
Abstract
The adaptive immune system and associated inflammation are vital in surveillance and host protection against internal and external threats, but can secondarily damage host tissues. The central nervous system is immune-privileged and largely protected from the circulating inflammatory pathways. However, T cell involvement and the disruption of the blood–brain barriers have been linked to several neurodegenerative diseases including Parkinson's disease, Alzheimer’s disease, and multiple sclerosis. Under normal physiological conditions, regulatory T cells (Treg cells) dampen the inflammatory response of effector T cells. In the pathological states of many neurodegenerative disorders, the ability of Treg cells to mitigate inflammation is reduced, and a pro-inflammatory environment persists. This perspective review provides current knowledge on the roles of T cell subsets (e.g., effector T cells, Treg cells) in neurodegenerative and ocular diseases, including uveitis, diabetic retinopathy, age-related macular degeneration, and glaucoma. Many neurodegenerative and ocular diseases have been linked to immune dysregulation, but the cellular events and molecular mechanisms involved in such processes remain largely unknown. Moreover, the role of T cells in ocular pathologies remains poorly defined and limited literature is available in this area of research. Adoptive transfer of Treg cells appears to be a vital immunological approach to control ocular pathologies. Similarities in T cell dysfunction seen among non-ocular neurodegenerative diseases suggest that this area of research has a great potential to develop better therapeutic agents for ocular diseases and warrants further studies. Overall, this perspective review article provides significant information on the roles of T cells in numerous ocular and non-ocular neurodegenerative diseases.
Similar content being viewed by others


Adaptive immune responses and pathological conditions
The immune system can be divided into innate and adaptive immune responses and is designed to protect the body from disease-causing pathogens. The innate immune system is the body's first line of defense and requires no lag time to mount a response to infection. Innate immunity is largely nonspecific. Adaptive immunity is much more specific and provides long-lasting protection against pathogens [5, 176]. The cells that carry out adaptive immune responses are called lymphocytes and are classified into B lymphocytes and T lymphocytes, which accomplish humoral and cell-mediated immune responses, respectively [279]. In the humoral immune response, a B cell receptor reacts with a specific antigen stimulating the cell to synthesize and secrete antibodies, also known as immunoglobulins. These antibodies play crucial roles in neutralizing pathogens [5, 289]. Antibodies recognize epitopes on a single specific molecule (e.g., protein or carbohydrate) called an antigen. When an antigen, such as a virus or microbial toxin (e.g., tetanus or diphtheria toxin), binds an antibody, it can no longer bind to receptors on host cells and is therefore neutralized [81]. T lymphocytes (T cells) arise in the bone marrow and mature in the thymus. They are classified into two major categories CD8 + and CD4 + cells based on their effector functions and recognition of different classes of MHC molecules. CD8 + cells defend the host against intracellular pathogens such as viruses and cancer, as they can detect surface antigens displayed by infected cells [152,153,154]. Cytotoxic CD8 + cells do not recognize free antigens but bind short peptide antigens expressed on the major histocompatibility complex (MHC) class I protein on the surface of most cells [131, 152,153,154]. MHC class I molecules are found on all nucleated cells and are important in T cell activity against viruses, but MHC class II molecules are only found on APCs, which help with the proliferation of B cells [152,153,154]. An antigen-presenting cell (APC; e.g., dendritic cells, macrophages, Langerhans cells, and B cells) must stimulate a cytotoxic T cell to activate it. A pathogen-activated APC will subsequently travel to secondary lymphoid tissues, such as lymph nodes [178]. After activation by an APC, the T cell will expand its population to eliminate the specific pathogen [152,153,154]. Once a population of T cells has been created to combat pathogens, memory T cells will provide lifelong immunity against the pathogen [91]. Mechanistically, CD8 + T cells bind to MHC class I, whereas CD4 + T cells bind to MHC class II [110]. Once a CD8 + T cell has been primed by an APC cell, it is activated and ready to kill infected or invading cells. When the CD8 + T cell binds to its target antigen on the surface of an infected cell, it will release lytic granules containing perforins and granzymes that create pores in the target cell membrane or it induces apoptosis via Fas ligand and caspase activation [362]. In addition, CD8 + T cells release cytokines including IFN-γ, TNF-α, and TNF-β. These cytokines will further inhibit viral replication, activate macrophages, and upregulate MHC class I expression. MHC class I expression increases the detection of an infected cell, as T cells cannot recognize cells without MHC molecules [121, 152,153,154].
CD4 + T cells were originally subdivided into two groups based on their effector functions: Th1 and Th2 cells. These Th1 and Th2 cells are antagonistic and maintain a balance under physiological conditions. Th1 cells, generally, secrete pro-inflammatory molecules such as IFN-γ, TNF-α, TNF-β, and IL-1β; while Th2 cells counteract inflammation with IL-4, IL-5, IL-6, IL-10, and IL-13 [25, 233, 361]. Until the mid-1990s, scientists were not aware of other subsets of CD4 + cells [63], but now CD4 + T cells have been expanded further into Th9, Th17, Th22, Treg cells, and T follicular cells [9, 110]. Th9, Th17, and Th22 T cell subsets secrete different pro-inflammatory cytokines and have been implicated in autoimmune and inflammatory diseases [193, 311, 328]. For example, in allergic asthma Th2, Th9, and Th17 cells play a role in the pathogenesis of the disease through cytokine secretion and the activation of mast cells and eosinophils, which leads to airway hyperactivity [174]. While the ability of T cells to cause inflammation may be necessary for proper immune function, a mechanism to regulate this response is also required. T regulatory (Treg) cells have been shown to play key roles in their regulation [14, 180, 244, 267, 342]. As evident from the name, Treg cells regulate and suppress the potentially dangerous effects of T cells and ultimately promote tolerance. Numerous functions of Treg cells have been documented including suppression of asthma and allergy [45, 51, 287], induction of tolerance to dietary antigens [12, 106], protection of commensal bacteria from elimination by the immune system [40], promotion of maternal–fetal tolerance [135], and prevention of autoimmune disease [63, 117, 333, 334]. The role of Treg cells is not fully understood, and it remains a key area of research, specifically in ocular pathology. However, it has become increasingly clear that Treg cells are required to maintain homeostasis. As referenced above, a hyperactive inflammatory immune response can harm host cells. Conversely, a lack of a properly functioning immune system can lead to life-threatening infections, so a balance must be struck between the two states. Treg cells maintain this balance by playing a suppressive role and preventing a pro-inflammatory response [189]. Numerous disease conditions such as systemic sclerosis [215], gestational diabetes [286], atherosclerosis [138], COPD [149], among many others have reported T cell subsets imbalance as a causative factor in disease pathogenesis.
Tolerance and autoimmunity
The immune system is designed to target foreign pathogens and leave host cells unharmed via a process known as tolerance. Autoimmunity occurs when a host loses self-tolerance. In other words, autoimmunity is a process of an immune response against the host leading to self-cell damage. Different autoimmune conditions can target specific areas of the body like pemphigus vulgaris and pemphigoid, which targets the skin [122], or systemic lupus erythematosus which is disseminated and targets the entire body [182]. An important aspect of autoimmunity is the presence of autoreactive lymphocytes. Auto-reactivity can be triggered through molecular mimicry, which is immunological cross-reactivity between host and foreign antigens. In this condition, foreign antigens closely resemble host antigens resulting in an erroneous self-directed attack on host tissue [67]. Autoreactive lymphocytes can also develop during the initial lymphocyte creation. When lymphocytes undergo genetic rearrangement, some are reactive against self-antigens, but under homeostatic conditions these lymphocytes undergo thymic deletion and undergo apoptosis [331]. Although up to 98% of T cells do not make it through the selection process [24], some autoreactive lymphocytes will escape the process of elimination even in healthy individuals [266]. One of the functions of Treg cells is to prevent the activation of these autoreactive lymphocytes and therefore protect the body from self-attack [297]. FoxP3 knockdown, a marker for Treg cells, leads to a loss of functional Treg cells in Scurfy mice [231]. The lack of Treg cells leads to a fatal autoimmune response by 3–4 weeks of age, demonstrating their critical role in immune tolerance. T cell activation suppression via the induction of anergy, a long-term state of T cell hypo-responsiveness, is the second mechanism of its modulation [43]. Inducing anergy will prevent T cells from undergoing clonal proliferation, providing another potential fail-safe against autoimmunity. Recent studies have shown a strong connection between Treg cells and anergy, anergic T cells can convert into Treg cells, suggesting that persistent anergic T cells may serve as a reservoir for Treg cells, though the mechanism remains largely unknown [246, 292].
Autoimmune diseases affect millions of people with increasing frequency in developing countries, making them an important area of study [265]. Some autoimmune conditions are hereditary, and many primarily affect women, but they typically require a trigger or underlying susceptibility [62]. Autoimmunity is multifactorial and involves genetic, environmental, hormonal, immunological, and unknown triggers [302]. Modifiable risk factors for autoimmune diseases include infection [185], vaccination [329], drugs [72], smoking [268], UV light exposure [166] and obesity [302]. Studies have also shown that people with autoimmune diseases are less active than the rest of the population, suggesting physical activity may play a regulatory role in autoimmunity [284].
Adaptive immunity and Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disorder and one of the most common causes of senile dementia worldwide. The loss of memory and cognitive decline was seen in AD is progressive, irreversible, and is usually seen in the elderly population. Characteristic features of the disease are the accumulation of β-amyloid plaque deposits as well as neurofibrillary tangles of hyper-phosphorylated tau in the brain [30, 78, 82, 339]. However, the familial AD (FAD) mouse models combined with frontotemporal dementia-linked human microtubule associated tau fail to show the extensive and progressive neurodegeneration reported in human patients [230]. Therefore, there is a major gap in our understanding of the steps leading to neurodegeneration and irreversible dementia. The evidence for immune system involvement in AD is compelling. Several studies have implicated both microglia and the innate immune system in the neurodegeneration seen in AD [29, 280]. Studies have also shown the involvement of the adaptive immune system in the pathology of AD. T cells have been identified in the brain parenchyma of postmortem Alzheimer’s patients [321] and T cell abnormalities have also been identified in the blood and cerebral spinal fluid of Alzheimer’s patients [104, 212]. Studies in a mouse model have also shown that β-amyloid promotes T cell infiltration, and it interferes with proper T cell functioning, including activation and antigen presentation, suggesting that failure to mount a protective immune response may contribute to AD pathology [95]. A breakdown in antigen presentation may contribute to an immune inability to clear β-amyloid. Alterations in the peripheral lymphocyte profiles have also been demonstrated in Alzheimer’s disease [49, 50, 184, 262]. Recently, studies have also shown an association between higher levels of CD 4 + cell counts and an increased risk of AD [90]. Most research has focused on T cells in AD, but a few recent findings have also highlighted the roles of B cells in the disease. One study suggests that B cell depletion may prevent disease progression, which is an interesting theory in light of recent identification of resident B cells in the dura matter [171, 275]. This data suggests that alterations in B lymphocyte number, subsets, and production of autoantibodies may all be involved in AD pathology and progression of the disease [33, 270, 271, 298].
Recent studies focusing on cerebrospinal fluid (CSF) biomarkers have consistently disregarded the cellular infiltrates, treating them as artifacts of collection. However, a careful examination of CSF by cutting-edge methods of mass cytometry, revealed a consistent increase in CD8+ T effector memory CD45RA+ (TEMRA) cells, and their association with cognitive impairment [104]. Additionally, single-cell RNA sequencing and artificial intelligence revealed an increase in T cell receptor (TCR) signaling, and further analysis identified clonally expanded CD8+ TEMRA cells targeting Epstein–Barr viral antigens. These studies show that T cells in the CSF play the same role as peripheral T cells in surveillance and maintenance of the intrathecal space. The existence of immune plasticity can be both beneficial and detrimental, depending on the target, extent of activity, and resolution of the inflammatory cascade [172, 278]. This complex pathway is consistent with findings showing that depletion of T cells prevented hippocampal infiltration and spatial memory deficits in AD models [186]. Acute inflammatory responses may be neuroprotective and promote plaque clearance, but chronic inflammatory responses may be detrimental due to excessive collateral tissue damage [4, 273]. Therefore, lymphocyte physiology is altered during AD pathogenesis and these changes likely play a role in the progression of the disease.
Immunotherapy is an area of high interest in AD. The recent approval of a monoclonal antibody called aducanumab that selectively targets aggregated forms of amyloid β in AD patients may help in preventing early disease progression [94, 277, 282]. This antibody is produced using selectively reactive B cells, and shows that the adaptive immune system may hold the key for better AD treatments and prognoses. However, the approval of aducanumab has been met with controversy and many questions arose about the efficacy of this drug [173, 282]. While the clinical success of aducanumab remains in question, immunotherapy using other antibodies will continue to be the focus of research for future drug development in this field. Overall, research involving monoclonal antibodies that recruit T cells (e.g., effector T cells and Tregs) to target amyloid β may hold promise in restoring the disrupted immune balance in AD.
Adaptive immunity and Parkinson’s disease
Parkinson’s disease (PD) is another example of an inflammatory disease process causing progressive neurodegeneration of the dopaminergic neurons in the substantia nigra of the brain [20]. Aggregates of misfolded α-synuclein (α-syn protein accumulate in the brain of PD patients, similar to the protein aggregates observed in Alzheimer’s disease. Patients suffering from PD experience motor symptoms such as tremors, muscle rigidity, and slowness of movement (bradykinesia but they can also experience cognitive impairment. The involvement of immune cells in PD has been suggested. For example, studies have shown activated microglia in the substantia nigra of postmortem Parkinson’s disease patients [207] and potential role of the adaptive immune system in PD has been shown [132, 163, 250].
Parkinson’s disease is considered to be a systemic inflammatory disorder because elevated pro-inflammatory cytokines are found in the blood of PD patients [27, 79, 103, 140, 257, 259, 304]. An increased level of inflammatory cytokines is believed to be due to T cell activation. Studies have shown that activation of T cells in response to Parkinsonian α-synuclein peptides and the inflammation observed in PD could be, partly, due to the involvement of autoreactive T cells [23, 307]. Autoreactive T cells are central to autoimmune pathology, so their presence may suggest the disease is autoimmune in nature. Autoimmune conditions are also characterized by the creation of autoantibodies by B cells against self-antigens which has been demonstrated in the peripheral blood of PD patients [83, 235, 347]. Recently, a study has shown an alteration in B cells population, which may play a role in PD [332]. Additionally, studies have also shown abnormal profiles of B cells and T follicular cells, indicating a polarization towards an inflammatory phenotype [195].
Regardless of the autoimmune nature of Parkinson’s disease, T cells, and the adaptive immune system are believed to contribute to the disease development [132, 163, 250]. Several studies reported a Th1 bias in PD patients and experimental animal models [15, 53, 181]. As Th1 cells are pro-inflammatory, a shift favoring their expansion is congruent with the theory that neuroinflammation plays a role in PD. Another study has shown that CD4 + cells were the primary mediator of dopaminergic damage [31], while other studies have shown a decrease in the circulating CD4 + cell population in PD [18, 301]. If CD4 + T cells are the main mediators of disease, decreased circulating levels seem contradictory. However, the overall decrease in CD4 + T cells may be attributed to a decrease in Th17, Th2, and Treg cells and not the Th1 lineage [181]. Maintenance of the Th1 lineage despite a decrease in the other subsets would still fit with the studies citing a Th1 bias, though not all studies are in agreement [229]. One study marked an observed increase in the proportional CD4 + and CD3 + T cells as well as the CD4 + /CD8 + ratio in PD patients [52]; whereas, another study showed a decrease in the CD3 + , CD8 + T cells and B lymphocyte subsets in addition to a decrease in CD4 + T cells [114, 229]. Reasons for a decrease in the CD4 + T cell population may be explained by a study done by Calopa et al. which found increased susceptibility to apoptosis in the CD4 + T cells in the peripheral blood of PD patients [34]. Other conflicting data exist on the Th17 subset. While Kustrimovic et al. found that Th17 cells were decreased in the blood of PD patients, other studies have found that peripheral Th17 cells were increased in PD patients [49, 50, 53, 349]. There is conflicting evidence on the relative prevalence of each subset level, but overall, many abnormalities in T cells subsets populations have been reported in PD in favor of an inflammatory phenotype. More research is needed to clarify how effector T cell populations are affected in PD.
Treg cells likely play an opposing role in PD to the inflammatory T cell subsets by suppressing their effector functions and preventing rampant inflammation [139]. This hypothesis is supported by studies that showed that the transfer of Treg cells could provide neuroprotection in mouse models of PD [260, 261]. The beneficial nature of Treg cells in PD may explain why global T cell deficiency worsened the motor deficits seen in a Parkinson mouse model by decreasing effector T cells and inadvertently reducing the protective effects of Treg cells [340]. However, as PD is plagued by neuroinflammation, it is evident that Treg cells cannot properly execute their job for unknown reasons, and this dysfunction may contribute to disease progression [165]. The idea that Treg cells are unable to adequately function is supported by a study that has shown an impaired ability of Treg cells to suppress effector T cells in PD [274]. This breakdown of functioning furthers the theory that an inflammatory imbalance is observed in PD. In spite of discrepancies relating to T cell subset numbers, a change in the ability of Treg cells to function properly would result in homeostatic deviations regardless of cell numbers. More research is needed to clarify the uncertainties in the field, but most of the literature available suggests that the inflammation observed during PD is partly due to a T cell subset imbalance, which favors inflammation [52, 196]. Better understanding of the mechanisms behind PD and how the immune system is involved will hopefully lend to the development of effective therapies for PD.
Adaptive immunity and multiple sclerosis
Multiple sclerosis (MS) is another neurodegenerative disease of the central nervous system that causes motor and sensory deficits [245]. The main hallmark of the disease is the presence of disseminated focal lesions or plaques in the CNS where demyelination and gliosis occur with relative axonal sparing [247]. Multiple sclerosis is an inflammatory disease similar to other neurodegenerative conditions, with macrophages and microglia contributing to the pathology. Other peripheral immune cells are also likely to be involved in the demyelination, including T cells (CD4 + and CD8 +), B lymphocytes, plasma cells, and dendritic cells [75, 129], and interactions between macrophages and lymphocytes may be part of the underlying pathogenesis [58].
Autoreactive T cells are an important part of autoimmune pathology in MS. Due to the extensive involvement of lymphocytes in MS plaques, the question has been raised as to whether the cause of the inflammation observed in MS may be autoimmune-dependent. A widely used animal model of MS, known as experimental autoimmune encephalomyelitis (EAE), is largely CD4 + T cell-driven [60]. Moreover, data have shown a link between CD4 + cytotoxic lymphocytes, disease severity, and plaque activity in MS patients [102, 241]. Despite haziness surrounding the underlying cause of the inflammation seen in MS, the evidence of T cell involvement is strong. It has been shown that activated T cells can induce experimental autoimmune encephalomyelitis in healthy mice [97, 177, 209] and that global reduction of most lymphocytes via alemtuzumab can improve MS pathology [161, 249]. Th1 cells have been implicated as important detrimental players in MS due to their ability to stimulate M1 macrophages through the secretion of TNF‐α and IFN‐γ, which are important mediators of inflammation and cellular damage [192]. Most of the earlier research in MS focused on the role of T cells, however, recently researchers have also acknowledged a cooperation between B cells and T cells in MS pathogenesis [155]. For example, B cells have been shown to work in tandem with T cells in human MS pathology [155]. Though more research is needed on the interplay between T cells and B cells in multiple sclerosis, a similar shift towards a pro-inflammatory B cell state has been suggested [85, 86, 193, 194]. The mechanisms involved in B cell-induced pathology in MS remains largely unknown. As of 2019, all approved MS disease-modifying therapies impact B cells in some way, such as depletion of CD20 + B cells [270]. However, not all B cell targeted therapies in MS have created positive results. One clinical trial using an experimental B cell depleting therapy for MS was terminated early due to worsening of disease progression [164]. This suggests that the role of B cells in MS is complex, and more research is needed to better understand the exact roles of B cells in MS pathology.
Unlike the harmful effects of the inflammatory T cell subsets discussed, Treg cells are likely protective in MS. For example, the transfer of myelin oligodendrocyte glycoprotein specific Treg cells displayed dose-dependent protection against experimental autoimmune encephalomyelitis [175, 258]. Inflammation in MS pathology may be in part due to a decrease in Treg cells, [136, 248] or functionality [92, 120, 179, 211, 326]. A reduced capacity of naïve CD4 + T cells to differentiate into Treg cells under pathological conditions has also been demonstrated [281]. A reduction in number or function of Treg cells would mean a decreased capacity for inflammatory suppression. On the other hand, some studies have shown relative increases in the levels of Treg cells in MS [92, 179], but reduced functionality [179]. This may suggest Treg cells functionality may be more consequential in MS pathology than the number itself. Although more research is needed to clarify how each subset of T cells is involved in MS, current data suggest that a failure in Treg cell number and or functioning combined with an upregulation of effector T cells contribute to the inflammation and CNS damage seen in MS [211]. A deeper understanding of T cells interaction within MS will hopefully lead to new therapy. Interventions that downregulate effector T cells or upregulate Treg cells may decrease the disease progression in MS patients.
Adaptive immunity and the eye
The eye is an “immune privileged” organ, which limits its inflammatory immune response so that vision is not harmed by swelling, infection, and other tissue changes. The eye is similar to the brain, testes, placenta, and fetus in regard to immune responses. Typically, even foreign antigens do not trigger immune responses in these organs. In addition, the blood–retinal barriers in the eye stop infiltration of blood-borne pathogens and immune cells under physiological conditions. However, under pathological conditions such as uveitis, glaucoma, diabetic retinopathy, and retinal ischemia, numerous immune cells can infiltrate the eye and may induce or facilitate autoimmunity that can lead to the development of autoimmunity [38].
In general, if an immune-privileged organ is damaged, previously insulated proteins will be exposed to peripheral immune cells that have not encountered these “novel” antigens. Having never come across these antigens before, peripheral immune cells have not learn to recognize them as self, allowing the generation of autoreactive lymphocytes [99]. The immune-privileged status of the eye is maintained by the blood–retinal barrier’s passive physical sequestration via tight junctions and the retinal pigment epithelium. The protective microenvironment of the eye is immunosuppressive and it expresses substances such as Qa-1, Fas ligand, and indolamine dioxidase (IDO) which function to prevent a damaging inflammatory reaction to ocular tissues [61]. The immunosuppressive environment is also influenced by the aqueous humor which dampens the activity of many immune responses including nitric oxide production by macrophages [310], complement activation [111], neutrophil activation [303], lymphocyte proliferation [162], and NK cell activity [10]. Treg cells are also involved in the immune privilege of the eye through anterior chamber-associated immune deviation [13], in which injection of a foreign antigen into the anterior chamber of the eye causes an antigen-dependent down regulation of delayed-type hypersensitivity [324]. Overall, these mechanisms create an environment that is sheltered from potential immune cell-induced injury [227].
Adaptive immunity and uveitis
Typically, uveitis is classified by the affected anatomical part of the eye (e.g., anterior uveitis, intermediate uveitis, posterior uveitis and panuveitis) [17, 191]. Often the cause of uveitis is idiopathic, but sometimes an infectious cause (e.g., toxoplasmosis, tuberculosis, onchocerciasis, cysticercosis, leprosy and leptospirosis) can be responsible for this disease [70, 71]. Non-infectious uveitis is immune mediated and can be limited to the eye (e.g., sympathetic ophthalmia and birdshot retinochoroidopathy) or part of a broader systemic disease (e.g., sero-negative HLA-B27-positive spondyloarthropathies, juvenile idiopathic arthritis, sarcoidosis, multiple sclerosis, inflammatory bowel disease, tubulointerstitial nephritis, Behçet disease, and Vogt–Koyanagi–Harada syndrome) [39, 71]. Ongoing research proposes that noninfectious uveitis may be an autoimmune condition through breakdown of self-tolerance and mobilization of autoreactive effector cells. However, there are some theories in the field suggesting some cases of idiopathic immune-mediated uveitis might be the result of reactivatable infectious agents concealed in ocular tissue rather than true autoimmunity [98]. Nevertheless, autoimmune pathogenesis is generally accepted as a contributing factor in uveitis [17]. Support for the autoimmune theory of uveitis is provided by the increased genetic susceptibility of people with certain HLA phenotypes, as HLA genes have long been linked to autoimmunity [205]. In addition, autoimmune conditions are characterized by autoreactive T cells targeting self-antigens, evidence of which has been demonstrated in patients with noninfectious uveitis with uveal melanin, retinal arrestin, and inter-photoreceptor retinoid binding protein (IRBP) [39]. The experimental autoimmune uveitis (EAU) mouse model of posterior uveitis has demonstrated reduced inflammation through anti-CD3-mediated T cell suppression [306]. Interestingly, another study that used an anti-CD3 antibody saw a decrease activation of effector T cells but enhanced activation of Treg cells [168]. The autoreactive T cells responsible for autoimmune pathology were thought to be Th1 CD4 + T cells [80, 100], but more recent studies have indicated both Th1 and Th17 cells can contribute [133, 337]. Most research implicates both Th1 and Th17 in uveitis pathology, but other subsets may also be involved as well, such as a small subset of T cells known as γδ T cells [66, 283]. Some studies have also reported the possibility of autoreactive CD8 + T cells involvement in uveitis [210, 283, 300]. It is also possible that the etiology may vary between the different conditions and the stages of uveitis. For example, in Behçet’s uveitis there is a greater number of CD8 + T cell in the aqueous humor [356], but in sarcoid uveitis the CD4 + /CD8 + ratio was increased [69]. These discrepancies suggest a possible difference in the pathogenesis between uveitis etiologies which could also affect their treatment approaches. There is still a lot to be done to fully clarify the role of CD8 + T cells in uveitis pathology, but there is mounting evidence that CD8 + T cells participate in autoimmune disease, making it plausible that they contribute significantly in uveitis pathology [76, 350]. Despite some remaining ambiguity, there is substantial support that Th1 cells and Th17 cells are implicated in autoimmune pathology [7, 115, 200, 308]. The specific role of each T cell subset in uveitis requires additional research that will fill in the gaps in our knowledge.
Most of the studies have focused on the roles of T cells, but B lymphocytes also likely play roles in uveitis. B lymphocytes may promote an inflammatory environment as well as promote T cell survival [296]. For example, depletion of B lymphocytes by rituximab and other monoclonal antibodies treatment in uveitis provides positive outcomes [68, 128, 214]. In contrast, some studies also have shown protective effects of B lymphocytes. For example, B lymphocytes suppressed intraocular inflammation and helped expand protective Treg cells in a mouse model [55]. Additionally, loss of a transcription factor in B cells caused suppression of both B regulatory and T regulatory cells, which resulted in worsening of the disease [232]. This suggests that certain B cell subsets may be contributing to uveitis pathology, while others may be protective.
Studies have shown that regulatory T (Treg) cells in uveitis have the ability to modulate inflammation and downregulate effector immune functions. The unregulated inflammation seen in uveitis suggests aberrant activation or a breakdown in proper Treg cells functionality or number. This idea was supported by a study done by Muhammad et al. that showed reduced ability to induce Treg cells in the experimental autoimmune uveoretinitis model [220, 333, 334]. In addition, a reduction in Treg cells has been shown in patients with active uveitis [269, 354] and Behçet’s disease before an ocular attack [222] in peripheral blood samples. Dysregulation of Treg cells has also been shown as a contributing factor for disease recurrence in recurrent experimental autoimmune uveitis [167]. Additionally, in a mouse model of experimental autoimmune uveitis adoptive transfer of Treg cells has been shown to suppress disease progression [290]. This idea is further supported by a study that claims a shift away from an inflammatory T cell phenotype in favor of Treg cells help mediate disease resolution [108]. Silver et al. also implicate Treg cells in the resolution and remission of uveitis pathology, although they claim Treg cells functionality is not impaired under pathologic condition [293]. This is a direct contradiction of what Ke et al. state when they claim that dysregulation and improper function of Treg cells contribute to disease reoccurrence [167]. Nevertheless, despite this discrepancy in whether Treg cells are decreased in number only or also in functionality, promoting the proliferation of Treg cells results in the suppression of pathology. These studies implicate Treg cells dysfunction during the development of uveitis. Moreover, a deficiency of Treg cells (e.g., function, numbers) may play a central role in disease pathogenesis. More research is needed to clarify how the population of Treg cells changed during the progression of uveitis.
Adaptive immunity and diabetic retinopathy
Diabetic retinopathy is one of the leading causes of blindness in human between the ages 27 and 75, and its estimated prevalence is 90% for patients who have had diabetes for over 20 years [41]. Chronic poor glucose control along with diabetes can lead to vascular complications such as macular edema, neovascularization, and microaneurysms which result in the loss of central vision in diabetic retinopathy patients [188]. As the incidence of diabetes continues to rise, the number of people suffering from diabetic retinopathy is expected to rise as well [295]. Many factors contribute to the development of diabetic retinopathy, but this perspective review will provide limited information for the role of T cells in the diabetic retinopathy.
Inflammation has been linked to obesity and metabolic disorders such as diabetes [134, 338]. Many pro-inflammatory cytokines have been shown to be elevated in the vitreous humor of patients with diabetic retinopathy including TNF-α [366], IL-8 and MCP-1 [130], IL-6 [119], IL-26 [333, 334] and IL-1β [366]. Studies have also shown that more Th1-dependent pro-inflammatory cytokines are secreted in diabetic retinopathy, suggesting an imbalance of lymphocytes [36]. An important part of diabetic retinopathy pathology is blood vessel angiogenesis which is promoted by vascular endothelial growth factor (VEGF). However, studies have shown that the Th1/Th2 ratio is an independent predictor of VEGF plasma levels in diabetic retinopathy [363]. Other studies suggest that Th17 cell-dependent IL-17 may be associated with the inflammation observed in diabetic retinopathy [46, 48, 148]. IL-17A has been shown to be an important detrimental cytokine in the progression of diabetic retinopathy [253, 254]. Studies have also shown that Th17 cells can infiltrate the retina in a diabetic retinopathy mouse model [291]. Moreover, elevated levels of IL-17 have been identified in the plasma [124], vitreous [151], and aqueous humor [93], of diabetic retinopathy patients. However, conflicting data exist on the level of IL-17 in the serum of diabetic retinopathy patients because studies have also shown a negative association with IL-17 and diabetic retinopathy [3, 221].
The overwhelming inflammation seen in diabetic retinopathy may be due to an imbalance of Treg cells. For example, studies have shown decreased numbers of Treg cells in type II diabetes and diabetic retinopathy patients [251, 351, 358]. Treg cells have also been shown to be beneficial in repairing abnormal angiogenesis in diabetic retinopathy [74]. In addition to the disturbance in the homeostasis of effector T cells and the population of Treg cells, Treg cells functions could also be altered by the surrounding environment. For instance, Treg cells may be unable to properly suppress inflammation in diabetic retinopathy due to elevated insulin levels [123]. Treg cells have been shown to have reduced suppressive capacity in type II diabetes mellitus patients [285]. Additional studies are required to clearly understand the neuroprotective role of Treg cells in diabetic retinopathy.
Adaptive immunity and age-related macular degeneration
Age-related macular degeneration (AMD) is a progressive disease-causing degeneration of the macula and is the leading cause of blindness among the elderly population in developed countries [344]. Two different pathologic components contribute to vision loss in AMD: focal atrophy of the retinal pigment epithelium (RPE) and photoreceptor loss (“dry” AMD) or choroidal neovascularization (“wet” AMD) [322]. Typically, dry AMD is a precursor for wet AMD, but not all patients will experience both [8]. In dry AMD, there is a gradual expansion of the atrophic lesions and a slow progressive vision loss. In wet AMD, the new blood vessels leak leading to edema, retinal damage, and can cause a rapid loss of visual acuity. Currently, there is no approved drug therapy to treat dry macular degeneration, though there are many ongoing clinical trials to address this problem [238]. Some clinical progress has been made to treat wet AMD, such as anti-VEGF therapy that targets VEGF, an important growth factor that facilitates angiogenesis [170]. However, anti-VEGF therapy only delays disease progression and upon treatment cessation relapse is a common problem [333, 334]. An important component of AMD is persistent inflammation [239]. Elevated levels of complement proteins have been detected in the blood samples of AMD patients, suggesting some sort of complement dysfunction may be contributing to the disease [126, 202, 263]. The current understanding of AMD is that local complement dysregulation is involved in the disease pathogenesis. One component of complement called complement factor H (CFH) is an important inhibitor of the alternative pathway of complement, and CFH polymorphisms have been linked to an increased risk of AMD development [88, 183]. Despite the abundance of genetic evidence linking complement to AMD, the exact role of how complement may be involved is still not clear.
Studies in an AMD mouse model have shown that T cells can contribute to AMD pathogenesis [65]. Patients with neovascularization AMD were also shown to have higher systemic lymphocyte counts, suggesting that lymphocytes may play an active role in initiating the neovascularization seen in AMD. [305]. Several studies found that the neutrophil-to-lymphocyte ratio (NLR), which are thought to be a marker of both inflammation and angiogenesis, were elevated in wet AMD [147, 225, 309]. These studies suggest that immune cells may be dysfunctional in neovascular AMD, however, the clinical relevance remains unclear [225]. In addition, increased recruitment of T cells in AMD has also been shown [216, 224]. Studies have also shown dysfunction or senescence of the immune system in the context of aging and neurodegenerative disease through the creation of a chronic state of low-grade inflammation referred to as “inflammaging” [73, 101]. This state of immune senescence has been implicated in AMD in which T cells are likely involved [190]. Additionally, several studies have shown an increased proportion of aged T cells in AMD patients, suggesting that immune dysfunction may play a role in disease pathogenesis [89, 305]. Dysregulation of follicular T cells has also been reported in AMD [345]. The idea of T cells dysregulation is further supported by a study showing alterations in systemic Th1 lymphocyte profiles in AMD patients [294]. More specifically, studies have shown that IL-17 may be an important part of AMD disease development [11, 288] since reducing IL-17 levels decreased the amount of choroidal neovascularization [125, 197]. However, unlike other diseases previously discussed in this review, the main source of IL-17 in AMD may not be from Th17 cells, but instead from γδ T cells [365]. These IL-17 producing γδ T cells have been shown to infiltrate the eye in a mouse model of choroidal neovascularization (CNV) [64]. Notably, IL-17 + cells have also been identified near areas of RPE atrophy [35]. Another mouse model study showed that Th2 cytokines, mainly IL-4, helped to decrease neovascularization in the disease process [346]. This supports the overall theory that an imbalance in the pro/anti-inflammatory phenotypes helps to drive AMD. Limited data have shown an increase in Th17 and Th1 cells in AMD patients [49, 50], whereas another study has shown a decrease in Th1 cells and no significant changes in Th17 cells [294]. Interestingly, one study found that decreased levels of CD4 + T cells correlated with the absence of subretinal fibrosis in AMD [187]. Overall, more studies are needed to better understand the role of adaptive immune cells in AMD.
Limited literature exists for the involvement of B lymphocytes in AMD, but anti-retinal antibodies have been identified in AMD patients [2, 118, 146, 217, 243]. It remains in question whether they are a consequence of disease-related damage or a contributory factor for the disease development [157, 158, 160]. In contrast, studies have also shown no difference in the number of circulating B lymphocytes in AMD patients when compared with healthy subjects, which does not rule out B lymphocytes involvement in AMD pathology because antibody production by B lymphocytes could still have a contributory effect [46, 48]. Studies have also suggested that autoantibodies could be used as biomarkers for future disease progression and prognostication [157, 158, 160, 218]. Another study found elevated levels of IL-17 correlated with response to anti-VEGF therapy [223]. Identification of a biomarker could have great clinical significance in AMD treatment, however more work is needed in this area.
Retina repair and reduced angiogenesis were observed through adoptive transfer of Treg cells and Treg cells expansion [74]. Currently, not many studies have looked at the role of Treg cells in AMD. One study did not find changes in the number of systemic Treg cells in AMD [203]. Like effector T cells and B cells, it is again possible that Treg cells dysfunction may play larger roles than Treg cells number [19]. Treg cells are a promising candidate to study in inflammatory diseases, and they may be implicated in AMD, though more research is needed.
Adaptive immunity and glaucoma
Glaucoma is the second leading cause of blindness worldwide in which retinal ganglion cells (RGCs) die slowly and progressively over a prolonged period of time. Glaucoma is now considered to be a multifactorial disease and molecular mechanisms involved in RGC death are complex and poorly understood [28, 109, 226, 325]. The primary risk factor for developing glaucoma is elevated intraocular pressure (IOP), but the pathophysiology of the disease is more complicated [22, 59, 84, 127, 299]. There is currently no known cure for glaucoma and treatment focuses on reducing IOP [47]. Unfortunately, some patients under IOP-lowering therapy still see progression of the disease, which clearly indicates that better therapeutic options are needed to fully cure the disease. Studies have shown that multiple factors play key roles in RGC degeneration including activation of caspases [44, 141, 142, 169, 208, 320], apoptosis [6, 116, 226, 255, 327], oxidative stress [56, 143, 150, 198, 219, 228, 272], ischemia and hypoxia [32, 57, 141, 141, 142, 142, 144, 234], epigenetic changes [105, 206, 242, 276, 353, 359, 359, 360, 360], Crosson 2010 #975, [145], alteration in the levels of pro-inflammatory cytokines [1, 312, 316, 341, 352, 357], and deprivation of neurotrophic factors [240, 256]. There is another opinion with limited evidence, which suggests the neurodegeneration observed in glaucoma could be vascular. This theory suggests that hemodynamic alterations and changes in local blood flow may cause unstable ocular perfusion to the optic nerve [42, 54, 96, 237, 355]. Recently, studies have also shown participation of T cells in glaucoma pathology. A study has shown that elevated IOP allowed for T cell infiltration of the retina and led to continued degeneration of RGCs after IOP was returned to a normal level [45, 51].
The innate immune system has long been tied to glaucoma through the action of glial cells and oxidative stress [201, 314, 319], but recent evidence provides additional support for the involvement of the adaptive immune system in glaucoma pathology [156]. Earlier studies have shown that glaucoma could be critically tied to the immune system because resistance of RGC death was shown to be correlated with immune potency [16]. This study suggested that immune dysfunction may be a prerequisite for developing glaucoma and would explain the disparities in degree of disease progression among patients treated with ocular hypertensive agents. Afterwards, numerous studies have shown the presence of autoantibodies to retinal and optic nerve proteins in the serum [21, 77, 159, 204, 264, 313, 317, 348], retina [112], and aqueous humor [157, 158, 160] of glaucoma patients. Studies have also shown the presence of autoantibodies to heat shock proteins [26, 107, 318, 323, 336] in glaucoma. Heat shock proteins are a large family of molecular chaperones that can be upregulated in times of stress [213], but also have significant potential to induce molecular mimicry resulting in the creation of autoantibodies to host proteins [335]. In glaucoma, elevated levels of heat shock proteins such as HSP72 [318], HSP60, and HSP27 [315], have been reported, some of which may help facilitate RGC death [318, 330]. Introduction of heat shock proteins to rats through immunization can induce glaucomatous injury [45, 51, 336], but other studies have shown that induction of heat shock proteins can provide neuroprotection for RGCs [37, 236, 252]. Overall, the role of autoantibodies and heat shock proteins in glaucoma remains unclear. It is not clear if they are involved in glaucoma pathology directly, indirectly, or play a role in protective autoimmunity [364].
The involvement of T cells in glaucoma pathology remains in question, but we believe that T cell subsets, specifically the T effector/Treg cells homoeostasis, play a critical role in determining the fate of RGCs during glaucoma progression. Additionally, studies have shown an adoptive transfer of lymphocytes from glaucomatous mice into healthy mice stimulated RGC loss [113]. This causative effect suggests that adaptive immune dysfunction plays a direct role in the pathophysiology of glaucoma. Studies have shown that CD4 + T cells facilitate RGC death in an acute ischemia/reperfusion injury model [137]. An imbalance in Treg cells/Th17 cells in experimental autoimmune optic neuritis (EAON) has been shown, which shares key characteristics of glaucoma pathology [199]. Recent studies have also shown that transient elevation of IOP can cause T cells infiltration to the retina, leading to RGC degeneration [45, 51]. However, the role of Treg cells in glaucoma remains fully unexplored. The authors’ perspective in this regard is “enhanced neuroinflammation during glaucoma could be due to low number and/or function of Treg cells”. In other systems, unchecked inflammation has been attributed due to improper functioning of Treg cells [343]. Adoptive transfer of Treg cells has shown promise in treating inflammatory conditions such as enteritis and multiple sclerosis suggesting it may prove an effective therapeutic option in glaucoma [87]. If under glaucomatous condition Treg cells are unable to differentiate, adoptive transfer of Treg cells may ameliorate the T cell imbalance and decrease inflammation. More work is needed in this field to better understand the involvement of T cells in glaucoma progression.
Conclusions
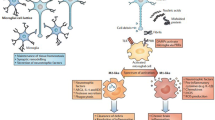
We provided a brief overview of adaptive immunity, autoimmunity, and tolerance and related them to neurodegenerative and ocular diseases. This perspective review article aims to emphasize the significance of adaptive immunity concerning neurodegenerative conditions and highlight the gaps in the field. It also highlights the pathological and neuroprotective roles of different subsets of lymphocytes in numerous neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and several ocular diseases. Figure 1 provides a framework of potential contributing factors to neurodegenerative diseases. Although the etiology of each neurodegenerative disease is different and complex, a few common players that may have crucial roles in the pathology have been shown. These factors include neuroinflammation, epigenetic changes, ischemia/hypoxia, oxidative stress, hemodynamic alterations, and changes in the immune cells. The focus of this perspective review is to provide information for the role of immune cells (e.g., effector T and Treg cells) in neurodegenerative diseases. Based on the literature in non-ocular and ocular neurodegenerative diseases, we provided our perspective that: (1) the number of effector T cells may be increased under ocular pathologies such as uveitis, diabetic retinopathy, AMD, and glaucoma. Elevated T cells can subsequently enhance the secretion of pro-inflammatory cytokines and expedite retinal degeneration similar to other neurodegenerative diseases, and (2) Treg cells are deficient in function and/or number rendering them unable to sufficiently regulate the function of effector T cells in ocular pathologies as seen in other neurodegenerative diseases. Evidence of lymphocyte involvement in ocular pathologies is promising and there is a vital need for more research in this field. We conclude that T cell subsets homeostasis is critical for the maintenance and regulation of neuroinflammation. If the homeostasis is lost and the balance of specialized subsets of immune cells breaks down, damage to the host’s tissues can lead to pathological conditions. The inability to yet discover a neuroprotective therapy for ocular pathologies (e.g., uveitis, diabetic retinopathy, AMD, and glaucoma) it is highly desirable to target immune cells for future research.

Schematic showing the roles of numerous factors and immune cells in the induction or protection in neurodegenerative diseases
Based on the literature provided in this perspective review article, we believe that in ocular neurodegenerative diseases, there may be an imbalance in the number of effector T cells as well as the number and or function of Treg cells, which might contribute to a pro-inflammatory state and facilitate neuronal death in such ocular diseases (e.g., uveitis, diabetic retinopathy, AMD, and glaucoma). This area of research is underdeveloped, hence more studies are needed to clearly understand how T cell subsets and dysfunction may play a role in developing ocular neurodegenerative diseases.
Availability of data and materials
Not applicable.
Abbreviations
- AMD:
-
Age-related macular degeneration
- APC:
-
Antigen presenting cell
- CFH:
-
Complement factor h
- CNS:
-
Central nervous system
- EAE:
-
Experimental autoimmune encephalomyelitis
- EAON:
-
Experimental autoimmune optic neuritis
- EAU:
-
Experimental autoimmune uveitis
- HLA:
-
Human leukocyte antigen
- HSP:
-
Heat shock protein
- IDO:
-
Indolamine dioxidase
- IFN-γ:
-
Interferon gamma
- IL-1β:
-
Interleukin-1-beta
- IL-4:
-
Interleukin-4
- IL-5:
-
Interleukin-5
- IL-6:
-
Interleukin-6
- IL-8:
-
Interleukin-8
- IL-10:
-
Interleukin-10
- IL-13:
-
Interleukin-13
- IL-17:
-
Interleukin-17
- IL26:
-
Interleukin-26
- IOP:
-
Intraocular pressure
- IRBP:
-
Inter-photoreceptor retinoid binding protein
- MCP-1:
-
Monocyte chemoattractant protein-1
- MHC:
-
Major histocompatibility complex
- MS:
-
Multiple sclerosis
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NK cells:
-
Natural killer cells
- PD:
-
Parkinson’s disease
- RGC:
-
Retinal ganglion cells
- TNF-α:
-
Tumor necrosis factor-alpha
- TNF-β:
-
Tumor necrosis factor-beta
- VEGF:
-
Vascular endothelial growth factor
- WBCs:
-
White blood cells
References
Abdul Y, Akhter N, Husain S. Delta-opioid agonist SNC-121 protects retinal ganglion cell function in a chronic ocular hypertensive rat model. Invest Ophthalmol Vis Sci. 2013;54(3):1816–28.
Adamus G, Chew EY, Ferris FL, Klein ML. Prevalence of anti-retinal autoantibodies in different stages of Age-related macular degeneration. BMC Ophthalmol. 2014;14:154.
Afzal N, Zaman S, Asghar A, Javed K, Shahzad F, Zafar A, Nagi AH. Negative association of serum IL-6 and IL-17 with type-II diabetes retinopathy. Iran J Immunol. 2014;11(1):40–8.
Agrawal A, Baulch J, Acharya M, Agrawal S. Identification of Peripheral Immune Mechanisms playing a protective role in Alzheimer’s Disease progression. J Immunol. 2019;202(1):182..136-182.136.
Alberts B, Johnson A, Lewis J, et al. The adaptive immune system. Molecular Biology of the Cell. New York, Garland Science. 2002.
Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012;31(2):152–81.
Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. 2007;13(6):711–8.
Ambati J, Fowler BJ. Mechanisms of age-related macular degeneration. Neuron. 2012;75(1):26–39.
Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. 2015;135(3):626–35.
Apte RS, Sinha D, Mayhew E, Wistow GJ, Niederkorn JY. Cutting edge: role of macrophage migration inhibitory factor in inhibiting NK cell activity and preserving immune privilege. J Immunol. 1998;160(12):5693–6.
Ardeljan D, Wang Y, Park S, Shen D, Chu XK, Yu CR, Abu-Asab M, Tuo J, Eberhart CG, Olsen TW, Mullins RF, White G, Wadsworth S, Scaria A, Chan CC. Interleukin-17 retinotoxicity is prevented by gene transfer of a soluble interleukin-17 receptor acting as a cytokine blocker: implications for age-related macular degeneration. PLoS ONE. 2014;9(4): e95900.
Arroyo Hornero R, Hamad I, Corte-Real B, Kleinewietfeld M. The impact of dietary components on regulatory T cells and disease. Front Immunol. 2020;11:253.
Ashour HM, Niederkorn JY. Gammadelta T cells promote anterior chamber-associated immune deviation and immune privilege through their production of IL-10. J Immunol. 2006;177(12):8331–7.
Attias M, Al-Aubodah T, Piccirillo CA. Mechanisms of human FoxP3(+) Treg cell development and function in health and disease. Clin Exp Immunol. 2019;197(1):36–51.
Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord. 2005;11(8):493–8.
Bakalash S, Kipnis J, Yoles E, Schwartz M. Resistance of retinal ganglion cells to an increase in intraocular pressure is immune-dependent. Invest Ophthalmol Vis Sci. 2002;43(8):2648–53.
Bansal S, Barathi VA, Iwata D, Agrawal R. Experimental autoimmune uveitis and other animal models of uveitis: an update. Indian J Ophthalmol. 2015;63(3):211–8.
Bas J, Calopa M, Mestre M, Mollevi DG, Cutillas B, Ambrosio S, Buendia E. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J Neuroimmunol. 2001;113(1):146–52.
Behnke V, Wolf A, Langmann T. The role of lymphocytes and phagocytes in age-related macular degeneration (AMD). Cell Mol Life Sci. 2020;77(5):781–8.
Beitz JM. Parkinson’s disease: a review. Front Biosci (Schol Ed). 2014;6:65–74.
Bell K, Funke S, Pfeiffer N, Grus FH. Serum and antibodies of glaucoma patients lead to changes in the proteome, especially cell regulatory proteins, in retinal cells. PLoS ONE. 2012;7(10): e46910.
Bell K, Und Hohenstein-Blaul NVT, Teister J, Grus F. Modulation of the immune system for the treatment of glaucoma. Curr Neuropharmacol. 2018;16(7):942–58.
Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE. 2008;3(1): e1376.
Berg JM, TJ, Stryer L. Immune responses against self-antigens are suppressed. Biochemistry. New York, W H Freeman. 2002.
Berger A. Th1 and Th2 responses: what are they? BMJ. 2000;321(7258):424.
Beutgen VM, Perumal N, Pfeiffer N, Grus FH. Autoantibody biomarker discovery in primary open angle glaucoma using serological proteome analysis (SERPA). Front Immunol. 2019;10:381.
Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett. 1994;172(1–2):151–4.
Boland MV, Quigley HA. Risk factors and open-angle glaucoma: classification and application. J Glaucoma. 2007;16(4):406–18.
Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28(16):4283–92.
Bondi MW, Edmonds EC, Salmon DP. Alzheimer’s disease: past, present, and future. J Int Neuropsychol Soc. 2017;23(9–10):818–31.
Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119(1):182–92.
Buchi ER. Cell death in the rat retina after a pressure-induced ischaemia-reperfusion insult: an electron microscopic study. I. Ganglion cell layer and inner nuclear layer. Exp Eye Res. 1992;55(4):605–13.
Bulati M, Buffa S, Martorana A, Gervasi F, Camarda C, Azzarello DM, Monastero R, Caruso C, Colonna-Romano G. Double negative (IgG+IgD-CD27-) B cells are increased in a cohort of moderate-severe Alzheimer’s disease patients and show a pro-inflammatory trafficking receptor phenotype. J Alzheimers Dis. 2015;44(4):1241–51.
Calopa M, Bas J, Callen A, Mestre M. Apoptosis of peripheral blood lymphocytes in Parkinson patients. Neurobiol Dis. 2010;38(1):1–7.
Camelo S, Lavelette S, Guillonneau X, Raoul W, Sennlaub F. Association of choroidal interleukin-17-producing T lymphocytes and macrophages with geographic atrophy. Ophthalmologica. 2016;236(1):53–8.
Cao YL, Zhang FQ, Hao FQ. Th1/Th2 cytokine expression in diabetic retinopathy. Genet Mol Res. 2016; 15(3).
Caprioli J, Ishii Y, Kwong JM. Retinal ganglion cell protection with geranylgeranylacetone, a heat shock protein inducer, in a rat glaucoma model. Trans Am Ophthalmol Soc. 2003;101:39–50.
Caspi R. Autoimmunity in the immune privileged eye: pathogenic and regulatory T cells. Immunol Res. 2008;42(1–3):41–50.
Caspi RR. A look at autoimmunity and inflammation in the eye. J Clin Invest. 2010;120(9):3073–83.
Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, Bry L, Kraj P, Kisielow P, Ignatowicz L. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature. 2013;497(7448):258–62.
Cecilia O-M, José Alberto C-G, José N-P, Ernesto Germán C-M, Ana Karen L-C, Luis Miguel R-P, Ricardo Raúl R-R, Adolfo Daniel R-C. Oxidative stress as the main target in diabetic retinopathy pathophysiology. J Diabetes Res. 2019;2019:8562408–8562408.
Chaphalkar RM, Stankowska DL, He S, Kodati B, Phillips N, Prah J, Yang S, Krishnamoorthy RR. Endothelin-1 mediated decrease in mitochondrial gene expression and bioenergetics contribute to neurodegeneration of retinal ganglion cells. Sci Rep. 2020;10(1):3571.
Chappert P, Schwartz RH. Induction of T cell anergy: integration of environmental cues and infectious tolerance. Curr Opin Immunol. 2010;22(5):552–9.
Chaudhary P, Ahmed F, Quebada P, Sharma SC. Caspase inhibitors block the retinal ganglion cell death following optic nerve transection. Brain Res Mol Brain Res. 1999;67(1):36–45.
Chen H, Cho KS, Vu THK, Shen CH, Kaur M, Chen G, Mathew R, McHam ML, Fazelat A, Lashkari K, Au NPB, Tse JKY, Li Y, Yu H, Yang L, Stein-Streilein J, Ma CHE, Woolf CJ, Whary MT, Jager MJ, Fox JG, Chen J, Chen DF. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat Commun. 2018;9(1):3209.
Chen H, Ren X, Liao N, Wen F. Th17 cell frequency and IL-17A concentrations in peripheral blood mononuclear cells and vitreous fluid from patients with diabetic retinopathy. J Int Med Res. 2016;44(6):1403–13.
Chen J, Runyan SA, Robinson MR. Novel ocular antihypertensive compounds in clinical trials. Clin Ophthalmol. 2011;5:667–77.
Chen JJ, Han BS, Xu SG, Vu H, Farrow JW, Rodman CL, Zhu Y, Wang WZ. Hypersensitivity toward bacterial stimuli in patients with age-related macular degeneration. APMIS. 2016;124(5):406–13.
Chen S, Liu Y, Niu Y, Xu Y, Zhou Q, Xu X, Wang J, Yu M. Increased abundance of myeloid-derived suppressor cells and Th17 cells in peripheral blood of newly-diagnosed Parkinson’s disease patients. Neurosci Lett. 2017;648:21–5.
Chen SH, Bu XL, Jin WS, Shen LL, Wang J, Zhuang ZQ, Zhang T, Zeng F, Yao XQ, Zhou HD, Wang YJ. Altered peripheral profile of blood cells in Alzheimer disease: a hospital-based case-control study. Medicine (Baltimore). 2017;96(21): e6843.
Chen T, Hou X, Ni Y, Du W, Han H, Yu Y, Shi G. The imbalance of FOXP3/GATA3 in regulatory T cells from the peripheral blood of asthmatic patients. J Immunol Res. 2018;2018:3096183.
Chen X, Feng W, Ou R, Liu J, Yang J, Fu J, Cao B, Chen Y, Wei Q, Shang H. Evidence for peripheral immune activation in Parkinson’s disease. Front Aging Neurosci. 2021;13: 617370.
Chen Y, Qi B, Xu W, Ma B, Li L, Chen Q, Qian W, Liu X, Qu H. Clinical correlation of peripheral CD4+cell subsets, their imbalance and Parkinson’s disease. Mol Med Rep. 2015;12(4):6105–11.
Chidlow G, Wood JPM, Casson RJ. Investigations into hypoxia and oxidative stress at the optic nerve head in a rat model of glaucoma. Front Neurosci. 2017;11:478.
Choi JK, Egwuagu CE. Analysis of regulatory B cells in experimental autoimmune uveitis. Methods Mol Biol. 2021;2270:437–50.
Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol. 2013;13(1):12–5.
Chung HS, Harris A, Evans DW, Kagemann L, Garzozi HJ, Martin B. Vascular aspects in the pathophysiology of glaucomatous optic neuropathy. Surv Ophthalmol. 1999;43(Suppl 1):S43-50.
Codarri L, Greter M, Becher B. Communication between pathogenic T cells and myeloid cells in neuroinflammatory disease. Trends Immunol. 2013;34(3):114–9.
Coleman AL, Miglior S. Risk factors for glaucoma onset and progression. Surv Ophthalmol. 2008;53(Suppl 1):S3-10.
Comabella M, Khoury SJ. Immunopathogenesis of multiple sclerosis. Clin Immunol. 2012;142(1):2–8.
Cone RE, Pais R. Anterior chamber-associated immune deviation (ACAID): an acute response to ocular insult protects from future immune-mediated damage? Ophthalmol Eye Dis. 2009;1:33–40.
Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev. 2003;2(3):119–25.
Corthay A. How do regulatory T cells work? Scand J Immunol. 2009;70(4):326–36.
Coughlin B, Schnabolk G, Joseph K, Raikwar H, Kunchithapautham K, Johnson K, Moore K, Wang Y, Rohrer B. Connecting the innate and adaptive immune responses in mouse choroidal neovascularization via the anaphylatoxin C5a and gammadeltaT-cells. Sci Rep. 2016;6:23794.
Cruz-Guilloty F, Saeed AM, Duffort S, Cano M, Ebrahimi KB, Ballmick A, Tan Y, Wang H, Laird JM, Salomon RG, Handa JT, Perez VL. T cells and macrophages responding to oxidative damage cooperate in pathogenesis of a mouse model of age-related macular degeneration. PLoS ONE. 2014;9(2): e88201.
Cui Y, Shao H, Lan C, Nian H, O’Brien RL, Born WK, Kaplan HJ, Sun D. Major role of gamma delta T cells in the generation of IL-17+ uveitogenic T cells. J Immunol. 2009;183(1):560–7.
Cunningham MW. Molecular mimicry, autoimmunity, and infection: the cross-reactive antigens of group a streptococci and their sequelae. Microbiol Spectr. 2019; 7(4).
Davatchi F, Shams H, Rezaipoor M, Sadeghi-Abdollahi B, Shahram F, Nadji A, Chams-Davatchi C, Akhlaghi M, Faezi T, Naderi N. Rituximab in intractable ocular lesions of Behcet’s disease; randomized single-blind control study (pilot study). Int J Rheum Dis. 2010;13(3):246–52.
Dave N, Chevour P, Mahendradas P, Venkatesh A, Kawali A, Shetty R, Ghosh A, Sethu S. Increased aqueous humor CD4+/CD8+ lymphocyte ratio in sarcoid uveitis. Ocul Immunol Inflamm. 2019;27(7):1033–40.
de-la--Torre A, Lopez-Castillo CA, Rueda JC, Mantilla RD, Gomez-Marin JE, Anaya JM. Clinical patterns of uveitis in two ophthalmology centres in Bogota, Colombia. Clin Exp Ophthalmol. 2009;37(5):458–66.
de Smet MD, Taylor SR, Bodaghi B, Miserocchi E, Murray PI, Pleyer U, Zierhut M, Barisani-Asenbauer T, LeHoang P, Lightman S. Understanding uveitis: the impact of research on visual outcomes. Prog Retin Eye Res. 2011;30(6):452–70.
Dedeoglu F. Drug-induced autoimmunity. Curr Opin Rheumatol. 2009;21(5):547–51.
Deleidi M, Jaggle M, Rubino G. Immune aging, dysmetabolism, and inflammation in neurological diseases. Front Neurosci. 2015;9:172.
Deliyanti D, Talia DM, Zhu T, Maxwell MJ, Agrotis A, Jerome JR, Hargreaves EM, Gerondakis S, Hibbs ML, Mackay F, Wilkinson-Berka JL. Foxp3(+) Tregs are recruited to the retina to repair pathological angiogenesis. Nat Commun. 2017;8(1):748.
Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–58.
Deng Q, Luo Y, Chang C, Wu H, Ding Y, Xiao R. The emerging epigenetic role of CD8+T cells in autoimmune diseases: a systematic review. Front Immunol. 2019;10:856.
Dervan EW, Chen H, Ho SL, Brummel N, Schmid J, Toomey D, Haralambova M, Gould E, Wallace DM, Prehn JH, O’Brien CJ, Murphy D. Protein macroarray profiling of serum autoantibodies in pseudoexfoliation glaucoma. Invest Ophthalmol Vis Sci. 2010;51(6):2968–75.
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):32.
Dobbs RJ, Charlett A, Purkiss AG, Dobbs SM, Weller C, Peterson DW. Association of circulating TNF-alpha and IL-6 with ageing and parkinsonism. Acta Neurol Scand. 1999;100(1):34–41.
Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6(4):329–33.
Dorner T, Radbruch A. Antibodies and B cell memory in viral immunity. Immunity. 2007;27(3):384–92.
Dos Santos Picanco LC, Ozela PF, de Fatima de Brito Brito M, Pinheiro AA, Padilha EC, Braga FS, de Paula da Silva CHT, Dos Santos CBR, Rosa JMC, da Silva Hage-Melim LI. Alzheimer’s disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr Med Chem. 2018;25(26):3141–59.
Double KL, Rowe DB, Carew-Jones FM, Hayes M, Chan DK, Blackie J, Corbett A, Joffe R, Fung VS, Morris J, Riederer P, Gerlach M, Halliday GM. Anti-melanin antibodies are increased in sera in Parkinson’s disease. Exp Neurol. 2009;217(2):297–301.
Drance S, Anderson DR, Schulzer M, G. Collaborative Normal-Tension Glaucoma Study. Risk factors for progression of visual field abnormalities in normal-tension glaucoma. Am J Ophthalmol. 2001;131(6):699–708.
Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, Kim HJ, Bar-Or A. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol. 2007;178(10):6092–9.
Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol. 2004;172(6):3422–7.
Duffy SS, Keating BA, Moalem-Taylor G. Adoptive transfer of regulatory T cells as a promising immunotherapy for the treatment of multiple sclerosis. Front Neurosci. 2019;13:1107.
Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–4.
Faber C, Singh A, Kruger Falk M, Juel HB, Sorensen TL, Nissen MH. Age-related macular degeneration is associated with increased proportion of CD56(+) T cells in peripheral blood. Ophthalmology. 2013;120(11):2310–6.
Fani L, Georgakis MK, Ikram MA, Ikram MK, Malik R, Dichgans M. Circulating biomarkers of immunity and inflammation, risk of Alzheimer’s disease, and hippocampal volume: a Mendelian randomization study. Transl Psychiatry. 2021;11(1):291.
Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14(1):24–35.
Feger U, Luther C, Poeschel S, Melms A, Tolosa E, Wiendl H. Increased frequency of CD4+ CD25+ regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol. 2007;147(3):412–8.
Feng S, Yu H, Yu Y, Geng Y, Li D, Yang C, Lv Q, Lu L, Liu T, Li G, Yuan L. Levels of inflammatory cytokines IL-1beta, IL-6, IL-8, IL-17A, and TNF-alpha in aqueous humour of patients with diabetic retinopathy. J Diabetes Res. 2018;2018:8546423.
Ferrero J, Williams L, Stella H, Leitermann K, Mikulskis A, O’Gorman J, Sevigny J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement (N Y). 2016;2(3):169–76.
Ferretti MT, Merlini M, Spani C, Gericke C, Schweizer N, Enzmann G, Engelhardt B, Kulic L, Suter T, Nitsch RM. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav Immun. 2016;54:211–25.
Flammer J, Orgul S, Costa VP, Orzalesi N, Krieglstein GK, Serra LM, Renard JP, Stefansson E. The impact of ocular blood flow in glaucoma. Prog Retin Eye Res. 2002;21(4):359–93.
Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162(1):1–11.
Forrester JV, Kuffova L, Dick AD. Autoimmunity, autoinflammation, and infection in uveitis. Am J Ophthalmol. 2018;189:77–85.
Forrester JV, Xu H. Good news-bad news: the Yin and Yang of immune privilege in the eye. Front Immunol. 2012;3:338.
Foxman EF, Zhang M, Hurst SD, Muchamuel T, Shen D, Wawrousek EF, Chan CC, Gery I. Inflammatory mediators in uveitis: differential induction of cytokines and chemokines in Th1- versus Th2-mediated ocular inflammation. J Immunol. 2002;168(5):2483–92.
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128(1):92–105.
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(Pt 5):1175–89.
Garretti F, Agalliu D, Lindestam Arlehamn CS, Sette A, Sulzer D. Autoimmunity in Parkinson’s disease: the role of alpha-synuclein-specific T cells. Front Immunol. 2019;10:303.
Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, Chen K, Lehallier B, Channappa D, De Los Santos MB, McBride A, Pluvinage J, Elahi F, Tam GK, Kim Y, Greicius M, Wagner AD, Aigner L, Galasko DR, Davis MM, Wyss-Coray T. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577(7790):399–404.
Gauthier AC, Liu J. Epigenetics and signaling pathways in glaucoma. Biomed Res Int. 2017;2017:5712341.
Geem D, Harusato A, Flannigan K, Denning TL. Harnessing regulatory T cells for the treatment of inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(6):1409–18.
Geyer O, Levo Y. Glaucoma is an autoimmune disease. Autoimmun Rev. 2020;102535.
Gilbert RM, Zhang X, Sampson RD, Ehrenstein MR, Nguyen DX, Chaudhry M, Mein C, Mahmud N, Galatowicz G, Tomkins-Netzer O, Calder VL, Lightman S. Clinical remission of sight-threatening non-infectious uveitis is characterized by an upregulation of peripheral T-regulatory cell polarized towards T-bet and TIGIT. Front Immunol. 2018;9:907.
Goldblum D, Mittag T. Prospects for relevant glaucoma models with retinal ganglion cell damage in the rodent eye. Vision Res. 2002;42(4):471–8.
Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel). 2016;8(3):36.
Goslings WR, Prodeus AP, Streilein JW, Carroll MC, Jager MJ, Taylor AW. A small molecular weight factor in aqueous humor acts on C1q to prevent antibody-dependent complement activation. Invest Ophthalmol Vis Sci. 1998;39(6):989–95.
Gramlich OW, Beck S, von Thun Und N, Hohenstein-Blaul N, Boehm A, Ziegler JM, Vetter NP, Grus FH. Enhanced insight into the autoimmune component of glaucoma: IgG autoantibody accumulation and pro-inflammatory conditions in human glaucomatous retina. PLoS ONE. 2013;8(2): e57557.
Gramlich OW, Ding QJ, Zhu W, Cook A, Anderson MG, Kuehn MH. Adoptive transfer of immune cells from glaucomatous mice provokes retinal ganglion cell loss in recipients. Acta Neuropathol Commun. 2015;3:56.
Gruden MA, Sewell RD, Yanamandra K, Davidova TV, Kucheryanu VG, Bocharov EV, Bocharova OA, Polyschuk VV, Sherstnev VV, Morozova-Roche LA. Immunoprotection against toxic biomarkers is retained during Parkinson’s disease progression. J Neuroimmunol. 2011;233(1–2):221–7.
Guedes MC, Borrego LM, Proenca RD. Roles of interleukin-17 in uveitis. Indian J Ophthalmol. 2016;64(9):628–34.
Guo L, Moss SE, Alexander RA, Ali RR, Fitzke FW, Cordeiro MF. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Invest Ophthalmol Vis Sci. 2005;46(1):175–82.
Guo Z, Wang G, Lv Y, Wan YY, Zheng J. Inhibition of Cdk8/Cdk19 activity promotes treg cell differentiation and suppresses autoimmune diseases. Front Immunol. 2019;10:1988.
Gurne DH, Tso MO, Edward DP, Ripps H. Antiretinal antibodies in serum of patients with age-related macular degeneration. Ophthalmology. 1991;98(5):602–7.
Gustavsson C, Agardh CD, Agardh E. Profile of intraocular tumour necrosis factor-alpha and interleukin-6 in diabetic subjects with different degrees of diabetic retinopathy. Acta Ophthalmol. 2013;91(5):445–52.
Haas J, Hug A, Viehover A, Fritzsching B, Falk CS, Filser A, Vetter T, Milkova L, Korporal M, Fritz B, Storch-Hagenlocher B, Krammer PH, Suri-Payer E, Wildemann B. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35(11):3343–52.
Halle S, Halle O, Forster R. Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo. Trends Immunol. 2017;38(6):432–43.
Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. 2016;11:175–97.
Han JM, Patterson SJ, Speck M, Ehses JA, Levings MK. Insulin inhibits IL-10-mediated regulatory T cell function: implications for obesity. J Immunol. 2014;192(2):623–9.
Hang H, Yuan S, Yang Q, Yuan D, Liu Q. Multiplex bead array assay of plasma cytokines in type 2 diabetes mellitus with diabetic retinopathy. Mol Vis. 2014;20:1137–45.
Hasegawa E, Sonoda KH, Shichita T, Morita R, Sekiya T, Kimura A, Oshima Y, Takeda A, Yoshimura T, Yoshida S, Ishibashi T, Yoshimura A. IL-23-independent induction of IL-17 from gammadeltaT cells and innate lymphoid cells promotes experimental intraocular neovascularization. J Immunol. 2013;190(4):1778–87.
Hecker LA, Edwards AO, Ryu E, Tosakulwong N, Baratz KH, Brown WL, Charbel Issa P, Scholl HP, Pollok-Kopp B, Schmid-Kubista KE, Bailey KR, Oppermann M. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum Mol Genet. 2010;19(1):209–15.
Heijl A, Leske MC, Bengtsson B, Hyman L, Bengtsson B, Hussein M, G. Early Manifest Glaucoma Trial. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial. Arch Ophthalmol. 2002;120(10):1268–79.
Heiligenhaus A, Miserocchi E, Heinz C, Gerloni V, Kotaniemi K. Treatment of severe uveitis associated with juvenile idiopathic arthritis with anti-CD20 monoclonal antibody (rituximab). Rheumatology (Oxford). 2011;50(8):1390–4.
Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015;14(4):406–19.
Hernandez C, Segura RM, Fonollosa A, Carrasco E, Francisco G, Simo R. Interleukin-8, monocyte chemoattractant protein-1 and IL-10 in the vitreous fluid of patients with proliferative diabetic retinopathy. Diabet Med. 2005;22(6):719–22.
Hirosue S, Dubrot J. Modes of antigen presentation by lymph node stromal cells and their immunological implications. Front Immunol. 2015;6:446.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–97.
Horai R, Caspi RR. Cytokines in autoimmune uveitis. J Interferon Cytokine Res. 2011;31(10):733–44.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7.
Hu M, Eviston D, Hsu P, Marino E, Chidgey A, Santner-Nanan B, Wong K, Richards JL, Yap YA, Collier F, Quinton A, Joung S, Peek M, Benzie R, Macia L, Wilson D, Ponsonby AL, Tang MLK, O’Hely M, Daly NL, Mackay CR, Dahlstrom JE, Group BISI, Vuillermin P, Nanan R. Decreased maternal serum acetate and impaired fetal thymic and regulatory T cell development in preeclampsia. Nat Commun. 2019;10(1):3031.
Huan J, Culbertson N, Spencer L, Bartholomew R, Burrows GG, Chou YK, Bourdette D, Ziegler SF, Offner H, Vandenbark AA. Decreased FOXP3 levels in multiple sclerosis patients. J Neurosci Res. 2005;81(1):45–52.
Huang P, Huo Y, Lou LX, Li H, Barnstable CJ, Zhang C, Zhang SS. CD4 positive T helper cells contribute to retinal ganglion cell death in mouse model of ischemia reperfusion injury. Exp Eye Res. 2013;115:131–9.
Huang Y, Hu H, Liu L, Ye J, Wang Z, Que B, Liu W, Shi Y, Zeng T, Shi L, Ji Q, Chang C, Lin Y. Interleukin-12p35 deficiency reverses the Th1/Th2 imbalance, aggravates the Th17/Treg imbalance, and ameliorates atherosclerosis in ApoE-/- Mice. Mediators Inflamm. 2019;2019:3152040.
Huang Y, Liu Z, Cao BB, Qiu YH, Peng YP. Treg cells attenuate neuroinflammation and protect neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol. 2020;15(2):224–37.
Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, Hirsch EC. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience. 1996;72(2):355–63.
Husain S, Abdul Y, Crosson CE. Preservation of retina ganglion cell function by morphine in a chronic ocular-hypertensive rat model. Invest Ophthalmol Vis Sci. 2012;53(7):4289–98.
Husain S, Abdul Y, Potter DE. Non-analgesic effects of opioids: neuroprotection in the retina. Curr Pharm Des. 2012;18(37):6101–8.
Husain S, Abdul Y, Singh S, Ahmad A, Husain M. Regulation of nitric oxide production by delta-opioid receptors during glaucomatous injury. PLoS ONE. 2014;9(10): e110397.
Husain S, Potter DE, Crosson CE. Opioid receptor-activation: retina protected from ischemic injury. Invest Ophthalmol Vis Sci. 2009;50(8):3853–9.
Husain S, Zaidi SAH, Singh S, Guzman W, Mehrotra S. Reduction of neuroinflammation by delta-opioids via STAT3-dependent pathway in chronic glaucoma model. Front Pharmacol. 2021;12: 601404.
Iannaccone A, Giorgianni F, New DD, Hollingsworth TJ, Umfress A, Alhatem AH, Neeli I, Lenchik NI, Jennings BJ, Calzada JI, Satterfield S, Mathews D, Diaz RI, Harris T, Johnson KC, Charles S, Kritchevsky SB, Gerling IC, Beranova-Giorgianni S, Radic MZ, A. B. C. s. Health. Circulating autoantibodies in age-related macular degeneration recognize human macular tissue antigens implicated in autophagy, immunomodulation, and protection from oxidative stress and apoptosis. PLoS ONE. 2015;10(12): e0145323.
Ilhan N, Daglioglu MC, Ilhan O, Coskun M, Tuzcu EA, Kahraman H, Keskin U. Assessment of neutrophil/lymphocyte ratio in patients with age-related macular degeneration. Ocul Immunol Inflamm. 2015;23(4):287–90.
Ip B, Cilfone NA, Belkina AC, DeFuria J, Jagannathan-Bogdan M, Zhu M, Kuchibhatla R, McDonnell ME, Xiao Q, Kepler TB, Apovian CM, Lauffenburger DA, Nikolajczyk BS. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFalpha production. Obesity (Silver Spring). 2016;24(1):102–12.
Ito JT, Cervilha DAB, Lourenco JD, Goncalves NG, Volpini RA, Caldini EG, Landman G, Lin CJ, Velosa APP, Teodoro WPR, Tiberio I, Mauad T, Martins MA, Macchione M, Lopes F. Th17/Treg imbalance in COPD progression: a temporal analysis using a CS-induced model. PLoS ONE. 2019;14(1): e0209351.
Izzotti A, Bagnis A, Sacca SC. The role of oxidative stress in glaucoma. Mutat Res. 2006;612(2):105–14.
Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, Nikolajczyk BS. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. 2011;186(2):1162–72.
Janeway CA Jr TP, Walport M et al. Principles of innate and adaptive immunity. Immunobiology: The Immune System in Health and Disease 5th edition. New York, Garland Science. 2001.
Janeway CA Jr TP, Walport M et al. T cell-mediated cytotoxicity. Immunobiology: The Immune System in Health and Disease. New York, Garland Science. 2001.
Janeway CA Jr TP, Walport M et al. Antigen Recognition by T cells. Immunobiology: The Immune System in Health and Disease New York, Garland Science. 2001.
Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, Madjovski A, Ruhrmann S, Faigle W, Frauenknecht K, Pinilla C, Santos R, Hammer C, Ortiz Y, Opitz L, Grönlund H, Rogler G, Boyman O, Reynolds R, Lutterotti A, Khademi M, Olsson T, Piehl F, Sospedra M, Martin R. Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell. 2018;175(1):85-100.e123.
Jiang S, Kametani M, Chen DF. Adaptive immunity: new aspects of pathogenesis underlying neurodegeneration in glaucoma and optic neuropathy. Front Immunol. 2020;11:65.
Joachim SC, Bruns K, Lackner KJ, Pfeiffer N, Grus FH. Analysis of IgG antibody patterns against retinal antigens and antibodies to alpha-crystallin, GFAP, and alpha-enolase in sera of patients with “wet” age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2007;245(5):619–26.
Joachim SC, Bruns K, Lackner KJ, Pfeiffer N, Grus FH. Antibodies to alpha B-crystallin, vimentin, and heat shock protein 70 in aqueous humor of patients with normal tension glaucoma and IgG antibody patterns against retinal antigen in aqueous humor. Curr Eye Res. 2007;32(6):501–9.
Joachim SC, Reichelt J, Berneiser S, Pfeiffer N, Grus FH. Sera of glaucoma patients show autoantibodies against myelin basic protein and complex autoantibody profiles against human optic nerve antigens. Graefes Arch Clin Exp Ophthalmol. 2008;246(4):573–80.
Joachim SC, Wuenschig D, Pfeiffer N, Grus FH. IgG antibody patterns in aqueous humor of patients with primary open angle glaucoma and pseudoexfoliation glaucoma. Mol Vis. 2007;13:1573–9.
Jones JL, Anderson JM, Phuah CL, Fox EJ, Selmaj K, Margolin D, Lake SL, Palmer J, Thompson SJ, Wilkins A, Webber DJ, Compston DA, Coles AJ. Improvement in disability after alemtuzumab treatment of multiple sclerosis is associated with neuroprotective autoimmunity. Brain. 2010;133(Pt 8):2232–47.
Kaiser CJ, Ksander BR, Streilein JW. Inhibition of lymphocyte proliferation by aqueous humor. Reg Immunol. 1989;2(1):42–9.
Kannarkat GT, Boss JM, Tansey MG. The role of innate and adaptive immunity in Parkinson’s disease. J Parkinsons Dis. 2013;3(4):493–514.
Kappos L, Hartung HP, Freedman MS, Boyko A, Radü EW, Mikol DD, Lamarine M, Hyvert Y, Freudensprung U, Plitz T, van Beek J. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014;13(4):353–63.
Karaaslan Z, Kahraman OT, Sanli E, Ergen HA, Ulusoy C, Bilgic B, Yilmaz V, Tuzun E, Hanagasi HA, Kucukali CI. Inflammation and regulatory T cell genes are differentially expressed in peripheral blood mononuclear cells of Parkinson’s disease patients. Sci Rep. 2021;11(1):2316.
Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, Ruiz-Irastorza G, Hughes G. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039.
Ke Y, Jiang G, Sun D, Kaplan HJ, Shao H. Ocular regulatory T cells distinguish monophasic from recurrent autoimmune uveitis. Invest Ophthalmol Vis Sci. 2008;49(9):3999–4007.
Ke Y, Jiang G, Sun D, Kaplan HJ, Shao H. Anti-CD3 antibody ameliorates experimental autoimmune uveitis by inducing both IL-10 and TGF-beta dependent regulatory T cells. Clin Immunol. 2011;138(3):311–20.
Kermer P, Klocker N, Labes M, Thomsen S, Srinivasan A, Bahr M. Activation of caspase-3 in axotomized rat retinal ganglion cells in vivo. FEBS Lett. 1999;453(3):361–4.
Khanna S, Komati R, Eichenbaum DA, Hariprasad I, Ciulla TA, Hariprasad SM. Current and upcoming anti-VEGF therapies and dosing strategies for the treatment of neovascular AMD: a comparative review. BMJ Open Ophthalmol. 2019;4(1): e000398.
Kim K, Wang X, Ragonnaud E, Bodogai M, Illouz T, DeLuca M, McDevitt RA, Gusev F, Okun E, Rogaev E, Biragyn A. Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nat Commun. 2021;12(1):2185.
Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci U S A. 2002;99(24):15620–5.
Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021;17(4):696–701.
Koch S, Sopel N, Finotto S. Th9 and other IL-9-producing cells in allergic asthma. Semin Immunopathol. 2017;39(1):55–68.
Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169(9):4712–6.
Koonin EV, Krupovic M. Evolution of adaptive immunity from transposable elements combined with innate immune systems. Nat Rev Genet. 2015;16(3):184–92.
Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205(7):1535–41.
Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. 2018;48(2):202–13.
Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D, Mueller N, Hardt C, Kreuzfelder E, Grosse-Wilde H. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol. 2006;180(1–2):178–84.
Kumar P, Saini S, Khan S, Surendra Lele S, Prabhakar BS. Restoring self-tolerance in autoimmune diseases by enhancing regulatory T-cells. Cell Immunol. 2019;339:41–9.
Kustrimovic N, Comi C, Magistrelli L, Rasini E, Legnaro M, Bombelli R, Aleksic I, Blandini F, Minafra B, Riboldazzi G, Sturchio A, Mauri M, Bono G, Marino F, Cosentino M. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J Neuroinflammation. 2018;15(1):205.
La Paglia GMC, Leone MC, Lepri G, Vagelli R, Valentini E, Alunno A, Tani C. One year in review 2017: systemic lupus erythematosus. Clin Exp Rheumatol. 2017;35(4):551–61.
Landowski M, Kelly U, Klingeborn M, Groelle M, Ding JD, Grigsby D, Bowes Rickman C. Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc Natl Acad Sci U S A. 2019;116(9):3703–11.
Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, Fulop T. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis. 2009;17(1):91–103.
Lardo S, Soesatyo MH, Juffrie J, Umniyati SR. The autoimmune mechanism in dengue hemorrhagic fever. Acta Med Indones. 2018;50(1):70–9.
Laurent C, Dorothee G, Hunot S, Martin E, Monnet Y, Duchamp M, Dong Y, Legeron FP, Leboucher A, Burnouf S, Faivre E, Carvalho K, Caillierez R, Zommer N, Demeyer D, Jouy N, Sazdovitch V, Schraen-Maschke S, Delarasse C, Buee L, Blum D. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. 2017;140(1):184–200.
Lechner J, Chen M, Hogg RE, Toth L, Silvestri G, Chakravarthy U, Xu H. Alterations in circulating immune cells in neovascular age-related macular degeneration. Sci Rep. 2015;5:16754.
Lechner J, O’Leary OE, Stitt AW. The pathology associated with diabetic retinopathy. Vision Res. 2017;139:7–14.
Lee GR. The balance of Th17 versus treg cells in autoimmunity. Int J Mol Sci. 2018;19(3):730.
Lee KS, Lin S, Copland DA, Dick AD, Liu J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J Neuroinflammation. 2021;18(1):32.
Lee RW, Nicholson LB, Sen HN, Chan CC, Wei L, Nussenblatt RB, Dick AD. Autoimmune and autoinflammatory mechanisms in uveitis. Semin Immunopathol. 2014;36(5):581–94.
Legroux L, Arbour N. Multiple sclerosis and T lymphocytes: an entangled story. J Neuroimmune Pharmacol. 2015;10(4):528–46.
Li J, Chen S, Xiao X, Zhao Y, Ding W, Li XC. IL-9 and Th9 cells in health and diseases-from tolerance to immunopathology. Cytokine Growth Factor Rev. 2017;37:47–55.
Li R, Rezk A, Ghadiri M, Luessi F, Zipp F, Li H, Giacomini PS, Antel J, Bar-Or A. Dimethyl fumarate treatment mediates an anti-inflammatory shift in B cell subsets of patients with multiple sclerosis. J Immunol. 2017;198(2):691–8.
Li R, Tropea TF, Baratta LR, Zuroff L, Diaz-Ortiz ME, Zhang B, Shinoda K, Rezk A, Alcalay RN, Chen-Plotkin A, Bar-Or A. Abnormal B-cell and Tfh-cell profiles in patients with parkinson disease: a cross-sectional study. Neurol Neuroimmunol Neuroinflamm. 2022;9(2):e1125.
Li W, Luo Y, Xu H, Ma Q, Yao Q. Imbalance between T helper 1 and regulatory T cells plays a detrimental role in experimental Parkinson’s disease in mice. J Int Med Res. 2021;49(4):300060521998471.
Liu B, Wei L, Meyerle C, Tuo J, Sen HN, Li Z, Chakrabarty S, Agron E, Chan CC, Klein ML, Chew E, Ferris F, Nussenblatt RB. Complement component C5a promotes expression of IL-22 and IL-17 from human T cells and its implication in age-related macular degeneration. J Transl Med. 2011;9:1–12.
Liu Q, Ju WK, Crowston JG, Xie F, Perry G, Smith MA, Lindsey JD, Weinreb RN. Oxidative stress is an early event in hydrostatic pressure induced retinal ganglion cell damage. Invest Ophthalmol Vis Sci. 2007;48(10):4580–9.
Liu Y, You C, Zhang Z, Zhang J, Yan H. Roles of Treg/Th17 cell imbalance and neuronal damage in the visual dysfunction observed in experimental autoimmune optic neuritis chronologically. Neuromolecular Med. 2015;17(4):391–403.
Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205(4):799–810.
Luo C, Yang X, Kain AD, Powell DW, Kuehn MH, Tezel G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Invest Ophthalmol Vis Sci. 2010;51(11):5697–707.
Machalinska A, Dziedziejko V, Mozolewska-Piotrowska K, Karczewicz D, Wiszniewska B, Machalinski B. Elevated plasma levels of C3a complement compound in the exudative form of age-related macular degeneration. Ophthalmic Res. 2009;42(1):54–9.
Madelung CF, Falk MK, Sorensen TL. The association between neovascular age-related macular degeneration and regulatory T cells in peripheral blood. Clin Ophthalmol. 2015;9:1147–54.
Maruyama I, Ohguro H, Ikeda Y. Retinal ganglion cells recognized by serum autoantibody against gamma-enolase found in glaucoma patients. Invest Ophthalmol Vis Sci. 2000;41(7):1657–65.
Mattapallil MJ, Sahin A, Silver PB, Sun SH, Chan CC, Remmers EF, Hejtmancik JF, Caspi RR. Common genetic determinants of uveitis shared with other autoimmune disorders. J Immunol. 2008;180(10):6751–9.
McDonnell F, Irnaten M, Clark AF, O’Brien CJ, Wallace DM. Hypoxia-induced changes in DNA methylation alter RASAL1 and TGFbeta1 expression in human trabecular meshwork cells. PLoS ONE. 2016;11(4): e0153354.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia Nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–91.
McKinnon SJ, Lehman DM, Tahzib NG, Ransom NL, Reitsamer HA, Liston P, LaCasse E, Li Q, Korneluk RG, Hauswirth WW. Baculoviral IAP repeat-containing-4 protects optic nerve axons in a rat glaucoma model. Mol Ther. 2002;5(6):780–7.
McPherson RC, Cambrook HE, O’Connor RA, Anderton SM. Induction of passive EAE using myelin-reactive CD4+ T cells. Methods Mol Biol. 2014;1193:187–98.
McPherson SW, Yang J, Chan CC, Dou C, Gregerson DS. Resting CD8 T cells recognize beta-galactosidase expressed in the immune-privileged retina and mediate autoimmune disease when activated. Immunology. 2003;110(3):386–96.
Mexhitaj I, Nyirenda MH, Li R, O’Mahony J, Rezk A, Rozenberg A, Moore CS, Johnson T, Sadovnick D, Collins DL, Arnold DL, Gran B, Yeh EA, Marrie RA, Banwell B, Bar-Or A. Abnormal effector and regulatory T cell subsets in paediatric-onset multiple sclerosis. Brain. 2019;142(3):617–32.
Mietelska-Porowska A, Wojda U. T lymphocytes and inflammatory mediators in the interplay between brain and blood in Alzheimer’s disease: potential pools of new biomarkers. J Immunol Res. 2017;2017:4626540.
Miller DJ, Fort PE. Heat shock proteins regulatory role in neurodevelopment. Front Neurosci. 2018;12:821.
Miserocchi E, Pontikaki I, Modorati G, Bandello F, Meroni PL, Gerloni V. Rituximab for uveitis. Ophthalmology. 2011;118(1):223–4.
Mo C, Zeng Z, Deng Q, Ding Y, Xiao R. Imbalance between T helper 17 and regulatory T cell subsets plays a significant role in the pathogenesis of systemic sclerosis. Biomed Pharmacother. 2018;108:177–83.
Mo FM, Proia AD, Johnson WH, Cyr D, Lashkari K. Interferon gamma-inducible protein-10 (IP-10) and eotaxin as biomarkers in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51(8):4226–36.
Morohoshi K, Goodwin AM, Ohbayashi M, Ono SJ. Autoimmunity in retinal degeneration: autoimmune retinopathy and age-related macular degeneration. J Autoimmun. 2009;33(3–4):247–54.
Morohoshi K, Ohbayashi M, Patel N, Chong V, Bird AC, Ono SJ. Identification of anti-retinal antibodies in patients with age-related macular degeneration. Exp Mol Pathol. 2012;93(2):193–9.
Mozaffarieh M, Grieshaber MC, Flammer J. Oxygen and blood flow: players in the pathogenesis of glaucoma. Mol Vis. 2008;14:224–33.
Muhammad F, Wang D, Montieth A, Lee S, Preble J, Foster CS, Larson TA, Ding K, Dvorak JD, Lee DJ. PD-1(+) melanocortin receptor dependent-Treg cells prevent autoimmune disease. Sci Rep. 2019;9(1):16941.
Nadeem A, Javaid K, Sami W, Zafar A, Jahan S, Zaman S, Nagi A. Inverse relationship of serum IL-17 with type-II diabetes retinopathy. Clin Lab. 2013;59(11–12):1311–7.
Nanke Y, Kotake S, Goto M, Ujihara H, Matsubara M, Kamatani N. Decreased percentages of regulatory T cells in peripheral blood of patients with Behcet’s disease before ocular attack: a possible predictive marker of ocular attack. Mod Rheumatol. 2008;18(4):354–8.
Nassar K, Grisanti S, Elfar E, Luke J, Luke M, Grisanti S. Serum cytokines as biomarkers for age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2015;253(5):699–704.
Newman AM, Gallo NB, Hancox LS, Miller NJ, Radeke CM, Maloney MA, Cooper JB, Hageman GS, Anderson DH, Johnson LV, Radeke MJ. Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med. 2012;4(2):16.
Niazi S, Krogh Nielsen M, Sorensen TL, Subhi Y. Neutrophil-to-lymphocyte ratio in age-related macular degeneration: a systematic review and meta-analysis. Acta Ophthalmol. 2019;97(6):558–66.
Nickells RW. Apoptosis of retinal ganglion cells in glaucoma: an update of the molecular pathways involved in cell death. Surv Ophthalmol. 1999;43(Suppl 1):S151-161.
Niederkorn JY. Immune privilege in the anterior chamber of the eye. Crit Rev Immunol. 2002;22(1):13–46.
Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev. 2016;2016:3164734.
Niwa F, Kuriyama N, Nakagawa M, Imanishi J. Effects of peripheral lymphocyte subpopulations and the clinical correlation with Parkinson’s disease. Geriatr Gerontol Int. 2012;12(1):102–7.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–21.
Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. 2015;45(2):344–55.
Oladipupo FO, Yu C-R, Olumuyide E, Jittaysothorn Y, Choi JK, Egwuagu CE. STAT3 deficiency in B cells exacerbates uveitis by promoting expansion of pathogenic lymphocytes and suppressing regulatory B cells (Bregs) and Tregs. Sci Rep. 2020;10(1):16188.
Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. 2000;117(4):1162–72.
Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23(1):91–147.
Papachroni KK, Ninkina N, Papapanagiotou A, Hadjigeorgiou GM, Xiromerisiou G, Papadimitriou A, Kalofoutis A, Buchman VL. Autoantibodies to alpha-synuclein in inherited Parkinson’s disease. J Neurochem. 2007;101(3):749–56.
Park KH, Cozier F, Ong OC, Caprioli J. Induction of heat shock protein 72 protects retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2001;42(7):1522–30.
Park Y, Cho KJ. Choroidal vascular index in patients with open angle glaucoma and preperimetric glaucoma. PLoS ONE. 2019;14(3): e0213336.
Patel HR, Hariprasad SM, Eichenbaum D. Geographic atrophy: clinical impact and emerging treatments. Ophthalmic Surg Lasers Imaging Retina. 2015;46(1):8–13.
Patel M, Chan CC. Immunopathological aspects of age-related macular degeneration. Semin Immunopathol. 2008;30(2):97–110.
Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41(3):764–74.
Peeters LM, Vanheusden M, Somers V, Van Wijmeersch B, Stinissen P, Broux B, Hellings N. Cytotoxic CD4+ T cells drive multiple sclerosis progression. Front Immunol. 2017;8:1160.
Pelzel HR, Schlamp CL, Nickells RW. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010;11:62.
Penfold PL, Provis JM, Furby JH, Gatenby PA, Billson FA. Autoantibodies to retinal astrocytes associated with age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 1990;228(3):270–4.
Pereira LMS, Gomes STM, Ishak R, Vallinoto ACR. Regulatory T cell and Forkhead box protein 3 as modulators of immune homeostasis. Front Immunol. 2017;8:605.
Pérez-Cerdá F, Sánchez-Gómez MV, Matute C. The link of inflammation and neurodegeneration in progressive multiple sclerosis. Mult Scler Demyelinating Disord. 2016;1(1):9.
Pletinckx K, Vaeth M, Schneider T, Beyersdorf N, Hunig T, Berberich-Siebelt F, Lutz MB. Immature dendritic cells convert anergic nonregulatory T cells into Foxp3- IL-10+ regulatory T cells by engaging CD28 and CTLA-4. Eur J Immunol. 2015;45(2):480–91.
Popescu BF, Pirko I, Lucchinetti CF. Pathology of multiple sclerosis: where do we stand? Continuum (Minneap Minn). 2013;19(4):901–21.
Praksova P, Stourac P, Bednarik J, Vlckova E, Mikulkova Z, Michalek J. Immunoregulatory T cells in multiple sclerosis and the effect of interferon beta and glatiramer acetate treatment on T cell subpopulations. J Neurol Sci. 2012;319(1–2):18–23.
Prosperini L, Annovazzi P, Boffa L, Buscarinu MC, Gallo A, Matta M, Moiola L, Musu L, Perini P, Avolio C, Barcella V, Bianco A, Farina D, Ferraro E, Pontecorvo S, Granella F, Grimaldi LME, Laroni A, Lus G, Patti F, Pucci E, Pasca M, Sarchielli P, G. Italian Alemtuzumab Study. No evidence of disease activity (NEDA-3) and disability improvement after alemtuzumab treatment for multiple sclerosis: a 36-month real-world study. J Neurol. 2018;265(12):2851–60.
Qian L, Flood PM, Hong JS. Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. J Neural Transm (Vienna). 2010;117(8):971–9.
Qiao YC, Shen J, He L, Hong XZ, Tian F, Pan YH, Liang L, Zhang XX, Zhao HL. Changes of regulatory T cells and of proinflammatory and immunosuppressive cytokines in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. J Diabetes Res. 2016;2016:3694957.
Qing G, Duan X, Jiang Y. Heat shock protein 72 protects retinal ganglion cells in rat model of acute glaucoma. Yan Ke Xue Bao. 2005;21(3):163–8.
Qiu AW, Bian Z, Mao PA, Liu QH. IL-17A exacerbates diabetic retinopathy by impairing Muller cell function via Act1 signaling. Exp Mol Med. 2016;48(12): e280.
Qiu AW, Liu QH, Wang JL. Blocking IL-17A alleviates diabetic retinopathy in rodents. Cell Physiol Biochem. 2017;41(3):960–72.
Quigley HA. Neuronal death in glaucoma. Prog Retin Eye Res. 1999;18(1):39–57.
Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, Kerrigan DF, Mitchell RS. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000;41(11):3460–6.
Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, Onofrj M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav Immun. 2009;23(1):55–63.
Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, Sobel RA, Wucherpfennig KW, Kuchroo VK. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2004;101(43):15434–9.
Rentzos M, Nikolaou C, Andreadou E, Paraskevas GP, Rombos A, Zoga M, Tsoutsou A, Boufidou F, Kapaki E, Vassilopoulos D. Circulating interleukin-15 and RANTES chemokine in Parkinson’s disease. Acta Neurol Scand. 2007;116(6):374–9.
Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol. 2007;82(5):1083–94.
Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol. 2010;184(5):2261–71.
Richartz-Salzburger E, Batra A, Stransky E, Laske C, Kohler N, Bartels M, Buchkremer G, Schott K. Altered lymphocyte distribution in Alzheimer’s disease. J Psychiatr Res. 2007;41(1–2):174–8.
Rohrer B, Frazer-Abel A, Leonard A, Ratnapriya R, Ward T, Pietraszkiewicz A, O’Quinn E, Adams K, Swaroop A, Wolf BJ. Association of age-related macular degeneration with complement activation products, smoking, and single nucleotide polymorphisms in South Carolinians of European and African descent. Mol Vis. 2019;25:79–92.
Romano C, Barrett DA, Li Z, Pestronk A, Wax MB. Anti-rhodopsin antibodies in sera from patients with normal-pressure glaucoma. Invest Ophthalmol Vis Sci. 1995;36(10):1968–75.
Rose NR. Prediction and prevention of autoimmune disease in the 21st century: a review and preview. Am J Epidemiol. 2016;183(5):403–6.
Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015;125(6):2228–33.
Rosenblum MD, Way SS, Abbas AK. Regulatory T cell memory. Nat Rev Immunol. 2016;16(2):90–101.
Rosso M, Chitnis T. Association between cigarette smoking and multiple sclerosis: a review. JAMA Neurol. 2019.
Ruggieri S, Frassanito MA, Dammacco R, Guerriero S. Treg lymphocytes in autoimmune uveitis. Ocul Immunol Inflamm. 2012;20(4):255–61.
Sabatino JJ Jr, Probstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci. 2019;20(12):728–45.
Sabatino JJ Jr, Zamvil SS, Hauser SL. B-cell therapies in multiple sclerosis. Cold Spring Harb Perspect Med. 2019;9(2):a032037.
Sacca SC, Izzotti A, Rossi P, Traverso C. Glaucomatous outflow pathway and oxidative stress. Exp Eye Res. 2007;84(3):389–99.
Salani F, Sterbini V, Sacchinelli E, Garramone M, Bossu P. Is innate memory a double-edge sword in Alzheimer’s disease? A reappraisal of new concepts and old data. Front Immunol. 2019;10:1768.
Saunders JA, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, Meza JL, Santamaria PM, Bertoni JM, Murman DL, Ali HH, Standaert DG, Mosley RL, Gendelman HE. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J Neuroimmune Pharmacol. 2012;7(4):927–38.
Schafflick D, Wolbert J, Heming M, Thomas C, Hartlehnert M, Borsch AL, Ricci A, Martin-Salamanca S, Li X, Lu IN, Pawlak M, Minnerup J, Strecker JK, Seidenbecher T, Meuth SG, Hidalgo A, Liesz A, Wiendl H, Meyer Zu Horste G. Single-cell profiling of CNS border compartment leukocytes reveals that B cells and their progenitors reside in non-diseased meninges. Nat Neurosci. 2021;24:1225.
Schmitt HM, Pelzel HR, Schlamp CL, Nickells RW. Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol Neurodegener. 2014;9:39.
Schneider L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020;19(2):111–2.
Schwartz M, Baruch K. Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: boosting autoimmunity to fight-off chronic neuroinflammation. J Autoimmun. 2014;54:8–14.
Sebina I, Pepper M. Humoral immune responses to infection: common mechanisms and unique strategies to combat pathogen immune evasion tactics. Curr Opin Immunol. 2018;51:46–54.
Serpente M, Bonsi R, Scarpini E, Galimberti D. Innate immune system and inflammation in Alzheimer’s disease: from pathogenesis to treatment. NeuroImmunoModulation. 2014;21(2–3):79–87.
Severin ME, Lee PW, Liu Y, Selhorst AJ, Gormley MG, Pei W, Yang Y, Guerau-de-Arellano M, Racke MK, Lovett-Racke AE. MicroRNAs targeting TGFbeta signalling underlie the regulatory T cell defect in multiple sclerosis. Brain. 2016;139(Pt 6):1747–61.
Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–6.
Shao H, Sun SL, Kaplan HJ, Sun D. Characterization of rat CD8+ uveitogenic T cells specific for interphotoreceptor retinal-binding protein 1177–1191. J Immunol. 2004;173(4):2849–54.
Sharif K, Watad A, Bragazzi NL, Lichtbroun M, Amital H, Shoenfeld Y. Physical activity and autoimmune diseases: get moving and manage the disease. Autoimmun Rev. 2018;17(1):53–72.
Sheikh V, Zamani A, Mahabadi-Ashtiyani E, Tarokhian H, Borzouei S, Alahgholi-Hajibehzad M. Decreased regulatory function of CD4(+)CD25(+)CD45RA(+) T cells and impaired IL-2 signalling pathway in patients with type 2 diabetes mellitus. Scand J Immunol. 2018;88(4): e12711.
Sheu A, Chan Y, Ferguson A, Bakhtyari MB, Hawke W, White C, Chan YF, Bertolino PJ, Woon HG, Palendira U, Sierro F, Lau SM. A proinflammatory CD4(+) T cell phenotype in gestational diabetes mellitus. Diabetologia. 2018;61(7):1633–43.
Shi YH, Shi GC, Wan HY, Ai XY, Zhu HX, Tang W, Ma JY, Jin XY, Zhang BY. An increased ratio of Th2/Treg cells in patients with moderate to severe asthma. Chin Med J (Engl). 2013;126(12):2248–53.
Shin JI, Bayry J. A role for IL-17 in age-related macular degeneration. Nat Rev Immunol. 2013;13(9):701.
Shishido SN, Varahan S, Yuan K, Li X, Fleming SD. Humoral innate immune response and disease. Clin Immunol. 2012;144(2):142–58.
Siepmann K, Biester S, Plskova J, Muckersie E, Duncan L, Forrester JV. CD4+CD25+ T regulatory cells induced by LPS-activated bone marrow dendritic cells suppress experimental autoimmune uveoretinitis in vivo. Graefes Arch Clin Exp Ophthalmol. 2007;245(2):221–9.
Sigurdardottir S, Zapadka TE, Lindstrom SI, Liu H, Taylor BE, Lee CA, Kern TS, Taylor PR. Diabetes-mediated IL-17A enhances retinal inflammation, oxidative stress, and vascular permeability. Cell Immunol. 2019;341: 103921.
Silva Morales M, Mueller D. Anergy into T regulatory cells: an integration of metabolic cues and epigenetic changes at the Foxp3 conserved non-coding sequence 2. F1000Res 2018;7.
Silver PB, Horai R, Chen J, Jittayasothorn Y, Chan CC, Villasmil R, Kesen MR, Caspi RR. Retina-specific T regulatory cells bring about resolution and maintain remission of autoimmune uveitis. J Immunol. 2015;194(7):3011–9.
Singh A, Subhi Y, Krogh Nielsen M, Falk MK, Matzen SMH, Sellebjerg F, Sorensen TL. Systemic frequencies of T helper 1 and T helper 17 cells in patients with age-related macular degeneration: a case-control study. Sci Rep. 2017;7(1):605.
Singh RP, Elman MJ, Singh SK, Fung AE, Stoilov I. Advances in the treatment of diabetic retinopathy. J Diabetes Complications. 2019;33(12): 107417.
Smith JR, Stempel AJ, Bharadwaj A, Appukuttan B. Involvement of B cells in non-infectious uveitis. Clin Transl Immunol. 2016;5(2):e63–e63.
Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression—a diverse arsenal for a moving target. Immunology. 2008;124(1):13–22.
Sollvander S, Ekholm-Pettersson F, Brundin RM, Westman G, Kilander L, Paulie S, Lannfelt L, Sehlin D. Increased number of plasma B cells producing autoantibodies against Abeta42 protofibrils in Alzheimer’s disease. J Alzheimers Dis. 2015;48(1):63–72.
Sommer A, Tielsch JM, Katz J, Quigley HA, Gottsch JD, Javitt J, Singh K. Relationship between intraocular pressure and primary open angle glaucoma among white and black Americans. The Baltimore Eye Survey. Arch Ophthalmol. 1991;109(8):1090–5.
Song L, Le J, Ye F, Shao H, Kaplan HJ, Sun D. Sequence 168 to 177 of interphotoreceptor retinoid-binding protein (IRBP) is an antigenic epitope for autoreactive CD8 T cells in the B10RIII mouse. J Neuroimmunol. 2008;193(1–2):68–76.
Stevens CH, Rowe D, Morel-Kopp MC, Orr C, Russell T, Ranola M, Ward C, Halliday GM. Reduced T helper and B lymphocytes in Parkinson’s disease. J Neuroimmunol. 2012;252(1–2):95–9.
Stojanovich L, Marisavljevich D. Stress as a trigger of autoimmune disease. Autoimmun Rev. 2008;7(3):209–13.
Streilein JW, Stein-Streilein J. Does innate immune privilege exist? J Leukoc Biol. 2000;67(4):479–87.
Stypula G, Kunert-Radek J, Stepien H, Zylinska K, Pawlikowski M. Evaluation of interleukins, ACTH, cortisol and prolactin concentrations in the blood of patients with Parkinson’s disease. NeuroImmunoModulation. 1996;3(2–3):131–4.
Subhi Y, Lykke Sorensen T. New neovascular age-related macular degeneration is associated with systemic leucocyte activity. Acta Ophthalmol. 2017;95(5):472–80.
Sugita S, Shimizu J, Makabe K, Keino H, Watanabe T, Takahashi M. Inhibition of T cell-mediated inflammation in uveitis by a novel anti-CD3 antibody. Arthritis Res Ther. 2017;19(1):176.
Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, Liong C, McMurtrey C, Hildebrand WH, Mao X, Dawson VL, Dawson TM, Oseroff C, Pham J, Sidney J, Dillon MB, Carpenter C, Weiskopf D, Phillips E, Mallal S, Peters B, Frazier A, Lindestam Arlehamn CS, Sette A. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature. 2017;546(7660):656–61.
Takeda A, Sonoda K-H, Ishibashi T. Regulation of Th1 and Th17 cell differentiation in uveitis. Inflamm Regen. 2013;33(5):261–8.
Tamhane UU, Aneja S, Montgomery D, Rogers EK, Eagle KA, Gurm HS. Association between admission neutrophil to lymphocyte ratio and outcomes in patients with acute coronary syndrome. Am J Cardiol. 2008;102(6):653–7.
Taylor AW, Yee DG, Streilein JW. Suppression of nitric oxide generated by inflammatory macrophages by calcitonin gene-related peptide in aqueous humor. Invest Ophthalmol Vis Sci. 1998;39(8):1372–8.
Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113.
Tezel G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog Brain Res. 2008;173:409–21.
Tezel G, Edward DP, Wax MB. Serum autoantibodies to optic nerve head glycosaminoglycans in patients with glaucoma. Arch Ophthalmol. 1999;117(7):917–24.
Tezel G, Fourth APORICWG. The role of glia, mitochondria, and the immune system in glaucoma. Invest Ophthalmol Vis Sci. 2009;50(3):1001–12.
Tezel G, Hernandez R, Wax MB. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch Ophthalmol. 2000;118(4):511–8.
Tezel G, Li LY, Patil RV, Wax MB. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. 2001;42(8):1787–94.
Tezel G, Seigel GM, Wax MB. Autoantibodies to small heat shock proteins in glaucoma. Invest Ophthalmol Vis Sci. 1998;39(12):2277–87.
Tezel G, Yang J, Wax MB. Heat shock proteins, immunity and glaucoma. Brain Res Bull. 2004;62(6):473–80.
Tezel G, Yang X, Luo C, Peng Y, Sun SL, Sun D. Mechanisms of immune system activation in glaucoma: oxidative stress-stimulated antigen presentation by the retina and optic nerve head glia. Invest Ophthalmol Vis Sci. 2007;48(2):705–14.
Thomas CN, Berry M, Logan A, Blanch RJ, Ahmed Z. Caspases in retinal ganglion cell death and axon regeneration. Cell Death Discov. 2017;3:17032.
Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, Oda T, Tsuchiya K, Kosaka K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol. 2002;124(1–2):83–92.
Toomey CB, Johnson LV, Bowes Rickman C. Complement factor H in AMD: bridging genetic associations and pathobiology. Prog Retin Eye Res. 2018;62:38–57.
Tsai T, Grotegut P, Reinehr S, Joachim SC. Role of heat shock proteins in glaucoma. Int J Mol Sci. 2019;20(20):5160.
Vendomele J, Khebizi Q, Fisson S. Cellular and molecular mechanisms of anterior chamber-associated immune deviation (ACAID): what we have learned from knockout mice. Front Immunol. 2017;8:1686.
Vidal-Sanz M, Salinas-Navarro M, Nadal-Nicolas FM, Alarcon-Martinez L, Valiente-Soriano FJ, de Imperial JM, Aviles-Trigueros M, Agudo-Barriuso M, Villegas-Perez MP. Understanding glaucomatous damage: anatomical and functional data from ocular hypertensive rodent retinas. Prog Retin Eye Res. 2012;31(1):1–27.
Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199(7):971–9.
Vrabec JP, Levin LA. The neurobiology of cell death in glaucoma. Eye (Lond). 2007;21(Suppl 1):S11-14.
Vyas SP, Goswami R. A decade of Th9 cells: role of Th9 cells in inflammatory bowel disease. Front Immunol. 2018;9:1139.
Wajih Ullah M, Qaseem A, Amray A. Post vaccination Guillain Barre syndrome: a case report. Cureus. 2018;10(4): e2511.
Wang GH, Xing YQ. Evaluation of heat shock protein (HSP-72) expression in retinal ganglion cells of rats with glaucoma. Exp Ther Med. 2017;14(2):1577–81.
Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. 2015;278(4):369–95.
Wang P, Luo M, Zhou W, Jin X, Xu Z, Yan S, Li Y, Xu C, Cheng R, Huang Y, Lin X, Yao L, Nie H, Jiang Q. Global characterization of peripheral B cells in Parkinson’s disease by single-cell RNA and BCR sequencing. Front Immunol. 2022;13: 814239.
Wang P, Wang WY, Zhang XD. Increased interleukin-26 expression in proliferative diabetic retinopathy. Int J Ophthalmol. 2019;12(11):1688–92.
Wang S, Wang X, Cheng Y, Ouyang W, Sang X, Liu J, Su Y, Liu Y, Li C, Yang L, Jin L, Wang Z. Autophagy dysfunction, cellular senescence, and abnormal immune-inflammatory responses in AMD: from mechanisms to therapeutic potential. Oxid Med Cell Longev. 2019;2019:3632169.
Wax MB. The case for autoimmunity in glaucoma. Exp Eye Res. 2011;93(2):187–90.
Wax MB, Tezel G, Yang J, Peng G, Patil RV, Agarwal N, Sappington RM, Calkins DJ. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J Neurosci. 2008;28(46):12085–96.
Weinstein JE, Pepple KL. Cytokines in uveitis. Curr Opin Ophthalmol. 2018;29(3):267–74.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–9.
Weller J, Budson A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res. 2018;7:1161.
Wheeler CJ, Seksenyan A, Koronyo Y, Rentsendorj A, Sarayba D, Wu H, Gragg A, Siegel E, Thomas D, Espinosa A, Thompson K, Black K, Koronyo-Hamaoui M, Pechnick R, Irvin DK. T-lymphocyte deficiency exacerbates behavioral deficits in the 6-OHDA unilateral lesion rat model for Parkinson’s disease. J Neurol Neurophysiol 2014;5(3).
Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, Smithies O, John SW. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355(6326):756–60.
Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11(1):7–13.
Wong M, Huang P, Li W, Li Y, Zhang SS, Zhang C. T-helper1/T-helper2 cytokine imbalance in the iris of patients with glaucoma. PLoS ONE. 2015;10(3): e0122184.
Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, Wong TY. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106-116.
Wu Q, Liu B, Yuan L, Peng Q, Cheng L, Zhong P, Yang X, Yu H. Dysregulations of follicular helper T cells through IL-21 pathway in age-related macular degeneration. Mol Immunol. 2019;114:243–50.
Wu WK, Georgiadis A, Copland DA, Liyanage S, Luhmann UF, Robbie SJ, Liu J, Wu J, Bainbridge JW, Bates DO, Ali RR, Nicholson LB, Dick AD. IL-4 regulates specific Arg-1(+) macrophage sFlt-1-mediated inhibition of angiogenesis. Am J Pathol. 2015;185(8):2324–35.
Yanamandra K, Gruden MA, Casaite V, Meskys R, Forsgren L, Morozova-Roche LA. alpha-synuclein reactive antibodies as diagnostic biomarkers in blood sera of Parkinson’s disease patients. PLoS ONE. 2011;6(4): e18513.
Yang J, Tezel G, Patil RV, Romano C, Wax MB. Serum autoantibody against glutathione S-transferase in patients with glaucoma. Invest Ophthalmol Vis Sci. 2001;42(6):1273–6.
Yang L, Guo C, Zhu J, Feng Y, Chen W, Feng Z, Wang D, Sun S, Lin W, Wang Y. Increased levels of pro-inflammatory and anti-inflammatory cellular responses in Parkinson’s disease patients: search for a disease indicator. Med Sci Monit. 2017;23:2972–8.
Yang M, Shi XQ, Peyret C, Oladiran O, Wu S, Chambon J, Fournier S, Zhang J. Effector/memory CD8(+) T cells synergize with co-stimulation competent macrophages to trigger autoimmune peripheral neuropathy. Brain Behav Immun. 2018;71:142–57.
Yang TT, Song SJ, Xue HB, Shi DF, Liu CM, Liu H. Regulatory T cells in the pathogenesis of type 2 diabetes mellitus retinopathy by miR-155. Eur Rev Med Pharmacol Sci. 2015;19(11):2010–5.
Yang X, Luo C, Cai J, Powell DW, Yu D, Kuehn MH, Tezel G. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol Vis Sci. 2011;52(11):8442–54.
Yang Z, Quigley HA, Pease ME, Yang Y, Qian J, Valenta D, Zack DJ. Changes in gene expression in experimental glaucoma and optic nerve transection: the equilibrium between protective and detrimental mechanisms. Invest Ophthalmol Vis Sci. 2007;48(12):5539–48.
Yeh S, Li Z, Forooghian F, Hwang FS, Cunningham MA, Pantanelli S, Lew JC, Wroblewski KK, Vitale S, Nussenblatt RB. CD4+Foxp3+ T-regulatory cells in noninfectious uveitis. Arch Ophthalmol. 2009;127(4):407–13.
Yokoyama Y, Aizawa N, Chiba N, Omodaka K, Nakamura M, Otomo T, Yokokura S, Fuse N, Nakazawa T. Significant correlations between optic nerve head microcirculation and visual field defects and nerve fiber layer loss in glaucoma patients with myopic glaucomatous disk. Clin Ophthalmol. 2011;5:1721–7.
Yu HG, Lee DS, Seo JM, Ahn JK, Yu YS, Lee WJ, Chung H. The number of CD8+ T cells and NKT cells increases in the aqueous humor of patients with Behcet’s uveitis. Clin Exp Immunol. 2004;137(2):437–43.
Yuan L, Neufeld AH. Tumor necrosis factor-alpha: a potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia. 2000;32(1):42–50.
Yuan N, Zhang HF, Wei Q, Wang P, Guo WY. Expression of CD4+CD25+Foxp3+ regulatory T cells, interleukin 10 and transforming growth factor beta in newly diagnosed type 2 diabetic patients. Exp Clin Endocrinol Diabetes. 2018;126(2):96–101.
Zaidi SAH, Guzman W, Singh S, Mehrotra S, Husain S. Changes in class I and IIb HDACs by delta-opioid in chronic rat glaucoma model. Invest Ophthalmol Vis Sci. 2020;61(14):4.
Zaidi SAH, Thakore N, Singh S, Guzman W, Mehrotra S, Gangaraju V, Husain S. Histone deacetylases regulation by delta-opioids in human optic nerve head astrocytes. Invest Ophthalmol Vis Sci. 2020;61(11):17.
Zhang JM, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. 2007;45(2):27–37.
Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35(2):161–8.
Zhang Q, Fang W, Ma L, Wang ZD, Yang YM, Lu YQ. VEGF levels in plasma in relation to metabolic control, inflammation, and microvascular complications in type-2 diabetes: a cohort study. Medicine (Baltimore). 2018;97(15): e0415.
Zhao W, Yin Z, Li J, Ma M, Li C. Autoantibodies associated with glaucoma. Biomed Res-Tokyo. 2017;28:4913–21.
Zhao Z, Xu P, Jie Z, Zuo Y, Yu B, Soong L, Sun J, Chen Y, Cai J. gammadelta T cells as a major source of IL-17 production during age-dependent RPE degeneration. Invest Ophthalmol Vis Sci. 2014;55(10):6580–9.
Zhou J, Wang S, Xia X. Role of intravitreal inflammatory cytokines and angiogenic factors in proliferative diabetic retinopathy. Curr Eye Res. 2012;37(5):416–20.
Acknowledgements
Not applicable.
Funding
This study was supported by NIH/NEI Grant EY-027355 (SH). This funding body helped in writing of this review article.
Author information
Authors and Affiliations
Contributions
AD contributed to collection of literature and organizing the manuscript. SH guided AD for the organization of manuscript with pertinent information, read, wrote, and edited the manuscript. SM read the manuscript and provided guidance for the immunological aspects of this manuscript. KS read the manuscript and provided guidance for the Alzheimer’s, Parkinson’s, and Multiple Sclerosis sections of this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
DeMaio, A., Mehrotra, S., Sambamurti, K. et al. The role of the adaptive immune system and T cell dysfunction in neurodegenerative diseases. J Neuroinflammation 19, 251 (2022). https://doi.org/10.1186/s12974-022-02605-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-022-02605-9