
Abstract
The success of many current vaccines relies on a formulation that incorporates an immune activating adjuvant. This will hold true for the design of a successful therapeutic HIV vaccine targeted at controlling reactivated virus following cessation of combined antiretroviral therapy (cART). The HIV accessory protein Nef functions by interfering with HIV antigen presentation through the major histocompatibility complex I (MHC-I) pathway thereby suppressing CD8+ cytotoxic T cell (CTL)-mediated killing of HIV infected cells. Thus, this important impediment to HIV vaccine success must be circumvented. This review covers our current knowledge of Nef inhibitors that may serve as immune adjuvants that will specifically restore and enhance CTL-mediated killing of reactivated HIV infected cells as part of an overall vaccine strategy to affect a cure for HIV infection.
Similar content being viewed by others

Background
Over the last 25 years, cART has evolved into a highly effective therapy that prolongs the lifespan of HIV infected individuals. However, it only targets actively replicating virus in permissive cells [1]. Indeed, infected individuals contain tissue reservoirs that replicate low levels of virus and may continually express low levels of Nef, one of HIV’s key pathogenicity-associated accessory proteins [2, 3]. The goals of current HIV cure research are to eliminate these reservoirs thereby purging infected patients of HIV. An integral portion of this strategy is the reactivation of latent virus (shock) and the subsequent elimination of the cells producing the reawakened HIV (kill) by the immune response. An IL-15 agonist, ALT-803, is currently in phase 2 trials due to its ability to enhance the CTL response [4]. However, shock and kill strategies will always be confronted with the ability of reactivated HIV-1 to produce a functional Nef protein that mediates critical HIV immune evasion effects. Indeed, HIV-1 Nef suppresses MHC-I antigen presentation in infected cells thereby blunting any therapeutic HIV vaccine efficacy.
Nef alters host cellular trafficking pathways to attenuate the immune response
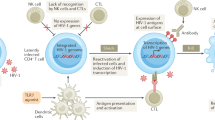
During the early stages of the HIV life cycle, the most heavily transcribed gene within infected cells is nef. This robust Nef expression affects infected cells in numerous ways, including downregulation of key cell surface receptors such as MHC-I and CD4 [3], enhancement of viral replication [5], alteration of T cell activation [6], and the subversion of the apoptotic machinery [7]. The downregulation of CD4 prevents superinfection of the cell and antibody-dependent cellular cytotoxicity [8, 9]. The downregulation of MHC-I attenuates the cytotoxic T-lymphocyte (CTL) recognition mechanism that seeks and destroys infected cells, allowing HIV-1-infected cells to evade the CTL immune response [10]. Nef’s ability to disrupt the CTL response is counter to the multiple shock and kill approaches currently used to target the latent reservoir of HIV-1 [1, 11] many of which include the use of a therapeutic vaccine prior to cessation of cART as part of a cure strategy. Thus, inhibition of Nef represents an essential arm of any anti-HIV shock and kill cure therapy that can restore and boost the efficacy of the anti-HIV CTL response (Fig. 1).

Shock and kill therapies to cure HIV infection require the inhibition of Nef activity. Shock therapies aim to reactivate latent HIV (shock and to eliminate virus-producing cells (kill). Viral reactivation will enhance Nef activity to evade the immune surveillance system by decreasing cell surface levels of MHC-I on CD4+ T cells (+Nef). Evasion: the interaction between Nef and a Src Family Kinase (SFK) results in the intracellular retention of MHC-I. Subsequently, a CD8+T lymphocyte (CTL) will fail to recognize an HIV infected cell. Killing: conversely, the inhibition of Nef’s activity using molecular adjuvants, such as 2c-like compounds (red hexagon) that block the interaction between Nef and SFKs (−Nef) will restore cell surface levels of MHC-I which in turn will promote HIV antigen presentation (purple dot) and enhance susceptibility to HIV specific CD8+-CTL
We have defined the mechanism used by Nef to downregulate cell surface MHC-I. This pathway, which ultimately mutes immune responses, requires the sequential use of multiple evolutionally conserved Nef motifs [3]. Indeed, conservation of these motifs in the pandemic M group of HIV-1, which is responsible for over 90% of AIDS cases worldwide, suggests they control essential pathways [12]. First, the Nef EEEE65 acidic cluster is required for trafficking Nef to paranuclear compartments, including an endosomal sub-population and the trans-Golgi network (TGN), through its interaction with the membrane trafficking regulator PACS-2 [13]. Second, the PxxP75 SH3 domain allows Nef to activate a TGN-localized Src Family Kinase (SFK) [14]. The activated Nef–SFK complex then recruits and activates the tyrosine kinase ZAP-70, which activates phosphatidylinositol-3 kinase (PI3K) triggering endocytosis of cell-surface MHC-I molecules which exhibit delayed recycling to the plasma membrane [15]. The membrane trafficking regulator PACS-1 would then contribute with the heterotetrameric adaptor protein-1 (AP-1) complex to sequester MHC-I away from the cell surface [13, 16, 17]. Thus, the Nef–SFK interaction attenuates the CTL response.
The Nef PxxP75 site
The importance of the Nef–SFK interaction has been confirmed in vivo using a mouse model expressing Nef or a mutated Nef PxxP75 deficient in SFK binding (Nef AxxA75) in CD4+ T-cells, macrophages, and dendritic cells from the CD4 gene promoter (CD4C/HIV) [18]. Transgenic mice expressing Nef AxxA75 were completely protected from the AIDS-like phenotype induced by wild-type Nef, indicating an intact PxxP75 domain is critical for Nef-induced pathogenesis [18]. Thus, the Nef AxxA75 transgenic mouse model suggested that disruption of Nef-PxxP75 interactions (such as Nef–SFK) largely abolishes the pathogenic potential of Nef and prevents the AIDS-like phenotype.
Along with the Nef–SFK requirement in MHC-I downregulation, the inverse ability of Nef to enhance viral replication is also dependent on Nef–SFK interactions. Cells expressing Nef AxxA75 were deficient in Nef-mediated enhancement of viral replication by fivefold [19]. In addition, we described how disrupting the Nef–SFK interaction with the small molecule 2c blocked Nef-mediated MHC-I downregulation in human primary CD4+ T-cells [13]. Subsequently, 2c was also reported to inhibit Nef PxxP75-dependent HIV-1 infectivity and replication in macrophages [19]. Similar results were obtained in human monocytes using diphenylfuropyriminde compounds, which block Nef PxxP75-dependent HIV replication by directly inhibiting SFK activity [20]. Thus, small molecule inhibitors of the Nef PxxP75 interaction can repress two key aspects of Nef activity by: (1) preventing the downregulation of MHC-I which is key to disrupting the CTL response and (2) blocking Nef’s ability to enhance HIV replication in the primary human cells HIV normally replicates in and can exist latently in. Both are key elements required of any shock and kill therapeutic vaccine strategy to be an effective long-term cure for HIV infection strategy. Eliminating HIV infected cells while minimizing infection of new T cells will enhance the possibilities of eliminating HIV reservoirs.
Conclusions
Due to Nef’s primary role in the pathogenesis of HIV-1, the identification of novel small molecules that block key aspects of Nef function creates an important opportunity for the generation of a new class of molecular adjuvants that will augment current therapeutic vaccine approaches such that maximal anti-HIV CTL activity can be achieved. Current efforts to generate and test a second generation of 2c-like Nef inhibitors with greater affinity for the Nef–SFK interface are under way. While 2c-like inhibitors block the Nef–SFK-mediated effects on MHC I surface expression and virus production, inhibitors that block the other functions of Nef still need to be found. Ultimately, this will lead the way to future non-human primate pre-clinical trials and subsequently to human trials. We believe a successful therapeutic HIV vaccine strategy applied pre-cART cessation necessitates the concomitant inhibition of Nef’s activity in order to maximize efficacy of shock and kill therapeutic strategies. Immunotherapies based on the MHC-I independent chimeric antigen receptor (CAR)-engineered T cells targeted specifically to HIV infected cells [21] will still benefit from the presence of 2c-like Nef inhibitors as they will ensure that virus production from reactivated T cells is kept to a minimum. In this way, vaccine- and CAR-based immunotherapies can be optimized for HIV-1 infected individuals such that the chances of a permanent cure may be realized.
References
Cary DC, Peterlin BM. Targeting the latent reservoir to achieve functional HIV cure. F1000Res. 2016;5.doi:10.12688/f1000research.8109.1
Ostalecki C, Wittki S, Lee J-H, Geist MM, Tibroni N, Harrer T, Schuler G, Fackler OT, Baur AS. HIV Nef- and Notch1-dependent endocytosis of ADAM17 induces vesicular TNF secretion in chronic HIV infection. EBioMedicine. 2016;13:294–304.
Pawlak EN, Dikeakos JD. HIV-1 Nef: a master manipulator of the membrane trafficking machinery mediating immune evasion. Biochem Biophys Acta. 2015;1850(4):733–41.
Seay K, Church C, Zheng JH, Deneroff K, Ochsenbauer C, Kappes JC, Liu B, Jeng EK, Wong HC, Goldstein H. In vivo activation of human NK cells by treatment with an interleukin-15 superagonist potently inhibits acute in vivo HIV-1 infection in humanized mice. J Virol. 2015;89(12):6264–74.
Papkalla A, Munch J, Otto C, Kirchhoff F. Nef enhances human immunodeficiency virus type 1 infectivity and replication independently of viral coreceptor tropism. J Virol. 2002;76(16):8455–9.
Skowronski J, Parks D, Mariani R. Altered T cell activation and development in transgenic mice expressing the HIV-1 nef gene. EMBO J. 1993;12(2):703.
Rasola A, Gramaglia D, Boccaccio C, Comoglio PM. Apoptosis enhancement by the HIV-1 Nef protein. J Immunol. 2001;166(1):81–8.
Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol. 2007;81(8):3877–90.
Veillette M, Désormeaux A, Medjahed H, Gharsallah N-E, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol. 2014;88(5):2633–44.
Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391(6665):397–401.
Jones RB, Walker BD. HIV-specific CD8(+) T cells and HIV eradication. J Clin Investig. 2016;126(2):455–63.
Kirchhoff F, Easterbrook PJ, Douglas N, Troop M, Greenough TC, Weber J, Carl S, Sullivan JL, Daniels RS. Sequence variations in human immunodeficiency virus type 1 Nef are associated with different stages of disease. J Virol. 1999;73(7):5497–508.
Dikeakos JD, Atkins KM, Thomas L, Emert-Sedlak L, Byeon IJ, Jung J, Ahn J, Wortman MD, Kukull B, Saito M, et al. Small molecule inhibition of HIV-1-induced MHC-I down-regulation identifies a temporally regulated switch in Nef action. Mol Biol Cell. 2010;21(19):3279–92.
Atkins KM, Thomas L, Youker RT, Harriff MJ, Pissani F, You H, Thomas G. HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation analysis using short interfering rna and knock-out mice. J Biol Chem. 2008;283(17):11772–84.
Hung CH, Thomas L, Ruby CE, Atkins KM, Morris NP, Knight ZA, Scholz I, Barklis E, Weinberg AD, Shokat KM, et al. HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe. 2007;1(2):121–33.
Dirk BS, Pawlak EN, Johnson AL, Van Nynatten LR, Jacob RA, Heit B, Dikeakos JD. HIV-1 Nef sequesters MHC-I intracellularly by targeting early stages of endocytosis and recycling. Sci Rep. 2016;6:37021.
Dikeakos JD, Thomas L, Kwon G, Elferich J, Shinde U, Thomas G. An interdomain binding site on HIV-1 Nef interacts with PACS-1 and PACS-2 on endosomes to down-regulate MHC-I. Mol Biol Cell. 2012;23(11):2184–97.
Hanna Z, Weng X, Kay DG, Poudrier J, Lowell C, Jolicoeur P. The pathogenicity of human immunodeficiency virus (HIV) type 1 Nef in CD4C/HIV transgenic mice is abolished by mutation of its SH3-binding domain, and disease development is delayed in the absence of Hck. J Virol. 2001;75(19):9378–92.
Chutiwitoonchai N, Hiyoshi M, Mwimanzi P, Ueno T, Adachi A, Ode H, Sato H, Fackler OT, Okada S, Suzu S. The identification of a small molecule compound that reduces HIV-1 Nef-mediated viral infectivity enhancement. PLoS ONE. 2011;6(11):e27696.
Emert-Sedlak L, Kodama T, Lerner EC, Dai W, Foster C, Day BW, Lazo JS, Smithgall TE. Chemical library screens targeting an HIV-1 accessory factor/host cell kinase complex identify novel antiretroviral compounds. ACS Chem Biol. 2009;4(11):939–47.
Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, Scharenberg AM, Rawlings DJ, Wagner TA. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T Cells. Mol Ther. 2017;25(3):570–9.
Authors’ contributions
JDD and GAD wrote the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
We thank Emily Pawlak for help with the artwork.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
Work in the Dikeakos lab is supported by the Canadian Institutes of Health Research (MOP-286719). Work in the Dekaban lab is supported by Canadian Institutes of Health Research (PJT-148651).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dekaban, G.A., Dikeakos, J.D. HIV-I Nef inhibitors: a novel class of HIV-specific immune adjuvants in support of a cure. AIDS Res Ther 14, 53 (2017). https://doi.org/10.1186/s12981-017-0175-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12981-017-0175-6