
Abstract
Background
Marine bacteriophages play key roles in the community structure of microorganisms, biogeochemical cycles, and the mediation of genetic diversity through horizontal gene transfer. Recently, traditional isolation methods, complemented by high-throughput sequencing metagenomics technology, have greatly increased our understanding of the diversity of bacteriophages. Oceanospirillum, within the order Oceanospirillales, are important symbiotic marine bacteria associated with hydrocarbon degradation and algal blooms, especially in polar regions. However, until now there has been no isolate of an Oceanospirillum bacteriophage, and so details of their metagenome has remained unknown.
Results
Here, we reported the first Oceanospirillum phage, vB_OliS_GJ44, which was assembled into a 33,786 bp linear dsDNA genome, which includes abundant tail-related and recombinant proteins. The recombinant module was highly adapted to the host, according to the tetranucleotides correlations. Genomic and morphological analyses identified vB_OliS_GJ44 as a siphovirus, however, due to the distant evolutionary relationship with any other known siphovirus, it is proposed that this virus could be classified as the type phage of a new Oceanospirivirus genus within the Siphoviridae family. vB_OliS_GJ44 showed synteny with six uncultured phages, which supports its representation in uncultured environmental viral contigs from metagenomics. Homologs of several vB_OliS_GJ44 genes have mostly been found in marine metagenomes, suggesting the prevalence of this phage genus in the oceans.
Conclusions
These results describe the first Oceanospirillum phage, vB_OliS_GJ44, that represents a novel viral cluster and exhibits interesting genetic features related to phage–host interactions and evolution. Thus, we propose a new viral genus Oceanospirivirus within the Siphoviridae family to reconcile this cluster, with vB_OliS_GJ44 as a representative member.
Similar content being viewed by others

Background
From the ocean surface to the hadal zones and from the Arctic to the Antarctic, viruses are the most abundant and diverse life forms in the ocean [1, 2]. They control the microbial community through infection and lysis of their hosts, which promote biogeochemical cycling through the “viral shunt” and “viral shuttle” [3]. Viruses also mediate the horizontal gene transfer and the evolution of their hosts and contribute to marine carbon sequestration through the “biological pump” and “microbial carbon pump” [4,5,6]. However, more than 90% of the viral population remains unknown [7]. Thus, an increase in phage identification will promote a better understanding of their evolution and their effects on microbial communities and biogeochemical cycles.
Oceanospirillum is the type genus of the family Oceanospirillaceae, in the order Oceanospirillales of the class Gammaproteobacteria. Members of this family have often been found in oil-contaminated habitats [1, 8,9,10], and are well known for their ability to degrade petroleum hydrocarbons [11]. They are also abundant in the Mariana Trench, suggesting potentially important roles in extreme environments [12]. Currently, six Oceanospirillum species have been identified from habitats including coastal areas, sediments, the deep-sea, putrid infusions of marine mussels and especially from oil-contaminated environments [13,14,15,16,17]. Despite the ecological importance of this bacteria lineage, our knowledge about the viruses infecting Oceanospirillaceae is quite few. Currently, only six phages infecting Oceanospirillaceae have been isolated so far, including five infecting Marinomonas and one infecting Nitrincola. Phages infecting other genera of Oceanospirillum have yet not been isolated.
In this study, we isolated and characterized the first bacteriophage infecting Oceanospirillum, vB_OliS_GJ44. It was found to possess novel genomic features and represented a novel siphoviral cluster. Combined with the eight environmental viral contigs from metagenomics, this study helps fill the gap in our understanding of the isolation, genomic and evolutionary development of Oceanospirillum bacteriophages and provides new insights into the interactions between hosts and bacteriophages for these important marine hydrocarbon-degrading microbial populations.
Materials and methods
Isolation of host Oceanospirillum sp. ZY01 and phage vB_OliS_GJ44
Oceanospirillum sp. ZY01 and its phage vB_OliS_GJ44 were both isolated from surface water samples in the Yellow Sea (35°23′59.582″N, 119°34′7.158″E) in October, 2019. 2216E media (peptone 5 wt.%, yeast extract 1 wt.%) dissolved in artificial seawater (Sigma) was used to culture and propagate the host. The host was able to be grown efficiently in shake cultivation at 28 °C and 120 rpm.
To obtain a concentrated sample of the phage, 50 L of coastal water was concentrated to 10 ml by tangential flow filtration with 50-kDa and 30-kDa cartridges, (Pellicon® XL Cassette, Biomax® 50 kDa; polyethersulfone, Millipore Corporation, Billerica, MA, USA), after passing through a 0.2-μm membrane filter (Isopore™ 0.2 μm GTTP; Merck, Ireland) [18]. A PE centrifuge tube was used to retain the concentrated viral stock, which was then stored in the dark place at 4 °C.
The double-layer plating method was used to isolate the phage. Briefly, 200 μl of concentrated viral stock was mixed with the host culture (approximately 10-h) and incubated for 20-min, allowing the absorption of the phages at room temperature. Then, 4 ml of the semi-solid culture at 45 °C was added into the mixture, pouring onto the plate after vortex. Plates were cultivated at 27 °C for 24-h and visible plaques were formed in the double layer culture.
Purification and concentration of vB_OliS_GJ44
A single plaque was picked, placed in SM buffer and shaken for 3-h at 120 rpm to dissociate the viral particles from the agar. The mixture was passed through a 0.22 μm membrane filter and allowed to infect the host, as described above. This step was repeated five times to obtain purified viral stock.
To concentrate the viral stock, 5 ml of purified viral stock was incubated with 50 ml of the exponentially growing host at 28 °C for 12-h. The mixture was filtered through a 0.22-μm membrane filter to harvest phages particles, and PFU (plaque-forming unit) was counted by flow cytometry to assess the efficiency of propagation.
The lysate was concentrated from 50 ml to 2 ml using an ultrafilter (Milipore® Amicon Ultra-15) under 5000 g. And the concentrated and purified viral stock was stored in the dark place at 4 °C.
Morphological identification, host range test and one-step growth of vB_OliS_GJ44
The morphology of vB_OliS_GJ44 was characterized by transmission electron microscopy (TEM) using established protocols of the negative staining method [19]. A drop of 20 μl concentrated, purified viral stock (~ 109 PFU/ml) was placed on the copper net, stained with 2 wt.% phosphotungstic acids (pH 7.5) for 5 min, and then observed under the TEM (JEOLJEM-1200EX, Japan) at 100 KV.
The host range test was performed using the double-layer plating method on 35 Oceanospirillales strains. In summary, different bacterial cultures were mixed with a series of viruses in multiples according to the optimal multiplicity of infection (MOI); the mixture was then spread on a soft agar layer. Plaque formation was observed after incubating overnight at 28 °C.
The one-step growth assay was conducted following Sillankorva S. et al. [20]. Briefly, the exponentially growing host culture (~ 108 CFU/L) was mixed with vB_OliS_GJ44 stock under the MOI 0.01 and incubated for 30 min. Then, the mixture was centrifuged (6000 g) for 5 min and the supernatant discarded to remove unabsorbed phages, the pellet was then resuspended in 1 ml of 2216E medium. This step was repeated three times and the sample was then transferred to 300 ml 2216E medium and shaken at 28 °C for 180-min. Sampling was conducted throughout the incubation at 10-min intervals. Each sample was immediately fixed with glutaraldehyde (final concentration: 0.5%), flash-frozen in the liquid nitrogen and stored at − 80 °C prior to analysis. Flow cytometry was used to count the viral particles of each sample, as described above (water bath for 10 min at 80 °C). Three parallel tests were conducted for this assay.
The phylogeny of Oceanospirillum sp. ZY01
A total of 121 Oceanospirillaceae reference sequences of 16S rRNA genes, including the host strain Oceanospirillum sp. ZY01, were retrieved from GenBank and aligned by mafft [21] using G-INS-1 of strategy with 1000 iterations. The phylogenic tree was calculated from multiple sequence alignments using IQ-tree2 [22], applying GTR + F + R4 as the suggested DNA model with 1000 iterations of bootstrap. The tree was visualized by iTOL v4 [23].
Genome sequencing and annotation of vB_OliS_GJ44
Sequencing was performed by Shanghai Biozeron Biotechnology Co., Ltd. (Shanghai, China.). The high-quality DNA sample was used to construct an Illumina pair-end library and then used for Illumina NovaSeq 6000 sequencing. The raw paired-end reads were trimmed and quality controlled by Trimmomatic (v. 0.3.6) with parameters: SLIDINGWINDOW:4:15, MINLEN:75 [24]. ABySS was used to assemble the viral genome after the quality control processes, multiple-Kmer parameters were chosen to obtain the optimal assembly results [25]. GapCloser software was subsequently applied to fill in the remaining local inner gaps and to correct the single base polymorphism for the final assembly and for further analysis [26].
Coding DNA sequences of phage vB_OliS_GJ44 were predicted using GeneMarkS [27], RAST [28], and Glimmer [29]. All open reading frames (ORFs) were annotated by BLASTp and Position-Specific Iterated BLAST (PSI-BLAST), against the nonredundant proteins (NR) NCBI database (e-value was set at 1e-5, identity > 30%). PSI-BLAST was used to identify the putative proteins in the structural gene cluster of the phage (non-default parameters: num_iterations 1000, e-value <1e-5, query coverage (qcov) > 50%). The InterPro database [30], the Conserved Domain Database suite (CDD/SPARCLE) [31], the UniProtKB database [32], and the HHpred server [33] were used to detect the conserved domain in every ORF. Possible inconsistencies, produced by different prediction and annotation tools, were checked manually. Easyfig v2.2.2 was used for genome visualization and tRNAscan-SE was used for tRNA gene detection [34, 35].
Moreover, GC skew analysis was performed on Webskew, which is the online version of Genskew (https://genskew.csb.univie.ac.at/webskew).
Phylogenetic analysis of vB_OliS_GJ44
The major capsid protein (MCP) was selected as the hallmark protein to be identified by BLASTp from the NR database. A total of 50 best hit sequences were selected and aligned using MUSCLE [36], with e- value 1E-150, 99% coverage and 65% identity cutoff. A maximum-likelihood phylogenetic tree was generated using MEGA v10 [37] and visualized with iTOL v4 [23]. Another phylogenetic tree constructed for the terminase large subunit (TerL) was undertaken in the same way as for MCP.
A proteomic tree based on the similarities of the whole genome was generated using VIPTree (https://www.genome.jp/viptree/) [38]. Each encoding nucleic sequences as a query were searched against the Virus-Host DB using tBLASTx. All viral sequences in Virus-Host DB were selected to generate a circular tree. The 461 related phages in the circular tree were automatically selected by VIPtree according to genomic similarity scores (SG) larger than 0.05, then used to generate a more accurate phylogenetic tree with vB_OliS_GJ44.
Three conserved genes (MCP, TerL, and portal protein) were selected as hallmark proteins to build a polygenic phylogenetic tree of the extended vB_OliS_GJ44. Homologous proteins of three hallmark proteins were identified using Diamond blastp (v0.9.4.105), with e-value 1 × 10− 5 and 85% qcov cut-off. These sequences were retrieved and aligned using MUSCLE [36]. Sixty-seven viral genomes with at least two of the three homologous hallmark proteins were selected. Gap were removed from the alignment with trimal [39] and connected with seqkit [40]. A maximum-likelihood phylogenetic tree was then calculated based on the concatenated alignment of all three proteins with IQ-tree2 with ultrafast bootstrap 1000 and VT + F + R4 as suggested by the model test as the best-fit substitution model [22]. The phylogenetic tree was visualized with iTOL v4 [23].
Phage vB_OliS_GJ44 homologs in IMG/VR
To expand the phage vB_OliS_GJ44 group, each coding sequence was queried against the IMG/VR [41] database using tBLASTx to search for homologous proteins and to map the contig ID (threshold: e-value <1e-5, idendity > 20, −max_target_seqs 1). Virus contigs with more than five homologous sequences were selected and removed the low-quality contigs according the information of IMG/VR [41]. Finally, 25 uncultivated high-quality virus contigs and 13 isolated sequences were selected as references, of which eight Brucella phages appeared as an outgroup. All these 27 sequences and the vB_OliS_GJ44 genome were used to construct the whole-genome phylogenetic tree using VIPTree [38].
Average nucleotide sequence identity was calculated by OAT software, which used the orthogonal method to determine the overall similarity between the two genomic sequences [42].
Environmental distribution of phage vB_OliS_GJ44
The relative abundance of vB_OliS_GJ44 was assessed through three marine viral metagenomic datasets, including Pacific Ocean Virome (POV) [43], Global Ocean Sampling (GOS) [44] (available at CAMERA (http://camera.calit2.net), and Malaspina (available at www.pangaea.de) viral metagenomes [45]. A total 67 viruses were retrieved from the three datasets and eciprocal best-hit BLASTp (RBB), as applied by Zhao et al. [46], was used to avoid potential false-positive homologies. To identify the homologs of vB_OliS_GJ44 proteins, BLAST nucleic acid libraries were built from each virome, and proteins of vB_OliS_GJ44 were compared against libraries by tBLASTn (non-default parameters -max_target_seqs 10,000,000, −max_hsps 1, −seg no, −outfmt 6). Then, subjects matched in the last step were extracted and were compared against the proteins of vB_OliS_GJ44 by BLASTx (non-default parameter: -max_target_seqs 10,000,000, −max_hsps 1, −seg no, −outfmt 6) Reciprocal best hits were retained as the final result. The relative abundance of each ORF was calculated by two normalizations, the total number of reads in each metagenome and the number of amino acids of each ORF.
Tetranucleotides (tetra) correlations analysis
Thirty-four fragments were sliced from the nucleic acid sequence of vB_OliS_GJ44 (10 kbp for window size, 1 kbp for step size), the sequence was extended to both sides to avoid the bias of uneven slicing. Thus, each 1 kbp of the genome could be presented by a corresponding 10 kbp fragment. Two hundred fifty-six combinations of tetra frequency (from “AAAA” to “TTTT”) were calculated for each fragment, and normalized by z-scoring. The Pearson’s correlation coefficient was calculated from either the array of each fragment and that of the host genome as a whole, or the array of each fragment and that of the viral genome as a whole [47].
Results and discussion
Isolation, morphology, host range, and one-step growth
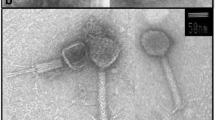
The bacteriophage vB_OliS_GJ44, infecting Oceanospirillum sp. ZY01 (accession: MW547060), was isolated from a surface seawater sample from the Yellow Sea; this is the first reported phage infecting this genus. Infection by vB_OliS_GJ44 formed clear and round (2–3 mm diameter average) plaques. The center of the plaque was more transparent than the rest (Fig. 1B). TEM results show that the vB_OliS_GJ44 viral particle possesses a siphoviral morphology. Measurements of 20 vB_OliS_GJ44 phage particles showed it had an icosahedral head, with an average diameter of 47 nm, and a long non-contractile tail, with an average length of 76 nm (Fig. 1A). The graph of TEM showed an interesting and special structure in the middle of the tail, which is similar to a tail filament. To the best of our knowledge, vB_OliS_GJ44 is the first phage where the tail filament is located in other positions of the tail.

Morphology and biological properties of phage vB_OliS_GJ44. A Electron micrographs of Oceanospirillum phage vB_OliS_GJ44. vB_OliS_GJ44 lysate was stained with 4% uranyl acetate on a copper grid and viewed with a Philips/FEI transmission electron microscope. B Phage plaques formed in double-layer agar plate after culturing 24 h. C Increase in phage titers during one-step growth. The data shown are averages from triplicate experiments, and error bars indicate SDs
The host cross-infection experiment showed that phage vB_OliS_GJ44 has a narrow host range. Of the 35 strains tested, it was found to only infect four strains of Oceanospirillum scanctuarii OLL623, OSL14, OSX334, and its propagating host bacterium ZY01 (Table 1). It could not infect Oceanospirillum scanctuarii 1A14960, even though they have a close evolutionarily relationship. This result is consistent with our understanding of the species specificity of siphoviruses. The one-step growth curve of phage vB_OliS_GJ44 showed the latent period was approximately 35 min and reached a growth plateau after 70 min. The burst size was approximately 107 viral particles released from each cell (Fig. 1C).
The phylogeny of Oceanospirillum sp. ZY01
From the phylogenic tree based on the 16S rRNA gene of Oceanospirillum sp. ZY01 and other 120 reference sequences of Oceanospirillaceae (Fig. 2), Oceanospirillum sp. ZY01 was the most closely related to Oceanospirillum sanctuarii strain AK56, but had farther distance length from the branch root (n = 0.047) than Oceanospirillum sanctuarii strain AK56 (n = 0.002), indicating that O. sp. ZY01 might represent a novel variant of Oceanospirillum sanctuarii.

The phylogenic tree based on the 16S rRNA gene of Oceanospirillum sp. ZY01 and other 120 reference 16S rRNA gene sequences of Oceanospirillaceae
Genomic features of Phage vB_OliS_GJ44
According to the sequencing and assembly results, vB_OliS_GJ44 had a 33,786-bp linear dsDNA genome with a GC content of 48.8%. No tRNA was found in the genome. The genome had a 92% encoding rate consisting of 60 predicted ORFs. There were 24 coding regions (40%) that did not match any homologous sequence under the restriction of e-value <1e-5 in all 60 coding DNA sequences (CDS). Among the remaining 36 CDS that matched homologous sequences, 32 identified specific functions, and 4 matched homologous sequences with proteins of unknown function. The 36 ORFs could be classified into six different modules: 19 ORFs for phage structure and packing proteins, seven for DNA replication and metabolism, six for recombination, two for lysis, and one auxiliary metabolic gene (AMG). The remaining ORFs were all classified into hypothetical proteins. Forty-eight genes are located on the sense strand, accounting for 80% of the total coding genes. There were few genes on the antisense strand, only twelve, eleven of which were continuous (ORF 38 - ORF 48), including all six recombination genes. In contrast, there were many and various genes on the sense strand (Fig. 3A, Additional file 1: Table S1). The cumulative GC skew analyses was performed in order to determine the origin and terminus of replication of the phage genome. The results (Fig. 3B) indicate that the origin of replication is at the position 500 nt, and a replication terminus could be located at the region 33,500 nt. Two inflection points were identified at above regions, indicating an asymmetric base composition, which are the lowest at the origin and the highest at the terminus [48]. The annotation of genome showed that the first gene encoded a replication protein (20–766 nt), which provided additional support to the origin of replication (Additional file 1: Table S1).

A Circularized genome map of vB_OliS_GJ44. The outer circle represents genes. Putative functional categories were defined according to annotation and are represented by different colors. The second circle shows the length of the genome, the green arc represents the length of the tail-related genes, and the third circle is a tetranucleotides correlation. The weaker correlations are circled by a red ellipse. B Cumulative GC skew analysis of the phage genome sequence. The global minimum and maximum are displayed in the cumulative graph were calculated by using a window size of 1,00 bp and a step size of 100 bp. The GC-skew and the cumulative GC-skew are represented by blue and red lines, respectively. The minimum and maximum of a GC-skew can be used to predict the origin of replication (500 nt) and the terminus location (33,500 nt)
Genes related to the DNA replication and metabolism
The DNA replication protein encoded by ORF 1, classified to the DNA replication and metabolism module, had a helix-turn-helix domain, a common denominator in basal and specific transcription factors in bacterial cells. They have been recruited to a wide range of functions, not only transcription regulation and DNA repair and replication, but also RNA metabolism, and protein-protein interactions [49]. KilA-N domain-containing protein (ORF 14) was a novel, conserved DNA-binding domain found at the N-terminus of the poxvirus D6R/NIR proteins, which may play a role as nuclease domains mediate additional and specific interactions with nucleic acids or proteins. Its homologs have been widely detected in large bacteria or eukaryotic DNA viruses and even in some protozoa and fungal DNA-binding APSES domains [50, 51].
Recombination module in the genome of phage vB_OliS_GJ44
Tetra correlations between per 10 kb genome fragments of vB_OliS_GJ44 and its whole genome are shown in Fig. 3A. The high score demonstrates the higher adaptive ability of the genes to their genome. In the red elliptical part of Fig. 3A, which includes seven fragments (from 26th to 32th), the tetra correlations drop significantly, indicating that this sequence was less adapted to its genome. These seven fragments correspond to six recombinant genes and one AMG. AMG, which is a group of genes that can modulate host cell metabolism, has a closer relationship with the host genome [52]. The 28th fragment (Fig. 3A) has the lowest tetranucleotide frequency correlation (0.83), further indicated that the recombination module was more closely related to the host.
The recombination module included six proteins. RusA can resolve Holliday intermediates and correct the defects in genetic recombination and DNA repair associated with the inactivation of RuvA, RuvB, or RuvC [53]. Following a previous report, the RecG pathway of junction resolution can be stimulated by the expression of RusA resolvase, whose gene resides on a cryptic prophage, such as prophage lambda [54]. The recombination enhancement function of RecA-dependent nuclease is a 21-kDa RecA-dependent HNH endonuclease that can be targeted to produce a double-strand break at any desired DNA sequence [55]. This gene was first reported in the genome of Escherichia phage P1, which is a prophage infection enterobacter. The unique signature of prophage P1 is the lysogenic strategy in the cell, which acts as a low copy of plasmid in the cell on its lysogenic stage [56]. Typically, both dsDNA and ssDNA could be bound by RecA-dependent nuclease, but will not produce cleavage to ssDNA. Cofactors or proteins, such as RecA, ATP, or Mg2+ are required for RecA-dependent nuclease degrading ssDNA [57]. The protein NinB is located in enterophage lambda, which is one of the components of NinR in ORF family recombinases of lambda, specifically binding to ssDNA [58]. The YqaJ viral recombinase protein family might play a similar role to exonuclease in enterophage lambda, that integrases to the chromosome of the host through recombination and which have been demonstrated to have a crucial role in viral replication. The ERF family protein was first reported in Salmonella phage P22, which also promotes homologous recombination like the Red system in phage lambda [59, 60]. ERF protein has been commonly observed in temperate bacteriophages infecting Gammaproteobacteria, and could promote circularization of the linear dsDNA viral genome upon entry into the host cell [61, 62]. The combination module carried by vB_OliS_GJ44 could play a vital role in its replication in a host cell. The ssDNA-binding protein located in this module might interact with multiple recombination genes, as RusA family crossover junction endodeoxyribonuclease, protein NinB, Yqajdomain-containing exonuclease, and ERF family protein could act on ssDNA under certain conditions. Many genes within this module might play a similar role to the recombination process in phage lambda. However, there has been no integrase annotated for vB_OliS_GJ44, and a homolog of recombinase ORF of phage lambda [63] was not observed in the genome of vB_OliS_GJ44. Also unexpected was the presence of two phage antirepressor proteins (ORF 7 and ORF 60) “Phage antirepressor KilAC domain-containing protein”, which prevents the repressor protein of the P22 434 and lambda-like moderate prophage from binding to its operators, turn on the transcription of phage genes and promote propagation [64, 65]. This indicates that vB_OliS_GJ44 has a different strategy from the mild lambda-like phages. Given this, the propagation pathway in its host bacterium is unclear; the mechanism of recombination and propagation in vB_OliS_GJ44 requires further in-depth study.
Tail-related genes of phage vB_OliS_GJ44
Compared with other siphoviruses that can infect Oceanospirillaceae (Marinomonas phage CPP1m 3, Marinomonas phage CB5A 3, Nitrincola phage 1 M3–16 3, Marinomonas phage P12026 1, and Marinomonas phage CPG1g 3), the number of tail-related proteins of vB_OliS_GJ44 was surprisingly high. A total of 13 genes were determined to be tail-related or cell adsorption and recognition proteins after PSI-BLAST analysis of all structural genes. The green line on the second circle of the genemap, accounting for 33% of the coding region (10,218 bp/31152 bp), represented the length of this region. These genes are tightly assembled into a continuous cluster.
ORF 29 was homologous with gene transfer agent family protein of Bordetella genomo sp. 7 with 98% coverage and 33% amino acid identity. In PSI-BLAST, most hits are of bacterial GTA (Gene transfer agent) proteins, which are derived from bacteria and archaea and are used to regulate horizontal gene transfer [66]. They are virus-like particles containing DNA fragments that can escape from mother cells and adhere to other cells to inject their DNA into the cytoplasm [67]. ORF 29 also hit tail protein sequences, except GTA protein in PSI-BLAST; it was speculated that ORF 29 mainly functions as a tail component in bacteriophages and identifying host cells.
ORF32 was annotated as a discoidin domain-containing protein, and homologues of this protein are widespread in bacteria proteins rather than phages in the NR database and usually play a role in cell adsorption. Discoidin domain-containing protein is a type of lectin, with an for galactose, that mediates cell adhesion and migration in the slime mould Dictostelial discoideum [68, 69]. The DS domain receptor family where the discoidin domain has usually been detected is in the cell outer membrane. It can bind to lipids such as glycans, polysaccharides, and collagens to regulates cell adhesion [70]. This protein is present in the phage genome and located in the tail protein cluster, so it may be related to the recognition of the receptor protein on the surface of the host cell.
The tails of siphoviruses are very efficient nanomachines, designed to infect the host, with extremely high specificity and effectiveness. They are essential for recognizing, attaching and piercing the host cell wall to ensure efficient delivery of genomic DNA to the host cytoplasm and determine the phage-specific characteristics, such as host range strategies [71]. The rich and diverse tail-related genes in the vB_OliS_GJ44 genome play an important role in the formation of the tail structure and the interaction between hosts.
AMG and lysis genes
The only AMG in the whole genome vB_OliS_GJ44 is the MazG-like family protein (ORF 44), which regulates host cell metabolism and promotes infection efficiency during the process of bacteriophage infection of the host, is found in bacteriophages but originated from bacteria [50]. Similarity has been observed with the dimeric 2-deoxyuridine 5′-triphosphate nucleotidohydrolase (dUTP pyrophosphatase or dUTPase) and NTP-PPase MazG proteins. However, members of this family consist of a single MazG-like domain that contains a well-conserved divalent ion-binding motif EXXE/D, different from the typical tandem-domain MazG proteins [72]. Studies have suggested that the viral MazG protein may reduce the content of guanosine 3′,5′-bispyrophosphate (ppGpp) in the host, deceive the host into maintaining a ‘hungry state’, and accelerate the metabolism of the host bacteria to promote their reproduction [73,74,75]. However, given that the gene of NTP pyrophosphohydrolase was located in the recombination module, it my alternatively have some unknown function in the recombination progress of vB_OliS_GJ44, which requires further investigation.
The genome also encoded N-acetylmuramoyl-L-alanine amidase, a phage lysin, which catalyzes the chemical bond between N-acetylmuramoyl residues and L-alanine residues in cell wall glycopeptides [76], has been shown to be highly similar to the same protein predicted in a Marinomonas phage P12026 genome [77]. Both the genera Marinomonas and Oceanospirillum are classified into the family Oceanospiraceae. The TMhelix containing protein, which is close behind in the genome encoded by ORF 17 homologated with Vibrio phages of Autographiviridae, is related to the transport of substances across cell membranes [78] and may be related to infecting and lysing host.
Phylogenetic analysis suggested that vB_OliS_GJ44 represents a novel viral cluster
To further understand the phylogenetic relationship of vB_OliS_GJ44 to other isolated phages, three different types of phylogenetic trees were generated: single-gene, multi-genes, and whole-genome. MCP and TerL phylogenetic trees were established using 98 and 50 sequences respectively, with the highest homology through BLASTp against the NR database. In the MCP tree, (Fig. 4A) 53 homologous bacterial sequences and 45 virus sequences were selected, vB_OliS_GJ44 presents a separate branch and is far from the other sequences. Twenty homologous bacterial sequences and 30 phage sequences were constructed by the TerL tree (Fig. 4B), although vB_OliS_GJ44 is grouped with some Vibrio protein sequences, the branch lengths are 0.35 and 0.75, respectively, so the evolutionary distances are also relatively far.

Phylogenetic trees of vB_OliS_GJ44 based on three different methods. A, B Unrooted maximum-likelihood dendrogram derived from amino acid sequences of the phage major capsid protein. and terminase large subunit respectively. The green branches represent that the protein sequences are from bacteria, and the black branches are from phages. C Phylogenetic analysis with other related phages identified using the genome-wide sequence similarity values computed by tBLASTx. D Maximum-likelihood phylogenetic tree of the vB_OliS_GJ44 inferred from a concatenated protein alignment of three hallmark proteins (MCP, TerL, and portal protein). Four shades of different colors indicate the boundaries of clades. Tree annotations from inside to the outside: (1) host lineage (2) assembly size
Seventy-six viruses were selected according to the sequence similarity and their MCP, TerL-related genes and portal protein were connected in series to establish a multi-genes phylogenetic tree. Among them, vB_OliS_GJ44 originates from the tree root and forms a separate clade (Fig. 4D). In the phylogenetic tree based on 461 viral whole-genomes, nine siphoviruses are clustered together, but the branch length was about 0.48; this further demonstrates that vB_OliS_GJ44 represents a novel siphoviral cluster (Fig. 4C).
These results show that the identification of vB_OliS_GJ44 not only expands the catalog of marine Oceanospirillum phages but also represents a new cluster of marine phages. As the first isolation of a phage from genus Oceanospirillum and classified into a novel viral cluster, we propose that vB_OliS_GJ44 represents a novel viral genus, named Oceanospirivirus, in the Siphoviridae.
The relationship between vB_OliS_GJ44 and uncultured phage contigs
During the last decade, through the application of metagenomics understanding of viral diversity has expanded rapidly, identifying 195,728 viral taxa from the global ocean. The was accomplished through a combination of isolation and genomic analyses, especially from the dominant and important bacterial groups, such as Synechococcus, Roseobacter, Pseudoalteromonas, Alteromonas, and Vibrio from coastal areas and Pelagibacter (SAR 11), Puniceispirillum (SAR 116) and Prochlorococcus from the open ocean [46, 79, 80].
vB_OliS_GJ44 lacks an obvious connection with the isolated virus strains in the NCBI virus database, perhaps because only a few phage isolates infect Oceanospirillum. Therefore, tBLASTx was used to search the IMG/VR [41] database in an attempt to expand the Oceanospirivirus database. The virus sequences in the IMG/VR [41] database is all derived from the assembly of metagenomic data. In total, 27 uncultured viruses were screened with at least 7 common genes. Thirteen isolated viruses together with vB_OliS_GJ44 were added to construct a genome-level phylogenetic tree. Phage vB_OliS_GJ44 and its closest relative, Station85_MES_COMBINED_FINAL_NODE_1213 (Station85_1213), are grouped into a diverse clade containing ten other marine phages, which shared the same node (Fig. 5A).

Comparisons of vB_OliS_GJ44 with other related uncultured phages in IMG/VR database. A Phylogenetic analysis with other related uncultured phages in IMG/VR database using the genome-wide sequence similarity values computed by tBLASTx. B Heat map based on OrthoANI values calculated using OAT software
The Bacterial and Archaeal Viruses Subcommittee (BAVS) of the International Committee on the Taxonomy of Viruses (ICTV) considers phages sharing ≥50% nucleotide sequence identity as members of the same genus [81]. In Fig. 5B, the highest average nucleotide identity (ANI, 81.44%) was between vB_OliS_GJ44 and IMGVR_UViG_3300019752_000029, and the smallest ANI is 58.82% with Station85_1213. This result provided further support for the suggestion that vB_OliS_GJ44 and the uncultured page contigs may represent a new cluster genus, Oceanospirivirus, which is likely to be widely distributed.
Comparative genomic analysis between vB_OliS_GJ44 and uncultured phages
In the comparative genomic analysis, vB_OliS_GJ44 showed some similarities to six uncultured phage contigs, most of which had similar genes that were continuous and concentrated in tail-related genes (Fig. 6). It is common to find some homologous genes encoding viral structural proteins among different Caudovirales genomes [82,83,84,85]. Tail-related genes are essential to the tail-phages for host adsorption and DNA ejection through the baseplate and the most effective gene arrangements. Unexpectedly, although synchronization was observed in all of the genome from tail-related virion proteins (ORF 24) to tail fiber proteins (ORF 35) (Fig. 6), there was almost no synchronization in other regions, except for Station85_1213, which had some synchronicity in the upstream area (terminase, MCP, and capsid assembly scaffolding protein) of the tail-related genes cassette. This lack of synchronization is probably due to the high genetic variability between these host recognition proteins. Indeed, a high level of variability among tail fibers has been reported several times [86].

Comparative genomic analysis of the tail-related genes cassette of vB_OliS_GJ44 and other uncultured phage contigs. Sequence comparisons performed using tBLASTx (10 bp minimum alignment) with percent identity shown as a black box (inset scale bar). Synteny was recognized when genomes featured a minimum of five consecutive syntenic genes within the same genomic area and separated by a maximum of four non syntenic genes
Distribution of ORF homologues of vB_OliS_GJ44 in marine viromes
Predicted ORFs from vB_OliS_GJ44 genomes were used to estimate relative abundances in quantitative POV, GOS, Malaspina viral metagenomes using a reciprocal best-BLAST approach with minor modifications. A total of 433 reads were successfully recruited at rates of approximately 10− 10 per amino acid pair in all three databases. In contrast, the ORF abundance was higher in GOS-estuary and POV-coastal areas, with 1.19E-07 and 1.33E-07 assigned reads per amino acid pair respectively (Table 2). Metagenomic analysis indicated that vB_OliS_GJ44 might be widely distributed in the ocean with low relative abundance. The relative abundances of vB_OliS_GJ44 in four viral metagenomes collected from the bathypelagic zone, > 4000 m, during the Malaspina Expedition (2011) were also investigated. Data shows that the abundance of phages in deep water is stable at 10− 9 reads per pair of amino acids.
Homologous sequences of each of the 60 ORFs were found in the database but the top five ORFs, with higher recruitment rates, were ORF 2 (AAA family ATPase, 2.84E-07 per pair), ORF 9 (DUF1289 domain-containing protein, 1.19E-07 per pair),
ORF 10 (N-acetylmuramoyl-L-alanine amidase, 4.68E-07 per pair), ORF 15 (Phage terminase small subunit, 2.90E-07 per pair), ORF 16 (Putative large terminase, 9.30E-08 per pair), which are mainly associated with phage replication, packaging, and lysis modules. A similar situation has also been found in other marine phages, such as Erythrobacter phage vB_EliS-R6L [87]. Several ORFs only have hits in a certain database, such as ORF39 (Hypothetical protein) and ORF53 (Hypothetical protein), that were only detected in the POV-open dataset. Similarly, ORF 22 (Hypothetical protein), ORF 25 (Putative head-tail joining protein), ORF 29 (Hypothetical protein), ORF 30 (Hypothetical protein), ORF 34 (Hypothetical protein), ORF 37 (Hypothetical protein), ORF 38 (Hypothetical protein), ORF 49 (Hypothetical protein), and ORF 58 (DNA-binding transcriptional regulator) were only detected in the POV-coast database (Fig. 7). The top five ORFs with the most recruitment were relatively abundant in each database. These results indicate that vB_OliS_GJ44 may represent a new and unknown ecological pedigree and provide a reference genome for the classification of environmental marine viral contigs in the future.

Relative abundance of homologs of vB_OliS_GJ44 phage genes in the metagenome datasets
Conclusions
Oceanospirillum has a very special niche and its phage will inevitably affect its community structure and metabolic efficiency. vB_OliS_GJ44 is the first isolated phage to infect Oceanospirillum. There are a large number of tail genes and a unique host-adapted recombination module in its genome architecture. Its evolutionary linage is novel and represents a cluster together with some uncultured virus sequences. This study has provided the first glimpse of the diversity, genomic evolution, abundance, and distribution of phages infecting Oceanospirillum. It provides a model interaction system and some new insights into interactions between Oceanospirivirus and Oceanspirillum phage-driven evolution and dynamics of their hosts, and the potential ecological significance of Oceanospirivirus. This study reinforces the importance of the combination of phage isolation and metagenomics to improve our knowledge of marine virus functions and diversity. Future isolation of phages infecting other Oceanospirillum species may disclose more novel phage clusters.
Availability of data and materials
The genome sequence of phage vB_OliS_GJ44 is available in the GenBank repository, https://www.ncbi.nlm.nih.gov/nuccore/MW560978
The 16S rRNA gene sequence of the host, Oceanospirillum sp. ZY01 is available in the GenBank repository, https://www.ncbi.nlm.nih.gov/nuccore/MW547060
Abbreviations
- % GC:
-
Percent guanine: cytosine
- qcov:
-
Query coverage
- AMG:
-
Auxiliary metabolic gene
- ANI:
-
Average nucleotide identity
- CDS:
-
Coding DNA sequences
- GOS:
-
Global Ocean Sampling
- GTA:
-
Gene transfer agent
- MCP:
-
Major capsid protein
- MOI:
-
Optimal multiplicity of infection
- NR:
-
Nonredundant proteins
- ORF:
-
Open reading frames
- PFU:
-
Plaque-forming unit
- POV:
-
Pacific Ocean Virome
- PSI-BLAST:
-
Position-Specific Iterated BLAST
- RBB:
-
Reciprocal best-hit BLASTp
- TEM:
-
Transmission electron microscopy
- TerL:
-
Terminase large subunit
- Malaspina_:
-
Malaspina viral metagenomes
- PFU:
-
Plaque forming unit
- CFU:
-
Colony-forming unit
References
Mugge RL, Brock ML, Salerno JL, Damour M, Church RA, Lee JS, et al. Deep-sea biofilms, historic shipwreck preservation and the Deepwater horizon spill. Front Mar Sci. 2019;6:48. https://doi.org/10.3389/fmars.2019.00048.
Aristegui J, Gasol JM, Duarte CM, Herndl GJ. Microbial oceanography of the dark ocean’s pelagic realm. Limnol Oceanogr. 2009;54(5):1501–29. https://doi.org/10.4319/lo.2009.54.5.1501.
Zimmerman AE, Howard-Varona C, Needham DM, John SG, Worden AZ, Sullivan MB, et al. Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nat Rev Microbiol. 2020;18(1):34–21. https://doi.org/10.1038/s41579-019-0270-x.
Weinbauer MG, Hornák K, Jezbera J, Nedoma J, Dolan JR, Šimek K. Synergistic and antagonistic effects of viral lysis and protistan grazing on bacterial biomass, production and diversity. Environ Microbiol. 2007;9(3):777–88. https://doi.org/10.1111/j.1462-2920.2006.01200.x.
Winter C, Herndl GJ, Weinbauer MG. Diel cycles in viral infection of bacterioplankton in the North Sea. Aquat Microb Ecol. 2004;35:207–16. https://doi.org/10.3354/ame035207.
Zhang R, Weinbauer MG, Qian PY. Viruses and flagellates sustain apparent richness and reduce biomass accumulation of bacterioplankton in coastal marine waters. Environ Microbiol. 2007;9(12):3008–18. https://doi.org/10.1111/j.1462-2920.2007.01410.x.
Gregory AC, Zayed AA, Conceição-Neto N, Temperton B, Bolduc B, Alberti A, et al. Marine DNA viral macro- and microdiversity from pole to pole. Cell. 2019;177(5):1109–23. https://doi.org/10.1016/j.cell.2019.03.040.
Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The prokaryotes: Gammaproteobacteria. Berlin, Heidelberg: Springer; 2013. p. 540–21. https://doi.org/10.1007/978-3-642-30123-0.
Coulon F, Chronopoulou PM, Fahy A, Païssé S, Goñi-Urriza M, Peperzak L, et al. Central role of dynamic tidal biofilms dominated by aerobic hydrocarbonoclastic bacteria and diatoms in the biodegradation of hydrocarbons in coastal mudflats. Appl Environ Microbiol. 2012;78(10):3638–48. https://doi.org/10.1128/AEM.00072-12.
Redmond MC, Valentine DL. Natural gas and temperature structured a microbial community response to the Deepwater horizon oil spill. Proc Natl Acad Sci U S A. 2012;109(50):20292–7. https://doi.org/10.1073/pnas.1108756108.
Kleindienst S, Paul JH, Joye SB. Using dispersants after oil spills: impacts on the composition and activity of microbial communities. Nat Rev Microbiol. 2015;13(6):388–96. https://doi.org/10.1038/nrmicro3452.
Liu J, Zheng Y, Lin H, Wang X, Li M, Liu Y, et al. Proliferation of hydrocarbon-degrading microbes at the bottom of the Mariana trench. Microbiome. 2019;7(1):1–13. https://doi.org/10.1186/s40168-019-0652-3.
Sidhu C, Thakur S, Sharma G, Tanuku NRS, Pinnaka AK. Oceanospirillum sanctuarii sp. Nov., isolated from a sediment sample. Int J Syst Evol Microbiol. 2017;67(9):3428–34. https://doi.org/10.1099/ijsem.0.002132.
Sass AM, Sass H, Coolen MJL, Cypionka H, Overmann J. Microbial communities in the chemocline of a hypersaline Deep-Sea basin (Urania Basin, Mediterranean Sea). Appl Environ Microbiol. 2001;67(12):5392–402. https://doi.org/10.1128/AEM.67.12.5392-5402.2001.
Terasaki Y. Transfer of five species and two subspecies of Spirillum to other genera (Aquaspirillum and Oceanospirillum), with emended descriptions of the species and subspecies. Int J Syst Bacteriol. 1979;29(2):130–44. https://doi.org/10.1099/00207713-29-2-130.
Coulon F, McKew BA, Osborn AM, McGenity TJ, Timmis KN. Effects of temperature and biostimulation on oil-degrading microbial communities in temperate estuarine waters. Environ Microbiol. 2007;9(1):177–86. https://doi.org/10.1111/j.1462-2920.2006.01126.x.
Voordouw G, Armstrong SM, Reimer MF, Fouts B, Telang AJ, Shen Y, et al. Characterization of 16s rRNA genes from oil field microbial communities indicates the presence of a variety of sulfate-reducing, fermentative, and sulfide-oxidizing bacteria. Appl Environ Microbiol. 1996;62(5):1623–9. https://doi.org/10.1128/aem.62.5.1623-1629.1996.
Yang Q, Gao C, Jiang Y, Wang M, Zhou X, Shao H, et al. Metagenomic characterization of the viral community of the south scotia ridge. Viruses. 2019;11(2):1–19. https://doi.org/10.3390/v11020095.
Deveau H, Labrie SJ, Chopin MC, Moineau S. Biodiversity and classification of lactococcal phages. Appl Environ Microbiol. 2006;72(6):4338–46. https://doi.org/10.1128/AEM.02517-05.
Sillankorva S, Neubauer P, Azeredo J. Isolation and characterization of a T7-like lytic phage for Pseudomonas fluorescens. BMC Biotechnol. 2008;8(1):80. https://doi.org/10.1186/1472-6750-8-80.
Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2018;20(4):1160–6. https://doi.org/10.1093/bib/bbx108.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37(5):1530–4. https://doi.org/10.1093/molbev/msaa015.
Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):256–9. https://doi.org/10.1093/nar/gkz239.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–23. https://doi.org/10.1101/gr.089532.108.
Xu M, Guo L, Gu S, Wang O, Zhang R, Peters BA, et al. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience. 2020;9(9):1–11. https://doi.org/10.1093/gigascience/giaa094.
Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29(12):2607–18. https://doi.org/10.1093/nar/29.12.2607.
Aziz RK, Bartels D, Best A, DeJongh M, Disz T, Edwards RA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1):57. https://doi.org/10.1186/1471-2164-9-75.
Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics. 2007;23(6):673–9. https://doi.org/10.1093/bioinformatics/btm009.
Blum M, Chang HY, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021;49(D1):344–54. https://doi.org/10.1093/nar/gkaa977.
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45(D1):200–3. https://doi.org/10.1093/nar/gkw1129.
Morgat A, Lombardot T, Coudert E, Axelsen K, Neto TB, Gehant S, et al. Enzyme annotation in UniProtKB using Rhea. Bioinformatics. 2020;36:1896–901.
Gabler F, Nam SZ, Till S, Mirdita M, Steinegger M, Söding J, et al. Protein sequence analysis using the MPI bioinformatics toolkit. Curr Protoc Bioinforma. 2020;72(1):1. https://doi.org/10.1002/cpbi.108.
Sullivan MJ, Petty NK, Beatson SA. Easyfig: A genome comparison visualizer. Bioinformatics. 2011;27(7):1009–10. https://doi.org/10.1093/bioinformatics/btr039.
Lowe TM, Chan PP. tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44(W1):W54–7. https://doi.org/10.1093/nar/gkw413.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;2(5):1792–7. https://doi.org/10.1093/nar/gkh340.
Stecher G, Tamura K, Kumar S. Molecular evolutionary genetics analysis (MEGA) for macOS. Mol Biol Evol. 2020;37(4):1237–9. https://doi.org/10.1093/molbev/msz312.
Nishimura Y, Yoshida T, Kuronishi M, Uehara H, Ogata H, Goto S. ViPTree: the viral proteomic tree server. Bioinformatics. 2017;33(15):2379–80. https://doi.org/10.1093/bioinformatics/btx157.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3. https://doi.org/10.1093/bioinformatics/btp348.
Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11(10):1–10. https://doi.org/10.1371/journal.pone.0163962.
Roux S, Páez-Espino D, Chen IMA, Palaniappan K, Ratner A, Chu K, et al. IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021;49(D1):D764–75. https://doi.org/10.1093/nar/gkaa946.
Lee I, Kim YO, Park SC, Chun J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol. 2016;66(2):1100–3. https://doi.org/10.1099/ijsem.0.000760.
Hurwitz BL, Sullivan MB. The Pacific Ocean Virome (POV): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS One. 2013;8:2.
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, et al. The sorcerer II Global Ocean sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5(3):0398–431. https://doi.org/10.1371/journal.pbio.0050077.
Duarte CM. Seafaring in the 21st century: the Malaspina 2010 circumnavigation expedition. Limnol Oceanography Bull. 2015;24(1):11–4. https://doi.org/10.1002/lob.10008.
Zhao Y, Temperton B, Thrash JC, Schwalbach MS, Vergin KL, Landry ZC, et al. Abundant SAR11 viruses in the ocean. Nature. 2013;494(7437):357–60. https://doi.org/10.1038/nature11921.
Duhaime MB, Wichels A, Waldmann J, Teeling H, Glöckner FO. Ecogenomics and genome landscapes of marine Pseudoalteromonas phage H105/1. ISME J. 2011;5(1):107–21. https://doi.org/10.1038/ismej.2010.94.
Uchiyama J, Rashel M, Takemura I, Wakiguchi H, Matsuzaki S. In silico and in vivo evaluation of bacteriophage φEF24C, a candidate for treatment of enterococcus faecalis infections. Appl Environ Microbiol. 2008;74(13):4149–63. https://doi.org/10.1128/AEM.02371-07.
Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev. 2005;29(2):231–62. https://doi.org/10.1016/j.femsre.2004.12.008.
Pritham EJ, Putliwala T, Feschotte C. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene. 2007;390(1-2):3–17. https://doi.org/10.1016/j.gene.2006.08.008.
Iyer LM, Koonin EV, Aravind L. Extensive domain shuffling in transcription regulators of DNA viruses and implications for the origin of fungal APSES transcription factors. Genome Biol. 2002;3(3):3. https://doi.org/10.1186/gb-2002-3-3-research0012.
Crummett LT, Puxty RJ, Weihe C, Marston MF, Martiny JBH. The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology. 2016;499:219–29. https://doi.org/10.1016/j.virol.2016.09.016.
Punatar RS, Martin MJ, Wyatt HDM, Chan YW, West SC. Resolution of single and double Holliday junction recombination intermediates by GEN 1. Proc Natl Acad Sci U S A. 2017;114(3):443–50. https://doi.org/10.1073/pnas.1619790114.
Sharples GJ, Curtis FA, McGlynn P, Bolt EL. Holliday junction binding and resolution by the rap structure-specific endonuclease of phage λ. J Mol Biol. 2004;340(4):739–51. https://doi.org/10.1016/j.jmb.2004.05.030.
Gruber AJ, Olsen TM, Dvorak RH, Cox MM. Function of the N-terminal segment of the RecA-dependent nuclease ref. Nucleic Acids Res. 2015;43(3):1795–803. https://doi.org/10.1093/nar/gku1330.
Ronayne EA, Wan YCS, Boudreau BA, Landick R, Cox MM. P1 ref endonuclease: a molecular mechanism for phage-enhanced antibiotic lethality. PLoS Genet. 2016;12(1):1. https://doi.org/10.1371/journal.pgen.1005797.
Gruenig MC, Lu D, Won SJ, Dulberger CL, Manlick AJ, Keck JL, et al. Creating directed double-strand breaks with the ref protein: a novel RecA-dependent nuclease from bacteriophage P1. J Biol Chem. 2011;28(10):8240–51. https://doi.org/10.1074/jbc.M110.205088.
Curtis FA, Malay AD, Trotter AJ, Wilson LA, Barradell-Black MMH, Bowers LY, et al. Phage Orf family recombinases: conservation of activities and involvement of the central channel in DNA binding. PLoS One. 2014;9(8):8. https://doi.org/10.1371/journal.pone.0102454.
Noirot P, Kolodner RD. DNA strand invasion promoted by Escherichia coli RecT protein. J Biol Chem. 1998;273(20):12274–80. https://doi.org/10.1074/jbc.273.20.12274.
Kowalczykowski EC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58(3):401–65. https://doi.org/10.1128/mr.58.3.401-465.1994.
Botstein D, Matz MJ. A recombination function essential to the growth of bacteriophage P22. J Mol Biol. 1970;54(3):417–40. https://doi.org/10.1016/0022-2836(70)90119-1.
Weaver S, Levine M. Recombinational circularization of Salmonella phage P22 DNA. Virology. 1977;76(1):29–38. https://doi.org/10.1016/0042-6822(77)90278-1.
Sawitzke JA, Stahl FW. Phage λ has an analog of Escherichia coli recO, recR and recF genes. Genetics. 1992;130(1):7–16. https://doi.org/10.1093/genetics/130.1.7.
Phizicky EM, Roberts JW. Kinetics of recA protein-directed inactivation of repressors of phage λ and phage P22. J Mol Biol. 1980;139(3):319–28. https://doi.org/10.1016/0022-2836(80)90133-3.
Silpe JE, Bassler BL. A host-produced quorum-sensing autoinducer controls a phage lysis-Lysogeny decision. Cell. 2019;176(1-2):268–80. https://doi.org/10.1016/j.cell.2018.10.059.
Grüll MP, Mulligan ME, Lang AS. Small extracellular particles with big potential for horizontal gene transfer: membrane vesicles and gene transfer agents. FEMS Microbiol Lett. 2018;365(19). https://doi.org/10.1093/femsle/fny192.
Lang AS, Westbye AB, Beatty JT. The distribution, evolution, and roles of gene transfer agents in prokaryotic genetic exchange. Annu Rev Virol. 2017;4(1):87–104. https://doi.org/10.1146/annurev-virology-101416-041624.
Baumgartner S, Hofmann K, Chiquet-Ehrismann R, Bucher P. The discoidin domain family revisited: new members from prokaryotes and a homology-based fold prediction. Protein Sci. 1998;7(7):1626–31. https://doi.org/10.1002/pro.5560070717.
Kiedzierska A, Smietana K, Czepczynska H, Otlewski J. Structural similarities and functional diversity of eukaryotic discoidin-like domains. Biochim Biophys Acta. 2007;1774(9):1069–78. https://doi.org/10.1016/j.bbapap.2007.07.007.
Villoutreix BO, Miteva MA. Discoidin domains as emerging therapeutic targets. Trends Pharmacol Sci. 2016;37(8):641–59. https://doi.org/10.1016/j.tips.2016.06.003.
Veesler D, Cambillau C. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol Mol Biol Rev. 2011;75(3):423–33. https://doi.org/10.1128/MMBR.00014-11.
Bryan MJ, Burroughs NJ, Spence EM, Clokie MRJ, Mann NH, Bryan SJ. Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS One. 2008;3(4):1–12. https://doi.org/10.1371/journal.pone.0002048.
Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010;12(11):3035–56. https://doi.org/10.1111/j.1462-2920.2010.02280.x.
Zhang J, Inouye M. MazG, a nucleoside triphosphate pyrophosphohydrolase, interacts with era, an essential GTPase in Escherichia coli. J Bacteriol. 2002;184(19):5323–9. https://doi.org/10.1128/JB.184.19.5323-5329.2002.
Gross M, Marianovsky I, Glaser G. MazG - a regulator of programmed cell death in Escherichia coli. Mol Microbiol. 2006;59(2):590–601. https://doi.org/10.1111/j.1365-2958.2005.04956.x.
Baker JR, Liu C, Dong S, Pritchard DG. Endopeptidase and glycosidase activities of the bacteriophage B30 lysin. Appl Environ Microbiol. 2006;72(10):6825–8. https://doi.org/10.1128/AEM.00829-06.
Kang I, Jang H, Oh H-M, Cho J-C. Complete genome sequence of Marinomonas bacteriophage P12026. J Virol. 2012;86(16):8909–10. https://doi.org/10.1128/JVI.01328-12.
Kauffman KM, Hussain FA, Yang J, Arevalo P, Brown JM, Chang WK, et al. A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nature. 2018;554(7690):118–22. https://doi.org/10.1038/nature25474.
Kang I, Oh HM, Kang D, Cho JC. Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. Proc Natl Acad Sci U S A. 2013;110(30):12343–8. https://doi.org/10.1073/pnas.1219930110.
Yang Y, Cai L, Ma R, Xu Y, Tong Y, Huang Y, et al. A novel roseosiphophage isolated from the oligotrophic South China Sea. Viruses. 2017;9(5):109. https://doi.org/10.3390/v9050109.
Gonzalez-Serrano R, Dunne M, Rosselli R, Martin-Cuadrado A-B, Grosboillot V, Zinsli LV, et al. Alteromonas Myovirus V22 Represents a New Genus of Marine Bacteriophages Requiring a Tail Fiber Chaperone for Host Recognition. mSystems. 2020;5:1–18.
Brüssow H, Hendrix RW. Phage genomics: small is beautiful. Cell. 2002;108(1):504–10. https://doi.org/10.1016/S0092-8674(01)00637-7.
Comeau AM, Bertrand C, Letarov A, Tétart F, Krisch HM. Modular architecture of the T4 phage superfamily: a conserved core genome and a plastic periphery. Virology. 2007;362(2):384–96. https://doi.org/10.1016/j.virol.2006.12.031.
Hatfull GF, Hendrix RW. Bacteriophages and their genomes. Curr Opin Virol. 2011;1(4):298–303. https://doi.org/10.1016/j.coviro.2011.06.009.
Brewer TE, Elizabeth Stroupe M, Jones KM. The genome, proteome and phylogenetic analysis of Sinorhizobium meliloti phage ΦM12, the founder of a new group of T4-superfamily phages. Virology. 2014;450:84–97. https://doi.org/10.1016/j.virol.2013.11.027.
Born Y, Fieseler L, Marazzi J, Lurz R, Duffy B, Loessner MJ. Novel virulent and broad-host-range Erwinia amylovora bacteriophages reveal a high degree of mosaicism and a relationship to Enterobacteriaceae phages. Appl Environ Microbiol. 2011;77(17):5945–54. https://doi.org/10.1128/AEM.03022-10.
Lu L, Cai L, Jiao N, Zhang R. Isolation and characterization of the first phage infecting ecologically important marine bacteria Erythrobacter. Virol J. 2017;14(1):104. https://doi.org/10.1186/s12985-017-0773-x.
Acknowledgments
We sincerely thank Jia Zhen, School of Computer Science and Technology, Guizhou University, for his help in data processing. We thank the three anonymous reviewers for their constructive comments and suggestions. We thank for the support of the high-performance servers of Center for High Performance Computing and System Simulation, Pilot National Laboratory for Marine Science and Technology (Qingdao), the Marine Big Data Center of Institute for Advanced Ocean Study of Ocean University of China, the IEMB-1, a high-performance computing cluster operated by the Institute of Evolution and Marine Biodiversity, and the high-performance servers of Frontiers Science Center for Deep Ocean Multispheres and Earth System.
Funding
The research was financially supported by the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao)(No.2018SDKJ0406–6), the National Key Research and Development Program of China (2018YFC1406704), the Fundamental Research Funds for the Central Universities (202072002, 201812002, Andrew McMinn), National Natural Science Foundation of China (No. 41976117, 41606153), and 973 Program (No. 2013CB429704).
Author information
Authors and Affiliations
Contributions
WZ performed main experiments, bioinformatic analyses and annotated the genome, and drafted the manuscript. YL (Yantao Liang) and MW planned, supervised, and coordinated the study and revised the manuscript. AM helped to modify the language of the manuscript. KZ performed the tetranucleotide correlations analysis and edited the manuscript. CG (Chengxiang Gu) and ZW conducted the biological characterization experiments and take TEM figures of phage vB_OliS_GJ44. YL (Yundan Liu) and XZ guided the physiological experiment. HS, YJ, CG (Cui Guo), HH, HW, YYS, WJM conceived and designed the experiments and critically evaluated the manuscript. YZ provided thirty-one different bacteria strains for the host range test. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file1: Table S1.
Genome annotation of phage vB_OliS_GJ44.
Additional file 2.
16S rRNA gene sequences of the bacteria used in host range test.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, W., Liang, Y., Zheng, K. et al. Characterization and genomic analysis of the first Oceanospirillum phage, vB_OliS_GJ44, representing a novel siphoviral cluster. BMC Genomics 22, 675 (2021). https://doi.org/10.1186/s12864-021-07978-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07978-4