

Abstract
Combination antiretroviral therapy (ART) can block multiple stages of the HIV-1 life cycle to prevent progression to AIDS in people living with HIV-1. However, owing to the persistence of a reservoir of latently infected CD4+ T cells, life-long ART is necessary to prevent viral rebound. One strategy currently under consideration for curing HIV-1 infection is known as ‘shock and kill’. This strategy uses latency-reversing agents to induce expression of HIV-1 genes, allowing for infected cells to be cleared by cytolytic immune cells. The role of innate immunity in HIV-1 pathogenesis is best understood in the context of acute infection. Here, we suggest that innate immunity can also be used to improve the efficacy of HIV-1 cure strategies, with a particular focus on dendritic cells (DCs) and natural killer cells. We discuss novel latency-reversing agents targeting DCs as well as DC-based strategies to enhance the clearance of infected cells by CD8+ T cells and strategies to improve the killing activity of natural killer cells.
Similar content being viewed by others

Introduction
Since the identification of HIV-1 in 1983 (ref.1), there have been major advances in the treatment and prevention of infection using antiretroviral therapy (ART). However, despite the effectiveness of ART, there is currently no cure for HIV-1 infection and life-long treatment is necessary to maintain suppression of HIV-1 replication owing to viral persistence in a latent reservoir2,3,4,5. The latent reservoir is a long-lived population of resting CD4+ T cells that harbour intact proviruses that are transcriptionally silent but retain the ability to produce infectious virions6,7,8. There are various methods used to quantify the size of the latent reservoir and the way in which reservoir size is estimated has implications for the interpretation of results (Box 1). Latently infected cells can persist through clonal expansion resulting from homeostatic, antigen-driven or integration site-driven proliferation9, which is a major barrier to the elimination of latently infected cells. Therefore, for most people living with HIV-1 (PLWH), the cessation of ART leads to viral rebound, typically after approximately 2 weeks10.
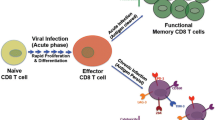
There are many proposed strategies for HIV-1 cure. These include approaches to achieve either control of viral replication without ongoing treatment or a complete elimination of infectious virus. The former strategy may involve therapeutic vaccines to promote immune responses against HIV-1 and thus allow for sustained immune-mediated suppression of viraemia. By contrast, the latter strategy will require a significant, multi-log reduction in the size of the latent reservoir to prevent viral rebound11. Recent efforts to eliminate the latent reservoir have focused on the ‘shock and kill’ strategy. This two-step strategy uses latency-reversing agents (LRAs) to ‘shock’ latently infected cells, thereby inducing the transcription of HIV-1 genes. This, in turn, results in the death of infected cells owing to viral cytopathic effects or it allows for immune cells to recognize and kill infected cells9,12 (Fig. 1).

Latently infected CD4+ T cells contain the HIV-1 genome integrated into the host cell genome and remain undetectable by other immune cells owing to the lack of HIV-1 gene expression. Antiretroviral therapy (ART) prevents active viral replication but is unable to eliminate latently infected cells. Toll-like receptor 7 (TLR7) agonists and dendritic cells presenting cognate antigen can help to reactivate (‘shock’) infected cells and induce the transcription of HIV-1 genes, leading to the production of viral proteins. The infected cells can then be recognized and ‘killed’ by cytolytic effectors such as cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. These effectors release granzymes and perforin to induce apoptotic cell death, leading to the elimination of infected cells. A reduction in the size of the latent reservoir is characterized by a decrease in the number of CD4+ T cells harbouring intact proviruses (Box 1).
Many LRAs have been identified in in vitro studies and several of these have been tested in vivo13. The LRAs tested in vivo have largely focused on reversing epigenetic blocks to HIV-1 gene expression using histone deacetylase inhibitors such as vorinostat, romidepsin and panobinostat14,15,16. Other latency-reversing strategies involve inducing some degree of T cell activation, for example, through protein kinase C agonists17. In resting CD4+ T cells, key host transcription factors necessary for HIV-1 gene expression are absent or sequestered in an inactive form. Activation of T cells allows for nuclear translocation of host transcription factors involved in the initiation of HIV-1 gene transcription, such as NF-κB and NFAT, as well as recruitment of the host transcription elongation factor P-TEFb18,19,20. In addition, recent evidence has shown that some LRAs might mediate their effects through stress response pathways involving the transcription factor HSF1, which can help to recruit transcription elongation factors to the HIV-1 promoter21,22. However, compared with global T cell activation, most candidate LRAs are ineffective at reversing latency ex vivo in cells from PLWH and, so far, they have failed to reduce the size of the latent reservoir in vivo23,24,25,26. Moreover, both HIV-1 infection27,28,29 and treatment with specific LRAs30,31 have been previously shown to impair the functions of CD8+ T cells and natural killer (NK) cells, both of which are involved in the clearance of HIV-1-infected cells. Therefore, novel therapeutic approaches are being explored for both reactivation and elimination of the latent reservoir of HIV-1 (Table 1).
The innate immune system is the first line of defence against foreign pathogens. However, the role of innate immunity during acute and chronic HIV-1 infection has not yet been fully explored. Importantly, recent studies show that both transmitted/founder viruses and viruses that are present in the plasma during initial rebound after cessation of ART have high levels of resistance to type I interferon responses32,33 (Box 2), which indicates that interferon-sensitive viral variants are inhibited and highlights the importance of targeting the innate immune system to achieve a cure. A better understanding of how HIV-1 escapes immune control could be achieved through further examination of the resistance of reservoir viruses to key elements of the innate and adaptive immune responses.
As the mechanism of recognition of HIV-1 infection by the innate immune system is not as well understood as recognition by the adaptive immune system, innate immunity has so far received less research attention in terms of HIV-1 cure strategies. However, it is becoming clear that innate immune pathways may contribute to both the reversal of HIV-1 latency and the subsequent elimination of infected cells. Indeed, some current trials of HIV-1 cure strategies are using therapeutics that engage aspects of the innate immune response to reactivate or eliminate latently infected cells (Table 1). In this Review, we discuss various approaches for engaging components of the innate immune system to eradicate the latent reservoir of HIV-1, focusing on the potential roles of dendritic cells (DCs) and NK cells.
Reactivating latently infected cells
DCs are a diverse population of innate immune cells with a unique ability to link innate and adaptive immune responses34. Based on the differential expression of key transcription factors, DCs are classified into three major subsets: plasmacytoid DCs (pDCs) and two types of myeloid conventional DCs (cDC1s and cDC2s)35,36. pDCs recognize viral nucleic acids in the form of single-stranded RNA or unmethylated CpG-rich DNA through their endosomal Toll-like receptors (TLR7 or TLR9, respectively). Activation of these TLRs in pDCs induces the transcription of type I interferon genes as well as of genes encoding other pro-inflammatory cytokines, such as CC-chemokine ligand 4 (CCL4), IL-6 and tumour necrosis factor (TNF), that contribute to the activation of other immune cell types37,38,39 (Fig. 2). By contrast, cDCs are highly efficient at capturing and processing antigens in peripheral tissues, trafficking to draining lymph nodes, and presenting the antigens to naive T cells together with costimulatory and polarizing signals40,41.

Monocytes from people living with HIV-1 (PLWH) are cultured with granulocyte–macrophage colony-stimulating factor (GM-CSF), IL-4, additional maturation-associated cytokines and sources of HIV-1 antigens to generate IL-12p70-producing mature monocyte-derived dendritic cells (moDCs). These DCs loaded with HIV-1 antigens ex vivo are returned to the donor to induce HIV-1-specific CD8+ T cell responses (cytotoxic T lymphocyte (CTL) responses) and overcome dysfunctional antigen presentation by conventional DCs (cDCs). Recent DC-based vaccination efforts have also involved the transduction of moDCs with CD40L as a strategy to enhance their maturation, increase IL-12p70 production and aid effective antigen presentation to T cells. Emerging studies also suggest that moDCs can induce the activation of CD4+ T cells and implicate moDCs in the induction of latency reversal. One recent study brought attention to canonical antigen presentation by DCs and CD40–CD40L signalling in latency reversal but the mechanism remains unexplained. Other strategies to reactivate latently infected CD4+ T cells involve the use of Toll-like receptor 7 (TLR7) agonists (such as GS-986 and GS-9620) to induce a type I interferon (IFN) response by plasmacytoid dendritic cells (pDCs) and the production of other pro-inflammatory cytokines associated with the antiviral response (such as CCL4 and IL-6) to activate other immune cells. TCR, T cell receptor.
The mechanisms by which HIV-1 avoids detection by DCs at multiple stages of infection have been described in detail elsewhere42,43,44. These reviews offer insights into approaches to enhance the innate sensing pathways of DCs and thereby improve adaptive immunity to HIV-1. Here, we describe strategies that engage innate immunity through manipulating DC responses to reactivate latently infected cells and improve the efficacy of HIV-1 cure efforts.
Toll-like receptor agonists
Of particular interest with respect to HIV-1 latency is TLR7, which recognizes single-stranded RNA in endosomes and can also be activated by small molecule agonists such as imidazoquinoline analogues or guanosine cyclic triphosphates45,46,47. Although, in humans, TLR7 is predominantly expressed by pDCs (Box 2) and B cells48,49, rather than by CD4+ T cells, several studies indicate that TLR7 agonists can affect the latent reservoir of HIV-1 by enhancing innate immune responses. In a study of peripheral blood mononuclear cells isolated from PLWH, treatment with the TLR7 agonist GS-9620 (vesatolimod) induced HIV-1 RNA production, which is suggestive of active transcription of HIV-1 genes from reactivated latently infected cells50. The depletion of pDCs from the culture, which decreased IFNα production in response to GS-9620, resulted in reduced activation of both CD4+ and CD8+ T cells. In addition, blocking the IFNα receptor in combination with GS-9620 treatment of CD4+ T cells did not result in increased HIV-1 RNA expression. Together, the results suggest that the latency reversal mediated by GS-9620 depends on the presence of pDCs and IFNα secretion50.
Rhesus macaques infected with simian immunodeficiency virus (SIV) establish a latent reservoir in resting CD4+ T cells, which makes these non-human primates an accepted in vivo model to study various aspects of the latent reservoir relevant to HIV-1 (ref.51). SIV-infected macaques on suppressive ART given repeated and escalating doses of the TLR7 agonist GS-986 (a close analogue of GS-9620) had transiently increased levels of SIV RNA in plasma into the detectable range52. In addition, treatment with GS-986 in combination with an adenovirus-based therapeutic vaccine (Ad26/MVA) expressing SIV gag, pol and env genes decreased levels of viral DNA in both peripheral blood mononuclear cells and lymph nodes, which suggests that there was a reduction in the size of the latent reservoir53. Furthermore, rhesus macaques that received both the Ad26/MVA vaccine and GS-986 not only had a significantly lower plasma SIV RNA setpoint (the value at which the viral load remains relatively stable) but also a 2.5-fold delay of viral rebound after cessation of ART53. The results suggest that the combination treatment generated SIV-specific cytotoxic T lymphocytes (CTLs) that recognize and kill reactivated latently infected cells. In a following study of macaques infected with simian–human immunodeficiency virus (SHIV) and treated with ART during acute infection54, GS-9620 was given in combination with a broadly neutralizing antibody (bNAb), PGT121, that recognizes N-linked glycans on the V3 loop of HIV-1 gp120 to enable antibody-dependent cellular cytotoxicity (ADCC). Macaques treated with both PGT121 and GS-9620 had significantly lower levels of viral DNA, indicating a potential reduction in the size of the latent reservoir; following discontinuation of ART, 5 of 11 animals receiving this combination treatment experienced a delay in viral rebound54. All three of these studies in rhesus macaques showed that treatment with TLR7 agonists can activate CD4+ T cells, CD8+ T cells and NK cells, and increase plasma levels of various cytokines and chemokines, including IFNα, IL-6 and IL-1β52,53,54. These results suggest that TLR7 agonists can induce a potent innate immune response and, in combination with an HIV-1-specific bNAb or therapeutic vaccine, could improve the elimination of latently infected cells. However, it remains unclear whether the beneficial effects of TLR7 agonists in these studies result from their activity as LRAs, their immunomodulatory capacity as vaccine adjuvants or both.
GS-9620 was also evaluated in two phase Ib clinical trials for its safety and effect on the latent reservoir of HIV-1. Treatment of PLWH on suppressive ART with GS-9620 was well tolerated but had minimal effects on plasma viraemia and reservoir size, with no significant changes from baseline measurements of plasma HIV-1 RNA observed55. However, treatment of ART-suppressed HIV-1 controllers with GS-9620 resulted in a modest delay in viral rebound after analytical treatment interruption compared with the placebo group56. Although there were no differences in total HIV-1 DNA between the placebo group and those receiving GS-9620, the intact proviral DNA assay (Box 1) detected a significant decrease in intact, replication-competent proviruses in the latent reservoir of GS-9620-treated HIV-1 controllers56 (Table 1). Although both studies55,56 observed increased expression of interferon-stimulated genes, serum cytokines and IFNα in response to GS-9620 treatment, only the treatment of HIV-1 controllers resulted in a decrease of intact proviruses in the latent reservoir56. The differing results between these two studies could be due to variations in participant HIV-1 controller status as HIV-1 controllers may have CD8+ T cells with enhanced polyfunctional responses compared with non-controllers. In addition, continued ART administration after GS-9620 treatment in one of the studies55 could have affected virus–host dynamics.
Treatment with a TLR7 agonist caused transient increases in SIV RNA in plasma that were suggestive of latency reversal in one study of non-human primates52; this latency-reversing activity may have been undetected or underestimated in cases where very early treatment with ART after infection resulted in a small latent reservoir53,54. Furthermore, differences in the challenge strain of SIV or SHIV, in dosing intervals and in the immune status of the macaques could affect the ability of TLR7 agonists to mediate latency reversal. The ability of TLR7 agonists to potently stimulate IFNα production by pDCs suggests a potential mechanism for indirectly activating HIV-1-infected CD4+ T cells50,57. In addition, the increased production of IFNα in response to TLR7 agonists may also indirectly enhance the cytolytic activity of NK cells (see later discussion), suggesting a role for DC–NK cell crosstalk in latency reversal56. Overall, TLR7 agonists, either in combination with a therapeutic vaccine to strengthen HIV-1-specific T cell responses or in combination with bNAbs to enable ADCC, have the potential to improve the elimination of HIV-1-infected cells. Agonists of other TLRs, such as TLR1/2, TLR5, TLR8 and TLR9, have also been shown to have modest latency-reversing effects in HIV-1-infected cell lines or HIV-1-infected central memory T cells58,59; these warrant further study, for example given the far broader effects of TLR8 agonists on innate immunity than those of TLR7 agonists. In summary, TLR agonists have proven to be potent activators of multiple immune functions and may aid HIV-1 cure strategies designed to reduce the size of the latent reservoir or maintain long-term virological control.
Using DCs for latency reversal
DCs could also contribute to the reactivation of latently infected cells during ‘shock and kill’ approaches through their antigen presentation function. For example, two vaccine studies using DCs expressing HIV-1 antigens have reported induction of CD4+ T cell activation and increases in viral gene expression in vivo60 and in vitro61. Through their effective presentation of antigen, DCs can contribute to the activation of latently infected CD4+ T cells that are specific for the presented antigen40,41,62. However, this approach of reactivating antigen-specific latently infected cells has several limitations, including the small fraction of infected cells specific for any given antigen and the fact that the antigen specificity of most latently infected cells is unknown.
The addition of autologous immature monocyte-derived DCs (iDCs) to a primary cell model of latent HIV-1 infection caused latency reversal by a contact-dependent mechanism that may have involved factors independent of canonical T cell receptor (TCR) stimulation63. Activation of infected CD4+ T cells through the TCR and subsequent stimulation with iDCs increased extracellular levels of HIV-1 RNA and induced more viral outgrowth compared with TCR stimulation or iDC stimulation alone. This DC-mediated latency reversal may have involved contact-induced activation of the PI3K–AKT–mTOR pathway in CD4+ T cells, which was necessary for potent viral reactivation. Furthermore, a separate study using the same primary cell model of HIV-1 latency to investigate the effects of different DC subsets and other immune cell types on latency reversal showed that tissue-resident and blood-derived myeloid DCs induce viral reactivation with different efficiencies64. However, these findings should be interpreted with caution considering that the latency model used in these studies was generated under conditions that do not reflect naturally occurring establishment of HIV-1 latency. In addition, these studies did not show whether DCs were presenting HIV-1 antigens and therefore affected HIV-1-specifc CD4+ T cells through an antigen-dependent mechanism62,63. Studies using primary cell latency models to investigate DC-induced reversal of HIV-1 latency have so far yielded inconclusive data owing to the fact that these model systems are more efficient to reactivate than latently infected cells from PLWH23,24. Thus, the role of DCs in latency reversal requires further investigation using cells from PLWH. One recent study using cells from ART-suppressed PLWH reported that using monocyte-derived DCs (moDCs) to present peptide pools of HIV-1 Gag or of cytomegalovirus to autologous CD4+ T cells resulted in latency reversal65. This effect was dependent on bidirectional crosstalk between moDCs and CD4+ T cells during their canonical antigen-driven interaction, including CD40–CD40 ligand (CD40L) signalling65. However, the latency reversal observed in this study might be influenced by the fact that prolonged cultures were maintained in the absence of antiretroviral drugs, which enabled viral spread from reactivated cells. Indeed, the authors acknowledge that moDC-mediated viral reactivation might have been an indirect result of a potent global antigen-specific response rather than a direct effect on HIV-1-infected cells in the reservoir; thus, the underlying mechanism of moDC-mediated latency reversal remains unclear. Further investigation of the antigen specificity of the latent reservoir may improve DC-based latency reversal strategies by allowing for the reactivation of latently infected cells through the presentation and recognition of cognate antigen.
DC vaccines to enhance CTL responses
Recent studies suggest that depleting the latent reservoir of HIV-1 may require a combination therapeutic approach with both ‘shock’ and ‘kill’ components. Latency reversal by itself may not necessarily be followed by the clearance of cells expressing HIV-1 antigens12,66,67. Novel candidate vaccines and immunotherapies designed to enhance the killing of infected cells following latency reversal have been tested in various clinical trials without any clear promising results68,69,70. Targeting DCs may offer a path to inducing HIV-1-specific CTLs as DCs naturally prime both CD4+ and CD8+ T cells62. DCs have been incorporated in immunotherapeutic strategies for both cancer and HIV-1 (refs71,72). However, there are multiple challenges associated with the effective targeting of infected cells by CTLs73, including spatial separation of CTLs and target cells74, CTL exhaustion28, impaired recognition of HIV-1 epitopes owing to escape mutations66,75, and intrinsic resistance of latently infected cells to cytolysis76. Another major obstacle for successful DC-based therapeutic vaccination is endogenous DC dysfunction during HIV-1 infection77. ART can restore the function of endogenous DCs although some functions, such as IL-12p70 secretion in response to certain stimuli, are only partially rescued78.
Vaccination with autologous antigen-loaded DCs
One approach to overcoming the problem of endogenous DC dysfunction and improving vaccine-induced immune responses has been to generate mature, antigen-loaded autologous DCs ex vivo and then reintroduce these DCs to PLWH as a form of adoptive immunotherapy. This has involved generating fully matured and functional type-1-polarized moDCs that express both CCR7, which is necessary for their migration to lymph nodes for antigen presentation to T cells, and IL-12p70, which promotes CD4+ T helper 1 cell responses79. Ex vivo generation of large numbers of autologous moDCs requires the culture of monocytes from PLWH with granulocyte–macrophage colony-stimulating factor (GM-CSF) and IL-4 (ref.80). The addition of IFNγ and a combination of maturation-associated cytokines to ex vivo-generated moDCs produces polarized DCs capable of increased IL-12p70 expression81,82. DCs of this phenotype are associated with sustained HIV-1-specific effector T cell responses83.
Reinfusion of autologous moDCs presenting HIV-1 antigens into PLWH has been shown to be safe and immunogenic in several trials68,69,70. For these studies, various HIV-1 antigens, including whole inactivated virus, recombinant envelope glycoproteins, viral peptides or recombinant viral vectors, were used to prime ex vivo-generated moDCs60,68,69,70,84,85 (Fig. 2). The most promising results were obtained by using whole inactivated autologous virus as the antigen source68,69,70. In one study, vaccination with moDCs pulsed with autologous heat-inactivated HIV-1 resulted in a small yet significant decrease in viraemia and increased HIV-1-specific T cell responses in ART-naive individuals at 24 weeks after vaccination69. In a follow-up study, the same DC vaccination strategy was tested in PLWH treated with ART who underwent several analytical treatment interruptions after vaccination70. A log10 decrease in HIV-1 RNA setpoint was achieved70, although viral rebound after stopping ART was detected in all study participants. However, the reservoir size at the time of the second treatment interruption after vaccination was not measured and the authors note that potential replenishment of the latent reservoir during the first analytical treatment interruption is a major problem with the study design.
It is important to also acknowledge the practical limitations of this vaccination approach for a larger scale-up, particularly with regard to obtaining large numbers of moDCs. In addition, the choice of immunogen can affect optimal maturation of moDCs and the propagation of IL-12p70-producing moDCs. Although using whole inactivated autologous virus as a source of antigen for loading DCs may have an advantage over other immunogens in terms of the presentation of relevant, donor-specific epitopes, the practicality of this approach on a large scale is a concern. Furthermore, there is the additional concern of ensuring that the virus preparation has been completely inactivated and is no longer infectious.
Novel approaches to DC-based vaccination
To facilitate the generation of IL-12p70-producing moDCs, new strategies have been developed for the ex vivo genetic manipulation of DCs. One such approach towards improving DC-based vaccines involves genetic delivery of CD40L to DCs86,87. CD40L, which is transiently expressed on the surface of activated T cells and B cells88,89, binds to its cognate CD40 receptor on the surface of DCs90. This CD40–CD40L interaction induces the upregulated expression of costimulatory molecules by DCs and their secretion of cytokines such as IL-12p70, which are important for the maturation of DCs into fully competent antigen-presenting cells91 and the effective priming of T cells. Exploiting the knowledge of this interaction has led to the development of a personalized DC-based immunotherapy, termed AGS-004, which consists of autologous moDCs co-electroporated with RNAs encoding human CD40L and autologous HIV-1 antigens (Gag, Vpr, Rev and Nef)92,93,94. In one clinical trial, this approach was shown to induce HIV-1-specific CD8+ T cell effector responses, although these responses were not effective compared with placebo in improving host control of viral replication as measured during an analytical treatment interruption92. A recent study (Table 1) investigated the combined effect of the LRA vorinostat and AGS-004 on the latent reservoir of HIV-1 (ref.94). Six individuals on suppressive ART received four doses of AGS-004 every 3 weeks over a 12-week period, followed by vorinostat treatment every 72 hours for 30 days. Although the treatment was well tolerated, there was no effect on the latent reservoir as measured using the quantitative viral outgrowth assay (Box 1). Furthermore, no increase in HIV-1-specific CD8+ T cell or NK cell responses was observed. There are several possible explanations for the absence of a treatment effect in this study. First, the study had a small sample size and lacked a control group. Second, although some in vivo studies have shown that vorinostat treatment causes a significant increase in HIV-1 RNA expression indicative of the reactivation of latently infected cells67,95, there is no evidence that treatment with vorinostat can induce sufficient expression of HIV-1 genes from replication-competent proviruses and effective antigen presentation to allow for the elimination of HIV-1-infected cells by autologous CD8+ T cells96. In addition, the absence of detectable HIV-1-specific CD8+ T cell responses could be a result of CTL escape variants of HIV-1 being dominant in the latent reservoir of the participants in this study75.
Overall, although DC-based immunotherapies for HIV-1 infection have proven safe, well tolerated and moderately immunogenic, they have had limited success in clinical trials in terms of reducing the size of the latent reservoir97. Methods of DC generation and preparation, choice of immunogen, immunological parameters to measure viral control, and the assessment of immunological and virological responses have varied between studies, which makes it difficult to compare results60,68,69,70,84,94. One possible explanation for a lack of clinical efficacy for DC-based therapies is the limited capacity of DCs to induce potent HIV-1-specific CD8+ T cell responses owing to CTL exhaustion. In this respect, the combination of DC-based vaccines with immune-checkpoint inhibitors, such as antibodies to PD1 or CTLA4, is currently being investigated in several clinical trials for solid tumours98,99, which could improve the effector functions of CD8+ T cells through reversal of exhaustion100. Furthermore, the incomplete maturation of DCs during vaccine production could result in DCs with a limited capacity to migrate to the lymph nodes and that have a tolerogenic phenotype101. As an alternative to the laborious process of generating DC-based vaccines in vitro, the cross-presentation capacity of native circulating cDC1s could be enhanced by targeted antigen delivery to these cells. For example, some studies have reported that tumour antigen-conjugated antibodies recognizing C-type lectin receptors, such as DEC-205 or CLEC9A, that are highly expressed on cDC1s elicit robust tumour-specific CTL responses99,102,103. Therefore, we anticipate that rather than DC-based vaccine monotherapies, combinatorial strategies that incorporate DC-based vaccination or approaches to improve the antigen presentation of native cDCs will be more effective for HIV-1 cure efforts. A better understanding of the factors leading to impairment of DC responses in PLWH may contribute to the design of effective adjuvants or vectors that can rescue DC function in vivo and improve current DC-based vaccine strategies for the treatment of HIV-1 infection.
Killing of infected cells by NK cells
NK cells are a subset of lymphoid cells that mount innate antiviral responses. In response to infection, NK cells can directly lyse infected cells and can also secrete pro-inflammatory cytokines that enhance the functions of other innate and adaptive immune cells. A protective role for NK cells in HIV-1 infection is suggested by their ability to drive viral escape mutations and by the existence of several conserved mechanisms that HIV-1 has evolved to avoid detection by NK cells. For example, one study proposed that the immune pressure generated by NK cell-mediated ADCC contributed to the selection of mutations in HIV-1 epitopes recognized by antibodies that mediate ADCC104. These mutations could not be attributed to escape from antibody-mediated neutralization as the antibodies involved did not have neutralizing activity105. Similarly, selective pressure may alter the repertoire of viral peptides presented on MHC class I molecules to favour the engagement of inhibitory members of the killer immunoglobulin-like receptor (KIR) family on NK cells105.
Another mechanism by which HIV-1-infected cells might be recognized by NK cells involves the G2 cell-cycle arrest activity of the HIV-1 protein Vpr. Vpr promotes the activation of DNA damage and stress response pathways, resulting in increased surface expression of the stress proteins ULBP1 and ULBP2 by infected cells, which are ligands for the activating NK cell receptor NKG2D and are sufficient to induce NK cell-mediated killing of target cells106,107. However, HIV-1 avoids the recognition of infected cells by NKG2D through Nef-mediated degradation of ULBP1, ULBP2 and an additional NKG2D ligand, MICA108. To avoid the killing of infected cells by CTLs, HIV-1 also downregulates surface expression of HLA-A and HLA-B in a Nef-dependent manner109,110. To counteract the loss of interaction with inhibitory KIRs on NK cells caused by downregulation of HLA-A and HLA-B, HIV-1 is thought to selectively maintain surface expression of HLA-C and HLA-E111,112, which interact with additional inhibitory KIRs and NKG2A on NK cells, respectively. It is, however, worth noting that most studies that have reported the maintenance of surface expression levels of HLA-C and HLA-E by HIV-1-infected cells have used laboratory-adapted HIV-1 strains, which may not reflect the activity of primary isolates. More recent studies have suggested that, in cells infected with primary HIV-1 strains, Vpu and Nef may cause the downregulation of HLA-C and HLA-E, respectively113,114,115.
Further support for a role of NK cells in inhibition of HIV-1 infection comes from evidence linking NK cell-mediated ADCC to protection from contracting HIV-1 infection and suppression of viraemia. Post hoc analysis of the clinical trial results of RV144, the only HIV-1 vaccine trial so far to have resulted in some degree of protective efficacy, showed that one of the strongest positive correlates of protection is a high titre of non-neutralizing antibodies to the V1V2 region of the HIV-1 Env protein that can mediate ADCC116,117. However, additional analysis suggests that such antibodies may have multiple Fc-mediated effector functions, such as antibody-dependent cellular phagocytosis, in addition to ADCC118,119. HIV-1-specific ADCC responses have also been detected in the acute phase of HIV-1 infection and may contribute to determining the setpoint viral load120,121. Finally, high levels of HIV-1-specific ADCC activity correlate with slower rates of disease progression122,123.
Although the data are controversial, there is some evidence that ADCC responses may be increased in HIV-1 controllers. In several cohorts, sera from HIV-1 controllers enhanced ADCC responses to a greater extent than did sera from viraemic progressors124,125,126. Furthermore, the breadth of ADCC-mediating antibodies may also contribute to viral control as HIV-1 controllers in one cohort had greater cross-reactivity of ADCC responses against multiple Env clades than did viraemic progressors125. By contrast, no differences in ADCC responses were detected between HIV-1 controllers and viraemic progressors in additional cohorts127,128,129. Definitive conclusions about the role of ADCC in HIV-1 controllers will likely require standardized definitions of HIV-1 control and standardized methodologies for detecting ADCC activity to allow for better comparison between studies.
Overall, the above studies suggest a role of NK cell-mediated cytolytic clearance of infected cells in inhibiting HIV-1 acquisition and disease progression. The ability of NK cells to potently kill HIV-1-infected CD4+ T cells in diverse biological contexts warrants further study into the design of strategies to augment the innate antiviral properties of NK cells for an HIV-1 cure. Immunotherapies that improve the existing capacity of NK cells to eliminate HIV-1-infected cells in vivo may be effective alone or in combination with CTL-based strategies to reduce the size of the latent reservoir.
Enhancing ADCC activity through passive antibody administration
Passive administration of antibodies to Env that are capable of mediating strong ADCC is a strategy under consideration to enhance the clearance of HIV-1-infected cells (Fig. 3). In vitro studies have shown that several well-characterized monoclonal bNAbs can facilitate the killing of HIV-1-infected cells through NK cell-mediated ADCC130,131,132. Evidence from treatment with bNAbs in models of HIV-1 or SHIV challenge suggests that bNAbs can also mediate the elimination of infected cells in vivo. bNAbs consistently protect from SHIV challenge in macaque models and potently reduce viraemia133,134,135,136,137,138,139. Some of this effect is clearly owing to the direct antibody-mediated neutralization of virus. However, mutation of the bNAb b12 to abrogate Fc receptor binding reduced its ability to prevent SHIV acquisition133,134, which implicates a role for Fc-mediated effector functions, such as ADCC, in this activity. Furthermore, mutation of the bNAb VRC07-523LS139 and the bNAb-derived bispecific antibody 117/1,400 (ref.140) to abrogate Fc receptor binding slows the rates at which these antibodies clear virus from the plasma of SHIV-infected macaques. An additional study in a humanized mouse model of HIV-1 challenge has shown that mutations in bNAbs that enhance Fc receptor binding result in greater protective activity, providing further evidence for the involvement of Fc-dependent mechanisms in bNAb-mediated protection against HIV-1 infection141. In PLWH, bNAb infusions have been well tolerated and have delayed rebound of viraemia following analytical treatment interruption142,143,144,145. In some individuals who receive bNAb monotherapy during analytical treatment interruption, the outgrowth of pre-existing bNAb-resistant escape variants of HIV-1 can occur142,143,144. However, this phenomenon was not observed when a combination of bNAbs targeting distinct epitopes of HIV-1 was administered during treatment interruption145. Disappointingly, however, bNAb treatment during analytical treatment interruption has not resulted in an observable reduction in size of the latent reservoir of HIV-1 (refs146,147). This may be owing to the infrequent reactivation of latently infected cells and, thus, pairing the administration of bNAbs with a strategy to reverse latency may improve the clearance of infected cells. It should also be noted that it is unclear to what extent ADCC or other Fc-dependent antibody effector functions may contribute to the observed delay in viral rebound after analytical treatment interruption in PLWH who received bNAb infusions because Fc-mutated variants of the bNAbs have not been evaluated in this context.

Various broadly neutralizing antibodies (bNAbs) and non-neutralizing antibodies (nNAbs) to HIV-1 Env protein bind the Fc receptor FcγRIIIa (CD16a) on the surface of natural killer (NK) cells through their Fc domains. In addition, bispecific antibodies (BsAbs) can bind both Env on the surface of HIV-1-infected CD4+ T cells and FcγRIIIa on NK cells. This interaction results in NK cell activation, release of cytolytic granules (containing perforin and granzymes) and pro-inflammatory cytokines (such as IFNγ and tumour necrosis factor (TNF)) and apoptosis of the infected CD4+ T cell through a process known as antibody-dependent cellular cytotoxicity. Binding of CD4 mimetic compounds to Env can stabilize it in an ‘open’ conformation, thereby exposing CD4-induced (CD4i) epitopes and rendering the infected cell more vulnerable to binding of serum and monoclonal antibodies to CD4i epitopes and subsequent antibody-dependent cellular cytotoxicity. Cytokine-based therapies, including pegylated IFNα and the IL-15 super-agonist ALT-803, sensitize NK cells to cytolytic killing of HIV-1-infected cells. IFNAR, interferon-α/β receptor; NKR, natural killer cell receptor.
Similarly to bNAbs, monoclonal non-neutralizing antibodies to the HIV-1 Env protein mediate ADCC in vitro, albeit with generally weaker activity130,148. Of particular interest are V1V2-targeting non-neutralizing antibodies, similar to those antibodies that were thought to be protective in the RV144 vaccine trial. One study found that V1V2-targeting non-neutralizing antibodies have highly potent ADCC activity, greater in magnitude than that observed with tested bNAbs149. However, in contrast to bNAbs, the administration of non-neutralizing antibodies failed to protect macaques from SHIV challenge150,151,152,153. Although the non-neutralizing antibodies were not protective against infection, treatment was associated with positive outcomes in some of these studies, including reductions in viral load150,153 and levels of cell-associated HIV-1 DNA153 as well as the clonal restriction of transmitted/founder viruses151. Given that non-neutralizing antibodies cannot inhibit infection by cell-free virions, these treatment effects may be attributed to Fc-mediated effector activity against infected cells.
Engineering antibodies for improved NK cell cytolytic activity
Advances in antibody engineering have made it possible to modify existing monoclonal antibodies to better elicit Fc-mediated effector functions. Mutational analysis of the IgG Fc domain has revealed combinations of point mutations that result in improved Fc receptor binding and ADCC activity compared with the wild-type Fc domain154,155,156. In addition to or as an alternative to modifying the protein sequence of the antibody Fc domain, alterations to the Fc glycan structure can enhance the ability of IgG antibodies to mediate ADCC. Compared with IgG antibodies expressed in commonly used cell lines (such as wild-type CHO or 293T cells), IgG antibodies expressed in cell lines that yield low Fc fucose content (such as Lec13, YB2/0 or FUT8 cells) have increased Fc receptor binding and ADCC activity157,158,159,160. The afucosylated forms of the bNAbs 2G12 and b12 have enhanced Fc receptor binding and ADCC activity compared with the fucosylated forms161,162. Unexpectedly, however, the afucosylated form of b12 did not improve protection from SHIV challenge compared with the fucosylated form162. It would be of considerable interest to compare an ADCC-enhanced bNAb to the wild-type version in the context of analytical treatment interruption for differences in efficacy in delaying viral rebound and reducing levels of cell-associated HIV-1 DNA.
In addition to these efforts to enhance the interaction between the IgG Fc domain and the Fc receptor FcγRIIIa (also known as CD16a), several groups have engineered bispecific antibodies that induce potent NK cell-mediated cytolytic activity (Fig. 3). Bispecific antibodies that engage Env on the surface of HIV-1-infected cells and CD3 on the surface of T cells have previously been shown to promote CTL-mediated killing of infected target cells163,164,165,166. Recent studies have shown that replacing the CD3-specific antibody variable fragment (Fv) of existing bispecific antibodies that engage CD3 and Env with a CD16-specific antibody Fv166 or fusing a CD16-specific antibody Fv to the carboxyl terminus of gp41-specific IgG antibodies167 enabled these molecules to effectively bind NK cells and enhance the killing of HIV-1-infected cells. The design of novel bispecific antibody reagents may contribute to more efficacious clearance of HIV-1-infected cells if incorporated into a ‘shock and kill’ strategy.
Sensitizing infected cells to ADCC
Recent work suggests that a specific class of small molecules may enhance the capacity of autologous antibodies to induce ADCC. Sera from PLWH contain a high concentration of antibodies that are potentially capable of mediating ADCC but that target Env epitopes that are conformationally occluded168. These epitopes, termed CD4-induced (CD4i) epitopes, are exposed only in the ‘open’ conformation of Env that is induced by CD4 binding168,169. The downregulation of CD4 expression in HIV-1-infected cells by Nef and Vpu is thought to minimize the interaction between Env and CD4 on the surface of infected cells, thus resulting in infrequent exposure of these CD4i epitopes and protection of the infected cells from ADCC mediated by CD4i-specific antibodies168,169. CD4 mimetic compounds can bind Env on the surface of infected cells in a similar manner to soluble CD4 and trigger the exposure of CD4i epitopes170, thereby enhancing the ADCC potential of serum antibodies against autologous infected CD4+ T cells171,172,173 (Fig. 3).
Cytokine-based therapies to enhance NK cell-mediated killing of infected cells
The incorporation of cytokine-based therapies into a ‘shock and kill’ regimen could be used to augment the clearance of HIV-1-infected cells by enhancing the cytolytic effector functions of innate immune cells. Of current interest are therapies based on IFNα or IL-15 (Table 1). The administration of pegylated IFNα2 to PLWH on ART has been shown to delay time to viral rebound upon analytical treatment interruption and reduce levels of cell-associated HIV-1 DNA as detected by Alu-Gag PCR174,175, prompting consideration for its use as part of a ‘shock and kill’ strategy. A growing body of evidence suggests that the ability of IFNα to decrease levels of cell-associated HIV-1 DNA results from increased NK cell-mediated cytotoxicity (Fig. 3). Low levels of cell-associated HIV-1 DNA in PLWH on ART who were administered IFNα2 correlated with changes in the expression of NK cell receptors, including an increased frequency of NK cells expressing the activating receptors NKp30 and NKG2D175. In addition, NK cells from individuals who maintained control of viral load throughout a 12-week analytical treatment interruption after receiving IFNα therapy had lower levels of expression of inhibitory KIRs and a superior capacity for cytolytic killing ex vivo176. In vitro treatment of NK cells from viraemic progressors and HIV-1 controllers with IFNα2 results in an increase of both cytokine production and cytolytic potential177.
Recently, there has been growing interest in using the IL-15 super-agonist ALT-803 in ‘shock and kill’ strategies for HIV-1 cure. IL-15 is naturally produced by macrophages and DCs and is presented by these cells in trans to neighbouring NK cells. IL-15 is known to stimulate various functions of NK cells that would be desirable in the context of a ‘shock and kill’ regimen, including cytotoxic activity, IFNγ production, ADCC and inhibition of HIV-1 outgrowth178. ALT-803 consists of a highly potent mutated form of IL-15 complexed with the sushi domain of IL-15 receptor-α fused to an IgG Fc domain179,180 (Fig. 3). In addition to improving the biodistribution and half-life of ALT-803, the Fc domain allows for binding to Fc receptors on the surface of macrophages and DCs, which mimics endogenous trans-presentation of IL-15 and further improves the potency of ALT-803 (ref.180). There is evidence from animal models of HIV-1 infection that ALT-803 enhances the activity of NK cells and CTLs. In a humanized mouse model of acute HIV-1 infection, ALT-803 inhibited viral replication and this inhibition was not observed in NK cell-depleted mice181. Furthermore, ALT-803 transiently reduced viral load in SIV-infected rhesus macaques, which was associated with an increased number of NK cells and CD8+ T cells in peripheral blood182. In SHIV-infected macaques, ALT-803 increased the migration of NK cells and SHIV-specific CD8+ T cells to lymph node follicles, sites that are known to harbour latent HIV-1 during ART and that normally exclude cytolytic effector cells183. In addition to its potential for enhancing the elimination of HIV-1-infected cells by cytolytic effector mechanisms, ALT-803 may also promote latency reversal. In SIV-infected or SHIV-infected macaques and HIV-1-infected humanized mice, the administration of ALT-803 paired with depletion of CD8+ T cells during ART led to detectable plasma viraemia184,185. The potential of this IL-15 super-agonist to function both in latency reversal and by enhancing the clearance of infected cells makes it an attractive candidate for ‘shock and kill’ strategies.
Concluding remarks
Emerging strategies for an HIV-1 cure include combination approaches that use LRAs coupled with methods to enhance immune effector functions. Although current efforts to reactivate latently infected cells are mainly focused on novel pharmacological LRAs, TLR agonists are promising candidates in this regard that may receive more attention in the future. Furthermore, recent studies reporting the potential of moDCs to reverse latency may offer insights into designing novel LRAs based on understanding the underlying mechanism of viral reactivation reported in those studies. The induction of a potent and broad immune response to HIV-1 infection might be achieved through the use of DC-based immunotherapies to enhance the killing of infected cells. However, there are many limitations to the use of DC-based therapies, including their large-scale production and the determination of which immune responses are the best measures of viral control. Future studies might focus, instead, on interventions that both activate and deliver antigen to DCs directly in situ. Another promising strategy to improve immune-mediated clearance of HIV-1-infected cells is to enhance the cytolytic capacity of innate immune effector cells such as NK cells. ADCC is a mechanism of cell lysis mediated by NK cells that could be exploited for the elimination of infected cells through the therapeutic administration of small-molecule pharmacological agents, monoclonal antibodies or recombinant proteins such as ALT-803. Clinical trials are already under way to study and apply innate immune-based therapies for HIV-1 cure (Table 1). With further continuation of clinical trials involving TLR7 agonists and ALT-803, we believe that it is of crucial importance to better understand the molecular mechanisms of latency reversal caused by these reagents and whether their effects are mediated by direct activation of latently infected cells or indirect engagement through an innate immune response. Better mechanistic understanding of these therapeutic agents may lead to more effective targeting of innate immune responses and inform the design of combinatorial approaches to an HIV-1 cure. We remain optimistic that progress will continue to be made in defining the role of innate immunity in shaping the biology of viral rebound and in contributing to spontaneous control of HIV-1. In summary, we expect future studies to focus on enhancing our understanding of the role of innate immunity in the progression and control of HIV-1 infection as well as on investigating innate immunity-enhancing therapeutics as tools for achieving HIV-1 cure.
References
Barre-Sinoussi, F. et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871 (1983).
Chun, T. W. et al. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1, 1284–1290 (1995).
Chun, T. W. et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387, 183–188 (1997).
Finzi, D. et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300 (1997).
Wong, J. K. et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295 (1997).
Siliciano, J. D. et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9, 727–728 (2003).
Ho, Y. et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155, 540–551 (2013).
Hosmane, N. N. et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: potential role in latent reservoir dynamics. J. Exp. Med. 214, 959–972 (2017).
Archin, N. M., Sung, J. M., Garrido, C., Soriano-Sarabia, N. & Margolis, D. M. Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 12, 750–764 (2014).
Li, J. Z. et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 30, 343–353 (2016).
Hill, A. L., Rosenbloom, D. I. S., Fu, F., Nowak, M. A. & Siliciano, R. F. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc. Natl Acad. Sci. USA 111, 13475–13480 (2014).
Kim, Y., Anderson, J. L. & Lewin, S. R. Getting the “kill” into “shock and kill”: strategies to eliminate latent HIV. Cell Host Microbe 23, 14–26 (2018). This review provides a thorough overview of the ‘shock and kill’ approach to an HIV-1 cure and putative candidates for latency reversal.
Spivak, A. M. & Planelles, V. Novel latency reversal agents for HIV-1 cure. Annu. Rev. Med. 69, 421–436 (2018).
Archin, N. M. et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485 (2012).
Søgaard, O. S. et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 11, e1005142 (2015).
Rasmussen, T. A. et al. Panobinostat, a histone deacetylase inhibitor, for latent virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 1, e13–e21 (2014).
Dechristopher, B. A. et al. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat. Chem. 4, 705–710 (2012).
Nabel, G. & Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326, 711–713 (1987).
Molitor, J. A., Walker, W. H., Doerre, S., Ballard, D. W. & Greene, W. C. NF-κB: a family of inducible and differentially expressed enhancer-binding proteins in human T cells. Proc. Natl Acad. Sci. USA 87, 10028–10032 (1990).
Karn, J. & Stoltzfus, C. M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2, a006916 (2012).
Pan, X. Y. et al. Heat shock factor 1 mediates latent HIV reactivation. Sci. Rep. 6, 26294 (2016).
Timmons, A. et al. HSF1 inhibition attenuates HIV-1 latency reversal mediated by several candidate LRAs in vitro and ex vivo. Proc. Natl Acad. Sci. USA 117, 15763–15771 (2020).
Bullen, C. K., Laird, G. M., Durand, C. M., Siliciano, J. D. & Siliciano, R. F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 20, 425–429 (2014).
Laird, G. M. et al. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Invest. 125, 1901–1912 (2015).
Margolis, D. M. Histone deacetylase inhibitors and HIV latency. Curr. Opin. HIV Aids 6, 25–29 (2011).
Swiecki, M. & Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 15, 471–485 (2015).
Mavilio, D. et al. Characterization of CD56-/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc. Natl Acad. Sci. USA 102, 2886–2891 (2005).
Day, C. L. et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443, 350–354 (2006).
Chew, G. M. et al. TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PLoS Pathog. 12, e1005349 (2016).
Jones, R. B. et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 10, e1004287 (2014).
Pace, M. et al. Histone deacetylase inhibitors enhance CD4 T cell susceptibility to NK cell killing but reduce NK cell function. PLoS Pathog. 12, e1005782 (2016).
Iyer, S. S. et al. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc. Natl Acad. Sci. USA 114, E590–E599 (2017).
Gondim, M. V. P. et al. Heightened resistance to host type 1 interferons characterizes HIV-1 at transmission and after antiretroviral therapy interruption. Sci. Transl. Med. 13, 8179 (2021).
Vatner, R. E. & Janssen, E. M. STING, DCs and the link between innate and adaptive tumor immunity. Mol. Immunol. 110, 13–23 (2019).
Collin, M., Mcgovern, N. & Haniffa, M. Human dendritic cell subsets. Immunology 140, 22–30 (2013).
Rhodes, J. W., Tong, O., Harman, A. N. & Turville, S. G. Human dendritic cell subsets, ontogeny, and impact on HIV infection. Front. Immunol. 10, 1088 (2019).
Schmidt, B., Ashlock, B. M., Foster, H., Fujimura, S. H. & Levy, J. A. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology 343, 256–266 (2005).
Di Domizio, J. et al. TLR7 stimulation in human plasmacytoid dendritic cells leads to the induction of early IFN-inducible genes in the absence of type I IFN. Blood 114, 1794–1802 (2009).
Gibson, S. J. et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell. Immunol. 218, 74–86 (2002).
Liu, Y.-J. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell 106, 259–262 (2001).
Lanzavecchia, A. & Sallusto, F. Regulation of T cell immunity by dendritic cells. Cell 106, 263–266 (2001).
Altfeld, M. & Gale, M. Jr Innate immunity against HIV-1 infection. Nat. Immunol. 16, 554–562 (2015).
Martín-Moreno, A. & Muñoz-Fernández, M. A. Dendritic cells, the double agent in the war against HIV-1. Front. Immunol. 10, 2485 (2019).
Luban, J. Innate immune sensing of HIV-1 by dendritic cells. Cell Host Microbe 12, 408–418 (2012).
Hemmi, H. et al. Small-antiviral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3, 196–200 (2002).
Lee, J. et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of toll-like receptor 7. Proc. Natl Acad. Sci. USA 100, 6646–6651 (2003).
Zhang, Z. et al. Structural analyses of toll-like receptor 7 reveal detailed RNA sequence specificity and recognition mechanism of agonistic ligands. Cell Rep. 25, 3371–3381.e5 (2018).
Kawasaki, T. & Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 5, 461 (2014).
Suthers, A. N. & Sarantopoulos, S. TLR7/TLR9- and B cell receptor-signaling crosstalk: promotion of potentially dangerous B cells. Front. Immunol. 8, 775 (2017).
Tsai, A. et al. Toll-like receptor 7 agonist GS-9620 induces HIV expression and HIV-specific immunity in cells from HIV-infected individuals on suppressive antiretroviral therapy. J. Virol. 91, e02166–16 (2017).
Dinoso, J. B. et al. A simian immunodeficiency virus-infected macaque model to study viral reservoirs that persist during highly active antiretroviral therapy. J. Virol. 83, 9247–9257 (2009).
Lim, S.-Y. et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci. Transl. Med. 10, eaao4521 (2018).
Borducchi, E. N. et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature 540, 284–287 (2016).
Borducchi, E. N. et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 563, 360–364 (2018). Combination treatment with GS-9620 (a TLR7 agonist) and PGT121 (a broadly neutralizing antibody) in SHIV-infected macaques delayed viral rebound and decreased levels of HIV-1 DNA.
Riddler, S. A. et al. Vesatolimod, a toll-like receptor 7 agonist, induces immune activation in virally suppressed adults living with human immunodeficiency virus–1. Clin. Infect. Dis. 72, e815–e824 (2021).
SenGupta, D. et al. The TLR7 agonist vesatolimod induced a modest delay in viral rebound in HIV controllers after cessation of antiretroviral therapy. Sci. Transl. Med. 13, eabg3071 (2021).
Van der Sluis, R. M. et al. Diverse effects of interferon alpha on the establishment and reversal of HIV latency. PLoS Pathog. 16, e1008151 (2020).
Novis, C. L. et al. Reactivation of latent HIV-1 in central memory CD4+ T cells through TLR-1/2 stimulation. Retrovirology 10, 119 (2013).
Thibault, S., Imbeault, M., Tardif, M. R. & Tremblay, M. J. TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4+ T cells. Virology 389, 20–25 (2009).
Norton, T. D., Miller, E. A., Bhardwaj, N. & Landau, N. R. Vpx-containing dendritic cell vaccine induces CTLs and reactivates latent HIV-1 in vitro. Gene Ther. 22, 227–236 (2015).
Macatangay, B. J. C. et al. Therapeutic vaccination with dendritic cells loaded with autologous HIV type 1-infected apoptotic cells. J. Infect. Dis. 213, 1400–1409 (2016).
Bousso, P. T-cell activation by dendritic cells in the lymph node: lessons from the movies. Nat. Rev. Immunol. 8, 675–684 (2008).
van Montfort, T. et al. Dendritic cells potently purge latent HIV-1 beyond TCR-stimulation, activating the PI3K-Akt-mTOR pathway. EBioMedicine 42, 97–108 (2019).
Van Der Sluis, R. M. et al. Dendritic cell type-specific HIV-1 activation in effector T cells: Implications for latent HIV-1 reservoir establishment. Aids 29, 1003–1014 (2015).
Kristoff, J. et al. Type 1-programmed dendritic cells drive antigen-specific latency reversal and immune elimination of persistent HIV-1. EBioMedicine 43, 295–306 (2019). The presentation of HIV-1 or cytomegalovirus antigens by monocyte-derived DCs to HIV-1-infected CD4+ T cells isolated from ART-treated PLWH induced latency reversal that was dependent on CD40–CD40L signalling.
Shan, L. et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36, 491–501 (2012).
Archin, N. M. et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Invest. 127, 3126–3135 (2017).
Lu, W., Arraes, L. C., Ferreira, W. T. & Andrieu, J. M. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nat. Med. 10, 1359–1365 (2004).
García, F. et al. A therapeutic dendritic cell-based vaccine for HIV-1 infection. J. Infect. Dis. 203, 473–478 (2011). A randomized, blinded-placebo-controlled vaccine trial of autologous monocyte-derived DCs loaded with heat-inactivated autologous HIV-1 in ART-treated PLWH. This study reports a ≥1 log decrease of the plasma viral load setpoint after ART interruption in 55% and 35% of individuals receiving therapeutic DC vaccination at weeks 12 and 24, respectively.
Garcia, F. et al. A dendritic cell-based vaccine elicits T cell responses associated with control of HIV-1 replication. Sci. Transl. Med. 5, 166ra2 (2013).
Kalinski, P. et al. Dendritic cells in cancer immunotherapy: vaccines or autologous transplants? Immunol. Res. 50, 235–247 (2011).
Ellebaek, E. et al. Metastatic melanoma patients treated with dendritic cell vaccination, interleukin-2 and metronomic cyclophosphamide: results from a phase II trial. Cancer Immunol. Immunother. 61, 1791–1804 (2012).
Jones, R. B. & Walker, B. D. HIV-specific CD8+ T cells and HIV eradication. J. Clin. Invest. 126, 455–463 (2016).
Koup, R. A. et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68, 4650–4655 (1994).
Deng, K. et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 517, 381–385 (2015). This study reports that the vast majority of latent HIV-1 genomes contain escape mutations that render them insensitive to CTL-mediated killing.
Ren, Y. et al. BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J. Clin. Invest. 130, 2542–2559 (2020).
Miller, E. & Bhardwaj, N. Dendritic cell dysregulation during HIV-1 infection. Immunol. Rev. 254, 170–189 (2013).
Miller, E. A. et al. Plasma factors during chronic HIV-1 infection impair IL-12 secretion by myeloid dendritic cells via a virus-independent pathway. J. Immune Defic. Syndr. 61, 535–544 (2012).
Mailliard, R. B. et al. α-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 64, 5934–5937 (2004).
Sapp, M. Dendritic cells generated from blood monocytes of HIV-1 patients are not infected and act as competent antigen presenting cells eliciting potent T-cell responses. Immunol. Lett. 66, 121–128 (1999).
Vieira, P. L., de Jong, E. C., Wierenga, E. A., Kapsenberg, M. L. & Kaliński, P. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J. Immunol. 164, 4507–4512 (2000).
Muthuswamy, R. et al. Ability of mature dendritic cells to interact with regulatory T cells is imprinted during maturation. Cancer Res. 68, 5972–5978 (2008).
Macatangay, B. J. C., Szajnik, M. E., Whiteside, T. L., Riddler, S. A. & Rinaldo, C. R. Regulatory T cell suppression of gag-specific CD8+ T cell polyfunctional response after therapeutic vaccination of HIV-1-infected patients on ART. PLoS One 5, e9852 (2010).
Allard, S. D. et al. A phase I/IIa immunotherapy trial of HIV-1-infected patients with Tat, Rev and Nef expressing dendritic cells followed by treatment interruption. Clin. Immunol. 142, 252–268 (2012).
Ide, F. et al. Peptide-loaded dendritic-cell vaccination followed by treatment interruption for chronic HIV-1 infection: a phase 1 trial. J. Med. Virol. 78, 711–718 (2006).
Flamar, A.-L. et al. HIV-1 T cell epitopes targeted to Rhesus macaque CD40 and DCIR: a comparative study of prototype dendritic cell targeting therapeutic vaccine candidates. PLoS One 13, e0207794 (2018).
Sabado, R. L., Balan, S. & Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 27, 74–95 (2017).
Armitage, R. J. et al. Molecular and biological characterization of a murine ligand for CD40. Nature 357, 80–82 (1992).
Clark, E. A. & Ledbetter, J. A. Activation of human B cells mediated through two distinct cell surface differentiation antigens, Bp35 and Bp50. Proc. Natl Acad. Sci. USA 83, 4494–4498 (1986).
Grewal, I. S. & Flavell, R. A. A central role of CD40 ligand in the regulation of CD4+ T-cell responses. Immunol. Today 17, 410–414 (1996).
Ma, D. Y. & Clark, E. A. The role of CD40 and CD154/CD40L in dendritic cells. Semin. Immunol. 21, 265–272 (2009).
Jacobson, J. M. et al. Dendritic cell immunotherapy for HIV-1 infection using autologous HIV-1 RNA: A randomized, double-blind, placebo-controlled clinical trial. J. Acquir. Immune Defic. Syndr. 72, 31–38 (2016).
Gay, C. L. et al. Immunogenicity of AGS-004 dendritic cell therapy in patients treated during acute HIV infection. AIDS Res. Hum. Retroviruses 34, 111–122 (2018).
Gay, C. L. et al. Assessing the impact of AGS-004, a dendritic cell-based immunotherapy, and vorinostat on persistent HIV-1 infection. Sci. Rep. 10, 1–13 (2020). Combined treatment with vorinostat and AGS-004 resulted in decreased levels of cell-associated HIV-1 RNA but no substantial impact on the replication-competent viral reservoir and no increase in HIV-1-specific immune responses.
Elliott, J. H. et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 10, e1004473 (2014).
Sung, J. A. et al. Vorinostat renders the replication-competent latent reservoir of human immunodeficiency virus (HIV) vulnerable to clearance by CD8 T cells. EBioMedicine 23, 52–58 (2017).
Coelho, A. et al. Dendritic cell-based immunotherapies to fight HIV: how far from a success story? A systematic review and meta-analysis. Int. J. Mol. Sci. 17, 1985 (2016).
van Willingen, W. W. et al. Dendritic cell cancer therapy: vaccinating the right patient at the right time. Front. Immunol. 9, 2265 (2018).
Bhardwaj, N. et al. Flt3 ligand augments immune responses to anti-DEC-205-NY-ESO-1 vaccine through expansion of dendritic cell subsets. Nat. Cancer 112, 1204–1217 (2020).
Velu, V. et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458, 206–210 (2009).
Dhodapkar, M. V. et al. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 193, 233–238 (2001).
Idoyaga, J. et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc. Natl Acad. Sci. USA 108, 2384–2389 (2011).
Masterman, K.-A. et al. Human CLEC9A antibodies deliver NY-ESO-1 antigen to CD141+dendritic cells to activate naïve and memory NY-ESO-1-specific CD8+ T cells. J. Immunother. Cancer 8, e000691 (2020).
Chung, A. W. et al. Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc. Natl Acad. Sci. USA 108, 7505–7510 (2011).
Alter, G. et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature 476, 96–101 (2011).
Richard, J., Sindhu, S., Pham, T. N. Q., Belzile, J. & Cohen, É. A. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 115, 1354–1363 (2010).
Ward, J. et al. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 5, e1000613 (2009).
Cerboni, C. et al. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J. Gen. Virol. 88, 242–250 (2007).
Collins, K. L., Chen, B. K., Kalams, S. A., Walker, B. D. & Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401 (1998).
Le Gall, S. et al. Nef interacts with the μ subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 8, 483–495 (1998).
Cohen, G. B. et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10, 661–671 (1999).
Ward, J. P., Bonaparte, M. I. & Barker, E. HLA-C and HLA-E reduce antibody-dependent natural killer cell-mediated cytotoxicity of HIV-infected primary T cell blasts. Aids 18, 1769–1779 (2004).
Apps, R. et al. HIV-1 Vpu mediates HLA-C downregulation. Cell Host Microbe 19, 686–695 (2016).
Körner, C. et al. HIV-1-mediated downmodulation of HLA-C impacts target cell recognition and antiviral activity of NK cells. Cell Host Microbe 22, 111–119.e4 (2017).
Van Stigt Thans, T. et al. Primary HIV-1 strains use Nef to downmodulate HLA-E surface expression. J. Virol. 93, e00719-19 (2019).
Haynes, B. F. et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366, 1275–1286 (2012).
Bonsignori, M. et al. Antibody-dependent cellular cytotoxicity-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. J. Virol. 86, 11521–11532 (2012).
Chung, A. W. et al. Polyfunctional Fc-effector profiles mediated by IgG subclass selection distinguish RV144 and VAX003 vaccines. Sci. Transl. Med. 6, 228ra38 (2014).
Chung, A. W. et al. Dissecting polyclonal vaccine-induced humoral immunity against HIV using systems serology. Cell 163, 988–998 (2015).
Chung, A. W. et al. Activation of NK cells by ADCC responses during early HIV infection. Viral Immunol. 24, 171–175 (2011).
Chen, X. et al. The early antibody-dependent cell-mediated cytotoxicity response is associated with lower viral set point in individuals with primary HIV infection. Front. Immunol. 9, 2322 (2018).
Baum, L. L. et al. HIV-1 gp120-specific antibody-dependent cell-mediated cytotoxicity correlates with rate of disease progression. J. Immunol. 157, 2168–2173 (1996).
Chung, A. W. et al. Activation of NK cells by ADCC antibodies and HIV disease progression. JAIDS 58, 127–131 (2011).
Lambotte, O. et al. High antibody-dependent cellular cytotoxicity responses are correlated with strong CD8 T cell viral suppressive activity but not with B57 status in HIV-1 elite controllers. PLoS One 8, e74855 (2013).
Madhavi, V. et al. Breadth of HIV-1 Env-specific antibody-dependent cellular cytotoxicity: Relevance to global HIV vaccine design. AIDS 28, 1859–1870 (2014).
Madhavi, V. et al. HIV-1 Env- and Vpu-specific antibody-dependent cellular cytotoxicity responses associated with elite control of HIV. J. Virol. 91, e00700-17 (2017).
Johansson, S. E. et al. NK cell function and antibodies mediating ADCC in HIV-1-infected viremic and controller patients. Viral Immunol. 24, 359–368 (2011).
Smalls-Mantey, A. et al. Antibody-dependent cellular cytotoxicity against primary HIV-infected CD4+ T cells is directly associated with the magnitude of surface IgG binding. J. Virol. 86, 8672–8680 (2012).
Ackerman, M. E. et al. Polyfunctional HIV-specific antibody responses are associated with spontaneous HIV control. PLoS Pathog. 12, e1005315 (2016).
von Bredow, B. et al. Comparison of antibody-dependent cell-mediated cytotoxicity and virus neutralization by HIV-1 Env-specific monoclonal antibodies. J. Virol. 90, 6127–6139 (2016).
Mujib, S. et al. Comprehensive cross-clade characterization of antibody-Mediated recognition, complement-mediated lysis, and cell-mediated cytotoxicity of HIV-1 envelope-specific antibodies toward eradication of the HIV-1 reservoir. J. Virol. 91, e00634-17 (2017).
Bruel, T. et al. Elimination of HIV-1-infected cells by broadly neutralizing antibodies. Nat. Commun. 7, 10844 (2016).
Hessell, A. J. et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104 (2007). Abrogation of Fc receptor binding by Fc domain mutation of the broadly neutralizing antibody b12 markedly reduced its ability to protect macaques from SHIV infection.
Hessell, A. J. et al. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat. Med. 15, 951–954 (2009).
Gautam, R. et al. A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature 533, 105–109 (2016).
Liu, J. et al. Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science 353, 1045–1049 (2016).
Nishimura, Y. et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature 543, 559–563 (2017).
Julg, B. et al. Broadly neutralizing antibodies targeting the HIV-1 envelope V2 apex confer protection against a clade C SHIV challenge. Sci. Transl. Med. 9, eaal1321 (2017).
Asokan, M. et al. Fc-mediated effector function contributes to the in vivo antiviral effect of an HIV neutralizing antibody. Proc. Natl Acad. Sci. USA 117, 18754–18763 (2020).
Wang, P. et al. Quantifying the contribution of Fc-mediated effector functions to the antiviral activity of anti-HIV-1 IgG1 antibodies in vivo. Proc. Natl Acad. Sci. USA 117, 18002–18009 (2020).
Bournazos, S. et al. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 158, 1243–1253 (2014).
Scheid, J. F. et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 535, 556–560 (2016).
Bar, K. J. et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N. Engl. J. Med. 375, 2037–2050 (2016).
Caskey, M. et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 23, 185–191 (2017).
Mendoza, P. et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 561, 479–484 (2018).
Salantes, D. B. et al. HIV-1 latent reservoir size and diversity are stable following brief treatment interruption. J. Clin. Invest. 128, 3102–3115 (2018).
Cohen, Y. Z. et al. Relationship between latent and rebound viruses in a clinical trial of anti-HIV-1 antibody 3BNC117. J. Exp. Med. 215, 2311–2324 (2018).
Bruel, T. et al. Lack of ADCC breadth of human nonneutralizing anti-HIV-1 antibodies. J. Virol. 91, e02440-16 (2017).
Mayr, L. M. et al. Non-neutralizing antibodies targeting the V1V2 domain of HIV exhibit strong antibody-dependent cell-mediated cytotoxic activity. Sci. Rep. 7, 12655 (2017).
Moog, C. et al. Protective effect of vaginal application of neutralizing and nonneutralizing inhibitory antibodies against vaginal SHIV challenge in macaques. Mucosal Immunol. 7, 46–56 (2014).
Santra, S. et al. Human non-neutralizing HIV-1 envelope monoclonal antibodies limit the number of founder viruses during SHIV mucosal infection in rhesus macaques. PLoS Pathog. 11, e1005042 (2015).
Astronomo, R. D. et al. Neutralization takes precedence Over IgG or IgA Isotype-related functions in mucosal HIV-1 antibody-mediated protection. EBioMedicine 14, 97–111 (2016).
Hessell, A. J. et al. Reduced cell-associated DNA and improved viral control in macaques following passive transfer of a single anti-V2 monoclonal antibody and repeated simian/human immunodeficiency virus challenges. J. Virol. 92, e02198-17 (2018).
Lazar, G. A. et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl Acad. Sci. USA 103, 4005–4010 (2006).
Stavenhagen, J. B. et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcγ receptors. Cancer Res. 67, 8882–8890 (2007).
Mimoto, F. et al. Novel asymmetrically engineered antibody Fc variant with superior FcγR binding affinity and specificity compared with afucosylated Fc variant. MAbs 5, 229–236 (2013).
Shields, R. L. et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J. Biol. Chem. 277, 26733–26740 (2002).
Shinkawa, T. et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 278, 3466–3473 (2003).
Yamane-Ohnuki, N. et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol. Bioeng. 87, 614–622 (2004).
Kanda, Y. et al. Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiology 17, 104–118 (2007).
Forthal, D. N. et al. Fc-glycosylation influences Fcγ receptor binding and cell-mediated anti-HIV activity of monoclonal antibody 2G12. J. Immunol. 185, 6876–6882 (2010).
Moldt, B. et al. A nonfucosylated variant of the anti-HIV-1 monoclonal antibody b12 has enhanced Fc riiia-mediated antiviral activity in vitro but does not improve protection against mucosal SHIV challenge in macaques. J. Virol. 86, 6189–6196 (2012).
Sung, J. A. M. et al. Dual-affinity re-targeting proteins direct T cell-mediated cytolysis of latently HIV-infected cells. J. Clin. Invest. 125, 4077–4090 (2015).
Pegu, A. et al. Activation and lysis of human CD4 cells latently infected with HIV-1. Nat. Commun. 6, 8447 (2015).
Sloan, D. D. et al. Targeting HIV reservoir in infected CD4 T cells by dual-affinity Re-targeting molecules (DARTs) that bind HIV envelope and recruit cytotoxic T cells. PLoS Pathog. 11, e1005233 (2015).
Pollara, J. et al. Redirection of cord blood T cells and natural killer cells for elimination of autologous HIV-1-infected target cells using bispecific DART® molecules. Front. Immunol. 11, 713 (2020).
Ramadoss, N. S. et al. Enhancing natural killer cell function with gp41-targeting bispecific antibodies to combat HIV infection. AIDS 34, 1313–1323 (2020).
Veillette, M. et al. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 89, 545–551 (2015).
Veillette, M. et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 88, 2633–2644 (2014).
Schön, A. et al. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry 45, 10973–10980 (2006).
Richard, J. et al. CD4 mimetics sensitize HIV-1-infected cells to ADCC. Proc. Natl Acad. Sci. USA 112, E2687–E2694 (2015).
Lee, W. S. et al. Antibody-dependent cellular cytotoxicity against reactivated HIV-1-infected cells. J. Virol. 90, 2021–2030 (2016).
Rajashekar, J. K. et al. Modulating HIV-1 envelope glycoprotein conformation to decrease the HIV-1 reservoir. Cell Host Microbe 29, 904–916.e6 (2021). In an IL15-transgenic humanized mouse model of HIV-1 infection, treatment with a CD4 mimetic and CD4i-directed antibodies limited viral replication, reduced levels of HIV-1 DNA and delayed time to viral rebound after ART interruption; these effects were dependent on both Fc-mediated antibody effector functions and the presence of NK cells.
Azzoni, L. et al. Pegylated interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J. Infect. Dis. 207, 213–222 (2013). Compared with untreated individuals, a larger proportion of PLWH who were treated with pegylated IFNα experienced delayed viral rebound after ART interruption, and the amount of HIV-1 DNA was decreased in those individuals who suppressed viraemia for the full duration of the study.
Hua, S. et al. Pegylated interferon-α-induced natural killer cell activation is associated with human immunodeficiency virus-1 DNA decline in antiretroviral therapy-Treated HIV-1/Hepatitis C virus-coinfected patients. Clin. Infect. Dis. 66, 1910–1917 (2018).
Papasavvas, E. et al. NK response correlates with HIV decrease in pegylated IFN-α2a–treated antiretroviral therapy–suppressed subjects. J. Immunol. 203, 705–717 (2019).
Kwaa, A. K. R., Talana, C. A. G. & Blankson, J. N. Interferon alpha enhances NK cell function and the suppressive capacity of HIV-specific CD8+ T cells. J. Virol. 93, 1–14 (2018).
Garrido, C. et al. Interleukin-15-stimulated natural killer cells clear HIV-1-infected cells following latency reversal ex vivo. J. Virol. 92, e00235–18 (2018).
Han, K. P. et al. IL-15:IL-15 receptor alpha superagonist complex: High-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 56, 804–810 (2011).
Rhode, P. R. et al. Comparison of the superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics in animal models. Cancer Immunol. Res. 4, 49–60 (2016).
Seay, K. et al. In vivo activation of human NK cells by treatment with an interleukin-15 superagonist potently inhibits acute in vivo HIV-1 infection in humanized mice. J. Virol. 89, 6264–6274 (2015). ALT-803 treatment during acute HIV-1 infection of humanized mice resulted in potent suppression of viral replication in an NK cell-dependent manner.
Ellis-Connell, A. L. et al. ALT-803 transiently reduces simian immunodeficiency virus replication in the absence of antiretroviral treatment. J. Virol. 92, e01748–17 (2018).
Webb, G. M. et al. The human IL-15 superagonist N-803 promotes migration of virus-specific CD8+ T and NK cells to B cell follicles but does not reverse latency in ART-suppressed, SHIV-infected macaques. PLoS Pathog. 16, e1008339 (2020).
McBrien, J. B. et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 578, 154–159 (2020). In both ART-suppressed SIV-infected macaques and HIV-1-infected humanized mice, ALT-803 treatment resulted in robust viral reactivation when combined with CD8+ T cell depletion.
McBrien, J. B. et al. Combination of CD8β depletion and interleukin-15 superagonist N-803 induces virus reactivation in simian-human immunodeficiency virus-infected, long-term ART-treated rhesus macaques. J. Virol. 94, e00755-20 (2020).
Laird, G. M., Rosenbloom, D. I. S., Lai, J., Siliciano, R. F. & Siliciano, J. D. In Methods in Molecular Biology Vol. 1354, 239–253 (Humana Press Inc., 2016).
Kwon, K. J. et al. Different human resting memory CD4+ T cell subsets show similar low inducibility of latent HIV-1 proviruses. Sci. Transl. Med. 12, eaax6795 (2020).
Wang, Z., Simonetti, F. R., Siliciano, R. F. & Laird, G. M. Measuring replication competent HIV-1: advances and challenges in defining the latent reservoir. Retrovirology 15, 21 (2018).
Bruner, K. M. et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 566, 120–125 (2019). The intact proviral DNA assay, a recently described droplet digital PCR assay, can accurately quantify and distinguish intact and defective proviruses in the latent reservoir of HIV-1.
Barron, M. A., Blyveis, N., Palmer, B. E., MaWhinney, S. & Wilson, C. C. Influence of plasma viremia on defects in number and immunophenotype of blood dendritic cell subsets in human immunodeficiency virus 1-infected individuals. J. Infect. Dis. 187, 26–37 (2003).
Donaghy, H. et al. Loss of blood CD11c+ myeloid and CD11c– plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood 98, 2574–2576 (2001).
Jacquelin, B. et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest. 119, 3544–3555 (2009).
Fernandez, S. et al. CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J. Infect. Dis. 204, 1927–1935 (2011).
Manion, M. et al. Interferon-alpha administration enhances CD8+ T cell activation in HIV infection. PLoS One 7, e30306 (2012).
Herbeuval, J. P. et al. Differential expression of IFN-α and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl Acad. Sci. USA 103, 7000–7005 (2006).
Martinson, J. A. et al. Dendritic cells from HIV-1 infected individuals are less responsive to toll-like receptor (TLR) ligands. Cell. Immunol. 250, 75–84 (2007).
Mitchell, J. L. et al. Plasmacytoid dendritic cells sense HIV replication before detectable viremia following treatment interruption. J. Clin. Invest. 130, 2845–2858 (2020).
Author information
Authors and Affiliations
Contributions
N.L.B., M.M. and F.W. contributed equally to all aspects of this article. R.F.S. and J.D.S. contributed substantially to the discussion of content and revision of the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks M. Altfeld and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrials.gov: https://clinicaltrials.gov/
Glossary
- Antiretroviral therapy
-
(ART). Treatment with drugs such as reverse transcriptase inhibitors, integrase inhibitors, protease inhibitors or entry inhibitors that prevent ongoing HIV-1 replication.
- Transmitted/founder viruses
-
The single or very few HIV-1 viral clones that establish productive infection of a new host during HIV-1 transmission.
- Broadly neutralizing antibody
-
(bNAb). An antibody that binds to a conserved region of HIV-1 Env and that can neutralize a large diversity of viral isolates.
- Antibody-dependent cellular cytotoxicity
-
(ADCC). The process by which a cytolytic immune effector cell (natural killer cell, monocyte, neutrophil or eosinophil) recognizes and lyses a target cell whose surface is bound by antibodies.
- HIV-1 controllers
-
A small subset of people living with HIV-1 who maintain a low but detectable level of viraemia and high CD4+ T cell counts without antiretroviral therapy.
- Analytical treatment interruption
-
A period during which people living with HIV-1 on ART temporarily stop therapy to evaluate the effects of a novel treatment on viral rebound.
- Killer immunoglobulin-like receptor
-
(KIR). A family of natural killer cell receptors that interact with MHC class I molecules on the surface of target cells to regulate natural killer cell activation.
- Antibody-dependent cellular phagocytosis
-
The process by which a phagocytic immune cell recognizes and engulfs a target cell whose surface is bound by antibodies, resulting in elimination of the target cell and MHC class II-dependent presentation of antigens derived from the target cell by the phagocyte.
- Bispecific antibody
-
An engineered antibody that recognizes two distinct epitopes or antigens.
- Alu-Gag PCR
-
A PCR method for measuring integrated HIV-1 DNA using primers that recognize Alu repeat elements in the host genome and the gag gene of the HIV-1 genome.
Rights and permissions
About this article

Cite this article
Board, N.L., Moskovljevic, M., Wu, F. et al. Engaging innate immunity in HIV-1 cure strategies. Nat Rev Immunol 22, 499–512 (2022). https://doi.org/10.1038/s41577-021-00649-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-021-00649-1
- Springer Nature Limited
This article is cited by
-
Bispecific antibodies promote natural killer cell-mediated elimination of HIV-1 reservoir cells
Nature Immunology (2024)
-
DNA framework signal amplification platform-based high-throughput systemic immune monitoring
Signal Transduction and Targeted Therapy (2024)
-
Pharmacokinetics, Disposition, and Biotransformation of [14C]Lenacapavir, a Novel, First-in-Class, Selective Inhibitor of HIV-1 Capsid Function, in Healthy Participants Following a Single Intravenous Infusion
Clinical Pharmacokinetics (2024)
-
Characteristics of blood immune cell profile and their correlation with disease progression in patients infected with HIV-1
BMC Infectious Diseases (2023)
-
Multimeric immunotherapeutic complexes activating natural killer cells towards HIV-1 cure
Journal of Translational Medicine (2023)