
Abstract
Background
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder, characterized by motor and non-motor symptoms, significantly affecting patients’ life. Pathologically, PD is associated with the extensive degeneration of dopaminergic neurons in various regions of the central nervous system (CNS), specifically the substantia nigra. This neuronal loss is accompanied by the aggregation of misfolded protein, named α-synuclein.
Main text
Recent studies detected several clues of neuroinflammation in PD samples using postmortem human PD brains and various PD animal models. Some evidence of neuroinflammation in PD patients included higher levels of proinflammatory cytokines in serum and cerebrospinal fluid (CSF), presence of activated microglia in various brain regions such as substantia nigra, infiltration of peripheral inflammatory cells in affected brain regions, and altered function of cellular immunity like monocytes phagocytosis defects. On the other side, Toll-like receptors (TLRs) are innate immune receptors primarily located on microglia, as well as other immune and non-immune cells, expressing pivotal roles in recognizing exogenous and endogenous stimuli and triggering inflammatory responses. Most studies indicated an increased expression of TLRs in the brain and peripheral blood cells of PD samples. Besides, this upregulation was associated with excessive neuroinflammation followed by neurodegeneration in affected regions. Therefore, evidence proposed that TLR-mediated neuroinflammation might lead to a dopaminergic neural loss in PD patients. In this regard, TLR2, TLR4, and TLR9 have the most prominent roles.
Conclusion
Although the presence of inflammation in acute phases of PD might have protective effects concerning the clearance of α-synuclein and delaying the disease advancement, the chronic activation of TLRs and neuroinflammation might lead to neurodegeneration, resulting in the disease progression. Therefore, this study aimed to review additional evidence of the contribution of TLRs and neuroinflammation to PD pathogenesis, with the hope that TLRs could serve as novel disease-modifying therapeutic targets in PD patients in the future.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by a wide variety of motor and non-motor symptoms [1]. The classical triad of motor symptoms in PD is bradykinesia, rest tremor, and rigidity. These cardinal symptoms might be preceded by different non-motor features, including sleep disorder, autonomic dysfunction, hyposmia, and depression [2]. Moreover, the disease could be accompanied by cognitive dysfunction, dementia, and poor balance in advanced stages [3]. The PD prevalence increases with age. In this regard, its prevalence rate is estimated to escalate from 1% in adults over 60 years of age to 3–4% in adults older than 85 [4, 5]. Therefore, considering the aging of the global population, the PD economic and societal burden is expected to increase in the following decades rapidly [4].
Pathologically, PD manifestations are associated with the extensive degradation of dopaminergic (DA) neurons in distinctive brain regions, spinal cord, and peripheral nerves [2]. These degenerative changes have been mostly observed in the midbrain’s substantia nigra pars compacta (SNc) [2]. It has been recently recognized that Lewy bodies, containing the aggregation of misfolded proteins named α-synuclein, accompany the neuronal loss in affected regions [6]. Scientists have historically made several hypotheses regarding the degeneration mechanisms of dopaminergic neurons in PD, among which oxidative stress, excitotoxic mechanisms, and environmental factors have a probable role [2]. However, the lack of disease-modifying treatments based on these mechanisms necessitates further exploration of the underlying causes of this disease. The evidence of neuroinflammation in PD in recent years has attracted much attention. This evidence includes increased proinflammatory cytokines in CSF and blood, detection of activated microglia and macrophages in SNc, infiltration of T lymphocytes in affected brain areas, and altered activity of T and B lymphocytes in PD patients [7, 8]. It has been recently found that Toll-like receptors (TLRs) might serve as significant immune receptors in PD, through which neuroinflammation could be triggered.
TLRs are innate immune system receptors, presenting in various human cells and tissues, including immune, epithelial, neural, and glial cells [9]. They are a member of the pattern recognition receptor (PRR) family and play different roles in recognizing pathogen-associated molecular patterns (PAMPs) expressed by microbial invaders and damage-associated molecular patterns (DAMPs), which are endogenous substances released during tissue damage [9]. TLRs, expressing primarily on microglia, can be stimulated during the course of PD through several molecules such as α-synuclein and heat shock proteins (HSPs) like HSP90 [10], HSP70 [11], and HSP60 [12] presented from affected neurons [9]. Their stimulation may activate several downstream pathways involving myeloid differentiation primary response 88 (MyD88) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), contributing to the production of several proinflammatory cytokines and oxidative stress molecules, leading to neuroinflammation [13]. In this way, TLRs may play pivotal roles in the neural loss in PD pathogenesis. Therefore, this review aims to explore the evidence of the involvement of TLRs in the pathophysiology of PD and their potential therapeutic efficacy.
An overview of TLRs
TLRs' structure and functions
TLRs are a group of pattern recognition receptors (PRRs) with central roles in initiating immune responses against different internal and external stimuli. The TLRs’ immune functions were firstly described in Drosophila mutants in TLR genes, showing high susceptibility to fungal infections [14]. Up to now, ten members of the TLR family have been recognized in the human species, compared with at least 13 members in rodents [15]. Accordingly, each species has a various number of TLR genes, as shown in Caenorhabditis elegance and Drosophila to have one and nine TLR genes, respectively [16]. All TLRs are class I transmembrane receptors, consisting of extracellular, transmembrane, and intracellular domains. The extracellular N-terminal domain of TLRs consists of numerous leucine-rich repeats (LRR) and cysteine-rich domains [15,16,17]. The intracellular section contains an evolutionary-conserved carboxy-terminal domain, denominated Toll/interleukin-1 receptor (TIR) domain, interacting with the TIR or death domain (DD) of adaptor molecules [17]. These receptors are expressed in a variety of immune and non-immune cells, including macrophages, lymphocytes, dendritic cells (DCs), granulocytes, hematopoietic stem cells (HSCs), epithelial cells, neuronal cells, and endothelial cells [18,19,20]. They usually function as homodimers, with the exception of TLR2, which forms heterodimers with TLR1, or TLR6, each showing a different ligand specificity [21]. In humans, four TLRs, including TLR3, TLR7, TLR8, and TLR9, are located within the endosomal membrane, while the other six TLRs reside in the cells' plasma membrane [20]. Generally, plasma membrane-resided TLRs (TLR 1, 2, 4, 5, 6, and 10) recognize bacterial-related lipid, protein, and lipoprotein compartments, including bacterial flagellin, lipoteichoic acid (LTA), lipopolysaccharide (LPS), di-acylated proteins, and tri-acylated lipoproteins [22]. Each TLR type is sensitive to a specific type of mentioned bacterial compartments; for example, TLR4 can specifically recognize Gram-negative bacterial LPS [22]. Moreover, for some TLRs, ligand binding is facilitated by coreceptors and accessory molecules, including the cluster of differentiation (CD) 12, CD14, and myeloid differentiation factor 2 (MD-2) [15, 23]. CD14 exists in two major forms, namely membrane CD14 (mCD14) and soluble CD14 (sCD14) [24]. In this regard, mCD14, expressed mainly on monocytes and macrophages, is more substantially involved in attaching to LPS. It should be noted that LPS is presented to mCD14 and TLR4 by Lipopolysaccharide binding protein (LBP) [25]. However, it has been shown that both sCD14 and LBP are crucial for LPS-induced TLR activation [26, 27]. Alternatively, endosome-associated TLRs (TLR 3, 7, 8, and 9) are specific to both self-derived and pathogen-related nucleic acids, like viral double- or single-stranded RNAs, and bacterial- or viral-related cytosine phosphate guanine (CpG) motif of DNA [28, 29].
TLRs' general signaling pathways
Activation of TLRs by binding to their specific ligands initiates downstream signaling cascades, ultimately leading to various cell responses, including the production of proinflammatory cytokines and interferons. Following engagement to their ligands, TLRs experience a conformational change and recruit intracellular adaptive molecules of myeloid differentiation primary response 88 (MyD88), or TIR-domain-containing adaptor-inducing interferon-β (TRIF). In this context, TRAM (TRIF-related adapter molecule) and TIRAP (TIR domain-containing adapter protein) could facilitate the recruitment of TRIF and MyD88 [30]. Depending on the recruited adaptor molecule, different downstream signaling pathways follow. In this regard, most TLRs utilize MyD88 as an adaptive molecule, except for TLR3, which uses the TRIF-dependent pathway. TLR4 can utilize both MyD88 and TRIF pathways [31]. Through the MyD88-dependent pathway, activated TLR recruits MyD88, followed by three members of the serine-threonine kinase interleukin-1 receptor-associated kinase (IRAK) family, including IRAK1, IRAK2, and IRAK4 [32], forming a complex called "Myddosome" [20]. Myddosome then recruits and stimulates the E3 ubiquitin ligase tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) by K63-related autoubiquitylation. Subsequently, TRAF6 recruits and activates the transformation growth factor beta-activated kinase 1 (TAK1) and IkB kinase (IKK) by ubiquitination of its IKKγ subunit. TAK1 then activates mitogen-activated protein kinases 3/6 (MKK3/6), MKK4/7, and the IKKb subunit of IKK. After that, MKK3/6, MKK4/7, and IKK activate P38 mitogen-activated protein kinase (MAPK), Jun N-terminal kinases (JNKs), and P65 NF-kB, respectively, leading ultimately to the transfer of these transcription factors to the nucleus and transcription and synthesis of proinflammatory cytokines such as IL-1, IL-6, and TNF-α [15, 20].
Alternatively, the activation of the endosome-resided TLR3 or cell surface TL4 recruits TRIF adaptor protein, which next attaches and activates TNF receptor-associated factor 3 (TRAF3) by using polyubiquitination. Activated TRAF3 recruits and stimulates IKKε and TANK-binding kinase 1 (TBK1). TBK1 then activates IFN regulatory factor 3 (IRF3), ultimately leading to type-I interferon production. It should be mentioned that TRIF also causes the MyD88-independent activation of TRAF6, leading to cytokine synthesis. Moreover, TRAF6 activated by the TLR4-MyD88 pathway can cause cellular inhibitor of apoptosis proteins (CIAPs) modification through K63-linked polyubiquitin. Modified CIAPs then contribute to proteasomal degeneration of TRAF3. The other endosome-resided TLRs, including TLR7, TLR8, and TLR9, signal through MyD88-dependent pathway and can thereby lead to cytokine production; besides, they can activate IFN regulatory factor 3 (IRF3), leading to interferon production [15, 20].
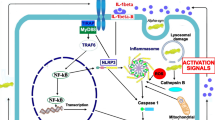
Besides, some inhibitor regulators have been found to negatively affect TLR signaling. For example, Fas-associated protein with death domain (FADD), Toll interacting protein (TOLLIP), and IRAK-M can prevent Myddosome from being activated. In this regard, FADD can antagonize MyD88 or IRAK molecules and their interaction, IRAK-M can inhibit IRAK1 molecule, and TOLLIP can inhibit IRAK phosphorylation and kinase activity [33, 34]. Other examples of TLR regulators are SH2-containing inositol-5′-phosphatase (SHIP) 1 and SHIP2, which contribute to dephosphorylation and inactivation of IRAK1 and TBK1. Furthermore, tumor necrosis factor α-induced protein 3 (TNFAIP3 or A20) can inhibit TRAF6 and IKK activation [15, 35]. The mentioned TLR regulator proteins are named as some bold examples of TLR signaling inhibitory adaptor molecules, which are naturally expressed by genes located in the human genome. These molecules integrate with the TLR signaling pathway to regulate inflammatory responses to different stimuli (Fig. 1).

By attaching to their ligands and experiencing conformational changes, all TLRs except for TLR3 recruit the MyD88 adaptive molecule. MyD88 then activates and recruits IRAK1, IRAK2, and IRAK4, forming a complex named Myddosome. Myddosome then recruits and activates TRAF6, followed by activation of TAK1. The activation of TAK1 resulted in the activation and transfer of different transcription factors, including NF-kB, JNK, and AP1, leading to proinflammatory cytokine production. Moreover, TRAF6 can activate IRF7 leading to interferons production. On the other side, TLR3 utilizes the TRIF adaptive molecule, which by activating TRAF3 and TBK leads to activation and transfer of IRF3 to the nucleus and interferons production. Moreover, TRIF can activate TRAF6 and result in MyD88-independent proinflammatory cytokine production. Furthermore, TLR4 can use both MyD88 and TRIF adaptor proteins. It should be mentioned that some TLRs like TLR4 can use coreceptors like CD14 and MD2 for attaching more efficiently to their ligands. Myddosome can be inhibited by several factors, including FADD, TOLLIP, IRAK-M, and SHIP-1. For instance, FADD can antagonize MyD88 or IRAK molecules and their interaction, IRAK-M can inhibit IRAK1 molecule, and TOLLIP can inhibit IRAK phosphorylation and kinase activity [33, 34]. Other examples of TLR regulators are SH2-containing inositol-5′-phosphatase (SHIP) 1 and SHIP2, which contribute to dephosphorylation and inactivation of IRAK1 and TBK1. TLR toll-like receptor, LPS lipopolysaccharide, CD14 cluster of differentiation 14, MD2 Myeloid Differentiation factor 2, MyD88 Myeloid differentiation primary response 88, IRAK4 Interleukin-1 receptor-associated kinase, TRAF6 TNF (tumor necrosis factor) receptor-associated factor 6, TAK1 transforming growth factor b-activated kinase 1, MKK mitogen-activated protein kinase kinase, JNK Jun N-terminal Kinase, IRF Interferon regulatory factor, P38MAPK P38 mitogen-activated protein kinase, AP1 Activating Protein-1, NF-kB nuclear factor kappa-light-chain-enhancer of activated B cells, TRIF TIR-domain-containing adaptor-inducing interferon-β, LBP Lipopolysaccharide binding protein, TIR Toll/interleukin-1 receptor, TANK binding kinase 1, FADD Fas-associated protein with death domain, TOLLIP toll interacting protein, SHIP SH2-containing inositol-5′-phosphatase, dsRNA double-stranded RNA, ssRNA single-stranded RNA
TLR roles in inflammation and neuroinflammation
Recent studies have questioned the common belief regarding considering the central nervous system (CNS) as a complete immune-privileged organ. In the CNS, there are various resident cell types such as microglia, astrocytes, and oligodendrocytes performing roles as innate immune cells [36]. The activation of these cells under several conditions can contribute to the secretion of inflammatory cytokines and reactive oxygen species (ROS), leading to neuroinflammation, which might disrupt the blood–brain barrier (BBB) and thereby allow and chemotactically attract several peripheral immune cells such as monocytes, macrophages, neutrophils, and T cells to join the CNS, aggravating the inflammation [37,38,39]. This inflammation might be triggered due to different CNS infections or sterile aggregation of toxic metabolites and proteins, as seen in neurodegenerative disorders. Though the presence of neuroinflammation is necessary for the clearance of pathological invaders or toxic metabolites accumulated in the CNS, the uncontrolled stimulation of this inflammation might propagate the condition like what can be seen during the courses of meningitis, PD, or Alzheimer’s disease [38]. TLRs expressed by the CNS-located immune or neural cells were shown to have substantial roles in inducing and maintaining neuroinflammation. Therefore, they could serve as double-edged swords in the pathogenesis of many CNS-related diseases [38].
Bacterial meningitis, viral infections, or sterile inflammatory disorders like Alzheimer's disease or cerebral ischemia can be named as examples of the involvement of TLRs in neuroinflammation. Bacterial meningitis can be caused by Gram-positive (G +) bacteria like Streptococcus pneumonia (S. pneumonia) and S. agalactiae, or Gram-negative (G−) bacteria like Neisseria meningitides (N. meningitides) and Escherichia coli (E. coli) [40]. The lipoteichoic acid (LTA), peptidoglycan (PGN), different lipoproteins or proteins of G + bacteria, and LPS of G- bacteria can serve as PAMPs to TLRs residing in the CNS, thereby can lead to excessive transcription of inflammatory cytokine-related genes that result in neuroinflammation followed by meningitis [41]. For example, the porin B protein of N. meningitides can cause the formation and stimulation of the TLR2/TLR2 homodimers on the CNS cells, thereby initiating downstream pathways resulting in the activation of NF-kB transcription factor and secretion of proinflammatory cytokines, leading to meningitis [42]. Alternatively, the LPS of G- bacteria and the bacterial DNA can trigger the inflammatory pathway by stimulating TLR4 and TLR9, respectively [38]. On the other hand, several viral infections can contribute to CNS inflammation. In this regard, double-stranded RNA (dsRNA), single-stranded RNA (ssRNA), and non-methylated CpG motifs of invader viruses can stimulate TLR3, TLR7, TLR8, and TLR9 as PAMPs. The stimulation of TLR3 causes the activation of the TRIF-dependent downstream pathway, while TLR7, TLR8, and TLR9 act via the MyD88-dependent pathway. Both of these pathways then contribute to the secretion of inflammatory cytokines and type-I interferon, leading to neuroinflammation [38, 43].
Unlike the mentioned infectious diseases, in plenty of sterile CNS conditions, including neurodegenerative disorders, the presence of toxic metabolites and proteins as intrinsic substances can trigger TLRs and initiate neuroinflammation. For instance, during the course of Alzheimer's disease, the most prevalent progressive neurodegenerative disorder characterized by memory loss and cognitive dysfunction, aggregation of extracellular plaques of amyloid-beta (Aβ) and the neurofibrillary tangles that consist of tau protein have the potential to exert neurotoxic effects and contribute to the pathogenesis of the disease [44]. These accumulated proteins can be recognized as DAMPs and thereby trigger TLRs presenting on different CNS-resided cells, which is necessary for the clearance of these accumulated proteins. However, the overactivation of TLR-dependent pathways by these proteins can detrimentally generate neuroinflammation, which is pathologic. As evidence of TLR involvement during AD, upregulation of TLR1, 2, 3, 4, 5, and 6 genes in the temporal cortex of AD patients can be mentioned [45]. It has been found that TLR2 is the primary receptor for the Aβ42 protein, responsible for neuroinflammation during AD, as its deficiency leads to subsided inflammation in this disease [45]. Cerebrovascular accidents might serve as another example of TLR-dependent inflammation in a sterile condition. As cerebral ischemia occurs, the lack of sufficient oxygen supply to the brain cells leads to the release of different intracellular factors, including hyaluronic acid, high-mobility group box 1, heat shock proteins like HSP70, and mRNAs. These released factors are recognized as DAMPs by TLRs residing on microglia, astrocytes, and other CNS cells, leading to proinflammatory cytokine secretion. For instance, heat shock protein 70 (HSP70) was shown to activate TLR2 and TLR4 downstream pathways [38]. The activation of the microglia-resided TLRs can also upregulate specific chemokines, including MCP-1 or CCL3, which are effective in the chemoattraction of peripheral immune cells to the injured area of the CNS [46]. Although some secreted factors such as neurotrophins caused by the activation of TLRs during cerebral ischemia were shown to have neuroprotective effects in animal studies, clinical trials on the efficacy of these neurotrophic factors in humans have failed so far [47, 48].
Evidence of neuroinflammation in PD
In 1988, McGeer et al. were the first to describe evidence of neuroinflammation in PD. They reported the presence of activated microglia and infiltrating lymphocytes in the substantia nigra of PD patients [7]. Although McGeer's way had not been continued for nearly 20 years, in 2008, Brochard et al. demonstrated and quantified the evidence of infiltrated T lymphocytes (CD8+ and CD4+) both in postmortem human brain specimens and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced mouse model of PD [49]. Since then, a significant body of literature has been accumulated on the substantial roles of neuroinflammation in PD. In this regard, evidence of increased levels of inflammatory cytokines in the blood, CSF, and brain of PD patients [8] and modifications in cellular immunity such as alterations in T regulatory/T effector ratio in the blood of PD patients [50, 51] potentially confirmed the previous observations. Besides, studies on animal models of PD and neuroimaging approaches in humans provided further clues on the role of neuroinflammation in PD [52, 53].
Qin et al. conducted a systematic review and meta-analysis, pooling data from 25 eligible studies covering 1547 patients with PD and 1107 controls, to determine PD patients' blood levels of cytokines. They found significantly higher peripheral concentrations of IL-6, TNF-α, IL-2, IL-10, IL-1β, C-reactive protein (CRP), and regulated on activation normal T-expressed and secreted (RANTES) in patients with PD compared with the control group [54]. However, this survey neglected the effects of some essential factors, including disease duration, disease severity, and concomitant therapies’ effects on the cytokine levels of PD patients [54]. Accordingly, a prospective cohort study on 230 incident PD patients and 93 age-matched controls demonstrated significantly higher levels of TNF-α, IL1-β, IL-2, and IL-10 in PD patients [55]. This study showed that higher proinflammatory cytokine levels and lower anti-inflammatory cytokine levels were associated with more rapid progression of motor symptoms over 36 months in PD patients [55]. Therefore, this study states the hypothesis that the cytokine level might have the potential to serve as a predictive factor of PD motor progression. On the other side, the CSF levels of IL-1β and IL-6 were shown to be higher in PD patients [56]. Also, another study reported a higher concentration of IL-1β, IL-2, IL-4, TGF-α, and IL-6 in ventricular CSF of PD patients compared to controls [55]. However, the correlation between CSF and blood levels of cytokines in PD patients should be elaborated on in future studies.
Alterations in the function of cellular immunity in PD have been reported in several studies. For example, memory T cells reacting to the α-synuclein epitopes have been detected in the serum of PD patients [57]. It has also been shown that these memory T cells can appear even before the start of clinical manifestations of the disease, and therefore it would be worthwhile to determine their efficacy as early PD biomarkers [58]. The other evidences of altered cellular immunity in PD patients were shown to be monocytes phagocytosis defection [59], dysregulation of peripheral monocytes and their hyperactivity in response to LPS [60], and the modified ratio of Teff/Treg in the serum of PD patients [51]. Moreover, a decreased concentration of naïve T cells, including CD45RA+ T cells, and an increased level of activated T cells like CD45RO+ T cells have been detected in the serum of PD patients, indicating a peripheral activation of lymphocytes [53] (Fig. 2).

In PD patients, proinflammatory cytokines, including IL-6, TNF-α, IL-2, IL-10, IL-1β, and RANTES in the blood, CSF, and the brain tissue, are higher than in the normal population. There are also more active microglia and astrocytes in the brain of PD patients than in normal people. Inflammatory factors secreted by such cells chemotactically attract peripheral inflammatory cells, including monocytes and T and B lymphocytes, which secrete proinflammatory cytokines and antibodies and contribute to neuroinflammation. The neuroinflammation ultimately leads to degeneration of dopaminergic neurons, worsening PD symptoms. Substances released by neuronal degradation, such as DAMPs and α-synuclein, further stimulate brain-resided inflammatory cells and reinforce neurodegeneration. TNF-α tumor necrosis factor α, RANTES regulated on activation normal T-expressed and secreted, IL interleukin, TLR toll-like receptor, DAMPs damage-associated molecular patterns, RBC red blood cell
Imaging studies provided further clues on the involvement of neuroinflammation in PD pathology. For example, Gerhard et al. used [11C] (R)-PK11195 marker, a positron emission tomography (PET) marker binding to peripheral benzodiazepine binding sites (PBBS), which is expressed mainly in mitochondria of activated microglia. They found that [11C] (R)-PK11195 was significantly more likely to bind to the basal ganglia, pons, and temporal and frontal cortical regions in patients with PD compared to the control group, suggesting the raised number of activated microglia in these regions [52]. However, by following the patients for two years, they could not find an alteration in the binding of the marker, indicating that it cannot be used as a monitoring tool for disease progression [52]. This result has been repeated by Terada et al. using [11C] DPA713 PET marker of microglial activation. In their small cohort, they found increased uptake of this tracer in the occipital, temporal, and parietal cortex of PD patients, which, unlike the Gerhard study, was increased in the second scan after 1 year [61]. Moreover, in 2019, a PET marker named [11C] CPPC was developed, which is a high-affinity ligand, attaching specifically to macrophage colony-stimulating factor 1 (CSF-1), the expression of which is limited to microglia. This marker demonstrated its efficacy for recognizing the microglia component of neuroinflammation in murine and non-human primate AD models. Therefore, it might be worthwhile to investigate its effectiveness in human subjects with PD in future studies [62].
Animal studies have also contributed to a better understanding of the pathophysiological and etiological roles of neuroinflammation in the disease. Animal models of PD are obtained through several methods using neurotoxin or inflammatory challenges. For obtaining neurotoxin-induced animal PD models, intracerebral injection of 6-hydroxydopamine (6OHDP) or peripheral injection of several types of complex-1 inhibitors such as MPTP, rotenone, and annonacin is most effective [53]. Moreover, polyinosinic: polycytidylic acid (poly I:C) and LPS were utilized for achieving inflammatory-induced PD animal models [63]. Also, transgenic mice overexpressing human α-synuclein genes are another useful animal model for investigating PD pathogenesis [64]. Microglial activation was the most experimental feature in the mentioned animal models of PD. For instance, microglial activation was detected following peripheral MPTP injection in mice and monkeys [65, 66]. MPTP injection chronicity also plays an essential role in the degree of microglial activation, as shown that chronic peripheral injection of MPTP caused less microglial activation than acute injection [53]. Moreover, microglial activation was also observed in human α-synuclein-overexpressing transgenic mice [67]. Therefore, the inducement of Parkinsonism through injection of neurotoxins, inflammogens [68], or manipulation of PD-associated genes could lead to microglial activation in animal studies. Several animal models have also demonstrated the association between this activation and neuronal degeneration [49, 69]. On the other side, infiltration of peripheral immune cells such as CD8+ or CD4+ T cells into the nigrostriatal system can further contribute to neuronal degeneration, as shown by a mice model of MPTP [49].
TLRs and PD
As mentioned earlier, accumulating evidence highlighted the role of neuroinflammation in PD pathogenesis. On the other side, with a better understanding of various inflammatory effects of TLRs over the last decade, scientists have begun to think about the possible involvement of TLRs in this disease. Although the precise mechanism of TLR contribution to PD neuroinflammation is still unclear, some theories pointed to the possible role of these receptors in recognizing α-synuclein aggregates as DAMPs, which can induce proinflammatory downstream pathways, leading to neuroinflammation [70]. Such hypotheses have led to designing several studies in recent years. These studies mainly compared the expression of different TLRs and the TLR-mediated responses between PD cases and control groups using in vivo or in vitro methods. In this regard, scientists primarily work on various PD animal models like neurotoxin- or inflammatory-induced mice models of PD, microglial cells extracted from PD mice models like BV2 microglial cell line using in vitro studies [71], postmortem human brains of patients with PD, or serum of PD patients. Researchers also used different TLR agonists or antagonists in PD animal models to observe the changes in PD-related motor behaviors or compare the severity of inflammatory responses and neurodegeneration between PD cases and controls. The results of most of these studies indicated the prominent roles of TLR2, TLR4, and TLR9 in the pathogenesis of PD, with scarce evidence of the possible involvement of other TLRs, including TLR7 and TLR8 [72]. For shedding more light on this issue, the following section firstly reviewed the strengths and weaknesses of different animal models of PD; then, we reported evidence of the contribution of TLR2, TLR4, TLR9, and other TLRs to PD pathophysiology (Table 1). Afterward, we presented the various effects of different drugs on TLRs in clinical, animal, and in vitro studies (Table 2).
Animal models of PD
Selection of a proper animal model in animal studies is crucial because the attribution of the results to humans depends highly on using appropriate animal models to imitate the human disease or condition. In this regard, along with our expanding knowledge of PD pathogenesis and manifestations, different PD animal models have been developed; each has its strengths and weaknesses. In other words, as the precise mechanism of PD is still unclear, development of a complete animal model is impossible, and all established PD animal models have disadvantages limiting their use as optimal PD models. To date, two main categories of PD animal models are applied in PD research, including neurotoxin models and genetic models.
Neurotoxin models are the most classic and most commonly used animal models of PD. The popularity of this model is due to its relatively simple way of establishment [73]. The most widely used neurotoxins for making these models are 6-OHDA and MPTP [73]. As 6-OHDA cannot penetrate BBB, it should be directly injected into the brain parenchyma. Depending on the target site of injection, including SN, striatum, or medial forebrain bundle, 6-OHDA can cause severe and quick or slow and progressive symptoms. 6-OHDA administration in the brain leads to the production of reactive oxygen species, which are thought to be responsible for the degeneration of DA neurons. Hence, this model lacks Lewy-related pathology [74]. On the other hand, MPTP can easily penetrate BBB and, therefore, it can be systematically administered. After entering the brain, MPTP is enzymatically converted to MPP + , absorbed by receptors of dopaminergic neurons. Within DA neurons, MPP + affects mitochondrial function leading to oxidative stress and degeneration of DA neurons [73, 75]. Despite other PD animal models, the MPTP model causes gradual appearance of induced symptoms, which can be considered as its strength point [73]. Although neurotoxin models are beneficial as they are easy to make, they cannot imitate any pathological features of PD; instead, they solely mimic PD symptoms. Hence these models cannot be appropriate for designing preventive treatment and clarifying the PD pathogenesis. Furthermore, they just demonstrate the late stages of PD, as the motor symptoms usually appear when severe dopaminergic degeneration has happened [74].
Genetic models are relatively new types of PD animal models established by manipulating several genes thought to be connected to PD pathogenesis. In fact, the establishment of transgenic animal models was preceded by identifying different human mutations responsible for familial cases of PD. In this regard, several genes causing mitochondrial dysregulation play their parts [76]. Moreover, specific genes, including α-synuclein, Parkin, PTEN-induced putative kinase 1 (PINK1), and DJ-1, thought to be causally linked to PD pathogenesis, were applied in developing different PD genetic models. By helping new technology, the knockout mice model of these genes could be generated, which could resemble more similarly human PD pathophysiology [77]. Moreover, different genetic models enabled us to study different genes whose expressions are changed in PD patients. Besides, genetic models of PD allow us to administer treatment before the onset of PD symptoms, having a significant role in developing preventive treatments. One of the disadvantages of genetic PD models is that many splicing events are not conserved between humans and mice. Therefore, the results of PD genetic models might not be fully applicable to humans [78]. Recently, a combined neurotoxin and genetic model has been developed, by which neurotoxins are administered to genetically manipulated animal models [79]. This model seems to be more promising and can resemble more closely human PD mechanisms. However, research on this combined model is in its infancy, and more studies are needed to examine its utility. In summary, none of the PD animal models can completely resemble human PD mechanisms, and each animal model's findings should be interpreted with caution considering its advantages and disadvantages.
TLR2
Several studies reported an upregulation of TLR2 expression in different regions of PD postmortem human brains. For example, Drouin-Ouellet et al. found an increased expression of TLR2 in the caudate and putamen of PD patients [80]. In line with this, Dzamko et al. showed that the increased expression of TLR2 on neurons located in the anterior cingulate cortex and striatum of PD human brains is positively correlated with pathological α-synuclein accumulation and the disease staging and burden [81]. They also found that activated TLR2 promotes endogenous α-synuclein aggregation, having roles in autophagy/lysosomal pathways [81]. As TLR2 is mainly expressed on microglia, it has been suggested that the neuronal expression of this receptor might have the potential to serve as a PD-specific biomarker [70]. On the other side, TLR2 expression was shown to positively correlate with the amoeboid type of microglia in the hippocampus and SN of PD patients, highlighting its role in the microglia-mediated inflammatory responses, leading to neuroinflammation in PD [82]. Although most studies reported the roles of microglial or neuronal TLR2 in PD, emerging evidence also underpinned the significant role of astrocytes-located TLR2 in the disease-induced neuroinflammation by intensifying the inflammatory responses of these cells to the disease stimulants, including α-synuclein [83].
To investigate features of different molecular forms of α-synuclein, Gustot et al., in a survey on human monocyte cell line treated with α-synuclein and anti-TLR2 antibodies, demonstrated that α-synuclein could activate TLR2 only when it folded as amyloid fibrils [84]. However, another study on human monocytes treated with anti-TLR2 before α-synuclein administration showed that both fibrillar and monomeric α-synuclein could activate TLR2 [85]. Although this discrepancy might arise from the lack of consistency in definitions of monomers, oligomers, and fibrils of α-synuclein across various studies, this issue remains to be further dissected in future clinical research.
The contribution of TLR2 to PD pathogenesis in human studies has also been confirmed by showing its higher expression on peripheral blood mononuclear cells (PBMCs) in the serum of PD patients compared with healthy controls [80, 86]. Furthermore, additional evidence of TLR2 involvement in PD pathogenesis in humans was obtained from a study indicating the correlation between TLR2 single nucleotide polymorphisms (SNPs) and the risk of PD development in a Chinese Han population [83].
In vitro and in vivo animal studies on PD also provided further evidence of the roles of TLR2, having the potential to be translated into human PD pathogenesis. In this regard, an in vitro study showed that stimulation of the murine BV2 microglial cell line with incubated recombinant human wild-type α-synuclein (SYNTR) leads to overexpression of the TLR2 and TLR3 genes [87]. This study also reported similar gene expression changes in primary microglia obtained from SYNTR-induced mice brain cortices [87]. Daniele et al. also indicated that oligomers of α-synuclein can activate microglia by engaging TLR2/TLR1 cell-membrane heterodimers, leading to translocation of NF-kB to the nucleus and proinflammatory cytokine production in BV2 mice microglia cell line [88].
A study on human α-synuclein-overexpressing transgenic mice showed that anti-TLR2 therapy reduced α-synuclein aggregation in neuronal and astroglial cells and alleviated neuroinflammation and neurodegeneration [89]. In vitro investigation in the same study uncovers the anti-TLR2 therapy efficacy through blocking neuron-to-astrocyte and neuron-to-neuron transmission of α-synuclein [89]. Another study has confirmed this result using the transgenic mice model of PD and confirmed that TLR2 activation leads to α-synuclein accumulation in neurons by inhibiting autophagy activity through the phosphatidylinositol-3-kinase mammalian target of rapamycin (AKT/mTOR) pathway [90]. In this regard, this study proposed that neuronal TLR2 regulates autophagy and TLR2 gene ablation results in reduced neuronal α-synuclein aggregation and improved motor functions [90]. In another study, pretreatment immunization with anti-TLR2 in wild-type (WT) acute mice model of PD led to alleviation in α-synuclein-induced memory deficits [91].
Moreover, several animal studies imply that MPTP-induced mice models showed an upregulation in TLR2 [92,93,94]. In their survey on WT and TLR2-/- mice, Kim et al. demonstrated that TLR2 acted as the receptor of neuron-secreted α-synuclein, thereby mediating microglial inflammatory responses, leading to dopaminergic neuron loss in this genetic animal model of PD [90].
TLR4
Human postmortem brain investigations by transcriptomic data analysis indicated a higher expression of TLR4 and its adaptor protein MyD88 in different brain regions of PD patients, with the most pronounced upregulation in SN and putamen [70, 95, 96]. In this regard, Kouli et al. found a concomitant upregulation of proinflammatory cytokine of IL-1β and TLR4 expression in the frontal cortex and SN of PD human postmortem brains [97]. In the same study, increased TLR4 expression in the amygdala was correlated with the increased CD4+ infiltration, higher number of activated microglia, and upregulated α-synuclein [97]. This increased TLR4 expression was also detected in peripheral immune cells of the PD patients [80, 96] as a study showed an enhanced expression of TLR4 in circulating monocytes and B cells of PD patients compared to controls [80]. Moreover, a study comparing the colonic biopsies of patients with PD and the control group detected a higher TLR4 expression in colonic samples of PD patients. This higher TLR4 expression was accompanied by a disruption in the intestinal barrier, increased microbial markers, and a higher proinflammatory gene profile in the colonic samples of PD patients [98]. Additionally, human studies found an association between SNPs of the TLR4 gene and sporadic PD in the Chinese Han population, serving as another evidence of TLR4 involvement in PD [99].
In vivo and in vitro studies using different animal models of PD provided further evidence of the contribution of TLR4 in these models. A study showed an increased vulnerability of WT mice brain than TLR4-deficient mice brain to DA neurodegeneration following MPTP injection [100]. In line with this, an in vivo study on the MPTP mice model reported that the absence of TLR4 inhibited dopamine depletion, reduced α-synuclein-positive neurons, alleviated PD-associated neuroinflammation modifications in inflammasome pathways, and induced changes in the performance of transcription factors such as NF-κB and activator protein 1 [101]. Moreover, a study on various PD mice models using the intra-striatal injection of neurotoxins or inflammogens, including 6-OHDA, rotenone, LPS, and poly I:C reported a significant upregulation in striatal TLR4 expression at day 28 after 6-OHDA, day four after LPS, and day 14 after poly I:C injections. This study showed that TLR4 upregulation occurred earlier following injection of inflammogens like LPS, or Poly I:C, and at a later time following neurotoxin challenge with 6-OHDA. These findings suggest that the induced neuroinflammation following neurotoxin challenge might be secondary to toxin-caused neurodegeneration and the release of different inflammatory stimulant molecules such as HSP-1 and chemokines [102]. On the other side, the triggered neuroinflammation after the inflammatory challenge was suggested to be caused by the rapid development of interactions between TLR4 and administered PAMPs (LPS, or Poly I:C) [102].
However, animal studies showed some controversy concerning the roles of TLR4 in a neuroprotective or detrimental manner in PD models. For example, a study showed that α-synuclein is upregulated in the brain of TLR4 knockout mice compared with WT mice [103]. This result was repeated in a multiple system atrophy mouse model, showing that TLR4 ablation exacerbates DA neuronal loss and motor deficits [104]. Moreover, another study comparing rotenone-induced WT and TLR4 knockout mice reported that TLR4 ablation improved the SN neuronal loss and motor deficits [98]. These findings may implicate the substantial roles of TLR4 in inducing phagocytosis, which can lead to α-synuclein clearance in the acute phase of the disease. To examine this theory, Stefanova et al. administered anti-TLR4 therapy in BV2 mice microglia cell line. They observed that TLR4 ablation inhibited α-synuclein phagocytosis by microglia leading to α-synuclein-associated neurodegeneration and exacerbation of motor deficits [105].
In contrast, MPTP-induced mice models of PD revealed an increased TLR4 expression (70). Moreover, ablating TLR4 through silencing its genes or administering small interfering RNAs (siRNAs) in these models improved MPTP-induced motor deficits, pointing out TLR4 detrimental effects on the disease [70]. For instance, it was shown that TLR4 knockout mice are less prone to MPTP-induced toxic effects, as they showed less DA neuronal damage and fewer activated nigral microglia following the MPTP challenge [70, 72]. Moreover, Zhou et al. demonstrated in an in vitro study that silencing TLR4 genes through siRNA led to a reduction in TLR4-dependent NF-kB activity and could thereby decrease microglial activation [106].
In addition, recent studies implicated the significant roles of TLR4 in recognizing α-synuclein and initiating inflammatory responses. In line with this discussion, the contribution of TLR4 to α-synuclein recognition was reported to be even more prominent than the contribution of TLR2. Hughes et al. revealed that oligomeric α-synuclein-induced microglial TNF-α production in WT mice was 10 to 100 times greater than in TLR4 knockout mice. They also showed that the reduction in TNF-α production in TLR2 knockout mice was not as much as in TLR4 knockout mice, suggesting the primary roles of TLR4 in α-synuclein recognition and initiation of inflammatory pathways [95]. Other studies have confirmed these results showing that TLR4-deficient mice produced lower amounts of TNF-α and ROS [13]. Moreover, another study revealed TLR4-mediated astroglial activation in mice following the exposure to purified human α-synuclein, leading to NF-kB and AP-1 pathways, Jun N-terminal kinase activation, and secretion of inflammatory cytokines and nitric oxide [107].
In line with the mentioned human study reporting higher TLR4 expression in colonic samples of PD patients, rotenone-induced TLR4 knockout mice showed reduced intestinal inflammation, intestinal dysfunction, and neurodegeneration compared with WT mice [98]. These results might point out the possible contribution of TLR4 in intestinal dysfunction and the roles of the microbiome and the gut–brain axis in disease development, which remain to be further investigated in forthcoming studies [98].
In summary, it is proposed that TLR4 has both detrimental and neuroprotective effects in PD pathogenesis, depending on the investigated time-point of the disease. In the acute phases of the disease, TLR4 contributes to α-synuclein recognition. It activates the phagocytosis response of glial cells, induces necessary inflammatory cytokines production, leading to α-synuclein clearance, and delays the disease progression. On the other side, in chronic phases of PD, excessive stimulation of TLR4-mediated proinflammatory cytokines production results in neuroinflammation, which in turn can lead to neurodegeneration, resulting in disease progression.
TLR9
Emerging evidence also suggested a partial involvement of TLR9 in the course of PD. For instance, Ros-Bernal et al. detected TLR9 upregulation in the striatum of PD patients [108]. Polymorphisms in the TLR9 gene also support the evidence of TLR9 involvement in PD. In addition, a study found that the rs352140 T allele of TLR9 is associated with a reduced risk of PD in the Northern Iranian population [109]. In line with this result, a Chinese study found that the mentioned allele is associated with a reduced risk of PD in the Chinese Han female population [110].
On the other side, an animal study demonstrated that MPTP-intoxication in mice could lead to TLR9 upregulation [108]. In another study, TLR9 ablation in mice showed its protective effects against intoxication with MPTP [111]. Furthermore, less dopaminergic neuron death occurred in the SN of TLR9 knockout mice compared with WT mice following MPTP infusion [111]. In the same study, CpG-ODN, a TLR9 agonist, induced degeneration of dopaminergic neurons in mice lacking glucocorticoid receptors in microglia. The authors of this study proposed that glucocorticoid receptors may have some protective effects in this PD model, which should be investigated as potential therapeutic targets in future studies [111].
Other TLRs
With respect to the evidence of the involvement of TLRs other than TLR2, 4, and 9 in PD, a human study showed that blood leukocytes' TLR7 and TLR8 responses are impaired in PD patients [72]. Moreover, an animal study using different rat models indicated a striatal upregulation of TLR3 expression following the injection of rotenone, LPS, Poly I:C, and 6-OHDA [102]. Another study showed that stimulation of murine BV2 microglial cells with α-synuclein leads to increased expression of TLR1 and TLR7 genes [87]. Nevertheless, research on the roles of TLRs other than TLR2, 4, 9 is in its infancy, and this subject is recommended to be dissected in detail through future studies.
Substances with potential effects on TLRs and neuroinflammation in PD
Cordycepin
Cordycepin, or 3'-deoxyadenosine, is a main bioactive constituent of a fungus species named Cordyceps militaris, extracted and used in traditional Chinese medicine [112]. The molecular structure of cordycepin is 9-(3-deoxy-β-d-ribofuranosyl) adenine, and its effects on various diseases have been extensively studied since its isolation from the C. militaris in 1951 [112, 113]. In this regard, cordycepin can exert a wide range of pharmacological effects, from which the anti-inflammatory, anti-cancer, neuroprotective, and anti-oxidant properties were its most observed features [112, 114]. For instance, cordycepin has shown its effects in upregulating IL-10 in human blood mononuclear cells, thereby suppressing the secretion of inflammatory cytokines [115]. Cordycepin was administered in different studies using either oral or intravenous routes [116, 117]. In this regard, the bioavailability of orally administered cordycepin can be significantly improved using nanoencapsulation of the drug combined with gastric acid inhibition strategies [118]. Cordycepin can reportedly efficiently protect BBB integrity by reducing local neuroinflammation, reorganizing tight junctions, and prohibiting the activity of NADPH oxidase (NOX) [119]. Although not completely clear, these protective effects on BBB suggest that Cordycepin probably has the ability to penetrate BBB [120]. Cheng et al. investigated Cordycepin's effects on PD rat models by administering Cordycepin one hour after the first MPTP injection to rats. Subsequently, they assessed motor function tests and other experiments after 15 days of MPTP injection. They demonstrated that cordycepin could alleviate MPTP-induced changes, including motor disorders, DA neuron loss, and activation of TLR/NF-kB. They also illustrated that cordycepin diminished the cytotoxic impacts of LPS on PC12 cell lines by microglia [113].
SA00025
Nuclear receptor-related 1 (Nurr-1) is a protein controlling the specific genes required for dopamine regulation in dopaminergic neurons in SNpc [121]. Eliminating Nurr-1 has led to dopaminergic phenotype loss in both in vitro and in vivo studies [122, 123]. Hence, Nurr-1 plays an essential role in the differentiation of dopaminergic neurons in embryonic phases and conservation of dopaminergic phenotype during the lifetime, suggesting its probable functions in PD. Additionally, Nurr-1 can reportedly mitigate inflammation, which is another risk factor for PD [124]. Therefore, increasing Nurr-1 or inducing it might exert beneficial effects on PD. SA00025 is a novel Nurr-1 agonist with molecular structure of 2-{3-[2-(4-chlorophenyl)imidazo[1,2-a]-pyridin-6-yl]phenyl}propan-2-ol, whose effectiveness was tested in an animal model of PD [124]. Smith et al. administered a daily 30 mg/kg dose of SA00025 by oral route to a 6-OHDA-induced model of rats. After seven consecutive days, the rats were killed. The pharmacokinetic and postmortem brain analysis showed that SA00025 entered the brain 1–48 h after the last oral administration on day 7 and induced transcriptional upregulation of Nurr-1 protein and modulated dopaminergic-related gens. This finding might suggest that SA00025 can penetrate the BBB. Furthermore, daily gavage administration of SA00025 for 32 days led to dopaminergic neuroprotective effects, induction of resting-state of microglia, and reduction of IBA-1 staining intensity of microglia, GFAP staining intensity of astrocytes, and IL-6 levels [124].
Peroxiredoxin 6
Peroxiredoxins (PRX) are highly conserved ubiquitous family enzymes with catalytic abilities. PRDX6 is the only member of this family capable of reducing phospholipid hydroperoxides and short-chain hydroperoxides [125]. Yeo et al. compared neural stem cells extracted from E15 days wild-type and PRDX6-overexpressing mice. They demonstrated that neurogenesis and expression of protein biomarkers are impaired in neural stem cells isolated from PRDX6 mice. They also showed that WD repeat and FYVE domain-containing protein 1 (WDFY1) expression was dramatically reduced in PRDX6 mice through a TLR4-dependent mechanism. In this regard, they showed the potential utility of introducing the plasmid of WDFY1 to neural stem cells of PRDX6 mice for improving neurogenesis and reversing TLR4 [126]. Collectively, this study showed that PRDX6 inhibitors might play beneficial roles in the future way of PD treatment.
Dihydrotestosterone
Dihydrotestosterone (DHT) is a 5α-reduced derivative of testosterone with potent androgenic activities, converted from testosterone in target tissues [127]. DHT is an injectable drug that can penetrate the BBB and affect the CNS as it has a lipophilic molecular structure [127, 128]. Yang et al. demonstrated the effects of DHT on neuroinflammation, impaired behavior, and neuronal damage in an LPS-induced mice model. They reported that DHT could reduce proinflammatory cytokine production in BV-2 cells and primary microglia by inhibiting TLR4-dependent NF-κB and MAPK p38 pathways, protecting neurons. Moreover, behavioral tests showed that DHT could ameliorate learning and spatial disability, motor incoordination, and locomotor abilities in LPS-induced mice models [129].
Hesperetin
Hesperetin is a derivative of flavanone named trihydroxyflavanone, which is found mainly in citrus fruits like oranges [130]. Hesperetin has been reported to have the ability to cross the BBB and reach the CNS [131]. Muhammad et al. investigated the effects of hesperetin on an LPS-induced mice model of neuroinflammation. They intraperitoneally administered seven doses of LPS in 14 days, and they orally gavaged hesperetin daily for 5 weeks starting 3 weeks before LPS injection. They observed that hesperetin treatment reduced proinflammatory cytokine secretion by reducing TLR4-dependent Iba1-GFAP expression. Furthermore, hesperetin improved synaptic integrity, cognition, and memory. These findings pointed out that hesperetin can exert neuroprotective effects using the TLR4/NF-κB signaling pathway [132].
Icariside II (ICS II)
Icariside II is an active flavonoid component derived from a Chinese herbal medicine named Epimedium brevicornum Maxim. This substance has been reported to exert neuroprotective effects in AD models. Moreover, it has been demonstrated that ICS II can cross the BBB [131]. ICS II has been administered orally in most animal studies [133]. Zhou et al. investigated the effects of ICS II on an LPS-induced rat model. They administered ICS II prophylactically seven days before intracerebroventricular injection of LPS. The findings demonstrated that pretreatment with ICS II could inhibit microglia and astrocyte activation, suppress proinflammatory cytokine production and reduce TLR4, MyD88, and TRAF expressions. Hence, ICS II could reportedly reverse LPS-induced neuroinflammation in rats, suggesting its potential therapeutic effects on neuroinflammation [134].
Monophosphoryl lipid A
Monophosphoryl lipid A (MPL), an LPS-derived substance, is a detoxified fraction of endotoxin lipid A, lacking a saccharide and phosphate group [134]. It can be obtained from bacteria like Salmonella Minnesota or by chemical synthesis from Lipid A obtained from Escherichia coli [135]. MPL is a potent, selective agonist of TLR4 and an inducer of phagocytosis without causing a severe inflammatory response compared to LPS [136]. MPL is also used in several vaccines [136]. This drug is administered via injection, and similar to LPS, it probably cannot cross the BBB [137]. Venezia et al. investigated the chronic systemic administration of MPLA in transgenic mice with proteolipid promoter-induced overexpressing α-synuclein (PLP-α-syn mice) model of multiple system atrophy. They concluded that this chronic treatment with MPLA led to motor improvement, higher-α-synuclein uptake by microglia, protection of nigral striatal and dopaminergic neurons, and decreased α-synuclein cytoplasmic inclusions without causing systemic inflammation [136]. These results might warrant the worth of further investigations of this drug in PD human patients.
Vinpocetine
Vinpocetine is a semisynthetic derivative of Vinca alkaloid vincamine, an alkaloid extracted from the periwinkle plant [138]. This substance can be administered either orally or intravenously and has the ability to penetrate the BBB and enter the CNS [139]. It has been known for its improving effects on memory, cerebral metabolism, and inducing vasodilation [140]. In a double-blind placebo-controlled trial, 89 patients with late-onset PD and 42 controls were recruited. PD patients were randomly allocated to traditional or vinpocetine PD treatment groups. However, in both groups, levodopa was administered from the first day to the last day of the study (the 14th day). Vinpocetine was administered daily to the vinpocetine treatment group. The researchers reported that vinpocetine administration could decrease mRNA levels of TLR2 and TLR4 and their downstream signaling molecules like MyD88 and NF-kB in peripheral blood monocytes. On the other side, it induced TLR3 and its downstream signaling molecules like TRIF-β and IRF-3 and serum anti-inflammatory cytokine concentrations. Additionally, vinpocetine reduced serum levels of proinflammatory cytokines. These findings may suggest that vinpocetine has beneficial anti-inflammatory effects and can improve cognitive impairment in PD patients [140]. However, it is not wholly evident whether this drug's anti-inflammatory effects or other induced mechanisms led to improved cognitive function, remaining to be elucidated in future studies.
CU-CPT22 and candesartan cilexetil
CU-CPT22 is a benzotropolone analog with potent inhibitory effects on TLR1/2, firstly developed by Cheng et al. in 2013 [141]. It has not yet been elucidated whether this molecule can cross the BBB or not. Daniele et al. demonstrated that inhibiting TLR1 and TLR2 with CU-CPT22 could reduce proinflammatory cytokine production in cultured BV2 microglia of mice [88]. On the other side, candesartan cilexetil is an approved angiotensin II receptor blocker used for treating hypertension. This drug poses inhibitory effects on TLRs expression, especially TLR2 and TLR4. Candesartan cilexetil is administered using the oral route and can cross the BBB. Candesartan cilexetil reversed the proinflammatory phenotype of primary mice microglia induced by oligomeric α-synuclein in Daniele’s study [88]. Moreover, candesartan cilexetil showed inhibitory effects on TLR2 and TLR4 expression and proinflammatory cytokine secretion like TNF-α, IL-6, and IL-1β, both in primary human monocyte and mice models of PD [142]. These promising effects suggest examining the translatability of candesartan effects to PD humans as an example of a drug-repurposing strategy in future clinical studies.
SIlibinin (a prothrombin kringle-2 inhibitor)
Prothrombin kringle-2 (PKr-2) is one of the domains of prothrombin that is cleaved and generated by active thrombin. Although PKr-2 does not directly exert a toxic effect on neurons, it could induce DA neuronal death in the SN of rats by TLR4-mediated activation of microglia [143]. In this regard, the levels of PKr-2 and TLR4 significantly increased in the SN of PD patients than in controls. These findings suggest that inhibition of PKr-2 might protect DA neurons from degeneration by suppressing TLR4-mediated microglial activation. In this regard, treatment with silibinin, a polyphenolic flavonoid mainly found in milk thistle, could reportedly reverse the pathological effects of PKr-2 in SN of rat brains, leading to DA neurons’ protection. Silibinin can cross the BBB and can be administered orally [144].
Asiatic acid
Asiatic acid is a pentacyclic triterpene found in several plant foods like Centella and olive [145]. AA was reported to have beneficiary effects on wound healing, cancer, and neurodegenerative diseases [146]. This substance can penetrate the BBB and enter the CNS [147]. AA has shown acceptable pharmacokinetics following oral or intraperitoneal administration [146]. In a study on the MPTP mice model of PD, the oral administration of AA reduced MPTP-induced apoptotic, inflammatory, and oxidative effects and also attenuated α-synuclein, TLR2, TLR4, and NF-kB expression, increased striatal levels of BDNF, dopamine, and neurotrophic factors in a dose-dependent manner [92]. However, this question has remained whether these effects can be translated to human PD.
Fecal microbial transplantation (FMT)
Evidence supports a connection between the gut microbiota composition and the onset or progression of PD [148]. In this regard, Sun et al. demonstrated that the microbial composition of the PD model of mice differs significantly from normal mice. However, fecal microbiota transplantation to the PD mice model showed significant improvements in physical impairment and elevation in striatal DA and 5-Ht components of the PD mice model. They also observed that FMT could reverse the microglia and astrocyte activation in SN and decreased TLR4 and TNF-αexpression in the gut and brain of the PD mice model [148].
Conclusion
Neuroinflammation is one of the key contributors to PD pathogenesis, resulting in dopaminergic neurodegeneration and progression of the disease. As shown through several studies, evidence of the role of neuroinflammation in PD patients are indicative of higher levels of proinflammatory cytokines in serum and cerebrospinal fluid, presence of activated microglia in different CNS areas such as substantia nigra, infiltration of CD4+ and CD8+ T cells in affected brain regions, and modified performance of the cellular immunity for example malfunction in monocytes phagocytosis.
Postmortem studies on human brains and different animal PD models indicated an upregulation in TLRs expression in different brain regions such as the striatum, putamen, and caudate. This increased expression was also observed in peripheral blood monocytes of the affected individuals. Moreover, different SNPs in some TLR genes demonstrated an association with susceptibility to PD. Most studies indicated that the increased TLR expression is associated with neuroinflammation and neural loss in PD cases. In this regard, TLR2, TLR4, and TLR9 play a more significant role compared to other contributors. The increased expression of TLR2 on neurons located in the anterior cingulate cortex and striatum of PD human brains was also shown to be correlated with the disease staging and burden. TLR2 and TLR4 are critical receptors of α-synuclein, performing crucial roles in its recognition and triggering inflammatory responses. Furthermore, TLR4 is necessary for the proper phagocytosis function of monocytes, resulting in α-synuclein clearance in the early stages of the disease.
However, despite extensive research on PD and its treatments, no effective curative therapy has yet been developed. This therapeutic failure might be due to the incomplete knowledge of the disease pathogenesis. Recently, the discovery of the involvement of TLR-mediated neuroinflammation in disease pathogenesis has attracted much attention in designing novel therapeutic approaches. Although the essential roles of TLR in PD onset and progression have been illustrated in several clinical and preclinical studies, some knowledge gaps still remain to be addressed regarding the precise mechanisms of TLRs’ contribution to PD. For instance, the molecular structure of all pathological molecules responsible for initiating TLRs activation in PD patients is still not entirely discovered. Moreover, the roles of TLRs expressed on CNS-residing cells other than microglia, including astroglial and neuronal TLRs, attracted inadequate attention, necessitating to be elucidated in future studies. In this regard, more research using combined genetic and neurotoxin animal models of PD might be helpful to elucidate the roles of TLRs in disease pathogenesis more accurately.
Although the exact molecular mechanisms of TLR contribution to PD and neuroinflammation are still to be elucidated, the information presented in this review underlines the importance of designing approved drugs targeting TLRs and their downstream signaling pathways as potential therapeutic approaches in future way of PD research. In this context, the future research path on PD should be devoted to designing more randomized clinical control trials examining the efficacy of TLRs-based neuroinflammation modulating substances that showed promising results in animal and in vitro studies. However, before this point is reached, establishing validated animal models of PD, based on our expanding knowledge of the disease pathogenesis, which more closely imitates the molecular mechanisms of PD in humans, would help. Moreover, the pharmacokinetic features and the ability to penetrate BBB are of undoubted importance in designing effective drugs targeting TLRs in PD. In this regard, nanoencapsulation of developed drugs for their effective delivery to their target sites could play essential roles. Hence, there is a substantial need to examine the translatability of the effects of TLRs-affecting compounds in human clinical research.
To draw a conclusion upon the discussion, neuroinflammation and activation of TLRs function as a double-edged sword, exerting both protective and detrimental effects on PD pathogenesis. In the early time points of the disease, they might lead to α-synuclein clearance, which delays the disease progression. However, in later time points, excessive induction of TLR-mediated neuroinflammation results in neurodegeneration, worsening the disease prognosis. In line with this, different factors responsible for the dual effects of TLRs on the disease prognosis should be more thoroughly explored in future studies, with the hope of establishing novel therapeutic strategies for turning detrimental responses of TLRs into beneficial ones.
Availability of data and materials
Not applicable.
Abbreviations
- PD:
-
Parkinson's disease
- TNF-α:
-
Tumor necrosis factor-α
- RANTES:
-
Regulated on activation normal T-expressed and secreted
- IL:
-
Interleukin
- TLR:
-
Toll-like receptor
- DAMPs:
-
Damage-associated molecular patterns
- RBC:
-
Red blood cell
- LPS:
-
Lipopolysaccharide
- CD:
-
Cluster of differentiation
- MD2:
-
Myeloid differentiation factor 2
- MyD88:
-
Myeloid differentiation primary response 88
- IRAK4:
-
Interleukin-1 receptor-associated kinase 4
- TRAF6:
-
TNF (tumor necrosis factor) receptor-associated factor 6
- TAK1:
-
Transforming growth factor β-activated kinase 1
- MKK:
-
Mitogen-activated protein kinase kinase
- JNK:
-
Jun N-terminal Kinase
- IRF:
-
Interferon regulatory factor
- MPL:
-
Monophosphoryl lipid A
- p38MAPK:
-
P38 mitogen-activated protein kinase
- AP1:
-
Activating protein-1
- NF-kB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- TRIF:
-
TIR-domain-containing adaptor-inducing interferon-β
- TIR:
-
Toll/interleukin-1 receptor
- TANK:
-
TRAF family member-associated NF-kappa-B activator
- WDFY1:
-
WD repeat and FYVE domain-containing protein 1
- LBP:
-
Lipopolysaccharide binding protein
- dsRNA:
-
Double-stranded RNA
- ssRNA:
-
Single-stranded RNA
- LRR:
-
Leucine-rich repeats
- PRRs:
-
Pattern recognition receptors
- PAMPs:
-
Pathogen-associated molecular patterns
- DAMPs:
-
Damage-associated molecular patterns
- SNc:
-
Substantia nigra pars compacta
- DD:
-
Death domain
- LTA:
-
Lipoteichoic acid
- MAPK:
-
Mitogen-activated protein kinase
- BBB:
-
Blood–brain barrier
- FADD:
-
Fas-associated protein with death domain
- TOLLIP:
-
Toll interacting protein
- SHIP:
-
SH2-containing inositol-5′-phosphatase
- CIAPs:
-
Cellular inhibitor of apoptosis proteins
- PBMC:
-
Peripheral blood mononuclear cell
- Aβ:
-
Amyloid beta
- mTOR:
-
Mammalian target of rapamycin
- PGN:
-
Peptidoglycan
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PET:
-
Positron emission tomography
- PBBS:
-
Peripheral benzodiazepine binding sites
- TBK1:
-
TANK-binding kinase 1
References
Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–76.
Hirsch EC, Standaert DG. Ten unsolved questions about neuroinflammation in Parkinson’s disease. Move Disord Off J Move Disord Soc. 2021;36(1):16–24.
Hawkes CH, Del Tredici K, Braak H. A timeline for Parkinson’s disease. Parkinsonism Relat Disord. 2010;16(2):79–84.
Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med. 2020;36(1):1–12.
Samii A, Nutt JG, Ransom BR. Parkinson’s disease. Lancet (London, England). 2004;363(9423):1783–93.
Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol. 2013;47(2):495–508.
McGeer PL, Itagaki S, Boyes BE, McGeer E. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285.
Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson’s disease. Adv Res Neurodegen. 2000:277–90.
Caputi V, Giron MC. Microbiome-gut-brain axis and toll-like receptors in Parkinson’s disease. Int J Mol Sci. 2018;19(6):1689.
Bohush A, Bieganowski P, Filipek A. Hsp90 and its co-chaperones in neurodegenerative diseases. Int J Mol Sci. 2019;20(20).
Mansilla MJ, Costa C, Eixarch H, Tepavcevic V, Castillo M, Martin R, et al. Hsp70 regulates immune response in experimental autoimmune encephalomyelitis. PLoS ONE. 2014;9(8): e105737.
Asea A. Heat shock proteins and toll-like receptors. Handb Exp Pharmacol. 2008;183:111–27.
Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. 2013;61(3):349–60.
Lemaitre B, Nicolas E, Michaut L, Reichhart J-M, Hoffmann JA. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86(6):973–83.
Lim K-H, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. 2013;5(1):a011247.
Anthoney N, Foldi I, Hidalgo A. Toll and Toll-like receptor signalling in development. Development. 2018;145(9):156018.
Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annu Rev Biochem. 2007;76:141–65.
McClure R, Massari P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. Front Immunol. 2014;5:386.
Fitzner N, Clauberg S, Essmann F, Liebmann J, Kolb-Bachofen V. Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin Vaccine Immunol. 2008;15(1):138–46.
Aluri J, Cooper MA, Schuettpelz LG. Toll-like receptor signaling in the establishment and function of the immune system. Cells. 2021;10(6):1374.
de Oliviera NL, Massari P, Wetzler LM. The role of TLR2 in infection and immunity. Front Immunol. 2012;3:79.
Figueroa-Hall LK, Paulus MP, Savitz J. Toll-like receptor signaling in depression. Psychoneuroendocrinology. 2020:104843.
Poltorak A, Smirnova I, He X, Liu M-Y, Van Huffel C, Birdwell D, et al. Genetic and physical mapping of the Lps Locus: identification of the Toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol Dis. 1998;24(3):340–55.
Pietruska M, Zak J, Pietruski J, Wysocka J. Evaluation of mCD14 expression on monocytes and the blood level of sCD14 in patients with generalized aggressive periodontitis. Adv Med Sci. 2006;51(Suppl 1):166–9.
Muta T, Takeshige K. Essential roles of CD14 and lipopolysaccharide-binding protein for activation of toll-like receptor (TLR)2 as well as TLR4 Reconstitution of TLR2- and TLR4-activation by distinguishable ligands in LPS preparations. Eur J Biochem. 2001;268(16):4580–9.
Pahwa R, Devaraj S, Jialal I. The effect of the accessory proteins, soluble CD14 and lipopolysaccharide-binding protein on Toll-like receptor 4 activity in human monocytes and adipocytes. Int J Obes. 2016;40(6):907–11.
Moreno C, Merino J, Ramírez N, Echeverría A, Pastor F, Sánchez-Ibarrola A. Lipopolysaccharide needs soluble CD14 to interact with TLR4 in human monocytes depleted of membrane CD14. Microbes Infect. 2004;6(11):990–5.
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
Bauer S, Kirschning CJ, Häcker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci. 2001;98(16):9237–42.
Nilsen NJ, Vladimer GI, Stenvik J, Orning MP, Zeid-Kilani MV, Bugge M, et al. A role for the adaptor proteins TRAM and TRIF in toll-like receptor 2 signaling. J Biol Chem. 2015;290(6):3209–22.
Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signaling. Nature. 2003;424:743–8.
Lin S-C, Lo Y-C, Wu H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465(7300):885–90.
Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277(9):7059–65.
Zhande R, Dauphinee SM, Thomas JA, Yamamoto M, Akira S, Karsan A. FADD negatively regulates lipopolysaccharide signaling by impairing interleukin-1 receptor-associated kinase 1-MyD88 interaction. Mol Cell Biol. 2007;27(21):7394–404.
Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. 2010;80(12):1981–91.
Heidari A, Sharif PM, Rezaei N. Potential target for therapy. Curr Pharmaceut Design. 2021.
Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol. 2017;17(3):179–94.
Kumar V. Toll-like receptors in the pathogenesis of neuroinflammation. J Neuroimmunol. 2019;332:16–30.
Heidari A, Rostam-Abadi Y, Rezaei N. The immune system and autism spectrum disorder: association and therapeutic challenges. Acta Neurobiol Exp. 2021;81:249–63.
Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci. 2011;121(9):367–87.
Klein M, Obermaier B, Angele B, Pfister H-W, Wagner H, Koedel U, et al. Innate immunity to pneumococcal infection of the central nervous system depends on toll-like receptor (TLR) 2 and TLR4. J Infect Dis. 2008;198(7):1028–36.
Massari P, Visintin A, Gunawardana J, Halmen KA, King CA, Golenbock DT, et al. Meningococcal porin PorB binds to TLR2 and requires TLR1 for signaling. J Immunol. 2006;176(4):2373–80.
Akira S. TLR signaling. From innate immunity to immunological memory. 2006:1–16.
Murphy MP, LeVine H III. Alzheimer’s disease and the amyloid-β peptide. J Alzheimers Dis. 2010;19(1):311–23.
Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J Immunol. 2012;188(3):1098–107.
Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic-ischemic injury induces macrophage inflammatory protein-1α expression in immature rat brain. Stroke. 2002;33(3):795–801.
Cai J, Hua F, Yuan L, Tang W, Lu J, Yu S, et al. Potential therapeutic effects of neurotrophins for acute and chronic neurological diseases. BioMed Res Int. 2014;2014.
Wu D. Neuroprotection in experimental stroke with targeted neurotrophins. NeuroRx. 2005;2(1):120–8.
Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Investig. 2008;119(1).
Hirsch EC, Standaert DG. Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov Disord. 2021;36(1):16–24.
Saunders JAH, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J Neuroimmune Pharmacol. 2012;7(4):927–38.
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21(2):404–12.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–97.
Qin X-Y, Zhang S-P, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: a systematic review and meta-analysis. JAMA Neurol. 2016;73(11):1316–24.
Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett. 1996;211(1):13–6.
Blum-Degena D, Müller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 1995;202(1–2):17–20.
Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. 2017;546(7660):656–61.
Arlehamn CSL, Dhanwani R, Pham J, Kuan R, Frazier A, Dutra JR, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun. 2020;11(1):1–11.
Gardai SJ, Mao W, Schüle B, Babcock M, Schoebel S, Lorenzana C, et al. Elevated alpha-synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson’s disease. PLoS ONE. 2013;8(8): e71634.
Grozdanov V, Bliederhaeuser C, Ruf WP, Roth V, Fundel-Clemens K, Zondler L, et al. Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropatholog. 2014;128(5):651–63.
Terada T, Yokokura M, Yoshikawa E, Futatsubashi M, Kono S, Konishi T, et al. Extrastriatal spreading of microglial activation in Parkinson’s disease: a positron emission tomography study. Ann Nucl Med. 2016;30(8):579–87.
Horti AG, Naik R, Foss CA, Minn I, Misheneva V, Du Y, et al. PET imaging of microglia by targeting macrophage colony-stimulating factor 1 receptor (CSF1R). Proc Natl Acad Sci. 2019;116(5):1686–91.
Liu M, Bing G. Lipopolysaccharide animal models for Parkinson's disease. Parkinson’s Dis. 2011;2011.
Hallett PJ, McLean JR, Kartunen A, Langston JW, Isacson O. Alpha-synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol Dis. 2012;47(2):258–67.
Depino AM, Earl C, Kaczmarczyk E, Ferrari C, Besedovsky H, Del Rey A, et al. Microglial activation with atypical proinflammatory cytokine expression in a rat model of Parkinson’s disease. Eur J Neurosci. 2003;18(10):2731–42.
McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Annal Neurol Off J Am Neurol Assoc Child Neurol Soc. 2003;54(5):599–604.
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging. 2008;29(11):1690–701.
Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep. 2019;9(1):1–12.
Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5(12):1403–9.
Kouli A, Horne C, Williams-Gray C. Toll-like receptors and their therapeutic potential in Parkinson’s disease and α-synucleinopathies. Brain Behav Immun. 2019;81:41–51.
Kim N, Yoo HS, Ju YJ, Oh MS, Lee KT, Inn KS, et al. Synthetic 3’,4’-dihydroxyflavone exerts anti-neuroinflammatory effects in BV2 microglia and a mouse model. Biomol Therapeut. 2018;26(2):210–7.
da Silva DJ, Borges AF, Souza PO, de Souza PR, de Barros Cardoso CR, Dorta ML, et al. Decreased toll-like receptor 2 and toll-like receptor 7/8-induced cytokines in Parkinson’s disease patients. NeuroImmunoModulation. 2016;23(1):58–66.
Kin K, Yasuhara T, Kameda M, Date I. Animal models for Parkinson's disease research: trends in the 2000s. Int J Mol Sci. 2019;20(21).
Kuruvilla KP, Nandhu MS, Paul J, Paulose CS. Oxidative stress mediated neuronal damage in the corpus striatum of 6-hydroxydopamine lesioned Parkinson’s rats: neuroprotection by serotonin, GABA and bone marrow cells supplementation. J Neurol Sci. 2013;331(1–2):31–7.
Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci USA. 1985;82(7):2173–7.
Smith GA, Isacson O, Dunnett SB. The search for genetic mouse models of prodromal Parkinson’s disease. Exp Neurol. 2012;237(2):267–73.
Moore DJ, Dawson TM. Value of genetic models in understanding the cause and mechanisms of Parkinson’s disease. Curr Neurol Neurosci Rep. 2008;8(4):288–96.
Yeo GW, Van Nostrand E, Holste D, Poggio T, Burge CB. Identification and analysis of alternative splicing events conserved in human and mouse. Proc Natl Acad Sci USA. 2005;102(8):2850–5.
Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci USA. 2005;102(14):5215–20.
Drouin-Ouellet J, St-Amour I, Saint-Pierre M, Lamontagne-Proulx J, Kriz J, Barker RA, et al. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int J Neuropsychopharmacol. 2015;18(6).
Dzamko N, Gysbers A, Perera G, Bahar A, Shankar A, Gao J, et al. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017;133(2):303–19.
Doorn KJ, Moors T, Drukarch B, van de Berg WD, Lucassen PJ, van Dam A-M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun. 2014;2(1):1–17.
Li X, Xue L, Sun J, Sun Y, Xie A. Single nucleotide polymorphisms in the toll-like receptor 2 (TLR2) gene are associated with sporadic Parkinson’s disease in the North-eastern Han Chinese population. Neurosci Lett. 2017;656:72–6.
Gustot A, Gallea JI, Sarroukh R, Celej MS, Ruysschaert J-M, Raussens V. Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem J. 2015;471(3):323–33.
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, et al. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE. 2013;8(1): e55375.
Ping Z, Xiaomu W, Xufang X, Liang S. Vinpocetine regulates levels of circulating TLRs in Parkinson’s disease patients. Neurol Sci. 2019;40(1):113–20.
Béraud D, Twomey M, Bloom B, Mittereder A, Neitzke K, Ton V, et al. α-Synuclein alters toll-like receptor expression. Front Neurosci. 2011;5:80.
Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal. 2015;8(376):ra45.
Kim C, Spencer B, Rockenstein E, Yamakado H, Mante M, Adame A, et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating α-synuclein transmission and neuroinflammation. Mol Neurodegener. 2018;13(1):1–18.
Kim C, Rockenstein E, Spencer B, Kim H-K, Adame A, Trejo M, et al. Antagonizing neuronal toll-like receptor 2 prevents synucleinopathy by activating autophagy. Cell Rep. 2015;13(4):771–82.
La Vitola P, Balducci C, Cerovic M, Santamaria G, Brandi E, Grandi F, et al. Alpha-synuclein oligomers impair memory through glial cell activation and via Toll-like receptor 2. Brain Behav Immun. 2018;69:591–602.
Chao P-c, Lee H-l, Yin M-c. Asiatic acid attenuated apoptotic and inflammatory stress in the striatum of MPTP-treated mice. Food Funct. 2016;7(4):1999–2005.
Drouin-Ouellet J, Gibrat C, Bousquet M, Calon F, Kriz J, Cicchetti F. The role of the MYD88-dependent pathway in MPTP-induced brain dopaminergic degeneration. J Neuroinflammation. 2011;8(1):1–12.
ChulJang Y, Hwang DJ, Koo JH, Um HS, Lee NH, Yeom DC, et al. Association of exercise-induced autophagy upregulation and apoptosis suppression with neuroprotection against pharmacologically induced Parkinson’s disease. J Exerc Nutr Biochem. 2018;22(1):1.
Hughes CD, Choi ML, Ryten M, Hopkins L, Drews A, Botía JA, et al. Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 2019;137(1):103–20.
Shin W-H, Jeon M-T, Leem E, Won S-Y, Jeong KH, Park S-J, et al. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: a potential pathogenic mechanism in Parkinson’s disease. Sci Rep. 2015;5(1):1–14.
Kouli A, Camacho M, Allinson K, Williams-Gray CH. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol Commun. 2020;8(1):1–19.
Perez-Pardo P, Dodiya HB, Engen PA, Forsyth CB, Huschens AM, Shaikh M, et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut. 2019;68(5):829–43.
Zhao J, Han X, Xue L, Zhu K, Liu H, Xie A. Association of TLR4 gene polymorphisms with sporadic Parkinson’s disease in a Han Chinese population. Neurol Sci. 2015;36(9):1659–65.
Conte C, Roscini L, Sardella R, Mariucci G, Scorzoni S, Beccari T, et al. Toll like receptor 4 affects the cerebral biochemical changes induced by MPTP treatment. Neurochem Res. 2017;42(2):493–500.
Campolo M, Paterniti I, Siracusa R, Filippone A, Esposito E, Cuzzocrea S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav Immun. 2019;76:236–47.
McCabe K, Concannon RM, McKernan DP, Dowd E. Time-course of striatal Toll-like receptor expression in neurotoxic, environmental and inflammatory rat models of Parkinson’s disease. J Neuroimmunol. 2017;310:103–6.
Mariucci G, Pagiotti R, Galli F, Romani L, Conte C. The potential role of toll-like receptor 4 in mediating dopaminergic cell loss and alpha-synuclein expression in the acute MPTP mouse model of Parkinson’s disease. J Mol Neurosci. 2018;64(4):611–8.
Stefanova N, Reindl M, Neumann M, Kahle PJ, Poewe W, Wenning GK. Microglial activation mediates neurodegeneration related to oligodendroglial α-synucleinopathy: implications for multiple system atrophy. Move Disord Off J Move Disord Soc. 2007;22(15):2196–203.
Stefanova N, Fellner L, Reindl M, Masliah E, Poewe W, Wenning GK. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am J Pathol. 2011;179(2):954–63.
Zhou Y, Ye L, Wan Q, Zhou L, Wang X, Li J, et al. Activation of Toll-like receptors inhibits herpes simplex virus-1 infection of human neuronal cells. J Neurosci Res. 2009;87(13):2916–25.
Rannikko EH, Weber SS, Kahle PJ. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015;16(1):1–11.
Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol J-C, Lu L, et al. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci. 2011;108(16):6632–7.
Miri NS, Saadat P, Azadmehr A, Oladnabi M, Daraei A. Toll-Like Receptor (TLR)-9 rs352140 Polymorphism is an Immunopathology Protective Factor in Parkinson’s Disease in the Northern Iranian Population. Iran J Immunol. 2020;17(4):313–23.
Zhu K, Teng J, Zhao J, Liu H, Xie A. Association of TLR9 polymorphisms with sporadic Parkinson’s disease in Chinese Han population. Int J Neurosci. 2016;126(7):612–6.
Maatouk L, Compagnion A-C, Carrillo-De Sauvage M-A, Bemelmans A-P, Leclere-Turbant S, Cirotteau V, et al. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat Commun. 2018;9(1):1–15.
Tan L, Song X, Ren Y, Wang M, Guo C, Guo D, et al. Anti-inflammatory effects of cordycepin: a review. Phytother Res PTR. 2020.
Cheng C, Zhu X. Cordycepin mitigates MPTP-induced Parkinson’s disease through inhibiting TLR/NF-κB signaling pathway. Life Sci. 2019;223:120–7.
Sun Y, Shao Y, Zhang Z, Wang L, Mariga AM, Pang G, et al. Regulation of human cytokines by Cordyceps militaris. J Food Drug Anal. 2014;22(4):463–7.
Tuli HS, Sharma AK, Sandhu SS, Kashyap D. Cordycepin: a bioactive metabolite with therapeutic potential. Life Sci. 2013;93(23):863–9.
Rodman LE, Farnell DR, Coyne JM, Allan PW, Hill DL, Duncan KL, et al. Toxicity of cordycepin in combination with the adenosine deaminase inhibitor 2′-deoxycoformycin in beagle dogs. Toxicol Appl Pharmacol. 1997;147(1):39–45.
Lee JB, Adrower C, Qin C, Fischer PM, de Moor CH, Gershkovich P. Development of cordycepin formulations for preclinical and clinical studies. AAPS PharmSciTech. 2017;18(8):3219–26.
Kengkittipat W, Kaewmalun S, Khongkow M, Iempridee T, Jantimaporn A, Bunwatcharaphansakun P, et al. Improvement of the multi-performance biocharacteristics of cordycepin using BiloNiosome-core/chitosan-shell hybrid nanocarriers. Colloids Surf B. 2021;197: 111369.
Yuan J, Wang A, He Y, Si Z, Xu S, Zhang S, et al. Cordycepin attenuates traumatic brain injury-induced impairments of blood–brain barrier integrity in rats. Brain Res Bull. 2016;127:171–6.
He MT, Lee AY, Kim JH, Park CH, Shin YS, Cho EJ. Protective role of Cordyceps militaris in Aβ(1-42)-induced Alzheimer’s disease in vivo. Food Sci and biotechnology. 2019;28(3):865–72.
Alavian KN, Jeddi S, Naghipour SI, Nabili P, Licznerski P, Tierney TS. The lifelong maintenance of mesencephalic dopaminergic neurons by Nurr1 and engrailed. J Biomed Sci. 2014;21(1):1–8.
Sonntag KC, Simantov R, Kim KS, Isacson O. Temporally induced Nurr1 can induce a non-neuronal dopaminergic cell type in embryonic stem cell differentiation. Eur J Neurosci. 2004;19(5):1141–52.
Baffi J, Palkovits M, Castillo S, Mezey E, Nikodem V. Differential expression of tyrosine hydroxylase in catecholaminergic neurons of neonatal wild-type and Nurr1-deficient mice. Neuroscience. 1999;93(2):631–42.
Smith GA, Rocha EM, Rooney T, Barneoud P, McLean JR, Beagan J, et al. A Nurr1 agonist causes neuroprotection in a Parkinson’s disease lesion model primed with the toll-like receptor 3 dsRNA inflammatory stimulant poly (I: C). PLoS ONE. 2015;10(3): e0121072.
Liao J, Zhang Y, Chen X, Zhang J. The roles of peroxiredoxin 6 in brain diseases. Mol Neurobiol. 2021;58(9):4348–64.
Yeo IJ, Park MH, Son DJ, Kim JY, Nam KT, Hyun BK, et al. PRDX6 inhibits neurogenesis through downregulation of WDFY1-mediated TLR4 signal. Mol Neurobiol. 2019;56(5):3132–44.
Swerdloff RS, Dudley RE, Page ST, Wang C, Salameh WA. Dihydrotestosterone: biochemistry, physiology, and clinical implications of elevated blood levels. Endocr Rev. 2017;38(3):220–54.
Borst SE, Yarrow JF. Injection of testosterone may be safer and more effective than transdermal administration for combating loss of muscle and bone in older men. Am J Physiol-Endocrinol Metab. 2015;308(12):E1035–42.
Yang L, Zhou R, Tong Y, Chen P, Shen Y, Miao S, et al. Neuroprotection by dihydrotestosterone in LPS-induced neuroinflammation. Neurobiol Dis. 2020;140: 104814.
Kim HK, Jeong T-S, Lee M-K, Park YB, Choi M-S. Lipid-lowering efficacy of hesperetin metabolites in high-cholesterol fed rats. Clin Chim Acta. 2003;327(1–2):129–37.
Gao J, Long L, Xu F, Feng L, Liu Y, Shi J, et al. Icariside II, a phosphodiesterase 5 inhibitor, attenuates cerebral ischaemia/reperfusion injury by inhibiting glycogen synthase kinase-3β-mediated activation of autophagy. Br J Pharmacol. 2020;177(6):1434–52.
Muhammad T, Ikram M, Ullah R, Rehman SU, Kim MO. Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-κB signaling. Nutrients. 2019;11(3):648.
Liu S, Li X, Gao J, Liu Y, Shi J, Gong Q. Icariside II, a phosphodiesterase-5 inhibitor, attenuates beta-amyloid-induced cognitive deficits via BDNF/TrkB/CREB Signaling. Cell Physiol Biochem. 2018;49(3):1010–25.
Zhou J, Deng Y, Li F, Yin C, Shi J, Gong Q. Icariside II attenuates lipopolysaccharide-induced neuroinflammation through inhibiting TLR4/MyD88/NF-κB pathway in rats. Biomed Pharmacother. 2019;111:315–24.
Xiao X, Sankaranarayanan K, Khosla C. Biosynthesis and structure-activity relationships of the lipid a family of glycolipids. Curr Opin Chem Biol. 2017;40:127–37.
Venezia S, Refolo V, Polissidis A, Stefanis L, Wenning GK, Stefanova N. Toll-like receptor 4 stimulation with monophosphoryl lipid A ameliorates motor deficits and nigral neurodegeneration triggered by extraneuronal α-synucleinopathy. Mol Neurodegener. 2017;12(1):1–13.
Vargas-Caraveo A, Sayd A, Maus SR, Caso JR, Madrigal JL, García-Bueno B, et al. Lipopolysaccharide enters the rat brain by a lipoprotein-mediated transport mechanism in physiological conditions. Sci Rep. 2017;7(1):1–15.
Zhang Y-S, Li J-D, Yan C. An update on vinpocetine: new discoveries and clinical implications. Eur J Pharmacol. 2018;819:30–4.
Gulyás B, Halldin C, Sandell J, Karlsson P, Sóvágó J, Kárpáti E, et al. PET studies on the brain uptake and regional distribution of [11C] vinpocetine in human subjects. Acta Neurol Scand. 2002;106(6):325–32.
Ping Z, Xiaomu W, Xufang X, Liang S. Vinpocetine regulates levels of circulating TLRs in Parkinson’s disease patients. Neurol Sci Off J Italian Neurol Soc Italian Soc Clin Neurophysiol. 2019;40(1):113–20.
Cheng K, Wang X, Zhang S, Yin H. Discovery of small-molecule inhibitors of the TLR1/TLR2 complex. Angew Chem. 2012;124(49):12412–5.
Kouli A, Horne CB, Williams-Gray CH. Toll-like receptors and their therapeutic potential in Parkinson’s disease and α-synucleinopathies. Brain Behav Immun. 2019;81:41–51.
Kim SR, Chung ES, Bok E, Baik HH, Chung YC, Won SY, et al. Prothrombin kringle-2 induces death of mesencephalic dopaminergic neurons in vivo and in vitro via microglial activation. J Neurosci Res. 2010;88(7):1537–48.
Pérez-Sánchez A, Cuyàs E, Ruiz-Torres V, Agulló-Chazarra L, Verdura S, González-Álvarez I, et al. Intestinal permeability study of clinically relevant formulations of silibinin in Caco-2 cell monolayers. Int J Mol Sci. 2019;20(7):1606.
Hashim P, Sidek H, Helan MHM, Sabery A, Palanisamy UD, Ilham M. Triterpene composition and bioactivities of Centella asiatica. Molecules. 2011;16(2):1310–22.
Nagoor Meeran MF, Goyal SN, Suchal K, Sharma C, Patil CR, Ojha SK. Pharmacological properties, molecular mechanisms, and pharmaceutical development of asiatic acid: a pentacyclic triterpenoid of therapeutic promise. Front Pharmacol. 2018;8:892.
Hanapi NA, Arshad ASM, Abdullah JM, Muhammad TST, Yusof SR. Blood-brain barrier permeability of asiaticoside, madecassoside and asiatic acid in porcine brain endothelial cell model. J Pharm Sci. 2021;110(2):698–706.
Sun M-F, Zhu Y-L, Zhou Z-L, Jia X-B, Xu Y-D, Yang Q, et al. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav Immun. 2018;70:48–60.
da Silva DJ, Borges AF, Souza PO, de Souza PR, Cardoso CR, Dorta ML, et al. Decreased toll-like receptor 2 and toll-like receptor 7/8-induced cytokines in Parkinson’s disease patients. NeuroImmunoModulation. 2016;23(1):58–66.
Drouin-Ouellet J, St-Amour I, Saint-Pierre M, Lamontagne-Proulx J, Kriz J, Barker RA, et al. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int J Neuropsychopharmacol. 2014;18(6):PYU103.
Dzamko N, Gysbers A, Perera G, Bahar A, Shankar A, Gao J, et al. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017;133(2):303–19.
Doorn KJ, Moors T, Drukarch B, van de Berg W, Lucassen PJ, van Dam AM. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun. 2014;2:90.
Huang M, Li Y, Tian T, Wang K, Wang Y, Yan W, et al. Knockdown of TLR4 represses the paraquat-induced neuroinflammation and microglial M1 polarization. Neurotox Res. 2020;38(3):741–50.
Miri NS, Saadat P, Azadmehr A, Oladnabi M, Daraei A. Toll-like receptor (TLR)-9 rs352140 polymorphism is an immunopathology protective factor in Parkinson’s disease in the Northern Iranian Population. Iran J Immunol IJI. 2020;17(4):313–23.
Campolo M, Paterniti I, Siracusa R, Filippone A, Esposito E, Cuzzocrea S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav Immun. 2019;76:236–47.
Shao QH, Chen Y, Li FF, Wang S, Zhang XL, Yuan YH, et al. TLR4 deficiency has a protective effect in the MPTP/probenecid mouse model of Parkinson’s disease. Acta Pharmacol Sin. 2019;40(12):1503–12.
Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562.
Kim C, Lee HJ, Masliah E, Lee SJ. Non-cell-autonomous neurotoxicity of α-synuclein through microglial toll-like receptor 2. Exp Neurobiol. 2016;25(3):113–9.
Smith GA, Rocha EM, Rooney T, Barneoud P, McLean JR, Beagan J, et al. A Nurr1 agonist causes neuroprotection in a Parkinson’s disease lesion model primed with the toll-like receptor 3 dsRNA inflammatory stimulant poly(I:C). PLoS ONE. 2015;10(3): e0121072.
Sun MF, Zhu YL, Zhou ZL, Jia XB, Xu YD, Yang Q, et al. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav Immun. 2018;70:48–60.
Venezia S, Refolo V, Polissidis A, Stefanis L, Wenning GK, Stefanova N. Toll-like receptor 4 stimulation with monophosphoryl lipid A ameliorates motor deficits and nigral neurodegeneration triggered by extraneuronal α-synucleinopathy. Mol Neurodegener. 2017;12(1):52.
Shin WH, Jeon MT, Leem E, Won SY, Jeong KH, Park SJ, et al. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: a potential pathogenic mechanism in Parkinson’s disease. Sci Rep. 2015;5:14764.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
AH conceptualized the title, prepared the initial draft, and designed the figures. NY helped in drafting the manuscript, revised the manuscript and prepared the final draft. NR helped in preparing the final draft, critically revised the manuscript, and supervised the project. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Heidari, A., Yazdanpanah, N. & Rezaei, N. The role of Toll-like receptors and neuroinflammation in Parkinson’s disease. J Neuroinflammation 19, 135 (2022). https://doi.org/10.1186/s12974-022-02496-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-022-02496-w