria ABS, Silva IHM, et al. Coronaviridae—old friends, new enemy! Oral Dis. 2021. https://doi.org/10.1111/odi.13447.
Pavlović-Lažetić GM, Mitić NS, Tomović AM, Pavlović MD, Beljanski MV. SARS-CoV genome polymorphism: a bioinformatics study. Genomics Proteomics Bioinf. 2005;3(1):18–35.
Naeem A, Hamed M, Alghoribi M, Aljabr W, Alsaran H, Enani M, et al. Molecular evolution and structural mapping of N-terminal domain in spike gene of middle east respiratory syndrome coronavirus (MERS-CoV). Viruses. 2020;12(5):502.
Cotten M, Watson SJ, Zumla AI, Makhdoom HQ, Palser AL, Ong SH, et al. Spread, circulation, and evolution of the middle east respiratory syndrome coronavirus. MBio. 2014;5(1):e01062-e1113.
Lau S, Wong A, Lau T, Woo P. Molecular evolution of MERS coronavirus: dromedaries as a recent intermediate host or long-time animal reservoir? IJMS. 2017;18(10):2138.
Forni D, Cagliani R, Mozzi A, Pozzoli U, Al-Daghri N, Clerici M, et al. Extensive positive selection drives the evolution of nonstructural proteins in lineage c betacoronaviruses. J Virol. 2016;90(7):3627–39.
AlBalwi MA, Khan A, AlDrees M, Gk U, Manie B, Arabi Y, et al. Evolving sequence mutations in the Middle East Respiratory Syndrome Coronavirus (MERS-CoV). J Infect Public Health. 2020;13(10):1544–50.
Hurdiss DL, Drulyte I, Lang Y, Shamorkina TM, Pronker MF, van Kuppeveld FJM, et al. Cryo-EM structure of coronavirus-HKU1 haemagglutinin esterase reveals architectural changes arising from prolonged circulation in humans. Nat Commun. 2020;11(1):4646.
Shirato K, Kawase M, Watanabe O, Hirokawa C, Matsuyama S, Nishimura H, et al. Differences in neutralizing antigenicity between laboratory and clinical isolates of HCoV-229E isolated in Japan in 2004–2008 depend on the S1 region sequence of the spike protein. J Gen Virol. 2012;93(9):1908–17.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–33.
Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 2020;5(11):1408–17.
Wu Z, Yang L, Ren X, Zhang J, Yang F, Zhang S, et al. ORF8-related genetic evidence for Chinese horseshoe bats as the source of human severe acute respiratory syndrome coronavirus. J Infect Dis. 2016;213(4):579–83.
Centers for Disease Control and Prevention. Update: outbreak of severe acute respiratory syndrome—Worldwide, 2003. CDC; 2003. https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5212a1.htm.
Drosten C, Günther S, Preiser W, van der Werf S, Brodt H-R, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–76.
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–20.
Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, et al. Close relative of human middle east respiratory syndrome coronavirus in Bat, South Africa. Emerg Infect Dis. 2013;19(10):1697–9.
Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, et al. Middle east respiratory syndrome coronavirus in Bats, Saudi Arabia. Emerg Infect Dis J. 2013;19(11):1819–23.
Anthony SJ, Gilardi K, Menachery VD, Goldstein T, Ssebide B, Mbabazi R, et al. Further evidence for bats as the evolutionary source of middle east respiratory syndrome coronavirus. MBio. 2017;8(2):e00373-e417.
Lau SKP, Zhang L, Luk HKH, Xiong L, Peng X, Li KSM, et al. Receptor usage of a novel bat lineage C betacoronavirus reveals evolution of middle east respiratory syndrome-related coronavirus spike proteins for human dipeptidyl peptidase 4 binding. J Infect Dis. 2018;218(2):197–207.
Annan A, Baldwin HJ, Corman VM, Klose SM, Owusu M, Nkrumah EE, et al. Human betacoronavirus 2c EMC/2012-related viruses in Bats, Ghana and Europe. Emerg Infect Dis. 2013;19(3):456–9.
Moreno A, Lelli D, de Sabato L, Zaccaria G, Boni A, Sozzi E, et al. Detection and full genome characterization of two beta CoV viruses related to Middle East respiratory syndrome from bats in Italy. Virol J. 2017;14(1):239.
McIntosh K, Dees JH, Becker WB, Kapikian AZ, Chanock RM. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. PNAS. 1967;57(4):933–40.
Hamre D, Procknow JJ. A new virus isolated from the human respiratory tract. Proc Soc Exp Biol Med. 1966;121(1):190–3.
Huynh J, Li S, Yount B, Smith A, Sturges L, Olsen JC, et al. Evidence supporting a zoonotic origin of human coronavirus strain NL63. J Virol. 2012;86(23):12816–25.
van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJM, Wolthers KC, et al. Identification of a new human coronavirus. Nat Med. 2004;10(4):368–73.
Fouchier RAM, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC, Simon JH, et al. A previously undescribed coronavirus associated with respiratory disease in humans. PNAS. 2004;101(16):6212–6.
Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182(5):1295-1310.e20.
Tang JW, Cheung JLK, Chu IMT, Sung JJY, Peiris M, Chan PKS. The large 386-nt deletion in SARS-associated coronavirus: evidence for quasispecies? J Infect Dis. 2006;194(6):808–13.
Guan Y. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science. 2003;302(5643):276–8.
Song H-D, Tu C-C, Zhang G-W, Wang S-Y, Zheng K, Lei L-C, et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci. 2005;102(7):2430–5.
Yuan Z, Nan Z, Pei H, Yang Z. Reconstruction of the most recent common ancestor sequences of SARS-Cov S gene and detection of adaptive evolution in the spike protein. Chin Sci Bull. 2004;49(12):1311–3.
Qu X-X, Hao P, Song X-J, Jiang S-M, Liu Y-X, Wang P-G, et al. Identification of two critical amino acid residues of the severe acute respiratory syndrome coronavirus spike protein for its variation in zoonotic tropism transition via a double substitution strategy*. J Biol Chem. 2005;280(33):29588–95.
Kleine-Weber H, Elzayat MT, Wang L, Graham BS, Müller MA, Drosten C, et al. Mutations in the spike protein of Middle East respiratory syndrome coronavirus transmitted in Korea increase resistance to antibody-mediated neutralization. J Virol. 2019;93(2):e01381-e1418.
Lau SKP, Lung DC, Wong EYM, Aw-Yong KL, Wong ACP, Luk HKH, et al. Molecular evolution of human coronavirus 229E in Hong Kong and a fatal COVID-19 case involving coinfection with a novel human coronavirus 229E genogroup. mSphere. 2021;6(1):e00819-e820.
National Center for Biotechnology Information. Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome. GenBank; 2020.
Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369(6508):1249–55.
Cornillez-Ty CT, Liao L, Yates JR, Kuhn P, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol. 2009;83(19):10314–8.
Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79(24):15189–98.
Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587(7835):657–62.
Sakai Y, Kawachi K, Terada Y, Omori H, Matsuura Y, Kamitani W. Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology. 2017;510:165–74.
Baruah C, Devi P, Sharma DK. In silico proteome analysis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Bioinformatics. 2020. https://doi.org/10.1101/2020.05.23.104919.
Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766–88.
Coelho C, Gallo G, Campos CB, Hardy L, Würtele M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS ONE. 2020;15(10):e0240079.
Snijder EJ, Decroly E, Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv Virus Res. 2016;96:59–126.
Jang K-J, Jeong S, Kang DY, Sp N, Yang YM, Kim D-E. A high ATP concentration enhances the cooperative translocation of the SARS coronavirus helicase nsP13 in the unwinding of duplex RNA. Sci Rep. 2020;10(1):4481.
Ogando NS, Ferron F, Decroly E, Canard B, Posthuma CC, Snijder EJ. The curious case of the nidovirus exoribonuclease: its role in RNA synthesis and replication fidelity. Front Microbiol. 2019;10:1813.
Senanayake SL. Overcoming nonstructural protein 15-nidoviral uridylate-specific endoribonuclease (nsp15/NendoU) activity of SARS-CoV-2. Future Drug Discov. 2020;2(3):FDD42.
Menachery VD, Debbink K, Baric RS. Coronavirus non-structural protein 16: evasion, attenuation, and possible treatments. Virus Res. 2014;194:191–9.
Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281-292.e6.
Siu K-L, Yuen K-S, Castaño-Rodriguez C, Ye Z-W, Yeung M-L, Fung S-Y, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33(8):8865–77.
Xu H, Chitre SA, Akinyemi IA, Loeb JC, Lednicky JA, McIntosh MT, et al. SARS-CoV-2 viroporin triggers the NLRP3 inflammatory pathway. bioRxiv. 2020. https://doi.org/10.1101/2020.10.27.357731.
Shi C-S, Nabar NR, Huang N-N, Kehrl JH. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov [Internet]. 2019 Jun 5 [cited 2021 Jan 18];5. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6549181/
Lam J-Y, Yuen C-K, Ip JD, Wong W-M, To KK-W, Yuen K-Y, et al. Loss of orf3b in the circulating SARS-CoV-2 strains. Emerg Microbes Infect. 2020;9(1):2685–96.
Castaño-Rodriguez C, Honrubia JM, Gutiérrez-Álvarez J, DeDiego ML, Nieto-Torres JL, Jimenez-Guardeño JM, et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. MBio. 2018;9(3):e02325-e2417.
Singh Tomar PP, Arkin IT. SARS-CoV-2 E protein is a potential ion channel that can be inhibited by Gliclazide and Memantine. Biochem Biophys Res Commun. 2020;530(1):10–4.
Voss D, Pfefferle S, Drosten C, Stevermann L, Traggiai E, Lanzavecchia A, et al. Studies on membrane topology, N-glycosylation and functionality of SARS-CoV membrane protein. Virol J. 2009;6:79.
Taylor JK, Coleman CM, Postel S, Sisk JM, Bernbaum JG, Venkataraman T, et al. Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J Virol. 2015;89(23):11820–33.
Pfefferle S, Krähling V, Ditt V, Grywna K, Mühlberger E, Drosten C. Reverse genetic characterization of the natural genomic deletion in SARS-Coronavirus strain Frankfurt-1 open reading frame 7b reveals an attenuating function of the 7b protein in-vitro and in-vivo. Virol J. 2009;6(1):131.
Jiang H-W, Zhang H-N, Meng Q-F, Xie J, Li Y, Chen H, et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol. 2020;17(9):998–1000.
Dominguez Andres A, Feng Y, Campos AR, Yin J, Yang C-C, James B, et al. SARS-CoV-2 ORF9c is a membrane-associated protein that suppresses antiviral responses in cells. bioRxiv. 2020;27:325.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–68.
European Centre for Disease Prevention and Control. Threat assessment brief: emergence of SARS-CoV-2 B.1.617 variants in India and situation in the EU/EEA. ECDC; 2021.
Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5(11):1403–7.
Acknowledgements
None.
Funding
SARS-CoV-2 studies in the Mossman lab are supported by a Canadian Institutes of Health Research COVID-19 rapid response grant to principal investigator K.M. and co-investigator A.B. A.B. was also supported by a Natural Sciences and Engineering Research Council of Canada fellowship. VIDO receives operational funding for its CL3 facility (InterVac) from the Canada Foundation for Innovation through the Major Science Initiatives. VIDO also receives operational funding from the Government of Saskatchewan through Innovation Saskatchewan and the Ministry of Agriculture.
Author information
Authors and Affiliations
Contributions
JS wrote the first draft. PP, AGM, AB, and KM edited the draft. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publication
This article is published with the permission of the Director of VIDO, journal series no. 937.
Competing interests
Authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Singh, J., Pandit, P., McArthur, A.G. et al. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol J 18, 166 (2021). https://doi.org/10.1186/s12985-021-01633-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-021-01633-w