
Abstract
Background
The study of bacteriophages continues to generate key information about microbial interactions in the environment. Many phenotypic characteristics of bacteriophages cannot be examined by sequencing alone, further highlighting the necessity for isolation and examination of phages from environmental samples. While much of our current knowledge base has been generated by the study of marine phages, freshwater viruses are understudied in comparison. Our group has previously conducted metagenomics-based studies samples collected from Lake Michigan - the data presented in this study relate to four phages that were extracted from the same samples.
Findings
Four phages were extracted from Lake Michigan on the same bacterial host, exhibiting similar morphological characteristics as shown under transmission electron microscopy. Growth characteristics of the phages were unique to each isolate. Each phage demonstrated a host-range spanning several phyla of bacteria – to date, such a broad host-range is yet to be reported. Genomic data reveals genomes of a similar size, and close similarities between the Lake Michigan phages and the Pseudomonas phage PB1, however, the majority of annotated genes present were ORFans and little insight was offered into mechanisms for host-range.
Conclusions
The phages isolated from Lake Michigan are capable of infecting several bacterial phyla, and demonstrate varied phenotypic characteristics despite similarities in host preference, and at the genomic level. We propose that such a broad host-range is likely related to the oligotrophic nature of Lake Michigan, and the competitive benefit that this characteristic may lend to phages in nature.
Similar content being viewed by others
Findings
In the last thirty years, studies of bacteriophages in the environment continue to demonstrate the importance of phages as drivers of bacterial mortality and diversity. Most of our knowledge with regard to phage diversity, in a wide range of niches, has come from metagenomic surveys [1, 2]. However, the majority of virally-related sequences generated by such surveys exhibit no amino acid similarity with annotated genes [3]. As technology improves, it has become clear that a severe paucity of genomic and phenotypic information exists for isolated environmental phages: this lack of information severely limits what can be achieved through deep-sequencing techniques alone. The high percentage of ORFans (open reading frames with no known match to coding DNA) identified in metagenomic studies [4] can be improved upon by the isolation and examination of phages directly from the environment. Comparatively, cultivation of phages allowing for the sequencing of clonal genomes: consequently, this improves the content of available databases. It also allows for the examination of physical features under high powered microscopy, such as transmission electron microscopy (TEM) and atomic force microscopy (AFM) [5, 6].
Current understanding of environmental phages has largely come from those which are aquatic in origin, however, freshwater phages are understudied in comparison to their marine counterpart and isolated phages are often cyanophages [7–9]. To date, few studies exist pertaining to specific characterization of freshwater phages that do not infect cyanobacteria [10], rendering them particularly under-represented in databases such as RefSeq. Representation of freshwater phages is of considerable importance when one considers the close proximity these environs often have to human activity: for example, the potential for anthropogenically-mediated pollution (e.g., sewage outfall), and, when considering Lake Michigan in particular, the use of freshwater as a source of drinking water and recreation. Furthermore, the heterogeneous nature of freshwater environments with regard to aspects such as water chemistry, and, crucially, bacterial diversity, suggest that viral diversity is likely to be greater than that observed in marine sample sets [11]. While there is evidence to suggest some evolutionary relationship between marine and freshwater phages [12, 13], conflicting results have also been reported [14] and a considerable degree of divergence is to be expected between the two groups.
Assessment of phage diversity in the environment should also take into account factors of phage lifestyle (e.g., lysis and lysogeny) and phage host-range. Regarding host-range, previous studies have assigned classification as a ‘generalist’ when a phage demonstrates capacity to infect more than one species of a bacterial genus [15], some restricting the definition further to include strains of a particular species [16, 17]. This is a difficult issue to address when considering concepts of bacterial species [18]. Here, we examine phages which are able to infect different bacterial phyla. In some instances, isolated phages that have been classified as generalists have been shown to demonstrate defined characteristics, such as reduction in infection efficiency compared to specialists [19]. This suggests that qualification as a true generalist may rely on more than just host-range. This notwithstanding, here we present four phages which are able to infect laboratory type strains, environmental and co-isolated environmental bacterial strains belonging to the phyla Proteobacteria, Actinobacteria and Bacteroidetes. Broad host-range is congruent with existing theory suggesting that generalism is of benefit of phages inhabiting an oligotrophic environment, such as Lake Michigan.
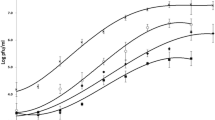
During this investigation, all phages were propagated on bacteria grown on/in lysogeny broth agar/lysogeny broth, at 37 °C. Four phages were isolated from the nearshore waters of Lake Michigan on E. coli (ATCC 8739), from the same samples used by the authors previously to investigate a metaviromic dataset generated from Lake Michigan [20]. Plaques originally isolated on E.coli were removed as single plugs and sequentially purified to generate clonal phage cultures. Host-range testing demonstrated lysis of laboratory strain P. aeruginosa (ATCC 15692), four strains of P. aeruginosa co-isolated from the same Lake Michigan samples, as well as strains of Arthrobacter, Chryseobacterium and Microbacterium (identified to the genus level via amplification and sequencing of the 16S rRNA gene) (Table 1). The phages were not able to infect E.coli K12, Achromobacter sp., Salmonella enterica, Shigella boydii or S. flexneri. Growth characteristics of the phages were assessed via one-step growth curve experiments on P.aeruginosa (ATCC 15692), performed at an m.o.i. of 0.1 with bacterial cells maintained at late exponential phase. Each isolate demonstrated differences in length of latent period (10–120 mins) and generation time (60–100 mins). Burst size for all four isolates remained within an order of magnitude.
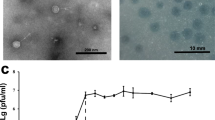
Transmission electron micrographs of the four phages (Fig. 1), all of which demonstrated morphological characteristics typical of Myoviridae, showed contractile tails (retracted tails were particularly evident in panel C) approximately 150 nm in length, and capsids at approximately 60 nm diameter, apart from ɸMoody (Fig. 1, panel c) which was slightly smaller at 50 nm.

TEM micrographs of phages ɸFenriz (a), ɸHabibi (b), ɸMoody (c) and ɸVader (d). Scale bar represents 50 nm
Infected cultures were saturated with chloroform, passed through a 0.22 μm filter and concentrated via tangential flow filtration. The final fraction was subject to DNase I treatment, and DNA extracted using the UltraClean® Microbial DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA). Library construction and sequencing was conducted at the University of Texas Medical Branch (Galveston, TX). Each of the five isolates was multiplexed and sequenced using the Illumina MiSeq platform via the MiSeq Reagent Kit v2 (500 cycle), producing paired-end reads each 250 nucleotides in length.
The genomes for the four Lake Michigan isolates were assembled and annotated using Velvet [21] (in Geneious, running VelvetOptimiser for selection of k), PhagePhisher (Hatzopoulos T, Watkins SC, Putonti C. PhagePhisher: a pipeline for the discovery of covert viral sequences in complex genomic datasets. submitted), and RAST [22]. While de novo assembly of ɸVader and ɸHabibi produced a single contig, four and five contigs for ɸMoody and ɸFenriz, were identified and subsequently closed via PCR. The data supporting the results of this article were deposited in NCBI (Genbank Accession numbers KT254130, KT254131, KT254132, and KT254133). Fundamentally, the four phages were similar in terms of genome length, GC content and number of predicted genes (Table 2). With regard to existing data, all four showed greatest similarity to Pseudomonas phage PB1 [23] and fell out into a distinct clade when compared to other PB1-like viruses (Fig. 2, panel a). Sequence identity to PB1 for each of the four isolates was 99 %, including mismatches as well as regions of homology between the Lake Michigan isolates absent from the PB1 genome (Fig. 2, panel b).ɸFenriz uniquely consisted of two frame shift mutations, which disturbed the coding regions of a putative minor head protein and putative structural protein. Between the isolates, variation was limited to five non-synonymous mutations at most: i.e., between ɸHabibi and ɸMoody, and ɸHabibi and ɸVader. ɸMoody and ɸVader differed by two non-synonymous mutations (Fig. 2, panel c).

Characterization of the four Lake Michigan isolates at the genomic level. Lake Michigan isolates were compared phylogenetically to other PB1-like viruses with a neighbor-joining tree (a), phage genomes were also directly compared to PB1 (b). Finally, genomes of the phages isolated as part of this study were closed and annotated (c)
The four phages examined in this study represent a group demonstrating a breadth of host-range capacity that has not been reported before. Host-range was assessed on ATCC strains of bacteria as well as those isolated from the environment recently. No conclusions with regard to host-range were offered from genomic level study of the phages, and in at least one of the isolated phages, the potential for lysogeny was observed (not shown) – however, again, no evidence was apparent for the repressor systems that directly mediate lysogeny in other phages. It is likely that synonymous mutations may be responsible for the breadth of phenotypic variations observed in the phages characterized here.
Clearly, the hosts identified here are very different from one another and likely the mode of infection for each candidate host is also very different. The determination of these mechanisms is now an important aspect of describing how broad host-range capacity is possible. That phages isolated from an oligotrophic lake would be able to infect across such a broad spectrum makes sense, fundamentally – in a system where host diversity may be high, but abundance low, this is a strong competitive strategy. That these phages demonstrate a host-range which extends beyond a single bacterial phyla also has clear implications for assigning taxonomical classification to viruses during metagenomic studies. This further emphasizes the importance of culture-based examinations which run parallel to those which are molecular-based.
References
Waller AS, Yamada T, Kristensen DM, Roat Kultima J, Sunagawa S, Koonin EV, et al. Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J. 2014;8:1391–402.
Adriaenssens EM, Zyl LV, De Maayer P, Rubagotti E, Rybicki E, Tuffin M, et al. Metagenomic analysis of the viral community in Namib Desert hypoliths. Environ Microbiol. 2014; doi: 10.1111/1462-2920.12528
Rosario K, Breitbart M. Exploring the viral world through metagenomics. Curr Opin Virol. 2011;1:289–97.
Yin Y, Fischer D. Identification and investigation of ORFans in the viral world. BMC Genomics. 2008;9:24.
Ackermann HW. Phage classification and characterisation. In: Clokie MRJ, Kropinski AM, editors. Bacteriophages: Methods and Protocols 1: Isolation, Characterization and Interactions. New York: Humana Press; 2009. p. 175–202.
Kuznetsov YG, Chang S-C, Credaroli A, Martiny J, McPherson A. An atomic force microscopy investigation of cyanophage structure. Micron. 2008;43:1336–42.
Watkins SC, Smith J, Hayes P, Watts J. Characterisation of host growth after infection with a broad-range freshwater cyanopodophage. PLoS One. 2014;9:e87339.
Dillon A, Parry J. Characterization of temperate cyanophages active against freshwater phycocyanin-rich Synechococcus species. Freshw Biol. 2008;53:1253–61.
Deng L, Hayes P. Evidence for cyanophages active against bloom-forming freshwater cyanobacteria. Freshw Biol. 2008;53:1240–52.
Maina A, Mwaura F, Oyugi J, Goulding D, Toribio A, Kariuki S. Characterization of Vibrio cholerae bacteriophages isolated from the environmental waters of the Lake Victoria region of Kenya. Curr Microbiol. 2013;68:67–70.
Maranger R, Bird D. Viral abundance in aquatic systems: a comparison between marine and fresh waters. Mar Ecol Prog Ser. 1995;121:217–26.
Breitbart M, Miyake J, Rohwer F. Global distribution of nearly identical phage‐encoded DNA sequences. FEMS Microbiol Lett. 2004;236:249–56.
Dreher T, Brown N, Bozarth C, Schwartz A, Riscoe E, Thrash C, et al. A freshwater cyanophage whose genome indicates close relationships to photosynthetic marine cyanomyophages. Environ Microbiol. 2011;13:1858–74.
Chénard C, Suttle C. Phylogenetic diversity of sequences of cyanophage photosynthetic gene psbA in marine and freshwaters. Appl Environ Microbiol. 2008;74:5317–24.
Czajkowski R, Ozymko Z, Zwirowski S, Lojkowska E. Complete genome sequence of a broad-host-range lytic Dickeya spp. bacteriophage ϕD5. Arch Virol. 2014, doi 10.1007/s00705-014-2170-8
Merabishvili M, Vandenheuvel D, Kropinski A, Mast J, Vos D, Verbeken G, et al. Characterization of newly isolated lytic bacteriophages active against Acinetobacter baumannii. PLoS One. 2014;9:e104853.
Bono L, Gensel C, Pfennig D, Burch C. Competition and the origins of novelty: experimental evolution of niche-width expansion in a virus. Biol Lett. 2013;9:2012–6.
Cohan F. What are bacterial species? Annu Rev Microbiol. 2002;56:457–87.
Holmfeldt K, Howard-Varona C, Solonenko N, Sullivan MB. Contrasting genomic patterns and infection strategies of two co-existing Bacteroidetes podovirus genera. Environ Microbiol. 2014;16:2501–13.
Watkins SC, Kuehnle N, Ruggeri CA, Malki K, Bruder K, Elayyan J, et al. Assessment of a metaviromic dataset generated from nearshore Lake Michigan. Mar Freshw Res. In press
Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9.
Overbeek R, Olson R, Pusch G, Olsen G, Davis J, Disz T, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nuc Acids Res. 2014;42:206–14.
Ceyssens P, Miroshnikov K, Mattheus W, Krylov V, Robben J, Noben J, et al. Comparative analysis of the widespread and conserved PB1‐like viruses infecting Pseudomonas aeruginosa. Environ Microbiol. 2009;11:2874–83.
Acknowledgements
This work was funded by the NSF (1149387) (CP).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KM, KB, and ES, performed bacterial isolations, host-range assays, phage growth experiments and all preparation and operation for/of the TEM. KM and AK performed host range assays. SS performed phage growth experiments. TH conducted computational analyses of genome data. SCW participated in the conception, design, and coordination of the study, performed data analysis and wrote the manuscript. CP participated in the conception, design and coordination of the study, and performed all computational analyses. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Malki, K., Kula, A., Bruder, K. et al. Bacteriophages isolated from Lake Michigan demonstrate broad host-range across several bacterial phyla. Virol J 12, 164 (2015). https://doi.org/10.1186/s12985-015-0395-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-015-0395-0